
Genetic Landscape of Multistep Hepatocarcinogenesis

 ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Genetic Landscape of HCC
2.1. Comprehensive Genetic Analysis of HCC
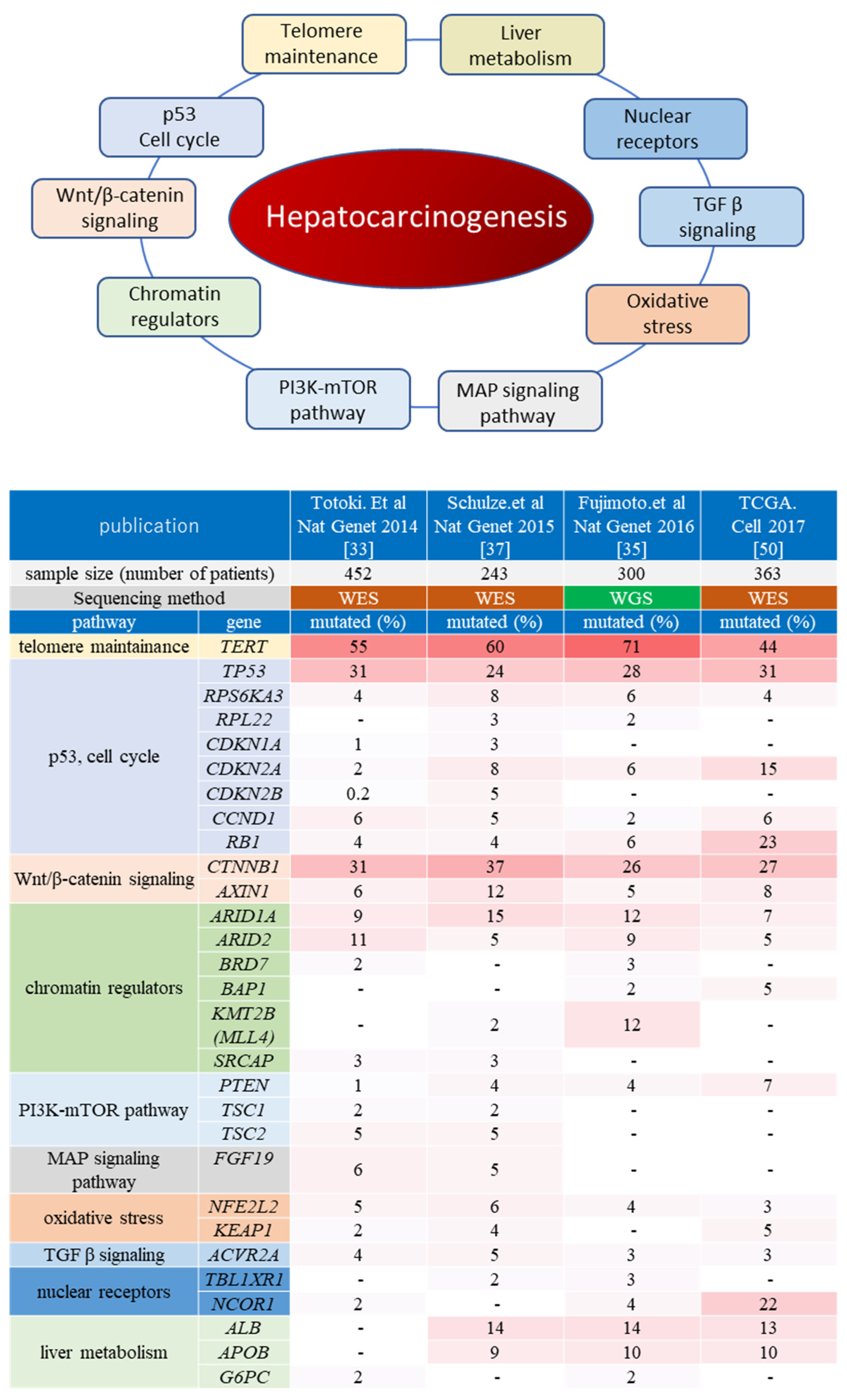
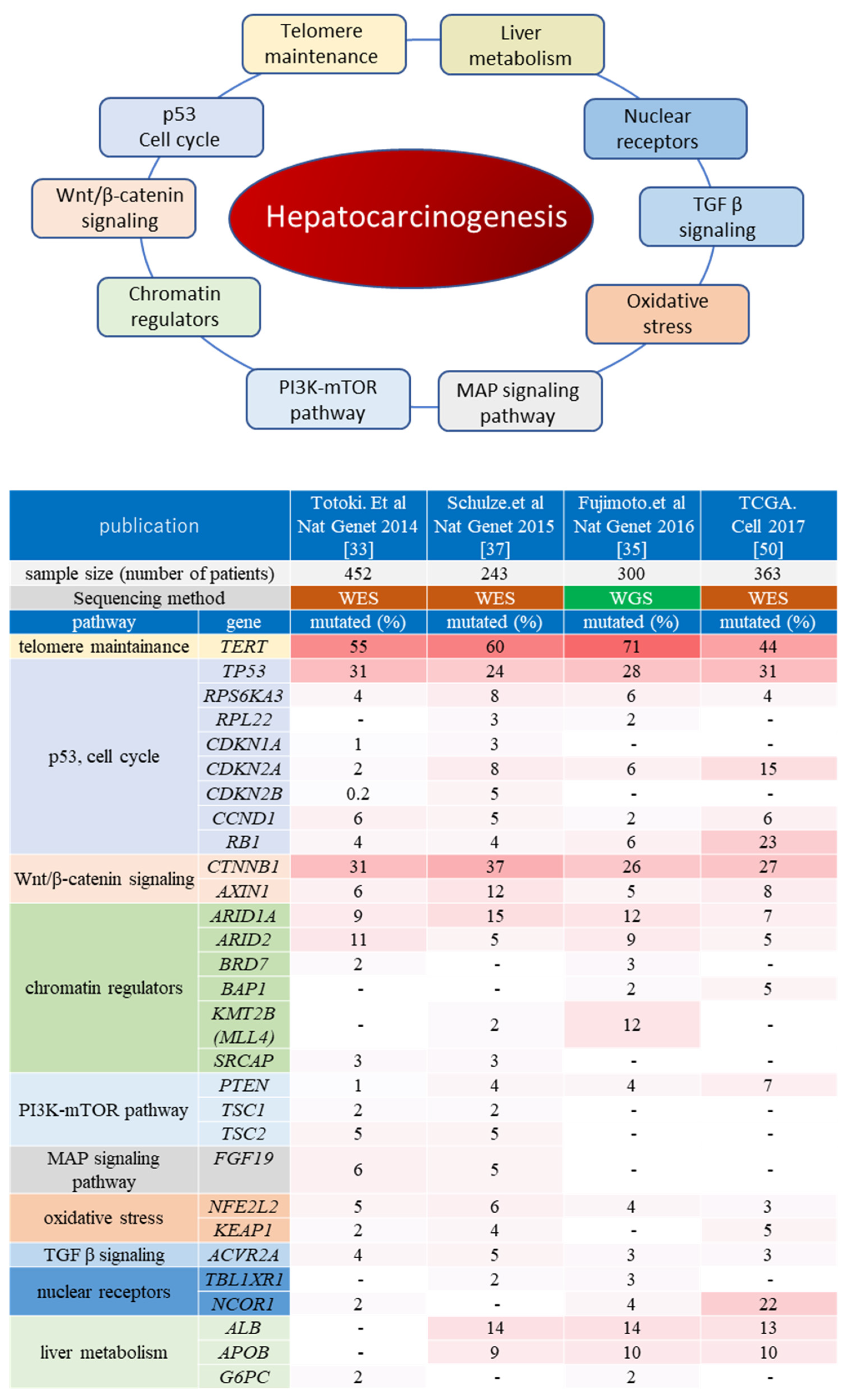
2.2. Molecular Pathways Associated with Hepatocarcinogenesis
2.2.1. Telomere Maintenance
2.2.2. Wnt/β-Catenin Pathway
2.2.3. p53/Cell Cycle
2.2.4. Chromatin Remodeling Factors
2.2.5. PI3K/Akt/mTOR and RAS/RAF/MAPK Pathways
2.2.6. Other Oncogenic Pathways in Hepatocarcinogenesis
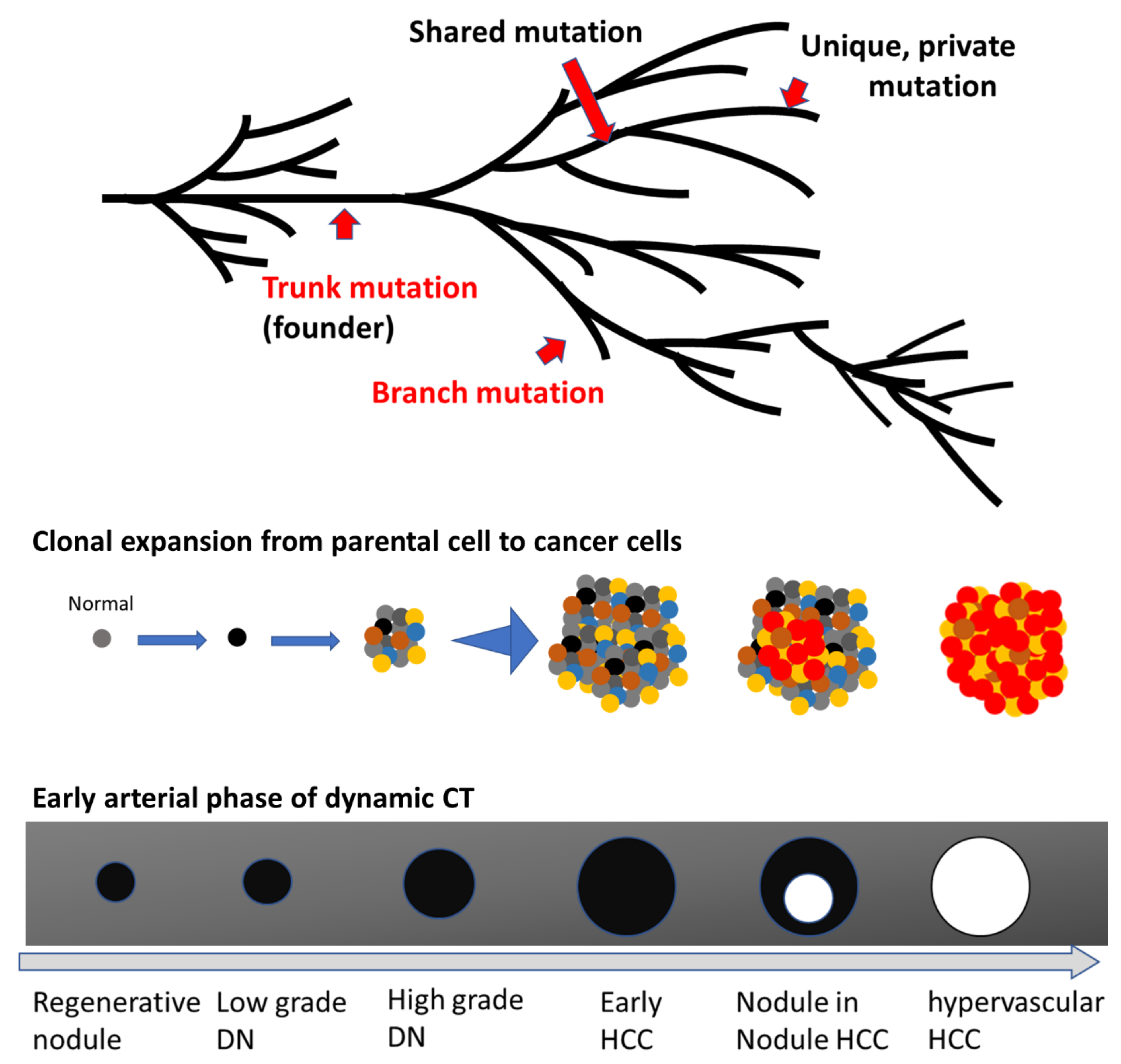
3. Intratumor Genetic Heterogeneity
3.1. Intratumoral Heterogeneity Revealed by NGS
3.2. Intratumoral Heterogeneity of HCC
4. Genetic Alterations in Early Hepatocarcinogenesis
4.1. Genetic Landscape of Early HCC
4.2. Genetic Landscape of Precancerous Liver Tissues
5. Multistep Acquisition of Genetic Aberrations during Hepatocarcinogenesis
5.1. The Significance of Comprehensive Analysis Using Clinical and Genetic Data
5.2. Whole-Genome Mutational Analysis Using Nodule-in-Nodule HCCs
5.3. Genetic Profiles with Tumor Progression
5.4. Multi-Omics Analyses of Multistep Hepatocarcinogenesis
6. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, H.; Takai, A.; Inuzuka, T.; Marusawa, H. Genetic basis of hepatitis virus-associated hepatocellular carcinoma: Linkage between infection, inflammation, and tumorigenesis. J. Gastroenterol. 2017, 52, 26–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Serag, H.B. Hepatocellular carcinoma. N. Engl. J. Med. 2011, 365, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012, 142, 1264–1273.e1261. [Google Scholar] [CrossRef] [Green Version]
- Bruix, J.; Sherman, M. Management of hepatocellular carcinoma: An update. Hepatology 2011, 53, 1020–1022. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Han, G.; Finn, R.S.; Poon, R.T.; Blanc, J.F.; Yan, L.; Yang, J.; Lu, L.; Tak, W.Y.; Yu, X.; et al. Brivanib as adjuvant therapy to transarterial chemoembolization in patients with hepatocellular carcinoma: A randomized phase III trial. Hepatology 2014, 60, 1697–1707. [Google Scholar] [CrossRef]
- Lencioni, R.; Llovet, J.M.; Han, G.; Tak, W.Y.; Yang, J.; Guglielmi, A.; Paik, S.W.; Reig, M.; Kim, D.Y.; Chau, G.Y.; et al. Sorafenib or placebo plus TACE with doxorubicin-eluting beads for intermediate stage HCC: The SPACE trial. J. Hepatol. 2016, 64, 1090–1098. [Google Scholar] [CrossRef] [Green Version]
- Mazzaferro, V.; Regalia, E.; Doci, R.; Andreola, S.; Pulvirenti, A.; Bozzetti, F.; Montalto, F.; Ammatuna, M.; Morabito, A.; Gennari, L. Liver transplantation for the treatment of small hepatocellular carcinomas in patients with cirrhosis. N. Engl. J. Med. 1996, 334, 693–699. [Google Scholar] [CrossRef]
- Ng, K.K.C.; Chok, K.S.H.; Chan, A.C.Y.; Cheung, T.T.; Wong, T.C.L.; Fung, J.Y.Y.; Yuen, J.; Poon, R.T.P.; Fan, S.T.; Lo, C.M. Randomized clinical trial of hepatic resection versus radiofrequency ablation for early-stage hepatocellular carcinoma. Br. J. Surg. 2017, 104, 1775–1784. [Google Scholar] [CrossRef] [PubMed]
- Shiina, S.; Teratani, T.; Obi, S.; Sato, S.; Tateishi, R.; Fujishima, T.; Ishikawa, T.; Koike, Y.; Yoshida, H.; Kawabe, T.; et al. A randomized controlled trial of radiofrequency ablation with ethanol injection for small hepatocellular carcinoma. Gastroenterology 2005, 129, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.D.; Tang, Z.Y.; Yang, B.H.; Lin, Z.Y.; Ma, Z.C.; Ye, S.L.; Wu, Z.Q.; Fan, J.; Qin, L.X.; Zheng, B.H. Experience of 1000 patients who underwent hepatectomy for small hepatocellular carcinoma. Cancer 2001, 91, 1479–1486. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kang, Y.K.; Yen, C.J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.; Finn, R.S.; Qin, S.; Han, K.; Ikeda, K.; Piscaglia, F.; Baron, A.D.; Park, J.W.; Han, G.; Jassem, J.; et al. Phase III trial of lenvatinib (LEN) vs. sorafenib (SOR) in first-line treatment of patients (pts) with unresectable hepatocellular carcinoma (uHCC). J. Clin. Oncol. 2017, 35, 116. [Google Scholar] [CrossRef]
- Kudo, M. Lenvatinib in Advanced Hepatocellular Carcinoma. Liver Cancer 2017, 6, 253–263. [Google Scholar] [CrossRef]
- Kudo, M. A New Era of Systemic Therapy for Hepatocellular Carcinoma with Regorafenib and Lenvatinib. Liver Cancer 2017, 6, 177–184. [Google Scholar] [CrossRef]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Llovet, J.M.; Villanueva, A.; Lachenmayer, A.; Finn, R.S. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat. Rev. Clin. Oncol. 2015, 12, 408–424. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M. Focal gains of VEGFA: Candidate predictors of sorafenib response in hepatocellular carcinoma. Cochrane Database Syst. Rev. 2018, 12, CD013188. [Google Scholar] [CrossRef]
- Horwitz, E.; Stein, I.; Andreozzi, M.; Nemeth, J.; Shoham, A.; Pappo, O.; Schweitzer, N.; Tornillo, L.; Kanarek, N.; Quagliata, L.; et al. Human and mouse VEGFA-amplified hepatocellular carcinomas are highly sensitive to sorafenib treatment. Cancer Discov. 2014, 4, 730–743. [Google Scholar] [CrossRef] [Green Version]
- Arao, T.; Ueshima, K.; Matsumoto, K.; Nagai, T.; Kimura, H.; Hagiwara, S.; Sakurai, T.; Haji, S.; Kanazawa, A.; Hidaka, H.; et al. FGF3/FGF4 amplification and multiple lung metastases in responders to sorafenib in hepatocellular carcinoma. Hepatology 2013, 57, 1407–1415. [Google Scholar] [CrossRef] [Green Version]
- Takeda, H.; Nishikawa, H.; Osaki, Y.; Tsuchiya, K.; Joko, K.; Ogawa, C.; Taniguchi, H.; Orito, E.; Uchida, Y.; Izumi, N.; et al. Clinical features associated with radiological response to sorafenib in unresectable hepatocellular carcinoma: A large multicenter study in Japan. Liver Int. Off. J. Int. Assoc. Study Liver 2015, 35, 1581–1589. [Google Scholar] [CrossRef] [PubMed]
- Kaibori, M.; Sakai, K.; Ishizaki, M.; Matsushima, H.; De Velasco, M.A.; Matsui, K.; Iida, H.; Kitade, H.; Kwon, A.H.; Nagano, H.; et al. Increased FGF19 copy number is frequently detected in hepatocellular carcinoma with a complete response after sorafenib treatment. Oncotarget 2016, 7, 49091–49098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudo, M. Recent Advances in Systemic Therapy for Hepatocellular Carcinoma in an Aging Society: 2020 Update. Liver Cancer 2020, 9, 640–662. [Google Scholar] [CrossRef]
- Llovet, J.M.; Pena, C.E.; Lathia, C.D.; Shan, M.; Meinhardt, G.; Bruix, J.; Group, S.I.S. Plasma biomarkers as predictors of outcome in patients with advanced hepatocellular carcinoma. Clin. Cancer Res. 2012, 18, 2290–2300. [Google Scholar] [CrossRef] [Green Version]
- Kudo, M. Biomarkers and Personalized Sorafenib Therapy. Liver Cancer 2014, 3, 399–404. [Google Scholar] [CrossRef]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.C.; Llovet, J.M. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 2015, 149, 1226–1239.e1224. [Google Scholar] [CrossRef] [Green Version]
- Totoki, Y.; Tatsuno, K.; Covington, K.R.; Ueda, H.; Creighton, C.J.; Kato, M.; Tsuji, S.; Donehower, L.A.; Slagle, B.L.; Nakamura, H.; et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet. 2014, 46, 1267–1273. [Google Scholar] [CrossRef]
- Fujimoto, A.; Furuta, M.; Shiraishi, Y.; Gotoh, K.; Kawakami, Y.; Arihiro, K.; Nakamura, T.; Ueno, M.; Ariizumi, S.; Nguyen, H.H.; et al. Whole-genome mutational landscape of liver cancers displaying biliary phenotype reveals hepatitis impact and molecular diversity. Nat. Commun. 2015, 6, 6120. [Google Scholar] [CrossRef]
- Fujimoto, A.; Furuta, M.; Totoki, Y.; Tsunoda, T.; Kato, M.; Shiraishi, Y.; Tanaka, H.; Taniguchi, H.; Kawakami, Y.; Ueno, M.; et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat. Genet. 2016, 48, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Deng, Q.; Wang, Q.; Li, K.Y.; Dai, J.H.; Li, N.; Zhu, Z.D.; Zhou, B.; Liu, X.Y.; Liu, R.F.; et al. Exome sequencing of hepatitis B virus-associated hepatocellular carcinoma. Nat. Genet. 2012, 44, 1117–1121. [Google Scholar] [CrossRef]
- Schulze, K.; Imbeaud, S.; Letouze, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Totoki, Y.; Tatsuno, K.; Yamamoto, S.; Arai, Y.; Hosoda, F.; Ishikawa, S.; Tsutsumi, S.; Sonoda, K.; Totsuka, H.; Shirakihara, T.; et al. High-resolution characterization of a hepatocellular carcinoma genome. Nat. Genet. 2011, 43, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Marusawa, H.; Endo, Y.; Chiba, T. Inflammation-mediated genomic instability: Roles of activation-induced cytidine deaminase in carcinogenesis. Cancer Sci. 2012, 103, 1201–1206. [Google Scholar] [CrossRef]
- Levrero, M.; Zucman-Rossi, J. Mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatol. 2016, 64, S84–S101. [Google Scholar] [CrossRef]
- Brunner, S.F.; Roberts, N.D.; Wylie, L.A.; Moore, L.; Aitken, S.J.; Davies, S.E.; Sanders, M.A.; Ellis, P.; Alder, C.; Hooks, Y.; et al. Somatic mutations and clonal dynamics in healthy and cirrhotic human liver. Nature 2019, 574, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Lu, T.; Jia, Y.; Luo, X.; Gopal, P.; Li, L.; Odewole, M.; Renteria, V.; Singal, A.G.; Jang, Y.; et al. Somatic Mutations Increase Hepatic Clonal Fitness and Regeneration in Chronic Liver Disease. Cell 2019, 177, 608–621.e612. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.K.; Takeda, H.; Takai, A.; Matsumoto, T.; Kakiuchi, N.; Yokoyama, A.; Yoshida, K.; Kaido, T.; Uemoto, S.; Minamiguchi, S.; et al. Comprehensive analysis of genetic aberrations linked to tumorigenesis in regenerative nodules of liver cirrhosis. J. Gastroenterol. 2019, 54, 628–640. [Google Scholar] [CrossRef] [PubMed]
- Midorikawa, Y.; Yamamoto, S.; Tsuji, S.; Kamimura, N.; Ishikawa, S.; Igarashi, H.; Makuuchi, M.; Kokudo, N.; Sugimura, H.; Aburatani, H. Allelic imbalances and homozygous deletion on 8p23.2 for stepwise progression of hepatocarcinogenesis. Hepatology 2009, 49, 513–522. [Google Scholar] [CrossRef]
- Midorikawa, Y.; Yamamoto, S.; Tatsuno, K.; Renard-Guillet, C.; Tsuji, S.; Hayashi, A.; Ueda, H.; Fukuda, S.; Fujita, T.; Katoh, H.; et al. Accumulation of Molecular Aberrations Distinctive to Hepatocellular Carcinoma Progression. Cancer Res. 2020, 80, 3810–3819. [Google Scholar] [CrossRef] [PubMed]
- Takeda, H.; Takai, A.; Kumagai, K.; Iguchi, E.; Arasawa, S.; Eso, Y.; Shimizu, T.; Ueda, Y.; Taura, K.; Uemoto, S.; et al. Multiregional whole-genome sequencing of hepatocellular carcinoma with nodule-in-nodule appearance reveals stepwise cancer evolution. J. Pathol. 2020, 252, 398–410. [Google Scholar] [CrossRef]
- Nault, J.C.; Calderaro, J.; Di Tommaso, L.; Balabaud, C.; Zafrani, E.S.; Bioulac-Sage, P.; Roncalli, M.; Zucman-Rossi, J. Telomerase reverse transcriptase promoter mutation is an early somatic genetic alteration in the transformation of premalignant nodules in hepatocellular carcinoma on cirrhosis. Hepatology 2014, 60, 1983–1992. [Google Scholar] [CrossRef]
- Hudson, T.J.; Anderson, W.; Artez, A.; Barker, A.D.; Bell, C.; Bernabe, R.R.; Bhan, M.K.; Calvo, F.; Eerola, I.; Gerhard, D.S.; et al. International network of cancer genome projects. Nature 2010, 464, 993–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, I.T.P.-C.A.o.W.G. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e1323. [CrossRef] [Green Version]
- Nault, J.C.; Datta, S.; Imbeaud, S.; Franconi, A.; Mallet, M.; Couchy, G.; Letouze, E.; Pilati, C.; Verret, B.; Blanc, J.F.; et al. Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas. Nat. Genet. 2015, 47, 1187–1193. [Google Scholar] [CrossRef]
- Fujimoto, A.; Totoki, Y.; Abe, T.; Boroevich, K.A.; Hosoda, F.; Nguyen, H.H.; Aoki, M.; Hosono, N.; Kubo, M.; Miya, F.; et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat. Genet. 2012, 44, 760–764. [Google Scholar] [CrossRef]
- Nault, J.C.; Mallet, M.; Pilati, C.; Calderaro, J.; Bioulac-Sage, P.; Laurent, C.; Laurent, A.; Cherqui, D.; Balabaud, C.; Zucman-Rossi, J. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat. Commun. 2013, 4, 2218. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Furuta, M.; Ueno, M.; Fujimoto, A.; Hayami, S.; Yasukawa, S.; Kojima, F.; Arihiro, K.; Kawakami, Y.; Wardell, C.P.; Shiraishi, Y.; et al. Whole genome sequencing discriminates hepatocellular carcinoma with intrahepatic metastasis from multi-centric tumors. J. Hepatol. 2016, 66, 363–373. [Google Scholar] [CrossRef]
- Shibata, T.; Aburatani, H. Exploration of liver cancer genomes. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Yoo, J.E.; Cho, J.Y.; Oh, B.K.; Yoon, Y.S.; Han, H.S.; Lee, H.S.; Jang, J.J.; Jeong, S.H.; Kim, J.W.; et al. Telomere length, TERT and shelterin complex proteins in hepatocellular carcinomas expressing “stemness”-related markers. J. Hepatol. 2013, 59, 746–752. [Google Scholar] [CrossRef]
- Castelo-Branco, P.; Choufani, S.; Mack, S.; Gallagher, D.; Zhang, C.; Lipman, T.; Zhukova, N.; Walker, E.J.; Martin, D.; Merino, D.; et al. Methylation of the TERT promoter and risk stratification of childhood brain tumours: An integrative genomic and molecular study. Lancet Oncol. 2013, 14, 534–542. [Google Scholar] [CrossRef]
- Lee, D.D.; Leao, R.; Komosa, M.; Gallo, M.; Zhang, C.H.; Lipman, T.; Remke, M.; Heidari, A.; Nunes, N.M.; Apolonio, J.D.; et al. DNA hypermethylation within TERT promoter upregulates TERT expression in cancer. J. Clin. Investig. 2019, 129, 223–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Weng, X.; Ye, J.; He, L.; Zhou, D.; Liu, Y. Promoter hypermethylation of TERT is associated with hepatocellular carcinoma in the Han Chinese population. Clin. Res. Hepatol. Gastroenterol. 2015, 39, 600–609. [Google Scholar] [CrossRef]
- Satoh, S.; Daigo, Y.; Furukawa, Y.; Kato, T.; Miwa, N.; Nishiwaki, T.; Kawasoe, T.; Ishiguro, H.; Fujita, M.; Tokino, T.; et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat. Genet. 2000, 24, 245–250. [Google Scholar] [CrossRef] [PubMed]
- De La Coste, A.; Romagnolo, B.; Billuart, P.; Renard, C.A.; Buendia, M.A.; Soubrane, O.; Fabre, M.; Chelly, J.; Beldjord, C.; Kahn, A.; et al. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc. Natl. Acad. Sci. USA 1998, 95, 8847–8851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abitbol, S.; Dahmani, R.; Coulouarn, C.; Ragazzon, B.; Mlecnik, B.; Senni, N.; Savall, M.; Bossard, P.; Sohier, P.; Drouet, V.; et al. AXIN deficiency in human and mouse hepatocytes induces hepatocellular carcinoma in the absence of β-catenin activation. J. Hepatol. 2018, 68, 1203–1213. [Google Scholar] [CrossRef] [PubMed]
- Désert, R.; Rohart, F.; Canal, F.; Sicard, M.; Desille, M.; Renaud, S.; Turlin, B.; Bellaud, P.; Perret, C.; Clément, B.; et al. Human hepatocellular carcinomas with a periportal phenotype have the lowest potential for early recurrence after curative resection. Hepatology 2017, 66, 1502–1518. [Google Scholar] [CrossRef]
- Boyault, S.; Rickman, D.S.; de Reyniès, A.; Balabaud, C.; Rebouissou, S.; Jeannot, E.; Hérault, A.; Saric, J.; Belghiti, J.; Franco, D.; et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology 2007, 45, 42–52. [Google Scholar] [CrossRef] [Green Version]
- Désert, R.; Nieto, N.; Musso, O. Dimensions of hepatocellular carcinoma phenotypic diversity. World J. Gastroenterol. 2018, 24, 4536–4547. [Google Scholar] [CrossRef]
- Bayard, Q.; Meunier, L.; Peneau, C.; Renault, V.; Shinde, J.; Nault, J.C.; Mami, I.; Couchy, G.; Amaddeo, G.; Tubacher, E.; et al. Cyclin A2/E1 activation defines a hepatocellular carcinoma subclass with a rearrangement signature of replication stress. Nat. Commun. Commun. 2018, 9, 5235. [Google Scholar] [CrossRef] [Green Version]
- Hoshida, Y.; Nijman, S.M.; Kobayashi, M.; Chan, J.A.; Brunet, J.P.; Chiang, D.Y.; Villanueva, A.; Newell, P.; Ikeda, K.; Hashimoto, M.; et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009, 69, 7385–7392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurent-Puig, P.; Legoix, P.; Bluteau, O.; Belghiti, J.; Franco, D.; Binot, F.; Monges, G.; Thomas, G.; Bioulac-Sage, P.; Zucman-Rossi, J. Genetic alterations associated with hepatocellular carcinomas define distinct pathways of hepatocarcinogenesis. Gastroenterology 2001, 120, 1763–1773. [Google Scholar] [CrossRef] [Green Version]
- Hussain, S.P.; Schwank, J.; Staib, F.; Wang, X.W.; Harris, C.C. TP53 mutations and hepatocellular carcinoma: Insights into the etiology and pathogenesis of liver cancer. Oncogene 2007, 26, 2166–2176. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.; Li, M.; Parsons, D.W.; Zhang, X.; Wesseling, J.; Kristel, P.; Schmidt, M.K.; Markowitz, S.; Yan, H.; Bigner, D.; et al. Somatic mutations in the chromatin remodeling gene ARID1A occur in several tumor types. Hum. Mutat. 2012, 33, 100–103. [Google Scholar] [CrossRef] [Green Version]
- Ji, X.; Zhang, Q.; Du, Y.; Liu, W.; Li, Z.; Hou, X.; Cao, G. Somatic mutations, viral integration and epigenetic modification in the evolution of hepatitis B virus-induced hepatocellular carcinoma. Curr. Genom. 2014, 15, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Sung, W.K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers. 2021, 7, 6. [Google Scholar] [CrossRef]
- Chiang, D.Y.; Villanueva, A.; Hoshida, Y.; Peix, J.; Newell, P.; Minguez, B.; LeBlanc, A.C.; Donovan, D.J.; Thung, S.N.; Sole, M.; et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res. 2008, 68, 6779–6788. [Google Scholar] [CrossRef] [Green Version]
- Shuai, S.; Suzuki, H.; Diaz-Navarro, A.; Nadeu, F.; Kumar, S.A.; Gutierrez-Fernandez, A.; Delgado, J.; Pinyol, M.; López-Otín, C.; Puente, X.S.; et al. The U1 spliceosomal RNA is recurrently mutated in multiple cancers. Nature 2019, 574, 712–716. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Zhai, W.; Lim, T.K.; Zhang, T.; Phang, S.T.; Tiang, Z.; Guan, P.; Ng, M.H.; Lim, J.Q.; Yao, F.; Li, Z.; et al. The spatial organization of intra-tumour heterogeneity and evolutionary trajectories of metastases in hepatocellular carcinoma. Nat. Commun. 2017, 8, 4565. [Google Scholar] [CrossRef] [Green Version]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.C.; Mayakonda, A.; Dinh, H.Q.; Huang, P.; Lin, L.; Liu, X.; Ding, L.W.; Wang, J.; Berman, B.; Song, E.; et al. Genomic and epigenomic heterogeneity of hepatocellular carcinoma. Cancer Res. 2017, 77, 2255–2265. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Lee, I.H.; Cho, H.J.; Park, C.K.; Jung, Y.S.; Kim, Y.; Nam, S.H.; Kim, B.S.; Johnson, M.D.; Kong, D.S.; et al. Spatiotemporal Evolution of the Primary Glioblastoma Genome. Cancer Cell 2015, 28, 318–328. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.K.; Wang, J.; Sa, J.K.; Ladewig, E.; Lee, H.O.; Lee, I.H.; Kang, H.J.; Rosenbloom, D.S.; Camara, P.G.; Liu, Z.; et al. Spatiotemporal genomic architecture informs precision oncology in glioblastoma. Nat. Genet. 2017; 49, 594–599. [Google Scholar] [CrossRef]
- Makohon-Moore, A.P.; Zhang, M.; Reiter, J.G.; Bozic, I.; Allen, B.; Kundu, D.; Chatterjee, K.; Wong, F.; Jiao, Y.; Kohutek, Z.A.; et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat. Genet. 2017, 49, 358–366. [Google Scholar] [CrossRef]
- Kumar, A.; Coleman, I.; Morrissey, C.; Zhang, X.; True, L.D.; Gulati, R.; Etzioni, R.; Bolouri, H.; Montgomery, B.; White, T.; et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat. Med. 2016, 22, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Uchi, R.; Takahashi, Y.; Niida, A.; Shimamura, T.; Hirata, H.; Sugimachi, K.; Sawada, G.; Iwaya, T.; Kurashige, J.; Shinden, Y.; et al. Integrated Multiregional Analysis Proposing a New Model of Colorectal Cancer Evolution. PLoS Genet. 2016, 12, e1005778. [Google Scholar] [CrossRef] [PubMed]
- Xue, R.; Li, R.; Guo, H.; Guo, L.; Su, Z.; Ni, X.; Qi, L.; Zhang, T.; Li, Q.; Zhang, Z.; et al. Variable Intra-Tumor Genomic Heterogeneity of Multiple Lesions in Patients With Hepatocellular Carcinoma. Gastroenterology 2016, 150, 998–1008. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Wu, L.; Lin, J.; Han, L.; Bian, J.; Wu, Y.; Robson, S.C.; Xue, L.; Ge, Y.; Sang, X.; et al. Whole-exome sequencing reveals the origin and evolution of hepato-cholangiocarcinoma. Nat. Commun. 2018, 9, 894. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lou, Y.; Yang, J.; Wang, J.; Feng, J.; Zhao, Y.; Wang, L.; Huang, X.; Fu, Q.; Ye, M.; et al. Integrated multiomic analysis reveals comprehensive tumour heterogeneity and novel immunophenotypic classification in hepatocellular carcinomas. Gut 2019, 68, 2019–2031. [Google Scholar] [CrossRef]
- Jeng, K.S.; Chang, C.F.; Jeng, W.J.; Sheen, I.S.; Jeng, C.J. Heterogeneity of hepatocellular carcinoma contributes to cancer progression. Crit. Rev. Oncol. Hematol. 2015, 94, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Guo, H.; Cao, C.; Li, X.; Hu, B.; Zhu, P.; Wu, X.; Wen, L.; Tang, F.; Huang, Y.; et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 2016, 26, 304–319. [Google Scholar] [CrossRef] [PubMed]
- Aihara, T.; Noguchi, S.; Sasaki, Y.; Nakano, H.; Imaoka, S. Clonal analysis of regenerative nodules in hepatitis C virus-induced liver cirrhosis. Gastroenterology 1994, 107, 1805–1811. [Google Scholar] [CrossRef]
- Gong, L.; Li, Y.H.; Su, Q.; Chu, X.; Zhang, W. Clonality of nodular lesions in liver cirrhosis and chromosomal abnormalities in monoclonal nodules of altered hepatocytes. Histopathology 2010, 56, 589–599. [Google Scholar] [CrossRef]
- Lin, W.R.; Lim, S.N.; McDonald, S.A.; Graham, T.; Wright, V.L.; Peplow, C.L.; Humphries, A.; Kocher, H.M.; Wright, N.A.; Dhillon, A.P.; et al. The histogenesis of regenerative nodules in human liver cirrhosis. Hepatology 2010, 51, 1017–1026. [Google Scholar] [CrossRef]
- Ochiai, T.; Urata, Y.; Yamano, T.; Yamagishi, H.; Ashihara, T. Clonal expansion in evolution of chronic hepatitis to hepatocellular carcinoma as seen at an X-chromosome locus. Hepatology 2000, 31, 615–621. [Google Scholar] [CrossRef]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, A.; Kakiuchi, N.; Yoshizato, T.; Nannya, Y.; Suzuki, H.; Takeuchi, Y.; Shiozawa, Y.; Sato, Y.; Aoki, K.; Kim, S.K.; et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature 2019, 565, 312–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuma, M.; Sakamoto, M.; Yamazaki, K.; Ohta, T.; Ohki, M.; Asaka, M.; Hirohashi, S. Expression profiling in multistage hepatocarcinogenesis: Identification of HSP70 as a molecular marker of early hepatocellular carcinoma. Hepatology 2003, 37, 198–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nault, J.C.; Martin, Y.; Caruso, S.; Hirsch, T.Z.; Bayard, Q.; Calderaro, J.; Charpy, C.; Copie-Bergman, C.; Ziol, M.; Bioulac-Sage, P.; et al. Clinical Impact of Genomic Diversity From Early to Advanced Hepatocellular Carcinoma. Hepatology 2020, 71, 164–182. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Treatment | Publications | Study Name | Objectives | 5-Year Survival Rate | 5-Year Recurrence Free Rate | OS | PFS (*;TTP) | ORR mRESICT/RECIST | |
|---|---|---|---|---|---|---|---|---|---|
| Curative | RFA | Siina et al., 2005 [14] | NA | Size < 3 cm and Tumor number < 3 | 74% (4-year) | NA | NA | NA | NA |
| Ng et al., 2017 [13] | NA | Size < 3 cm and Tumor number < 3 | 66.4% | 18.3% | NA | NA | NA | ||
| Hepatectomy | Ng et al., 2017 [13] | NA | Size < 3 cm and Tumor number < 3 | 66.5% | 28.7% | NA | NA | NA | |
| Zhou et al., 2001 [15] | NA | Small HCC Large HCC | 62.7%/37.1% | NA | NA | NA | NA | ||
| Liver transplantation | Mazzaferro, et al. [12] | NA | Within Milan criteria | 92% (4-year) | 85% (4-year) | NA | NA | NA | |
| Non-curative | TACE | Lencioni et al., 2016 [11] | SPACE | Intermediate | NA | NA | NR | 5.5 * | NA |
| Kudo et al., 2014 [10] | BRISK-TA | Intermediate | NA | NA | 26.1 | 4.9 * | 42% | ||
| Sorafenib | Llovet et al., 2008 [17] | SHARP | Advanced: Unresectable HCC (1st line) | NA | NA | 10.7–13.4 | 3.7–4.3 | NA/2% | |
| Kudo et al., 2018 [20] | REFLECT | ||||||||
| Finn et al., 2020 [9] | IMbrave150 | ||||||||
| Lenvatinib | Kudo et al., 2018 [20] | REFLECT | Unresectable HCC (1st line) | NA | NA | 13.6 | 7.4 | 24.1%/18.8% | |
| Regorafenib | Bruix et al., 2017 [8] | RESORCE | Unresectable HCC (2nd line) | NA | NA | 10.6 | 3.1 | 11%/7% | |
| Cabozantinib | Abou-Alfa et al., 2018 [7] | CELESTIAL | Unresectable HCC (2nd line) | NA | NA | 10.2 | 5.2 | NA/4% | |
| Ramucirumab | Zhu et al., 2019 [16] | REACH-2 | Unresectable HCC (2nd line) | NA | NA | 8.5 | 2.8 | NA/5% | |
| Atezolizumab plus bevacizumab | Finn et al., 2020 [9] | IMbrave150 | Unresectable HCC (1st line) | NA | NA | 19.2 | 6.8 | 35.4%/29.8% |
| Author, Year | Methodology | Genetic Characteristics According to Each Phase through Multistep Hepatocarcinogenesis from Precancerous Liver Tissues to Advanced HCCs | |||||
|---|---|---|---|---|---|---|---|
| Normal Liver | Cirrhosis | Dysplastic Nodule | Early HCC NIN-HCC Outer | Classical (Progressed) HCC NIN-HCC Inner | Advanced HCC | ||
| Brunner et al., 2019 [41] | WGS | CNV and SV: rare | TERTp mut not detected Mut in ACVR2A, ARID2, ARID1A, TSC2, ALB, etc. CNV and SV can be detected chromothripsis (3/9) | - | - | - | - |
| Zhu et al., 2019 [42] | WES, target seq | (F0 samples) low mutational burden HCC-related mut: not detected | PKD1, KMT2D PPARGC1B, ARID1A mut. liver protective mutation | - | - | - | - |
| Kim et al., 2019 [43] | WES, target seq | - | TERTp mut not detected (0/205) ARID1A(7/205), ARID2(4/205) TP53(6/205), ATM(4/205) etc. no CNV detected | - | - | TERTp mut (80%, 8/10) | - |
| Midorikawa et al., 2009 [44] | CGH array | - | 1q gain, 1p LOH and 18p LOH: not detected | - | (NIN-HCC outer and eHCC) Gain on 1q (12/19) LOH on 1p and 18p (2/19) | (NIN-HCC inner and pHCC) Gain on 5q11.1-35.3 and 8q11.1-24.3 (14/25 and 16/25) LOH on 4q11-34.3 and 8p11.21-23.3 (10/25 and 12/25) | - |
| Midorikawa et al., 2020 [45] | WES, RNAseq, Methylation array | - | - | - | (eHCCs, N = 52) TERTp mut (65.3%) TERT overexpression (84.6%) TP53/RB1 mut (34.6%) WNT mut (23.0%) | (overt or progressed HCC, n = 108) TERTp mut (58.3%) TERT overexpression (74.0%) TP53/RB1 mut (42.5%) WNT mut (43.5%) CDH inactivation, EZH2 upregulation, 4q loss, 16q loss etc. (categorized into 4 groups according to the methylation status) | - |
| Takeda et al., 2020 [46] | WGS, target seq, CNV array | - | - | - | (NIN-HCC outer) TERT overexpression (8/8) due to HBV integration/gain, translocation, methylation and promoter mutation SV, chromothripsis detected | (NIN-HCC inner) TERT overexpression (4/5) due to the genetic aberrations as same as eHCC Wnt, MAPK, and mTOR associated gene mutations (case specific) | - |
| Nault et al., 2014 [47] | Sanger seq. | - | TERTp mut; not detected (0%, 0/172) | TERTp mut (6–19%, 5/48) no other mutations | TERTp mut (61%, 14/23) no other mutations | TERTp mut (42%, 7/17) other HCC-related mut (CTNNB1, TP53) (28%, 2/7) | TERTp mut (64%, 60/94) other HCC-related mut (CTNNB1, TP53) (56%, 20/35) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takeda, H.; Takai, A.; Eso, Y.; Takahashi, K.; Marusawa, H.; Seno, H. Genetic Landscape of Multistep Hepatocarcinogenesis. Cancers 2022, 14, 568. https://doi.org/10.3390/cancers14030568
Takeda H, Takai A, Eso Y, Takahashi K, Marusawa H, Seno H. Genetic Landscape of Multistep Hepatocarcinogenesis. Cancers. 2022; 14(3):568. https://doi.org/10.3390/cancers14030568
Chicago/Turabian StyleTakeda, Haruhiko, Atsushi Takai, Yuji Eso, Ken Takahashi, Hiroyuki Marusawa, and Hiroshi Seno. 2022. "Genetic Landscape of Multistep Hepatocarcinogenesis" Cancers 14, no. 3: 568. https://doi.org/10.3390/cancers14030568
APA StyleTakeda, H., Takai, A., Eso, Y., Takahashi, K., Marusawa, H., & Seno, H. (2022). Genetic Landscape of Multistep Hepatocarcinogenesis. Cancers, 14(3), 568. https://doi.org/10.3390/cancers14030568