1. Introduction
An increased relative risk of renal cell carcinoma associated with chronic kidney disease (CKD) is well documented among populations with sub-Saharan African ancestry, most notably African-Americans. The estimated proportion of renal cell carcinoma attributable to CKD within these populations accounts for 10%, while it is negligible among Caucasians. However, it is not known whether the APOL1 risk variants for CDK elevate the risk of renal cell carcinoma in these populations [
1].
Apolipoprotein L1 (APOL1) is a circulating protein component of HDL (high-density lipoprotein) encoded by the APOL1 gene in humans and certain primate species [
2,
3,
4]. In the kidney, APOL1 is expressed in podocytes and vascular components of the glomerulus, suggesting that it might play a role in renal diseases that affect the structure and function of these cells [
2,
5]. Indeed, APOL1 is associated with an increased risk of CKD, including end-stage kidney disease (ESKD), focal segmental glomerulosclerosis (FSGS), and HIV-associated nephropathy (HIVAN), specifically in individuals with sub–Saharan African ancestry [
6,
7,
8]. The high CKD risk in this case is attributed to the high frequency of two APOL1 risk variants (RV) APOL1 G1 and APOL1 G2, which are associated with kidney disease, as opposed to the wild-type APOL1 G0 allele [
6,
9]. The high frequency of G1 and G2 alleles emanated from the protection they confer against African sleeping sickness caused by either
Trypanosoma brucei,
Trypanosoma gambiense, or
Trypanosoma rhodesiense [
10,
11]. The expression of APOL1 is regulated in a cell-specific mode by specific protein 1 (Sp1) and interferon regulatory factors 1 and 2 (IRF1 and IRF2) [
12]. Various roles were proposed for APOL1 proteins, including in autophagy [
13,
14,
15], inflammatory cell death (pyroptosis) [
15], impairment of vacuolar acidification [
16], ER stress [
17], lysosomal membrane permeability [
18], mitochondrial dysfunction [
19,
20,
21,
22], and cationic channel activity at the plasma membrane [
23,
24]. To date, APOL1 function has been investigated either in cellular or organismal platforms that overexpress the APOL1 variants [
15,
16,
20].
Attempts to fully elucidate the intricate mechanisms of APOL1-mediated renal injury to date have been inconclusive and sometimes contradictory. In part, this is attributable to the use of ectopic APOL1 variant constructs overexpressed in cell-culture platforms. In the current study, we utilize renal cell carcinoma (RCC) cells as a research model in which intrinsic high expression levels of APOL1 GO protein are tolerated and compatible with robust cell proliferation. Accordingly, to investigate the role of APOL1 and its risk alleles G1 and G2 in renal carcinoma cell growth, we set up tissue culture-based studies using the RCC 786-O cell line, a primary renal cell adenocarcinoma of a Caucasian male expressing APOL1-G0, thus circumventing the toxicity of ectopic overexpression. To further understand the specific contribution of the G1 and G2 APOL1 risk variants in kidney disease, we generated isogenic RCC clones expressing these variants. We took advantage of CRISPR/Cas9 genome editing to generate cells with levels of expression for APOL1 G1 and G2, comparable to those for G0, as well as APOL1 knock-out RCC. This methodology enables the expression of APOL1 G0, G1, and G2 variants on the same genetic background, avoiding the known modifying effects of varying haplotype backgrounds [
25]. We demonstrate that the substitution of one nucleotide for expressing the APOL1 G1 or deletion of six nucleotides for expressing the APOL1 G2 led to a significant alteration in cell metabolism and tumorigenic potential in RCC cancer cells, as did deleting the endogenous gene.
2. Materials and Methods
2.1. Mice, Tumor Formation, and Tissue Handling
SCID/beige mice were purchased from Harlan Laboratories, Jerusalem, Israel. The mice were housed and maintained under specific pathogen-free conditions as instructed by the Committee for Oversight of Animal Experimentation at the Technion-Israel Institute of Technology, Haifa, Israel. The study was approved by the Institutional Animal Care Use Committee of the Technion (Protocol # IL-055-04-21). and 4 × 106 cells were injected into the mices’ hind limb musculature. Mice were weighed and screened for tumor formation once a week. Mice were sacrificed using CO2 inhalation at 55 days following injection. Tumors were harvested, fixed in 10% neutral buffered formalin, transferred into 70% ethanol, and processed using a routine wax-embedding procedure for histological examination. Paraffin sections of six-micrometers were mounted on Super Frost Plus microscope slides (Menzel-Glaser, Braunschweig, Germany) and stained with hematoxylin/eosin.
2.2. Cell Lines and Tissue Culture
RCC 786-O (CRL-1932, ATCC) were grown at 37 °C in 5% CO2 in RPMI supplemented with 10% FCS, glutamine, and antibiotics. Human immortalized podocytes were grown under similar incubator conditions in DMEM supplemented with 10% FCS, glutamine, and antibiotics.
2.3. Antibodies
Anti-APOL1 (1:2500 HPA018885, Sigma, St. Louis, MO, USA); Anti-APOL1 5.17D12 (1 µg/mL, kindly provided by Genentech, San Francisco, CA, USA); Anti-APOL1 oligoclonal 3.7D6/3.1C1 (0.05 µg/mL, kindly provided by Genentech); Anti-Tubulin (1:5000 T5168, Sigma, St. Louis, MO, USA); Anti-LC3 (1:1000, PM036, MBL, Chicago, MA, USA); Anti-Calreticulin (1:1000, 12238, Cell Signaling, Danvers, MA, USA); anti-Cytochrome C (1:1000, sc-13156, Santa Cruz, Dallas, TX, USA); Anti-Calnexin (1:2000, ADI-SPA-860 Enzo, New York, NY, USA); Anti-SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B, 1:2500, LS C497529, LSbio); Anti-rabbit HRP (1:20,000; 111-035-144, Jackson ImmunoResearch, Baltimore, PN, USA); Anti-mouse HRP (1:10,000; 115-035-166, Jackson ImmunoResearch); Alexa Fluor 488 conjugated Donkey anti Rabbit (1:600; 711-545-152, Jackson ImmunoResearch).
2.4. Quantitative Real-Time RT-PCR
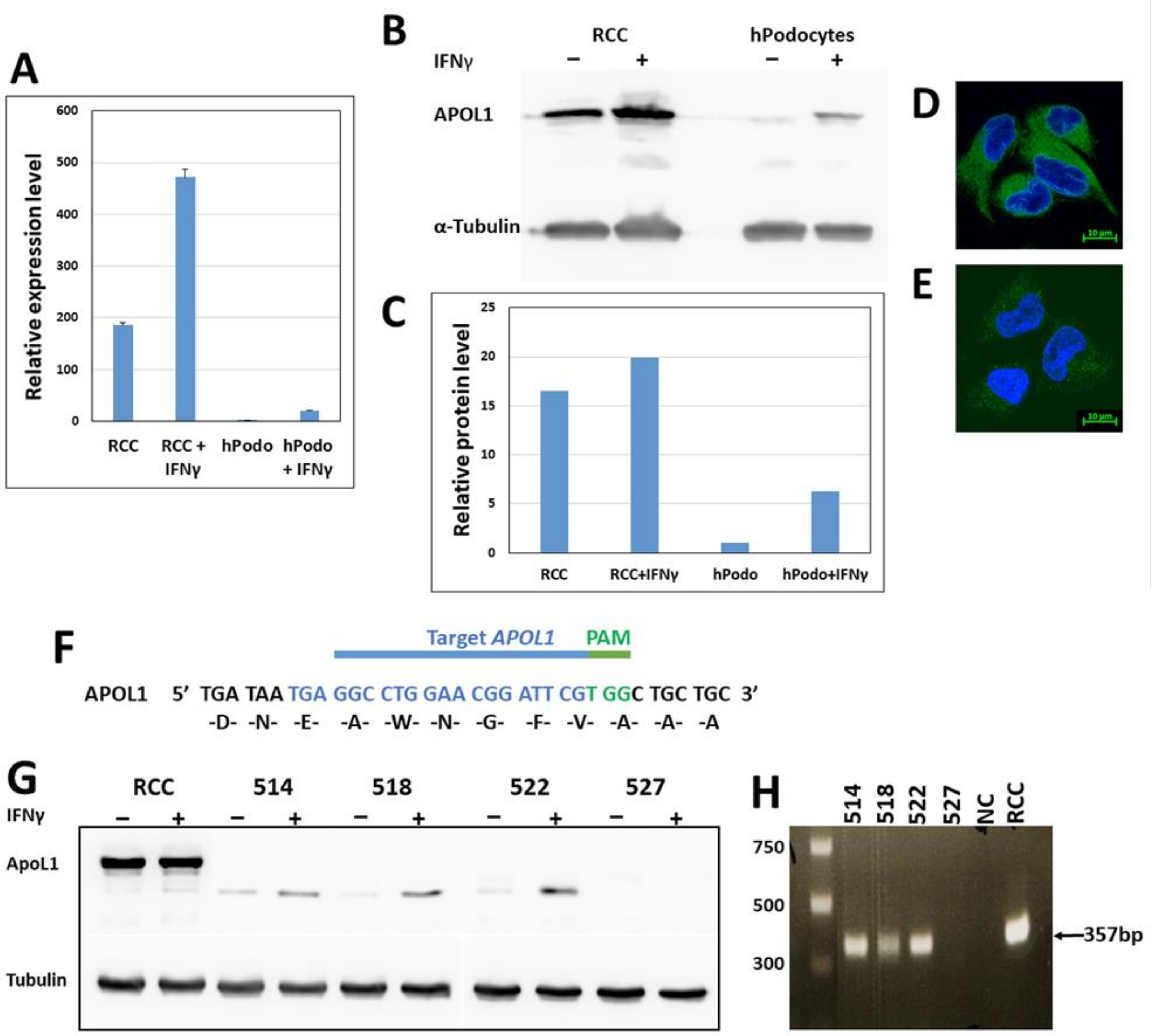
Total RNA was extracted from in vitro cultures, using the RNeasy® Mini Kit (Qiagen, Germantown, MD, USA, 74104) and reverse transcribed with the 5X All-In-One RT MasterMix (Applied Biological Materials, Richmond, BC, Canada, G486) according to the manufacturer’s instructions. Quantitative real-time PCR was performed in triplicate, using the Applied Biosystems StepOnePlus Real-Time PCR system with Fast SYBR Green Master Mix (Applied Biosystems, Waltham, MA, USA, 4385612). All reactions were performed as follows: initial denaturation for 20 s at 95 °C, followed by 40 cycles of 95 °C for 15 s and 60 °C for 30 s. APOL1 gene-specific primer sequences for PCR were as follows: Fwd 5′-GCTGAACTGCCCAGGAATGA, Rev 5′-TTATCGTGCCAGTTTTTGTCTTTC. The relative expression levels of the target genes were compared with the endogenous control Hypoxanthine Phosphoribosyl transferase-1 (HPRT1) using the ΔΔCt method.
2.5. CRISPR/cas9 Methodology
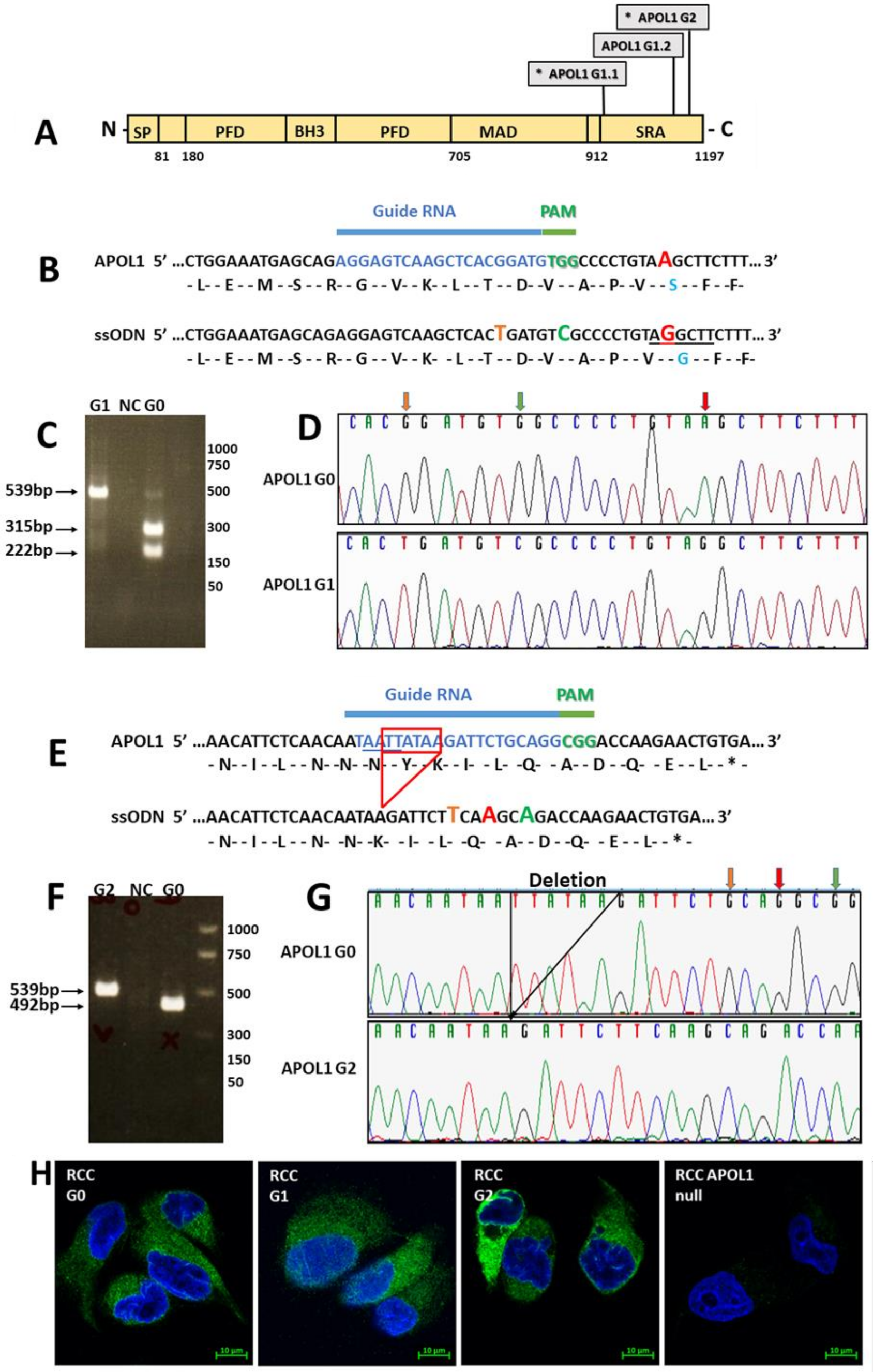
CRISPR/cas9 mediated knock-out of APOL1: a guide RNA (gRNA) targeting a coding region in exon 5 of APOL1 (5′-TGAGGCCTGGAACGGATTCGTGG-3′), was designed using the Optimized CRISPR Design online tool (
http://crispr.mit.edu, accessed on 13 December 2021) [
26], and cloned into pSpCas9(BB)−2A-GFP (PX458), a gift from Feng Zhang (Addgene plasmid #48138) [
26]. This plasmid was transfected into RCC cells using Lipofectamine™ 3000 Transfection Reagent (Invitrogen). GFP-positive single cells were collected into a 96-well plate using the FACS Melody cell sorter (BD Biosciences, San Jose, CA, USA) and expanded to obtain individual clones. Positive clones were identified by Western blot analysis to confirm the loss of APOL1 expression.
CRISPR/cas9 mediated homology-directed repair (HDR) for inserting the sequences of the G1 and G2 APOL1 alleles: For the generation of the APOL1 G1 variant: gRNA (5′ AGGAGTCAAGCTCACGGATGTGG-3′) and a ssODN template (5′-CTGGAAATGAGCAGAGGAGTCAAGCTCACTGATGTCGCCCCTGTAGGCTTCTTT-3′), were designed for an A to G single nucleotide substitution at position 1024 bp of the APOL1 coding sequence. For the generation of APOL1 G2 variant: sgRNA (5′-TAATTATAAGATTCTGCAGGCGG-3′) and ssODN (5′-AACATTCTCAACAATAAGATTCTTCAAGCAGACCAAGAACTGTGA-3′) were designed to generate an in-frame two amino acids deletion (Asn388 and Tyr389). gRNAs, cloned in the pSpCas9(BB)−2A-GFP (PX458) vector, were introduced together with the appropriate ssODN (100 µM) into RCC cells using Lipofectamine™ 3000 Transfection Reagent. GFP-positive single cells were collected as described above. Individual clones were screened by restriction fragment length polymorphism (RFLP). Briefly, genomic DNA was extracted from each clone and subjected to Polymerase chain reaction (PCR) using ApoL1 specific primers (Fwd 5-ACAAGCCCAAGCCCACGACC-3′ and Rev 5′-CCTGGCCCCTGCCAGGCATA-3′). The PCR reaction was as follows: 95 °C for 3 min; 35 cycles at 95 °C for 20 s, 63 °C for 20 s, and 72 °C for 30 s; 72 °C for 5 min. PCR products were digested with HindIII for identifying the G1 variant and with MlucI for identifying the G2 variant. The sequence of positive clones was confirmed by Sanger sequencing.
2.6. Western Blot
Cell proteins were extracted using extraction buffer: 150 mM NaCl, 50 mM Tris pH 8, 1 mM EDTA, 1% triton, and protease inhibitor cocktail (Sigma, St. Louis, MO, USA, P8340). Proteins (20–50 µg) were separated by SDS-PAGE on a 10–15% gel and blotted onto Western Bright NC Nitrocellulose Membranes (Amersham, St. Louis, MO, USA). The membranes were blocked with 5% nonfat dry milk (Santa Cruz sc-2325) and incubated with primary antibodies overnight at 4 °C. The membranes were then incubated with the appropriate secondary antibodies at room temperature for 1 h. The membranes were washed three times with TBST, and the proteins were visualized using chemiluminescence reagents (Advansta, San Jose, CA, USA, K-12045-D20). Fusion FX Spectra (VILBER) densitometry analysis was performed using TotalLab Quant software (TotalLab, Newcastle-Upon-Tyne, UK).
2.7. Immunofluorescence
For immunofluorescence, cells were seeded on fibronectin-coated coverslips in a 24 well plate. Cells were fixed in 4% paraformaldehyde, permeabilized with 0.15% TX-100 and Tween20 and incubated in a blocking solution (4% BSA, 0.15% Triton 0.15% Tween20). Cells were incubated with primary antibodies overnight at 4 °C, and then with a secondary antibody for 1 h at room temperature. Stained cells were mounted in Antifade Mounting Medium with DAPI (Vectashield H-1200) and visualized using a Confocal LSM 880 Upright Fluorescent Microscope (Zeiss, Berlin, Germany).
2.8. Fluorescent In Situ Hybridization (FISH)
RCC cells were treated with colcemid, harvested by trypsinization, treated with a hypotonic solution, and fixed with methanol/acetic acid (3:1). Cells were dropped on slides and hybridized by a standard FISH protocol to a probe generated from an APOL1-region BAC clone (RP1-6802). The probe was labeled with dUTP-digoxigenin. Hybridization was detected with anti-Dig-Rhodamine antibodies. DNA was stained with DAPI. Nuclei and chromosomes were visualized on a BX50 microscope (Olympus, Hamburg, Germany). Images were captured with an Olympus DP70 camera controlled by DP controller software (Olympus, Hamburg, Germany).
2.9. RCC Cell Fractionation
Cell fractionation was performed as described previously [
27]. In brief, cells at 80–90% confluence were harvested by trypsinization. Cells were then manually homogenized in 10 mL of IB cells-1 buffer (225 mM mannitol, 75 mM sucrose, 30 mM Tris-HCl, 0.1 mM EGTA, pH 7.4) with about 100 strokes of a conical glass homogenizer, and sonicated (8 s × 3 times, wheel set to 3.5 in a Misonix Sonicator XL2020 Ultrasonic Liquid Processor sonicator). For separation of endoplasmic reticulum (ER), mitochondria, and mitochondria-associated membrane (MAM) fractions. Homogenized cells were first centrifuged at 600×
g to yield total lysate extract. The total lysate was then centrifuged at 10,000×
g to yield the crude mitochondria extract. Crude mitochondria fraction was separated from the MAM by fractionation on a 30% Percoll gradient (Sigma, P4937) at 95,000×
g. The ER fraction was collected from the mitochondrial supernatant by centrifugation at 100,000×
g. 10 µg of each fraction were loaded on a 4–20% Tris-Glycine gel. The various obtained protein fractions were immunoblotted into a nitrocellulose membrane and subjected to immune detection using the anti-APOL1 oligoclonal 3.7D6/3.1C1 specific rabbit monoclonal antibody. As a control, immune detection was also performed with various cell compartment-specific antibodies. These include: (1) Anti-Calreticulin that reacts with Calreticulin stored in the endoplasmic reticulum (ER); (2) Anti-Cytochrome C that reacts with Cytochrome C localized to the mitochondrial intermembrane space in mammalian cells; (3) Anti-SDHB that reacts with succinate dehydrogenase complex subunit B, iron sulfur Complex II of the respiratory chain, which is specifically localized to the mitochondrial inner membrane; (4) Anti-Tubulin that reacts with Tubulin localized to the cytoplasm; (5) Anti-Calnexin that reacts with Calnexin localized to the endoplasmic reticulum.
2.10. Cell Proliferation
Cells were cultured in a 96 well plate with a regular medium (5000 cells per well). At 24 h, the growing medium was replaced with a medium containing glucose or galactose, and the plates were placed in the Incucyte zoom 2016B system (Essen Bioscience, Ann Arbor, MI, USA). Cells images were captured every three hours for three days. Proliferation analysis was performed using the Incucyte Zoom software. Alternatively, cell proliferation was analyzed using the Cell Titerglow (Promega, Fitchburg, WI, USA) according to the manufacturer’s instructions.
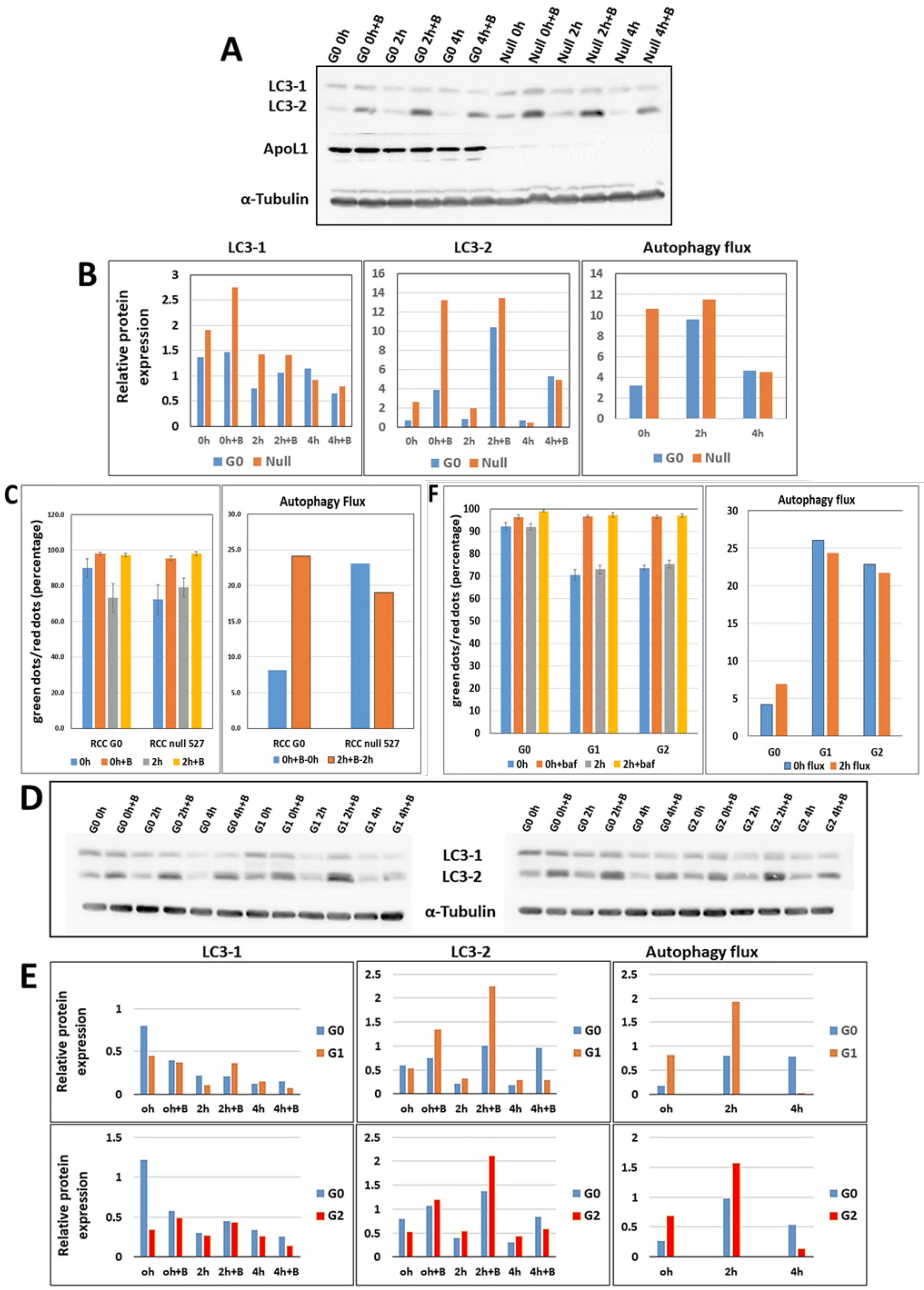
2.11. Autophagy Flux Measurements
Cells (1.5 × 106 cells/10 cm dish) were grown in a regular medium or Earle’s Balanced Salt Solution, EBSS (Biological Industries, Bet-Haemek, Israel) for starvation conditions, with or without Bafilomycin A1 (Santa Cruz, sc-201550A). For Western blot analysis, cells were harvested at 0, 3, 6, and 9 h following starvation for protein extraction. Protein samples were separated on 15% SDS-PAGE gel, transferred to a membrane, and LC3 protein was detected using anti LC3 antibodies. For the Tandem Sensor RFP-GFP-LC3B methodology, the Premo™ Autophagy Tandem Sensor RFP-GFP-LC3B Kit (Invitrogene, Carlsbad, CA, USA, P36239) was used according to manufacturer instructions. In brief, cells grown in regular medium or under starvation conditions, with or without Bafilomycin A1, were transduced with the BacMam 2.0 RFP-GFP-LC3B reagent and visualized using standard GFP (green fluorescent protein) and RFP (red fluorescent protein) settings of the Confocal LSM 880 Upright Fluorescent Microscope (Zeiss, Berlin, Germany). For image-based analyses of autophagy, red and green dots were quantified using the Imaris Image Analysis Software, and the ratio of green dots to red dots was determined.
2.12. Bioenergetic Measurements
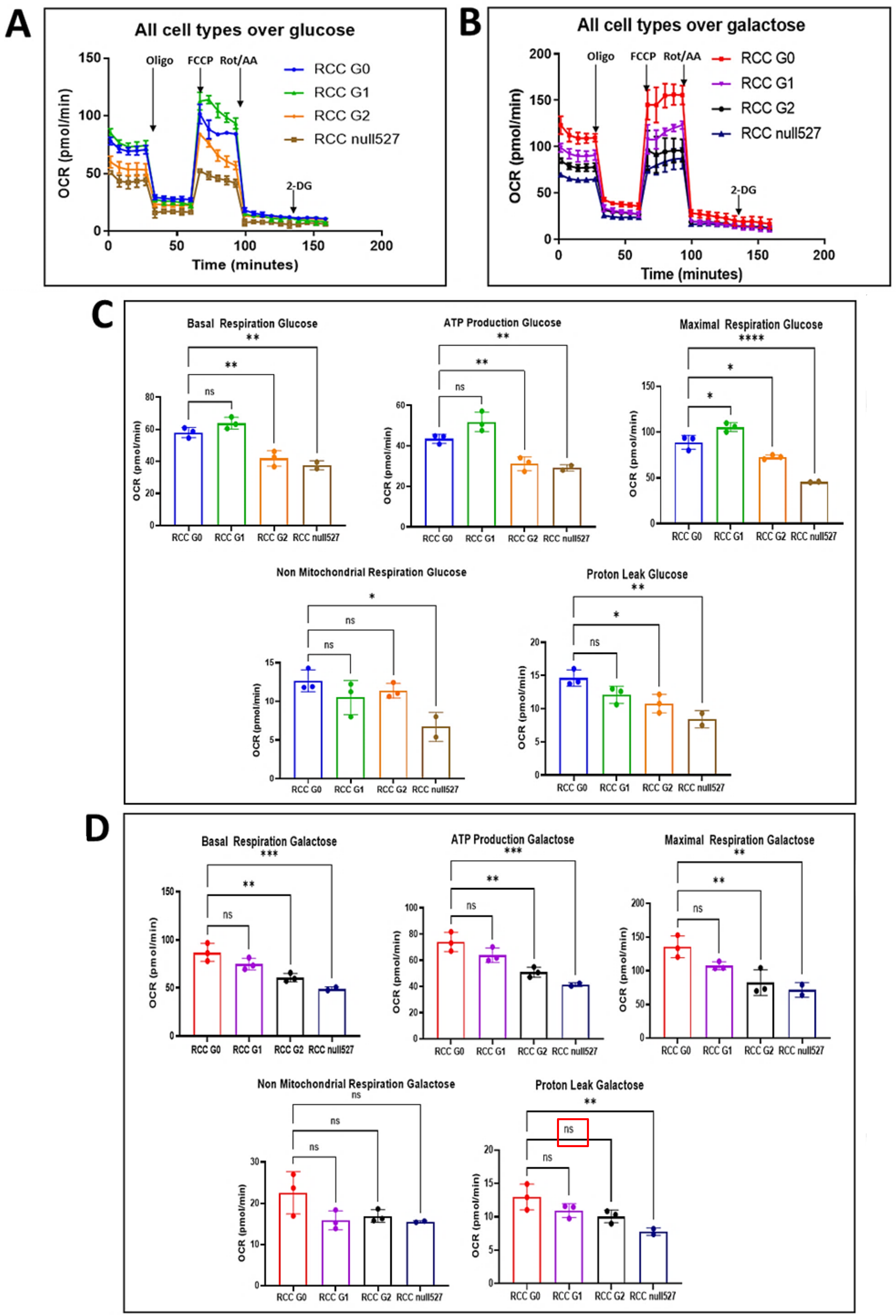
Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured using the Seahorse XFe96 analyzer (Agilent, Santa Clara, CA, USA). Cells were seeded into Seahorse XFe96 plates at 2 × 104 cells/well 24 h prior to measurements. The following day, the medium was replaced with 180 μL of unbuffered assay media (Sigma, St. Louis, MO, USA, D5030) supplemented with 5 mM glucose (Sigma, G8270,)or 5 mM galactose (Sigma, G0625), 1 mM pyruvate (Sigma, P8754), and 2 mM Glutamine (pH 7.4) (Sigma, G3126). Cells were then incubated at 37 °C in a non-CO2-incubator for 45 min. During the steady state, the basal OCR and basal ECAR were determined. Then, respiratory and glycolytic rates were measured in response to sequential injections of 2 µM oligomycin (Complex V inhibitor) (Sigma, 75351), 1.0 μM carbonyl cyanide-4-(trifluoromethoxy)-phenylhydrazone (FCCP, an uncoupling agent of mitochondrial respiration to achieve the maximal respiration rate) (Sigma, C2920), and 50 μM rotenone and antimycin A mixture (Complex I/III inhibitor) (Sigma, R8875 and A8674). Cells were immediately lysed using 30 μL/well Lysis Reagent A (Bio-Rad, Hercules, CA, USA), and proteins were quantified using the Laury protein assay kit (Sigma, 47641). Glycolytic rates in individual wells were normalized to milligrams of total protein, as quantified by a standard curve. Two independent experiments were performed, and all data were presented as M ± S.D.
2.13. Retroviral Infections
APOL1 gene variants G0, G1, and G2 cDNAs were cloned into pBABE-puromycin retroviral vectors using the restriction sites of EcoRI and BamHI. To generate retroviral particles, the viral packaging cell line GP2- 293 (stably expressing the gag and pol genes) was cotransfected with the pBABE constructs and a plasmid containing the vesicular stomatitis virus glycoprotein gene (VSVG). Transfection was performed using the Lipofectamine™ 3000 Reagent (Invitrogen, Carlsbad, CA, USA, L3000-008). 48 h post-transfection, the supernatant containing the viruses was collected and filtered through a 0.45-μm PVDF filter. Viruses were used to infect the RCC null cells in the presence of 6 µg/mL polybrene (Millipore, TR-1003-G).
4. Discussion
The relative risk of renal cell carcinoma associated with chronic kidney disease (CKD) is particularly high among black populations. The estimated incidence of RCC attributable to CKD within black populations accounts for 10%, while it is negligible among Caucasians. However, it is unknown whether the APOL1 risk variants for CKD also contribute to the risk for RCC in black populations [
1].
Previously, the mechanisms underlying APOL1 function were investigated mostly in experimental platforms involving ectopic overexpression (conditional or ubiquitous) of APOL1 G0 or APOL1 RV, either in cell lines (HEK293, normal podocytes) or in transgenic mice [
15,
16,
36,
37]. Over-expression of APOL1 leads to cytotoxicity via yet unclear mechanisms. The RCC 786-O cell line naturally expresses very high levels of WT
APOL1 (G0), both at the RNA and protein levels (~200 fold compared with normal cells), but interestingly, no signs of APOL1 toxicity are evident. This raised the question of how these cancer cells tolerate high APOL1 levels, and whether they could tolerate high levels of the APOL1 G1 and G2 RV, as well. Additionally, understanding how APOL1 cytotoxicity is abolished in RCC cancer cells could potentially shed light on the mechanism/s by which high levels of APOL1 induce toxicity in normal cells. Here, we describe tissue culture-based studies in RCC 786-O cells that highly express APOL1 G0, G1, or G2 from an endogenous gene copy, achieved by using CRISPR mediated gene editing methodology. Such an approach guarantees that all APOL1 variants are highly expressed in identical genetic backgrounds, thus ensuring that identified phenotypes are attributed specifically to the APOL1 variants. In addition, we used this methodology to knock out the expression of APOL1 and generate APOL1 deficient RCC cells (RCC null). Initial observations indicated slight differences in the proliferation capacity of RCC G0, RCC G1, RCC G2, and RCC null, which suggest that phenotypes related to respiration and energy metabolism are associated with APOL1.
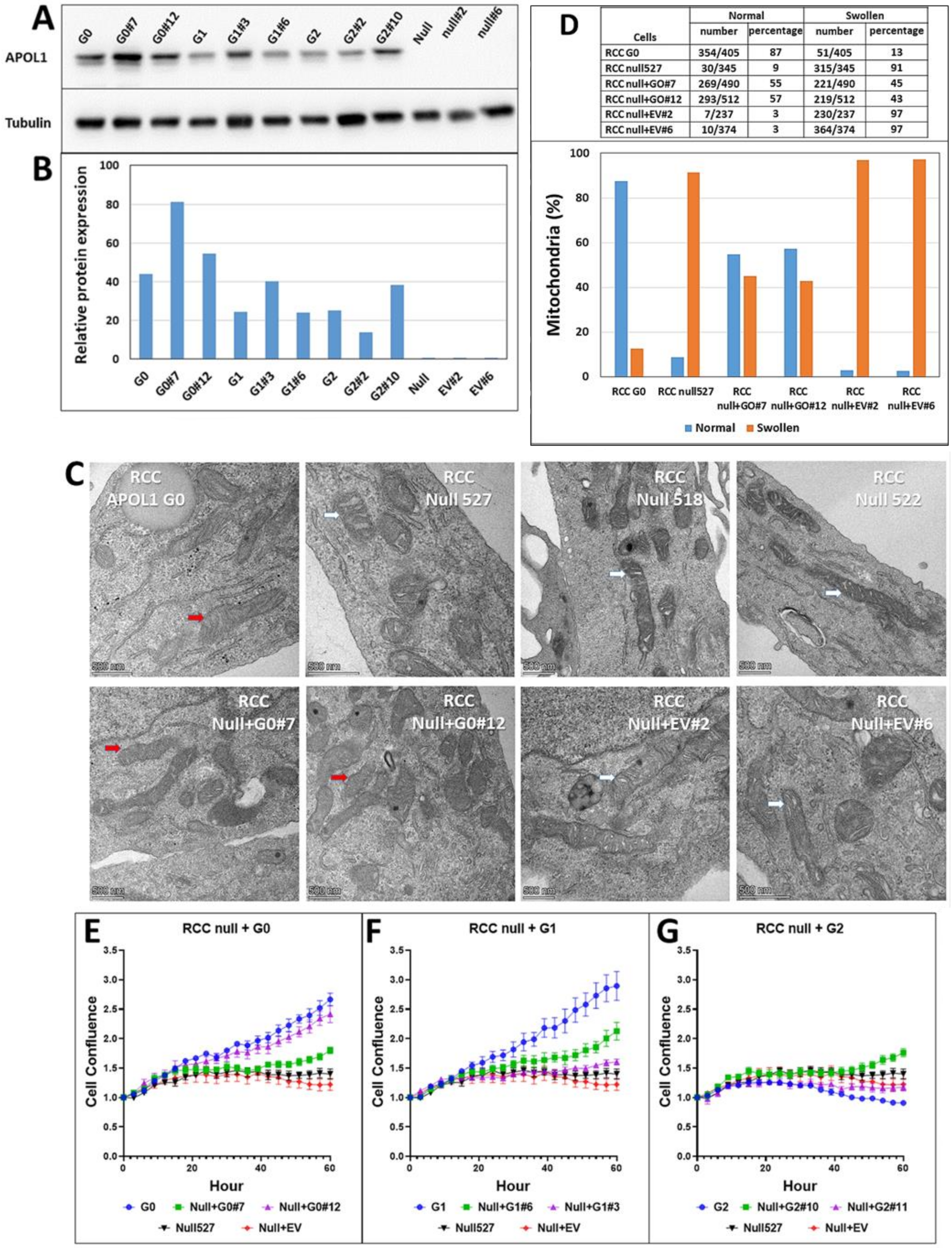
Ultrastructure studies indicated an abnormal mitochondrial phenotype in RCC cells following knock-out or mutations in
APOL1. This phenotype is characterized by the opening of the mitochondrial cristae (swollen cristae) in RCC null, RCC G1, and RCC G2, as opposed to clear cristae observed in RCC G0 parental cells [
38]. Mitochondrial swollen cristae were previously observed in normal podocytes extracted from individuals carrying APOL1 G1 and G2 alleles [
20], indicating that in RCC G1 and G2, this phenotype can be unequivocally attributed to the editing of APOL1 and not to an indirect consequence of the CRISPR methodology. These findings suggest a major role for APOL1 in maintaining and controlling the integrity and functionality of the mitochondria in RCC cancer cells and indicate that APOL1 RV may act through a loss of function needed for tumorigenic activity. In addition, we found that the ultrastructure phenotype of RCC G2 and RCC null exhibits short mitochondria containing swelling cristae. On the other hand, RCC G1 exhibits elongated mitochondria with swollen cristae, suggesting a fusion process in RCC G1 cells that can explain the higher proliferation rate and OCR values in this clone compared with RCC null and RCC G2. The differences in mitochondria lengths observed in RCC cells that express APOL1 G1 and G2 and in RCC null cells can be explained by a differential rate of mitochondrial fission and fusion in response to APOL1-related changes in cell metabolism. Mitochondria fusion can occur due to stress that drives the cells to extensively utilize oxidative phosphorylation as a source for ATP production, specifically by withdrawing glucose as a carbon source for cell metabolism [
33]. Fusion can also compensate for mitochondrial defects by complementing respiratory and ATP production functions [
33].
High levels of cellular stress can additionally lead to apoptosis, which can also culminate in excessive mitochondrial fission [
33]. Nevertheless, we could not detect a significant difference in the mRNA levels of the mitochondrial fission and fusion genes FIS1 and MFN1, as was observed in normal podocytes displaying a similar phenotype (data not shown) [
20]. Overall, the modulation of the mitochondrial structure and the abnormal phenotypic appearance of the mitochondrial cristae point to physiological stress induced in RCC cells following the alterations we generated in the APOL1 gene, and suggest that abnormal mitochondrial metabolism levels are linked to APOL1 risk variants or lack of APOL1 expression.
Since RCC are high energy demanding cancer cells, as indicated by their proliferation rates in vitro and in vivo, it was important to examine the mitochondrial function of APOL1 variants by an analysis that directly measures mitochondrial respiration. Mitochondrial dysfunction is difficult to study in a high glucose medium as rapidly proliferating cancer cells are highly glycolytic in such medium [
28]. The production of pyruvate via glycolytic metabolism of glucose yields two net ATP molecules, whereas the production of pyruvate via glycolytic metabolism of galactose yields no net ATP molecules. Therefore, galactose-dependent metabolism forces cells to rely on mitochondrial respiration via oxidative phosphorylation (OXPHOS) for energy production [
29]. Therefore, to reliably measure proliferation rates and mitochondrial metabolism, we substituted galactose for glucose in the growth medium [
29]. Indeed, RCC G0, G1, G2, and RCC null exhibit similar proliferation rates in vitro when grown in high glucose media, as they can compensate for mitochondrial dysfunction by utilizing glycolysis for ATP production. In contrast, G0 and G1 cells reached higher confluence when grown in galactose supplemented medium (as the sole carbohydrate source) compared to G2 and null cells. The differences in the mitochondrial structure and in the proliferation rates observed in RCC cells as a result of eliminating or editing APOL1 were integrated with bioenergetics values obtained by the metabolic flux technology of the Seahorse bioanalyzer. This analysis supports the notion that the genetic manipulation of APOL1 in RCC cancer cells leads to an alteration in mitochondrial metabolism, which in turn influences downstream gene expression, signal transduction, and the tumorigenic capacity of the cells [
39]. The most significant bioenergetic consequence of APOL1 manipulation was the loss of ATP-coupled respiration in APOL1 null cells, and to a lesser extent in G2 isoform expressing cells.
In high energy demanding cells, mitochondria cristae packaging is tightly regulated to ensure proper respiratory processes [
40]. The swollen mitochondrial cristae in RCC G1, G2, and in RCC null are consistent with a deregulated process wherein mitochondrial inner membrane unfolding leads to rupture of the outer membrane and to the release of pro-apoptotic molecules that eventuate in cell death [
41]. It has been demonstrated that swollen mitochondria cristae are associated with increased calcium concentrations and can disrupt mitochondrial function [
41,
42]. A recent study indicates that while all three APOL1 variants traffic to the plasma membrane (PM), only G1 and G2 RV, but not G0, have an enhanced capacity to form cation channels that enable Na
+, K
+, and Ca
2+ conductance [
23]. Thereby, we postulate that APOL1 G1 and G2 channel activity can augment cytotoxic influx of Na
+ and Ca
2+ that leads to uncontrolled gene expression, deregulated signaling pathways, cell swelling, and eventually to APOL1 associated cell death [
23]. APOL1 G1 and G2 cation channel-mediated cytotoxicity were also demonstrated when overexpressed in Human Embryonic Kidney 293 (HEK293) cells [
36]. These observations suggest that intracellular changes in Ca
2+ concentration additionally affect intra mitochondrial Ca
2+, leading to mitochondrial cristae swelling, disturbed metabolism, and eventually to cell cytotoxicity. In any case, the rise in cytosolic Ca
2+ needs to be considered and measured as part of future studies to unravel the serial steps leading to observed mitochondrial swelling. The fact that swelling mitochondria and disturbed metabolism are also observed in RCC APOL1-null cells implies that APOL1 RV cytotoxicity is mediated via additional unknown mechanisms linked to mitochondria function. Yet, we demonstrated that re-expression of APOL1 G0 in RCC null cells could significantly rescue the swelling cristae phenotype in these cells and improve their proliferation capacity, suggesting an APOL1 specific loss of function. The rescue of the mitochondrial phenotype by APOL1 G0 additionally supports the conclusion that the mitochondrial phenotype apparent in RCC null cells is directly associated with APOL1 loss of function and not an off-target effect of the editing procedure [
43].
Autophagy, which was also proposed as a mechanism responsible for APOL1 RV cytotoxicity, is associated with increasing levels of APOL1 expression in normal and cancer cells [
13,
14,
15]. Nevertheless, autophagy as a mechanism that mediates APOL1 associated cytotoxicity appears cell-dependent [
13,
18]. As high energy demanding cells, RCC cancer cells require high autophagy activity to remove damaged organelles via the lysosomal pathway and recycle them for cell growth, energy production, and nutrient consumption during starvation or stress [
44]. In the current study, we observed a significant elevation in autophagic flux in APOL1-null RCC cells, as well as in RCC cells that express APOL1 G1 and G2 RV. Hence, we suggest that these RCC cells, which exhibit mitochondrial dysfunction at both the morphological and functional levels, induce autophagy (and mitophagy) to remove damaged mitochondria. A previous study suggested that changes in Ca
+2 can lead to mitochondria swelling cristae and increased autophagy in the cells [
42]. However, elevation in autophagic flux both in the absence of APOL1 expression and in the presence of APOL1 RV might indicate that APOL1 RV-induced attenuation in respiration and growth capacity is independent of autophagy. These observations agree with a previous observation that autophagy is a response to APOL1 RV cytotoxicity rather than a mediator [
36]. The fact that elimination of APOL1 expression and the expression of G1 and G2 RV elicit similar effects in RCC cells might suggest that APOL1 loss of function in the cells can be mediated by more than one pathway, which eventually lead to cell death.
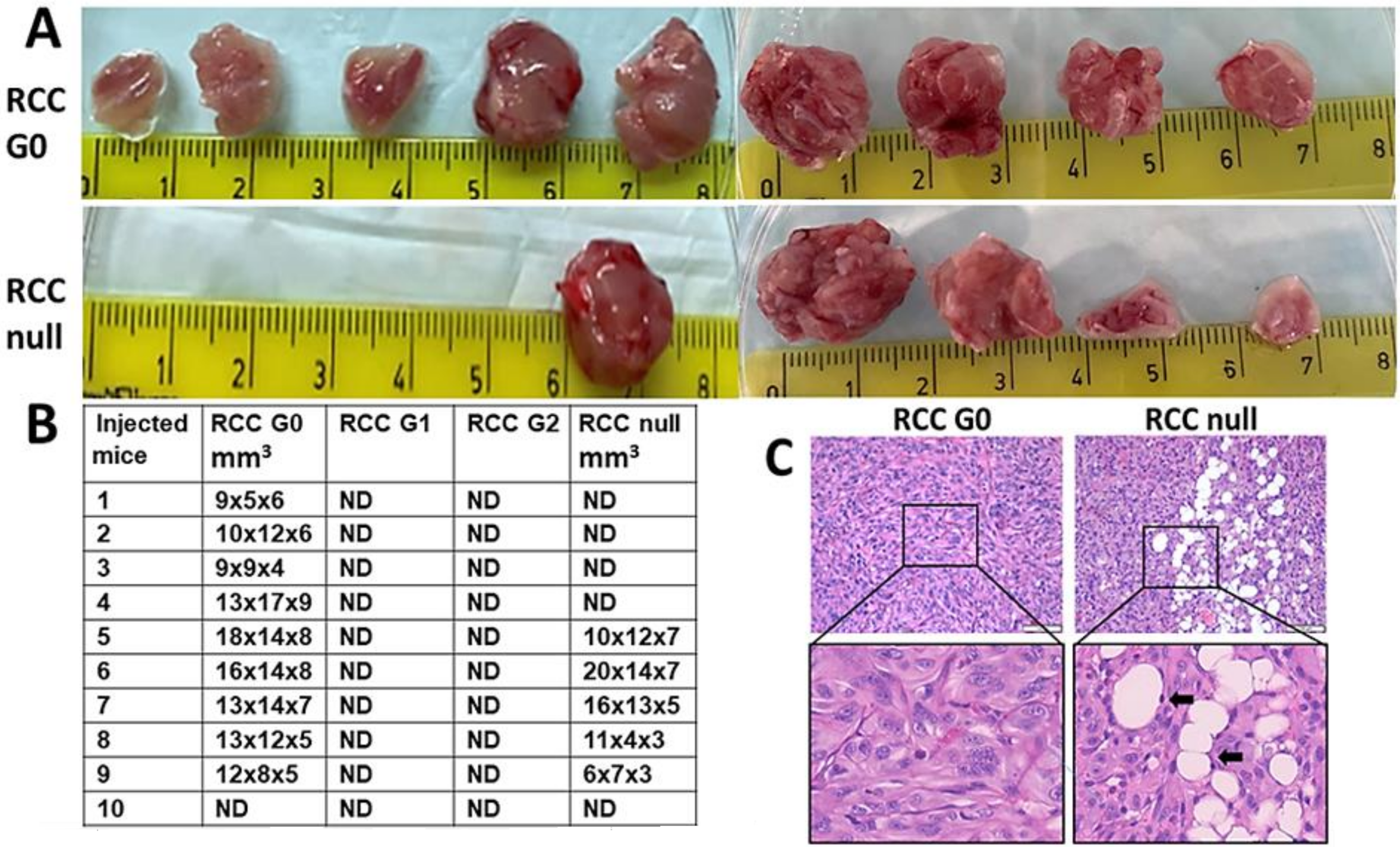
Proliferation and autophagy are considered two major hallmarks of cancer [
34]. Our findings that elimination or alteration of APOL1 in RCC leads to attenuated cell growth and metabolism and increased autophagy prompted us to examine whether these changes play a role in the tumorigenic capacity. Using in vivo tumor generation as the gold standard for tumorigenic potential, we observed that RCC cancer cells which express the APOL1 G1 and G2 RV fail to generate tumors in mice. Such an absolute elimination of RCC cell tumorigenic potential is puzzling given that RCC null cells succeeded in generating tumors, albeit at lower rates than G0 RCC cells. We postulate that the extreme elimination of the tumorigenic potential might result from APOL1 G1 and G2 cytotoxicity, which arises under conditions specific to in vivo growth, such as relatively low oxygen levels. However, under in vitro growth conditions that include a high glucose containing medium, APOL1 G1 and G2 are less toxic to RCC cells. The reduced in vivo tumorigenic potential of RCC G1 and G2 predicts that human carriers of these variants may be relatively protected from RCC and suggests that the high incidence of RCC in the black population is unrelated to the APOL1 RV for CKD. Future studies are necessary to determine the association of APOL1 G1 and G2 RV with kidney cancer in Sub-Saharan and African-American populations compared to populations of European ancestry.
The RCC G0 parental cells generated aggressive tumors with a condensed tissue appearance typical to RCC. Interestingly, the tumors generated by RCC null cells displayed a less condensed tumor tissue, with many foci of differentiation into adipocytes, carrying typical RCC cell nuclei. This observation might indicate an epithelial to mesenchymal transition (EMT) process occurring in RCC null cells growing in murine musculature and implies that APOL1 G0 inhibits EMT. Future studies related to such a role executed by APOL1 may shed light on the regulation of EMT in kidney cancer and be useful for devising therapeutic avenues for kidney-related cancers.


 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}