
Dual Inhibition of Myc Transcription and PI3K Activity Effectively Targets Colorectal Cancer Stem Cells

 ,
,  , ,
, ,  , , , ,
, , , ,  , ,
, ,  , , ,
, , ,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Isolation and Maintenance of CR-CSphCs and CAFs
2.2. CR-CSphCs In Vitro Treatment
2.3. Clonogenic, Sphere-Forming and Colony-Formation Assays
2.4. Cell Viability
2.5. Lentiviral Production and Transduction
2.6. Flow Cytometry and Cell Sorting
2.7. Western Blot
2.8. RNA Extraction and Gene Expression Analysis
2.9. Immunofluorescence/Immunohistochemistry
2.10. In Vivo Experiments
2.11. Statistical Analysis
3. Results
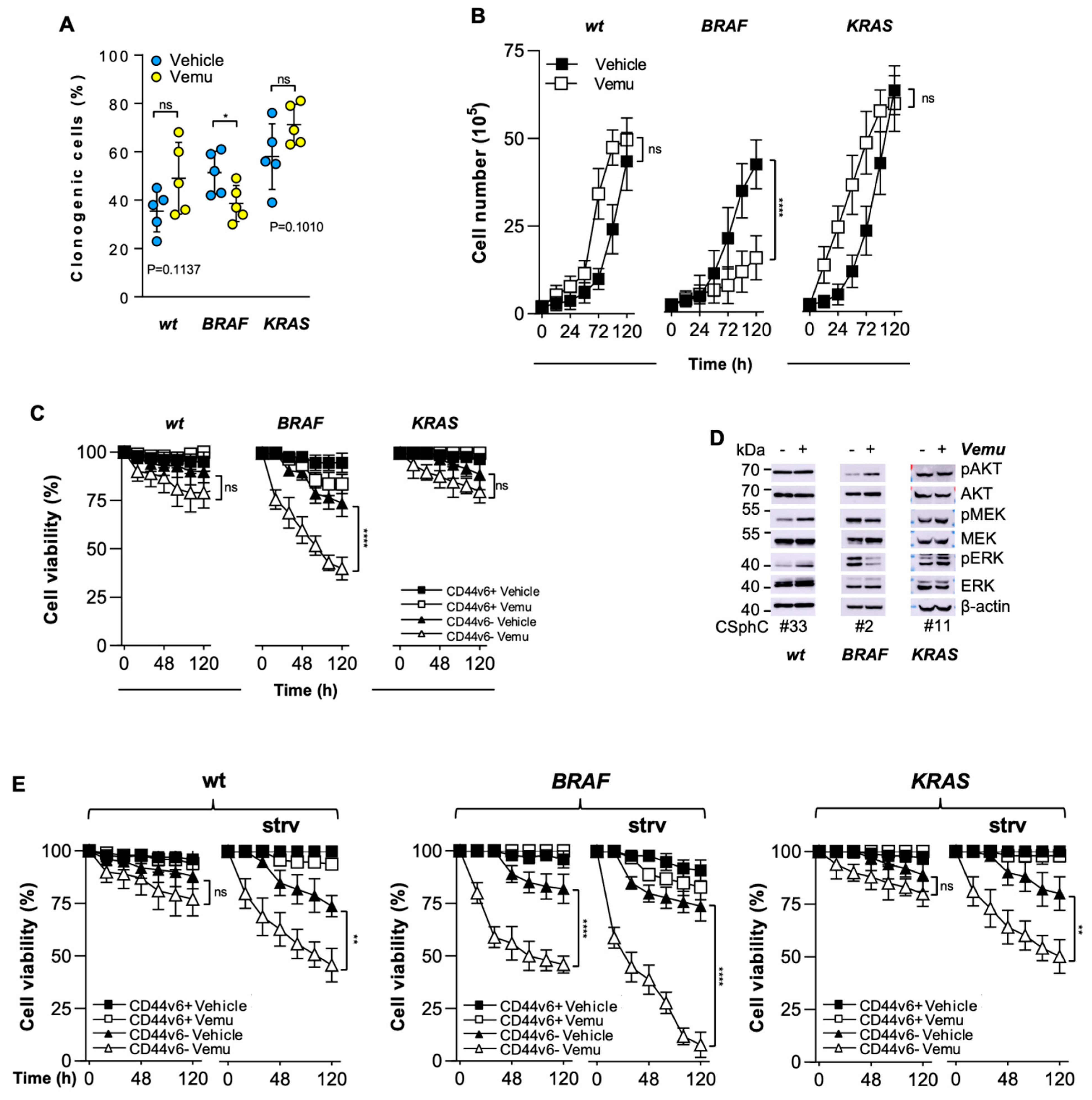
3.1. CD44v6-Positive CR-CSphCs Are Refractory toward BRAF Inhibition
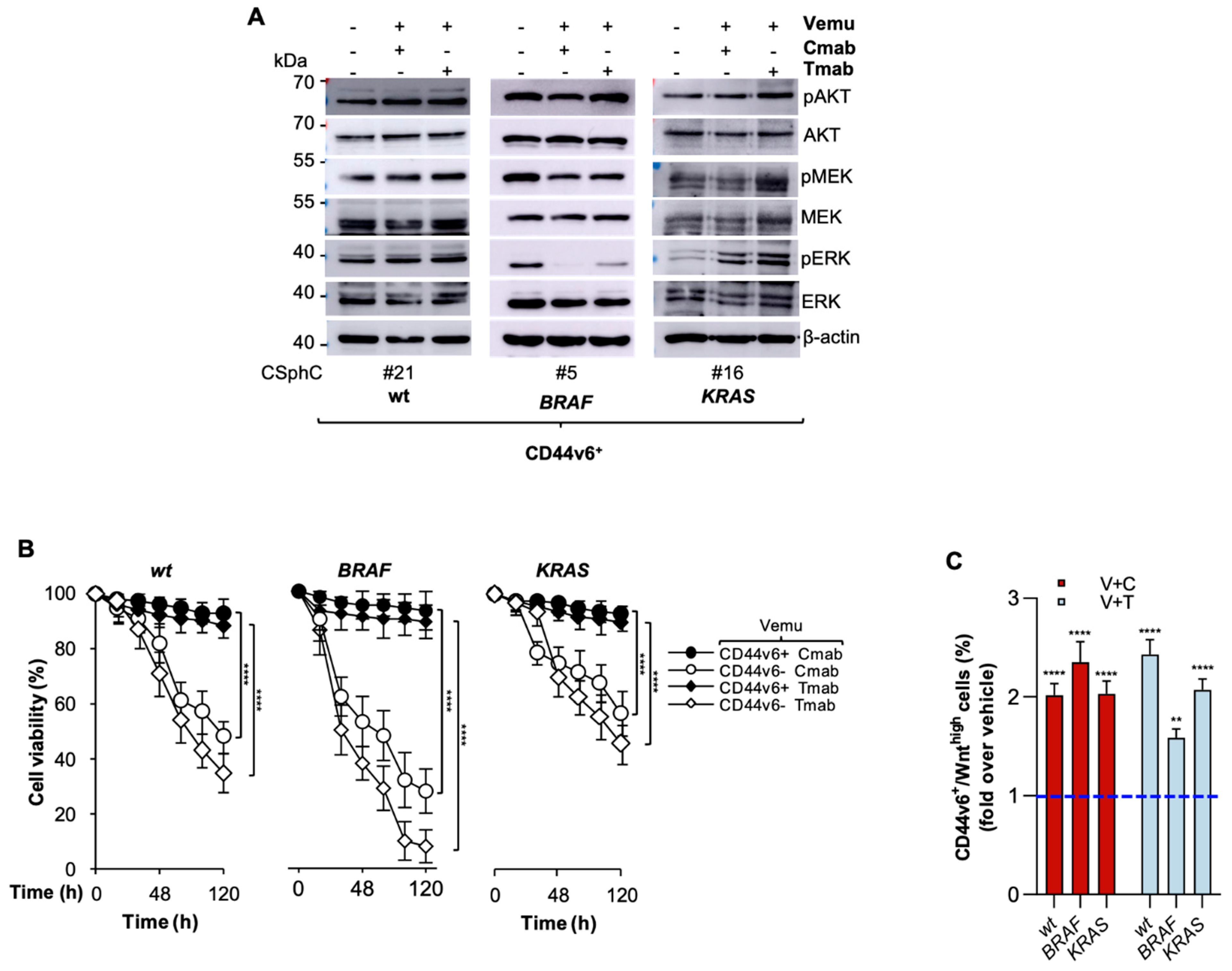
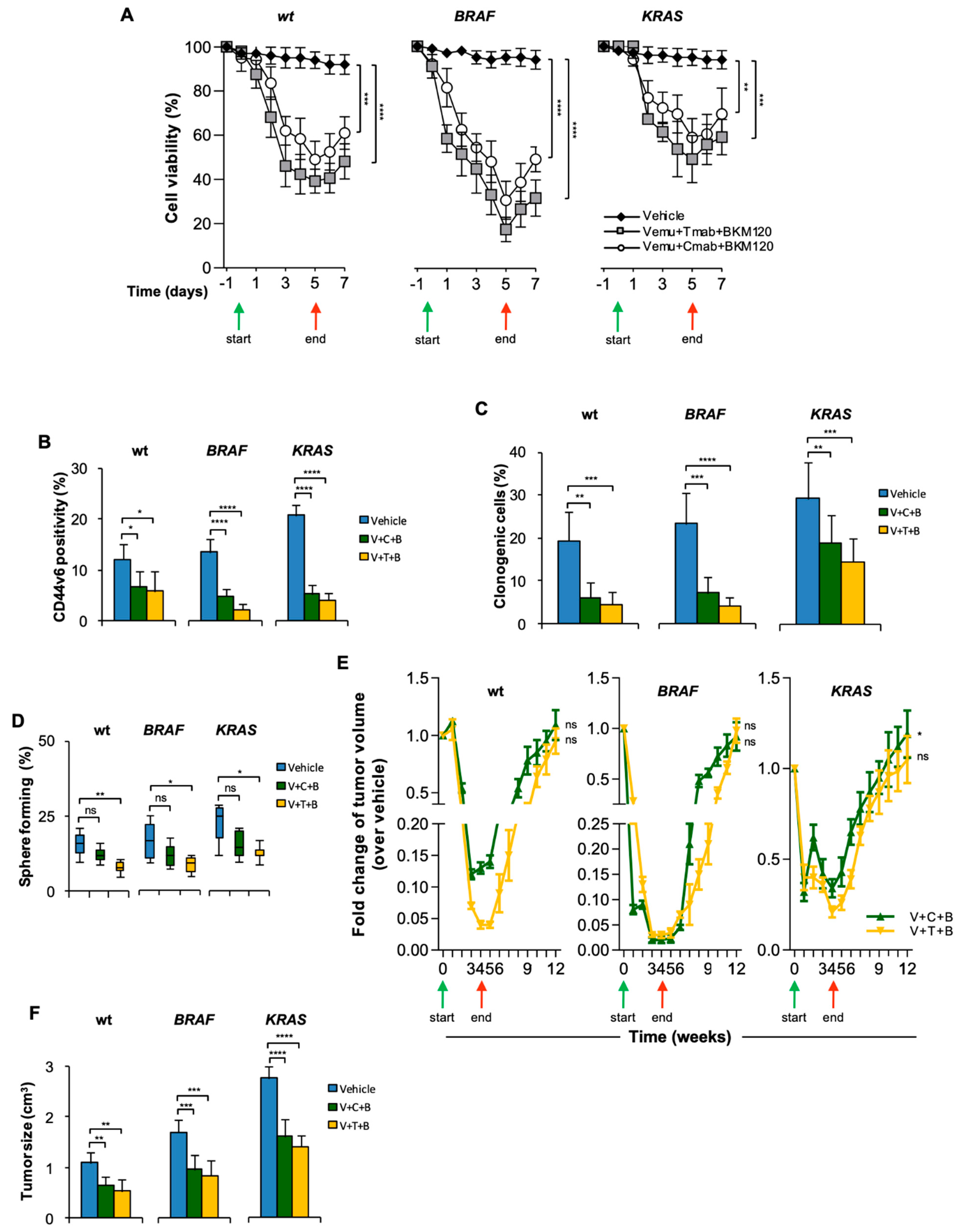
3.2. CR-CSCs Emerge after Inhibition of HER2, BRAF and PI3K
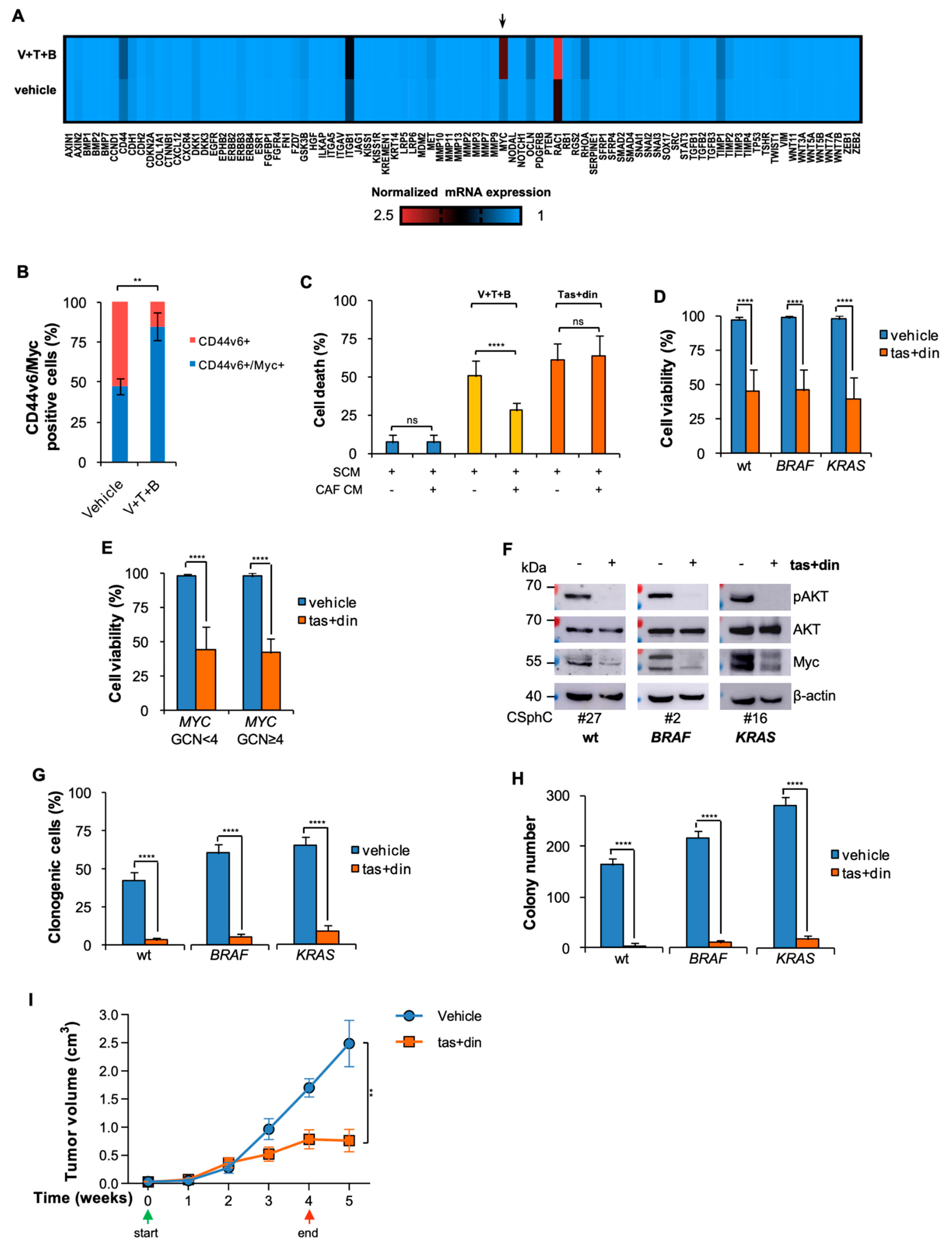
3.3. Resistance to BRAF-Based Combination Therapy Is Sustained by Myc Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.-H.; Chen, Y.-X.; Fang, J.-Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 1–30. [Google Scholar] [CrossRef]
- Raskov, H.; Søby, J.H.; Troelsen, J.T.; Bojesen, R.D.; Gögenur, I. Driver Gene Mutations and Epigenetics in Colorectal Cancer. Ann. Surg. 2020, 271, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Matano, M.; Date, S.; Shimokawa, M.; Takano, A.; Fujii, M.; Ohta, Y.; Watanabe, T.; Kanai, T.; Sato, T. Modeling colorectal cancer using CRISPR-Cas9–mediated engineering of human intestinal organoids. Nat. Med. 2015, 21, 256–262. [Google Scholar] [CrossRef]
- Gaggianesi, M.; Di Franco, S.; Pantina, V.D.; Porcelli, G.; D’Accardo, C.; Verona, F.; Veschi, V.; Colarossi, L.; Faldetta, N.; Pistone, G.; et al. Messing Up the Cancer Stem Cell Chemoresistance Mechanisms Supported by Tumor Microenvironment. Front. Oncol. 2021, 11, 2847. [Google Scholar] [CrossRef]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Turdo, A.; Veschi, V.; Gaggianesi, M.; Chinnici, A.; Bianca, P.; Todaro, M.; Stassi, G. Meeting the Challenge of Targeting Cancer Stem Cells. Front. Cell Dev. Biol. 2019, 7, 16. [Google Scholar] [CrossRef]
- Di Franco, S.; Todaro, M.; Dieli, F.; Stassi, G. Colorectal cancer defeating? Challenge accepted! Mol. Asp. Med. 2014, 39, 61–81. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- De Sousa e Melo, F.; Vermeulen, L. Wnt Signaling in Cancer Stem Cell Biology. Cancers 2016, 8, 60. [Google Scholar] [CrossRef]
- Todaro, M.; Gaggianesi, M.; Catalano, V.; Benfante, A.; Iovino, F.; Biffoni, M.; Apuzzo, T.; Sperduti, I.; Volpe, S.; Cocorullo, G.; et al. CD44v6 Is a Marker of Constitutive and Reprogrammed Cancer Stem Cells Driving Colon Cancer Metastasis. Cell Stem Cell 2014, 14, 342–356. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; Felipe de Sousa, E.M.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Bol. 2010, 12, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Douillard, J.-Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab–FOLFOX4 Treatment and RAS Mutations in Colorectal Cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Sorich, M.J.; Wiese, M.; Rowland, A.; Kichenadasse, G.; McKinnon, R.; Karapetis, C. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: A meta-analysis of randomized, controlled trials. Ann. Oncol. 2014, 26, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Liao, R.-Y.; Qiu, L.-X.; Wang, X.-W.; Ding, H.; Chen, Q. BRAF V600E mutation and resistance to anti-EGFR monoclonal antibodies in patients with metastatic colorectal cancer: A meta-analysis. Mol. Biol. Rep. 2010, 38, 2219–2223. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Garcia, E.; Argiles, G.; Elez, E.; Tabernero, J. BRAF mutant colorectal cancer: Prognosis, treatment, and new perspectives. Ann. Oncol. 2017, 28, 2648–2657. [Google Scholar] [CrossRef]
- Mangiapane, L.R.; Nicotra, A.; Turdo, A.; Gaggianesi, M.; Bianca, P.; Di Franco, S.; Sardina, D.S.; Veschi, V.; Signore, M.; Beyes, S.; et al. PI3K-driven HER2 expression is a potential therapeutic target in colorectal cancer stem cells. Gut 2021, 71, 119–128. [Google Scholar] [CrossRef]
- Hansen, T.; Qvortrup, C.; Pfeiffer, P. Angiogenesis Inhibitors for Colorectal Cancer. A Review of the Clinical Data. Cancers 2021, 13, 1031. [Google Scholar] [CrossRef]
- Overman, M.J.; Ernstoff, M.S.; Morse, M.A. Where We Stand with Immunotherapy in Colorectal Cancer: Deficient Mismatch Repair, Proficient Mismatch Repair, and Toxicity Management. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 239–247. [Google Scholar] [CrossRef]
- Jácome, A.A.; Eng, C. Role of immune checkpoint inhibitors in the treatment of colorectal cancer: Focus on nivolumab. Expert Opin. Biol. Ther. 2019, 19, 1247–1263. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Loupakis, F.; Argilés, G.; Prager, M.G. Challenging chemoresistant metastatic colorectal cancer: Therapeutic strategies from the clinic and from the laboratory. Ann. Oncol. 2016, 27, 1456–1466. [Google Scholar] [CrossRef] [PubMed]
- Sabnis, A.J.; Bivona, T.G. Principles of Resistance to Targeted Cancer Therapy: Lessons from Basic and Translational Cancer Biology. Trends Mol. Med. 2019, 25, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-M.; Giltnane, J.M.; Balko, J.M.; Schwarz, L.J.; Guerrero-Zotano, A.L.; Hutchinson, K.E.; Nixon, M.J.; Estrada, M.V.; Sánchez, V.; Sanders, M.E.; et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab. 2017, 26, 633–647.e7. [Google Scholar] [CrossRef] [PubMed]
- Strippoli, A.; Cocomazzi, A.; Basso, M.; Cenci, T.; Ricci, R.; Pierconti, F.; Cassano, A.; Fiorentino, V.; Barone, C.; Bria, E.; et al. c-MYC Expression Is a Possible Keystone in the Colorectal Cancer Resistance to EGFR Inhibitors. Cancers 2020, 12, 638. [Google Scholar] [CrossRef]
- Shivakumar, B.M.; Chakrabarty, S.; Rotti, H.; Seenappa, V.; Rao, L.; Geetha, V.; Tantry, B.V.; Kini, H.; Dharamsi, R.; Pai, C.G.; et al. Comparative analysis of copy number variations in ulcerative colitis associated and sporadic colorectal neoplasia. BMC Cancer 2016, 16, 1–11. [Google Scholar] [CrossRef]
- Dong, Y.; Tu, R.; Liu, H.; Qing, G. Regulation of cancer cell metabolism: Oncogenic MYC in the driver’s seat. Signal Transduct. Target. Ther. 2020, 5, 1–11. [Google Scholar] [CrossRef]
- Elbadawy, M.; Usui, T.; Yamawaki, H.; Sasaki, K. Emerging Roles of C-Myc in Cancer Stem Cell-Related Signaling and Resistance to Cancer Chemotherapy: A Potential Therapeutic Target Against Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 2340. [Google Scholar] [CrossRef]
- Shen, A.; Wang, L.; Huang, M.; Sun, J.; Chen, Y.; Shen, Y.-Y.; Yang, X.; Wang, X.; Ding, J.; Geng, M. c-Myc Alterations Confer Therapeutic Response and Acquired Resistance to c-Met Inhibitors in MET-Addicted Cancers. Cancer Res. 2015, 75, 4548–4559. [Google Scholar] [CrossRef]
- Wang, Q.; Tan, R.; Zhu, X.; Zhang, Y.; Tan, Z.; Su, B.; Li, Y. Oncogenic K-ras confers SAHA resistance by up-regulating HDAC6 and c-myc expression. Oncotarget 2016, 7, 10064–10072. [Google Scholar] [CrossRef]
- Veschi, V.; Mangiapane, L.R.; Nicotra, A.; di Franco, S.; Scavo, E.; Apuzzo, T.; Sardina, D.S.; Fiori, M.E.; Benfante, A.; Colorito, M.L.; et al. Targeting chemoresistant colorectal cancer via systemic administration of a BMP7 variant. Oncogene 2019, 39, 987–1003. [Google Scholar] [CrossRef]
- Di Franco, S.; Zhang, L.; Gaggianesi, M.; Iacono, M.L.; Medema, J.P.; Stassi, G. FACS-based protocol to assess cytotoxicity and clonogenic potential of colorectal cancer stem cells using a Wnt/β-catenin signaling pathway reporter. STAR Protoc. 2021, 2, 100880. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Nam, S.K.; Seo, S.H.; Park, K.U.; Oh, H.-K.; Kim, D.-W.; Kang, S.-B.; Kim, W.H.; Lee, H.S. Digital polymerase chain reaction for detecting c-MYC copy number gain in tissue and cell-free plasma samples of colorectal cancer patients. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ianevski, A.; He, L.; Aittokallio, T.; Tang, J. SynergyFinder: A web application for analyzing drug combination dose–response matrix data. Bioinformatics 2017, 33, 2413–2415. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010, 464, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Heidorn, S.J.; Milagre, C.; Whittaker, S.; Nourry, A.; Niculescu-Duvas, I.; Dhomen, N.; Hussain, J.; Reis-Filho, J.S.; Springer, C.J.; Pritchard, C.; et al. Kinase-Dead BRAF and Oncogenic RAS Cooperate to Drive Tumor Progression through CRAF. Cell 2010, 140, 209–221. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Ebi, H.; Turke, A.B.; Coffee, E.M.; Nishino, M.; Cogdill, A.; Brown, R.D.; Della Pelle, P.; Dias-Santagata, D.; Hung, K.E.; et al. EGFR-Mediated Reactivation of MAPK Signaling Contributes to Insensitivity of BRAF-Mutant Colorectal Cancers to RAF Inhibition with Vemurafenib. Cancer Discov. 2012, 2, 227–235. [Google Scholar] [CrossRef]
- Di Franco, S.; Bianca, P.; Sardina, D.S.; Turdo, A.; Gaggianesi, M.; Veschi, V.; Nicotra, A.; Mangiapane, L.R.; Iacono, M.L.; Pillitteri, I.; et al. Adipose stem cell niche reprograms the colorectal cancer stem cell metastatic machinery. Nat. Commun. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Dang, C.V.; Reddy, E.P.; Shokat, K.M.; Soucek, L. Drugging the ‘undruggable’ cancer targets. Nat. Cancer 2017, 17, 502–508. [Google Scholar] [CrossRef]
- Carey, J.P.; Karakas, C.; Bui, T.; Chen, X.; Vijayaraghavan, S.; Zhao, Y.; Wang, J.; Mikule, K.; Litton, J.K.; Hunt, K.K.; et al. Synthetic Lethality of PARP Inhibitors in Combination with MYC Blockade Is Independent of BRCA Status in Triple-Negative Breast Cancer. Cancer Res. 2017, 78, 742–757. [Google Scholar] [CrossRef]
- Lee, C.S.; Song, I.H.; Lee, A.; Kang, J.; Lee, Y.S.; Lee, I.K.; Song, Y.S.; Lee, S.H. Enhancing the landscape of colorectal cancer using targeted deep sequencing. Sci. Rep. 2021, 11, 1–26. [Google Scholar] [CrossRef]
- Tie, J.; Gibbs, P.; Lipton, L.; Christie, M.; Jorissen, R.; Burgess, A.W.; Croxford, M.; Jones, I.; Langland, R.; Kosmider, S.; et al. Optimizing targeted therapeutic development: Analysis of a colorectal cancer patient population with the BRAFV600E mutation. Int. J. Cancer 2010, 128, 2075–2084. [Google Scholar] [CrossRef] [PubMed]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, R.B.; André, T.; Atreya, C.E.; Schellens, J.H.; Yoshino, T.; Bendell, J.C.; Hollebecque, A.; McRee, A.J.; Siena, S.; Middleton, G.; et al. Combined BRAF, EGFR, and MEK Inhibition in Patients with BRAFV600E-Mutant Colorectal Cancer. Cancer Discov. 2018, 8, 428–443. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef]
- Turdo, A.; Glaviano, A.; Pepe, G.; Calapà, F.; Raimondo, S.; Fiori, M.; Carbone, D.; Basilicata, M.; Di Sarno, V.; Ostacolo, C.; et al. Nobiletin and Xanthohumol Sensitize Colorectal Cancer Stem Cells to Standard Chemotherapy. Cancers 2021, 13, 3927. [Google Scholar] [CrossRef]
- Kotelevets, L.; Chastre, E. Rac1 Signaling: From Intestinal Homeostasis to Colorectal Cancer Metastasis. Cancers 2020, 12, 665. [Google Scholar] [CrossRef]
- Muellner, M.K.; Uras, I.Z.; Gapp, B.V.; Kerzendorfer, C.; Smida, M.; Lechtermann, H.; Craig-Mueller, N.; Colinge, J.; Duernberger, G.; Nijman, S.M.B. A chemical-genetic screen reveals a mechanism of resistance to PI3K inhibitors in cancer. Nat. Chem. Biol. 2011, 7, 787–793. [Google Scholar] [CrossRef]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET Bromodomain Inhibition as a Therapeutic Strategy to Target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef]
- Shorstova, T.; Foulkes, W.D.; Witcher, M. Achieving clinical success with BET inhibitors as anti-cancer agents. Br. J. Cancer 2021, 124, 1478–1490. [Google Scholar] [CrossRef]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef]
- Gregory, G.; Hogg, S.; Kats, L.; Vidacs, E.; Baker, A.J.; Gilan, O.; Lefebure, M.; Martin, B.P.; Dawson, M.A.; Johnstone, R.; et al. CDK9 inhibition by dinaciclib potently suppresses Mcl-1 to induce durable apoptotic responses in aggressive MYC-driven B-cell lymphoma in vivo. Leukemia 2014, 29, 1437–1441. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, M.T. Long path to MYC inhibition approaches clinical trials. Nat. Rev. Drug Discov. 2019. [Google Scholar] [CrossRef]
- Risom, T.; Wang, X.; Liang, J.; Zhang, X.; Pelz, C.; Campbell, L.G.; Eng, J.; Chin, K.; Farrington, C.; Narla, G.; et al. Deregulating MYC in a model of HER2+ breast cancer mimics human intertumoral heterogeneity. J. Clin. Investig. 2019, 130, 231–246. [Google Scholar] [CrossRef]
- Pickering, K.A.; Gilroy, K.; Cassidy, J.W.; Fey, S.K.; Najumudeen, A.K.; Zeiger, L.B.; Vincent, D.F.; Gay, D.M.; Johansson, J.; Fordham, R.P.; et al. A RAC-GEF network critical for early intestinal tumourigenesis. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Amodio, V.; Yaeger, R.; Arcella, P.; Cancelliere, C.; Lamba, S.; Lorenzato, A.; Arena, S.; Montone, M.; Mussolin, B.; Bian, Y.; et al. EGFR Blockade Reverts Resistance to KRASG12C Inhibition in Colorectal Cancer. Cancer Discov. 2020, 10, 1129–1139. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CD44v6 expression | ||||||||||||||||||||||||||||||||||||||
| MYC GCN | ||||||||||||||||||||||||||||||||||||||
| MSI status | NA | |||||||||||||||||||||||||||||||||||||
| Site | S | L | R | R | R | R | R | R | S | L | R | R | R | R | L | R | L | S | R | R | M | M | R | NA | L | R | R | R | R | S | S | R | S | R | S | L | L | M |
| BRAF | ||||||||||||||||||||||||||||||||||||||
| KRAS | ||||||||||||||||||||||||||||||||||||||
| CSphC # | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 19 | 20 | 21 | 22 | 23 | 24 | 25 | 27 | 29 | 30 | 33 | 37 | 44 | 49 | 52 | 53 | 55 | 56 | 57 | 58 | 59 |
| CD44v6 expression levels (%) | MYC/EIF2C1 ratio | MSI status | Mutation Type | |||||||||||||||||||||||||||||||||||
| high | >70% | MYC GCN < 4 | stable | wt | ||||||||||||||||||||||||||||||||||
| medium | 30–70% | MYC GCN ≥ 4 | low | Missense | ||||||||||||||||||||||||||||||||||
| low | <30% | high | ||||||||||||||||||||||||||||||||||||
| NA | ||||||||||||||||||||||||||||||||||||||

| Name | Type | Target |
|---|---|---|
| Vemurafenib | Small molecule | BRAFV600E |
| Cetuximab | Recombinant human/mouse chimeric IgG(1) monoclonal antibody | EGFR |
| Trastuzumab | Recombinant humanized monoclonal antibody | HER2 |
| BKM120 | dimorpholino pyrimidine derivative | class I PI3K |
| Dinaciclib | small molecule | CDK1/2/5/9 |
| Taselisib | small molecule | PIK3CA |
| Gene Name | Assay ID | Gene Name | Assay ID | Gene Name | Assay ID |
|---|---|---|---|---|---|
| AXIN1 | qHsaCID0010131 | ILKAP | qHsaCID0011777 | SERPINE1 | qHsaCID0006432 |
| AXIN2 | qHsaCID0017930 | ITGA5 | qHsaCID0021495 | SFRP1 | qHsaCID0015548 |
| BMP1 | qHsaCID0010875 | ITGAV | qHsaCID0006233 | SFRP4 | qHsaCED0043151 |
| BMP2 | qHsaCID0015400 | ITGB1 | qHsaCED0005248 | SMAD2 | qHsaCID0022031 |
| BMP7 | qHsaCID0011038 | JAG1 | qHsaCID0006831 | SMAD4 | qHsaCID0015670 |
| CCND1 | qHsaCID0013833 | KISS1 | qHsaCED0004976 | SNAI1 | qHsaCED0002998 |
| CD44 | qHsaCID0013679 | KISS1R | qHsaCID0011504 | SNAI2 | qHsaCID0011342 |
| CDH1 | qHsaCID0015365 | KREMEN1 | qHsaCED0047626 | SNAI3 | qHsaCED0047761 |
| CDH2 | qHsaCID0015189 | KRT14 | qHsaCED0047868 | SOX17 | qHsaCED0001884 |
| CDKN2A | qHsaCED0023006 | LRP5 | qHsaCED0045974 | SRC | qHsaCED0004489 |
| COL1A1 | qHsaCED0002181 | LRP6 | qHsaCID0010231 | STAT3 | qHsaCID0010912 |
| CTNNB1 | qHsaCID0010363 | MDM2 | qHsaCID0011000 | TGFB1 | qHsaCID0017026 |
| CXCL12 | qHsaCID0012398 | MET | qHsaCED0002004 | TGFB2 | qHsaCID0018360 |
| CXCR4 | qHsaCED0002020 | MMP2 | qHsaCID0015623 | TGFB3 | qHsaCID0022239 |
| DKK1 | qHsaCED0002060 | MMP3 | qHsaCID0006170 | TIMP1 | qHsaCID0007434 |
| DKK3 | qHsaCED0001115 | MMP7 | qHsaCID0011537 | TIMP2 | qHsaCID0022953 |
| EGFR | qHsaCID0007564 | MMP9 | qHsaCID0011597 | TIMP3 | qHsaCID0015238 |
| EPHB2 | qHsaCID0009881 | MMP10 | qHsaCED0046399 | TIMP4 | qHsaCID0016129 |
| ERBB2 | qHsaCID0012766 | MMP11 | qHsaCID0022136 | TP53 | qHsaCID0013658 |
| ERBB3 | qHsaCID0018397 | MMP13 | qHsaCID0008487 | TSHR | qHsaCID0009606 |
| ERBB4 | qHsaCID0017862 | MYC | qHsaCID0012921 | TWIST1 | qHsaCED0003856 |
| ESR1 | qHsaCED0033920 | NODAL | qHsaCID0006123 | VIM | qHsaCID0012604 |
| FGFBP1 | qHsaCID0015948 | NOTCH1 | qHsaCID0011825 | WNT3A | qHsaCED0045634 |
| FGFR4 | qHsaCED0045915 | OCLN | qHsaCED0038290 | WNT5A | qHsaCID0012240 |
| FN1 | qHsaCID0012349 | PDGFRB | qHsaCID0013272 | WNT5B | qHsaCID0038673 |
| FZD7 | qHsaCED0019290 | PTEN | qHsaCED0036796 | WNT7A | qHsaCID0018523 |
| GAPDH | qHsaCED0038674 | RAC1 | qHsaCED0001330 | WNT7B | qHsaCED0003528 |
| GSK3B | qHsaCID0010097 | RB1 | qHsaCID0007095 | WNT11 | qHsaCID0011927 |
| HGF | qHsaCID0011441 | RGS2 | qHsaCED0001744 | ZEB1 | qHsaCID0009210 |
| HPRT1 | qHsaCID0016375 | RHOA | qHsaCID0008947 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaggianesi, M.; Mangiapane, L.R.; Modica, C.; Pantina, V.D.; Porcelli, G.; Di Franco, S.; Lo Iacono, M.; D’Accardo, C.; Verona, F.; Pillitteri, I.; et al. Dual Inhibition of Myc Transcription and PI3K Activity Effectively Targets Colorectal Cancer Stem Cells. Cancers 2022, 14, 673. https://doi.org/10.3390/cancers14030673
Gaggianesi M, Mangiapane LR, Modica C, Pantina VD, Porcelli G, Di Franco S, Lo Iacono M, D’Accardo C, Verona F, Pillitteri I, et al. Dual Inhibition of Myc Transcription and PI3K Activity Effectively Targets Colorectal Cancer Stem Cells. Cancers. 2022; 14(3):673. https://doi.org/10.3390/cancers14030673
Chicago/Turabian StyleGaggianesi, Miriam, Laura Rosa Mangiapane, Chiara Modica, Vincenzo Davide Pantina, Gaetana Porcelli, Simone Di Franco, Melania Lo Iacono, Caterina D’Accardo, Francesco Verona, Irene Pillitteri, and et al. 2022. "Dual Inhibition of Myc Transcription and PI3K Activity Effectively Targets Colorectal Cancer Stem Cells" Cancers 14, no. 3: 673. https://doi.org/10.3390/cancers14030673
APA StyleGaggianesi, M., Mangiapane, L. R., Modica, C., Pantina, V. D., Porcelli, G., Di Franco, S., Lo Iacono, M., D’Accardo, C., Verona, F., Pillitteri, I., Turdo, A., Veschi, V., Brancato, O. R., Muratore, G., Pistone, G., Bongiorno, M. R., Todaro, M., De Maria, R., & Stassi, G. (2022). Dual Inhibition of Myc Transcription and PI3K Activity Effectively Targets Colorectal Cancer Stem Cells. Cancers, 14(3), 673. https://doi.org/10.3390/cancers14030673