
Emerging Role of Epigenetic Alterations as Biomarkers and Novel Targets for Treatments in Pancreatic Ductal Adenocarcinoma


Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
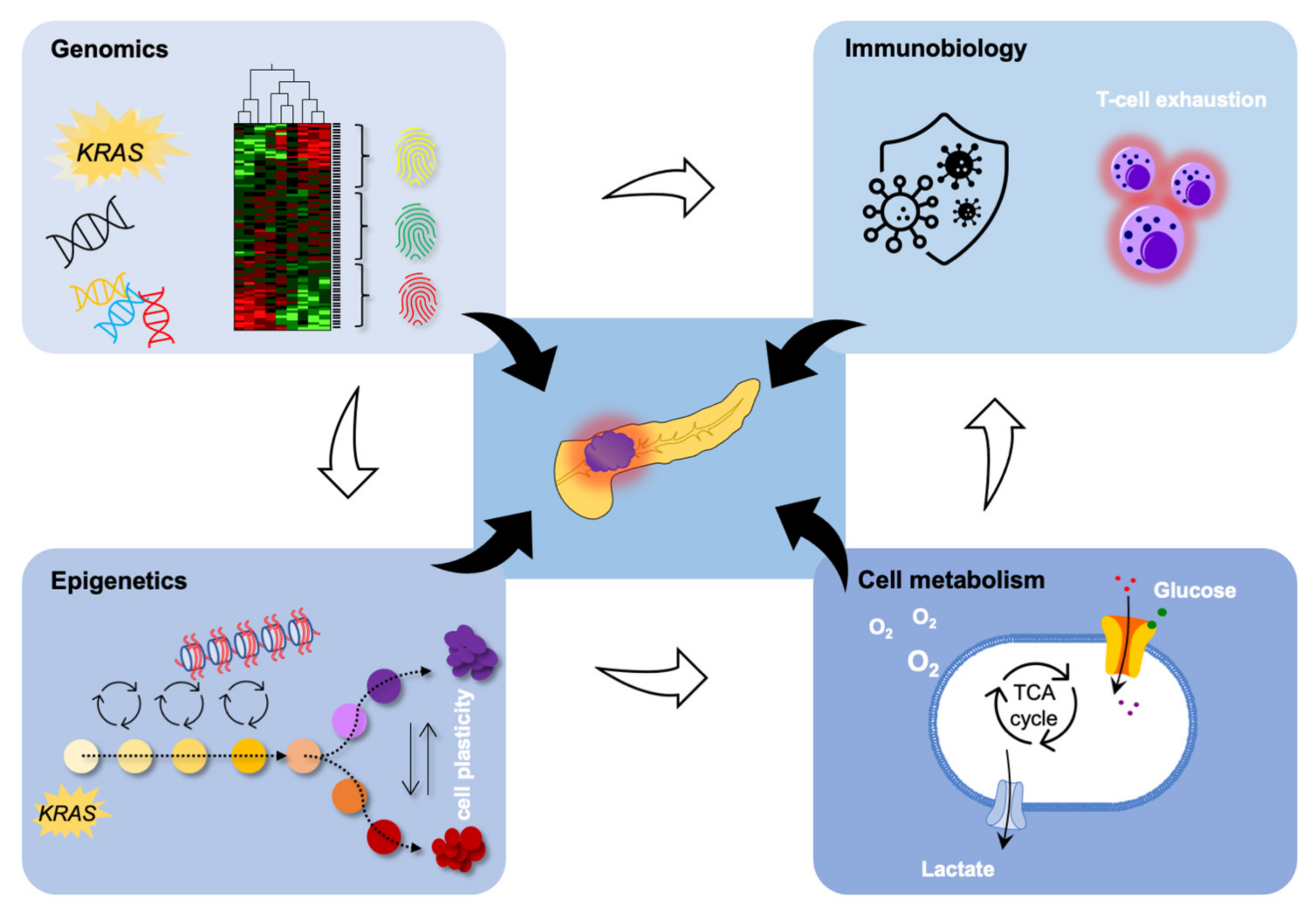
3. Background
3.1. Epigenetic Changes in PDAC
3.2. Tumor Microenvironment
3.3. Metastasis
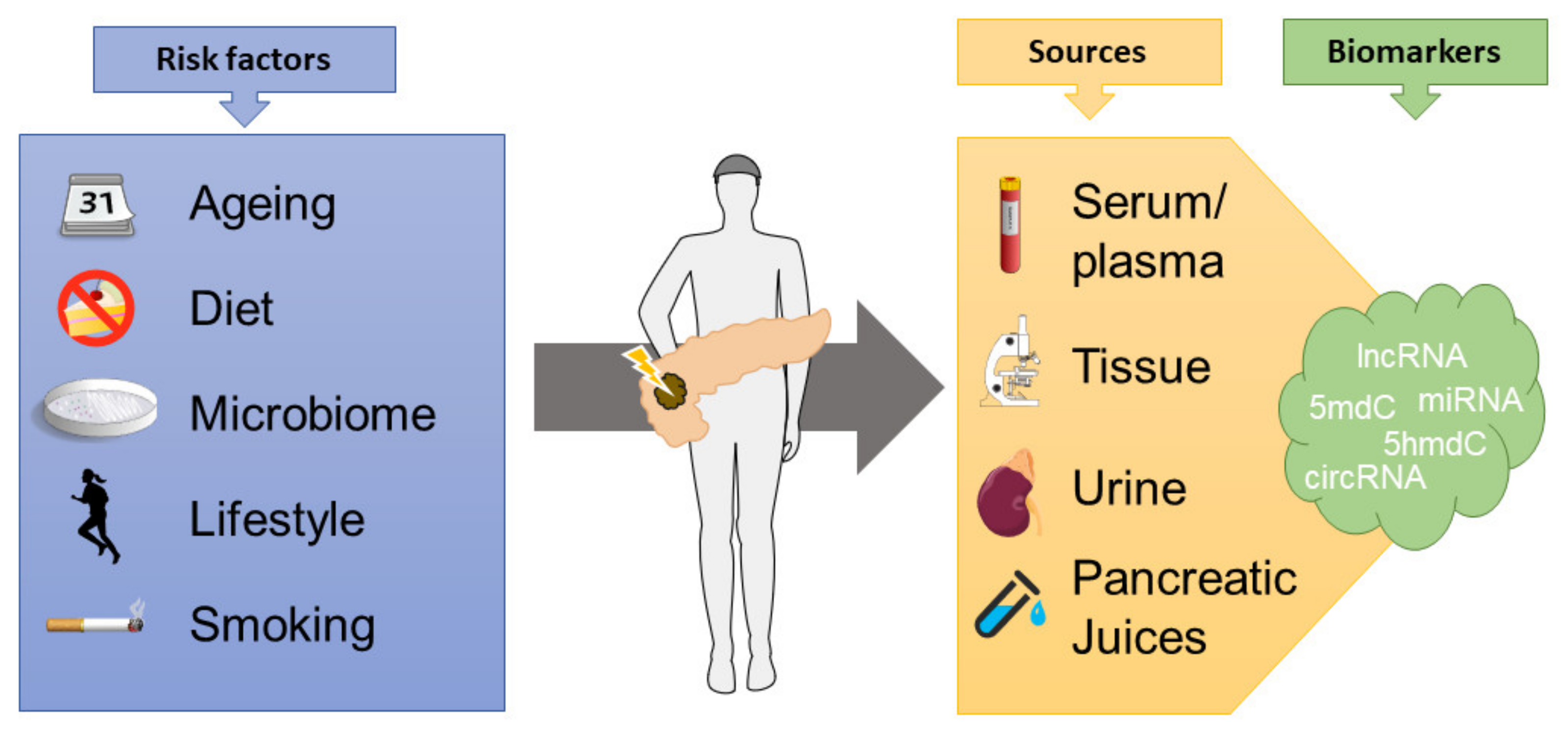
4. Epigenetic Biomarkers in PDAC
5. Epigenetic Therapeutic Options
5.1. Tumor Microenvironment
5.2. Immune Therapy
6. Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Søreide, K.; Roalsø, M.; Aunan, J.R. Is There a Trojan Horse to Aggressive Pancreatic Cancer Biology? A Review of the Trypsin-PAR2 Axis to Proliferation, Early Invasion, and Metastasis. J. Pancreat. Cancer 2020, 6, 12–20. [Google Scholar] [CrossRef]
- Dennaoui, R.; Shrestha, H.; Wagner, K.U. Models of pancreatic ductal adenocarcinoma. Cancer Metastasis Rev. 2021, 40, 803–818. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Zhang, C.; Xie, K.P. Therapeutic resistance of pancreatic cancer: Roadmap to its reversal. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188461. [Google Scholar] [CrossRef]
- Drake, T.M.; Soreide, K. Cancer epigenetics in solid organ tumours: A primer for surgical oncologists. Eur. J. Surg. Oncol. 2019, 45, 736–746. [Google Scholar] [CrossRef]
- Grady, W.M.; Yu, M.; Markowitz, S.D. Epigenetic Alterations in the Gastrointestinal Tract: Current and Emerging Use for Biomarkers of Cancer. Gastroenterology 2021, 160, 690–709. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, Y. N6-methylandenosine-related lncRNAs play an important role in the prognosis and immune microenvironment of pancreatic ductal adenocarcinoma. Sci. Rep. 2021, 11, 17844. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, F.; Liu, Y.; Wang, H.; Ni, B. N(6)-methyladenosine (m(6)A) in pancreatic cancer: Regulatory mechanisms and future direction. Int. J. Biol. Sci. 2021, 17, 2323–2335. [Google Scholar] [CrossRef]
- Li, J.; Yuan, S.; Norgard, R.J.; Yan, F.; Sun, Y.H.; Kim, I.K.; Merrell, A.J.; Sela, Y.; Jiang, Y.; Bhanu, N.V.; et al. Epigenetic and Transcriptional Control of the Epidermal Growth Factor Receptor Regulates the Tumor Immune Microenvironment in Pancreatic Cancer. Cancer Discov. 2021, 11, 736–753. [Google Scholar] [CrossRef] [PubMed]
- Del Poggetto, E.; Ho, I.L.; Balestrieri, C.; Yen, E.Y.; Zhang, S.; Citron, F.; Shah, R.; Corti, D.; Diaferia, G.R.; Li, C.Y.; et al. Epithelial memory of inflammation limits tissue damage while promoting pancreatic tumorigenesis. Science 2021, 373, eabj0486. [Google Scholar] [CrossRef]
- Alonso-Curbelo, D.; Ho, Y.J.; Burdziak, C.; Maag, J.L.V.; Morris, J.P.t.; Chandwani, R.; Chen, H.A.; Tsanov, K.M.; Barriga, F.M.; Luan, W.; et al. A gene-environment-induced epigenetic program initiates tumorigenesis. Nature 2021, 590, 642–648. [Google Scholar] [CrossRef]
- Kato, H.; Tateishi, K.; Fujiwara, H.; Nakatsuka, T.; Yamamoto, K.; Kudo, Y.; Hayakawa, Y.; Nakagawa, H.; Tanaka, Y.; Ijichi, H.; et al. MNX1-HNF1B axis is indispensable for intraductal papillary mucinous neoplasm lineages. Gastroenterology 2021. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Useros, J.; Martin-Galan, M.; Florez-Cespedes, M.; Garcia-Foncillas, J. Epigenetics of Most Aggressive Solid Tumors: Pathways, Targets and Treatments. Cancers 2021, 13, 3209. [Google Scholar] [CrossRef]
- Roalsø, M.; Hald, Ø.H.; Ansari, D.; Andersson, R.; Søreide, K. The Role of Epigenetics in Pancreatic Ductal Adenocarcinoma. In Textbook of Pancreatic Cancer: Principles and Practice of Surgical Oncology; Søreide, K., Stättner, S., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 321–336. [Google Scholar] [CrossRef]
- Jamieson, N.B.; Steele, C.W. Immuno-Oncology in Pancreatic Cancer. In Textbook of Pancreatic Cancer: Principles and Practice of Surgical Oncology; Søreide, K., Stättner, S., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 287–304. [Google Scholar] [CrossRef]
- Gonda, T.A.; Fang, J.; Salas, M.; Do, C.; Hsu, E.; Zhukovskaya, A.; Siegel, A.; Takahashi, R.; Lopez-Bujanda, Z.A.; Drake, C.G.; et al. A DNA Hypomethylating Drug Alters the Tumor Microenvironment and Improves the Effectiveness of Immune Checkpoint Inhibitors in a Mouse Model of Pancreatic Cancer. Cancer Res. 2020, 80, 4754–4767. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Bates, S.E. Epigenetic Therapies for Cancer. N. Engl. J. Med. 2020, 383, 650–663. [Google Scholar] [CrossRef]
- McDonald, O.G.; Li, X.; Saunders, T.; Tryggvadottir, R.; Mentch, S.J.; Warmoes, M.O.; Word, A.E.; Carrer, A.; Salz, T.H.; Natsume, S.; et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat. Genet. 2017, 49, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Khoshchehreh, R.; Totonchi, M.; Carlos Ramirez, J.; Torres, R.; Baharvand, H.; Aicher, A.; Ebrahimi, M.; Heeschen, C. Epigenetic reprogramming of primary pancreatic cancer cells counteracts their in vivo tumourigenicity. Oncogene 2019, 38, 6226–6239. [Google Scholar] [CrossRef]
- Juiz, N.A.; Iovanna, J.; Dusetti, N. Pancreatic Cancer Heterogeneity Can Be Explained Beyond the Genome. Front. Oncol. 2019, 9, 246. [Google Scholar] [CrossRef]
- Nicolle, R.; Blum, Y.; Marisa, L.; Loncle, C.; Gayet, O.; Moutardier, V.; Turrini, O.; Giovannini, M.; Bian, B.; Bigonnet, M.; et al. Pancreatic Adenocarcinoma Therapeutic Targets Revealed by Tumor-Stroma Cross-Talk Analyses in Patient-Derived Xenografts. Cell Rep. 2017, 21, 2458–2470. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Malinova, A.; Veghini, L.; Real, F.X.; Corbo, V. Cell Lineage Infidelity in PDAC Progression and Therapy Resistance. Front. Cell Dev. Biol. 2021, 9, 795251. [Google Scholar] [CrossRef]
- Lomberk, G.; Blum, Y.; Nicolle, R.; Nair, A.; Gaonkar, K.S.; Marisa, L.; Mathison, A.; Sun, Z.; Yan, H.; Elarouci, N.; et al. Distinct epigenetic landscapes underlie the pathobiology of pancreatic cancer subtypes. Nat. Commun. 2018, 9, 1978. [Google Scholar] [CrossRef]
- Bugter, J.M.; Fenderico, N.; Maurice, M.M. Mutations and mechanisms of WNT pathway tumour suppressors in cancer. Nat. Rev. Cancer 2021, 21, 5–21. [Google Scholar] [CrossRef]
- Yuan, C.; Bao, Y.; Wu, C.; Kraft, P.; Ogino, S.; Ng, K.; Qian, Z.R.; Rubinson, D.A.; Stampfer, M.J.; Giovannucci, E.L.; et al. Prediagnostic body mass index and pancreatic cancer survival. J. Clin. Oncol. 2013, 31, 4229–4234. [Google Scholar] [CrossRef]
- Carbone, C.; Piro, G.; Gaianigo, N.; Ligorio, F.; Santoro, R.; Merz, V.; Simionato, F.; Zecchetto, C.; Falco, G.; Conti, G.; et al. Adipocytes sustain pancreatic cancer progression through a non-canonical WNT paracrine network inducing ROR2 nuclear shuttling. Int. J. Obes. 2018, 42, 334–343. [Google Scholar] [CrossRef]
- Huang, J.; Fan, X.; Wang, X.; Lu, Y.; Zhu, H.; Wang, W.; Zhang, S.; Wang, Z. High ROR2 expression in tumor cells and stroma is correlated with poor prognosis in pancreatic ductal adenocarcinoma. Sci. Rep. 2015, 5, 12991. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Mir, R.; Galande, S. Epigenetic Regulation of the Wnt/beta-Catenin Signaling Pathway in Cancer. Front. Genet. 2021, 12, 681053. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Hao, H.X.; Growney, J.D.; Woolfenden, S.; Bottiglio, C.; Ng, N.; Lu, B.; Hsieh, M.H.; Bagdasarian, L.; Meyer, R.; et al. Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 12649–12654. [Google Scholar] [CrossRef] [PubMed]
- Hosein, A.N.; Dangol, G.; Okumura, T.; Roszik, J.; Rajapakshe, K.; Siemann, M.; Zaid, M.; Ghosh, B.; Monberg, M.; Guerrero, P.A.; et al. Loss of Rnf43 accelerates Kras-mediated neoplasia and remodels the tumor immune microenvironment in pancreatic adenocarcinoma. Gastroenterology 2021. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Koldobskiy, M.A.; Gondor, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef]
- Mishra, N.K.; Southekal, S.; Guda, C. Survival Analysis of Multi-Omics Data Identifies Potential Prognostic Markers of Pancreatic Ductal Adenocarcinoma. Front. Genet. 2019, 10, 624. [Google Scholar] [CrossRef] [PubMed]
- Wald, P.; Liu, X.S.; Pettit, C.; Dillhoff, M.; Manilchuk, A.; Schmidt, C.; Wuthrick, E.; Chen, W.; Williams, T.M. Prognostic value of microRNA expression levels in pancreatic adenocarcinoma: A review of the literature. Oncotarget 2017, 8, 73345–73361. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yuan, H.; Liu, J.; Zhao, L.; Wu, P.; Chen, G.; Chen, Q.; Shen, P.; Yang, T.; Fan, S.; Xiao, B.; et al. Prognostic Risk Model and Tumor Immune Environment Modulation of m5C-Related LncRNAs in Pancreatic Ductal Adenocarcinoma. Front. Immunol. 2021, 12, 800268. [Google Scholar] [CrossRef]
- Botla, S.K.; Savant, S.; Jandaghi, P.; Bauer, A.S.; Mucke, O.; Moskalev, E.A.; Neoptolemos, J.P.; Costello, E.; Greenhalf, W.; Scarpa, A.; et al. Early Epigenetic Downregulation of microRNA-192 Expression Promotes Pancreatic Cancer Progression. Cancer Res. 2016, 76, 4149–4159. [Google Scholar] [CrossRef] [PubMed]
- Syren, P.; Andersson, R.; Bauden, M.; Ansari, D. Epigenetic alterations as biomarkers in pancreatic ductal adenocarcinoma. Scand. J. Gastroenterol. 2017, 52, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Kirkegard, J.; Mortensen, F.V.; Cronin-Fenton, D. Chronic Pancreatitis and Pancreatic Cancer Risk: A Systematic Review and Meta-analysis. Am. J. Gastroenterol. 2017, 112, 1366–1372. [Google Scholar] [CrossRef]
- Valkenburg, K.C.; de Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef]
- Garcia-Gomez, A.; Rodriguez-Ubreva, J.; Ballestar, E. Epigenetic interplay between immune, stromal and cancer cells in the tumor microenvironment. Clin. Immunol. 2018, 196, 64–71. [Google Scholar] [CrossRef]
- Whatcott, C.J.; Diep, C.H.; Jiang, P.; Watanabe, A.; LoBello, J.; Sima, C.; Hostetter, G.; Shepard, H.M.; Von Hoff, D.D.; Han, H. Desmoplasia in Primary Tumors and Metastatic Lesions of Pancreatic Cancer. Clin. Cancer Res. 2015, 21, 3561–3568. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Haber, P.S.; Darby, S.J.; Rodgers, S.C.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells are activated by proinflammatory cytokines: Implications for pancreatic fibrogenesis. Gut 1999, 44, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Dangi-Garimella, S.; Krantz, S.B.; Barron, M.R.; Shields, M.A.; Heiferman, M.J.; Grippo, P.J.; Bentrem, D.J.; Munshi, H.G. Three-dimensional collagen I promotes gemcitabine resistance in pancreatic cancer through MT1-MMP-mediated expression of HMGA2. Cancer Res. 2011, 71, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Jurisica, I.; Do, T.; Hedley, D.W. Hypoxia predicts aggressive growth and spontaneous metastasis formation from orthotopically grown primary xenografts of human pancreatic cancer. Cancer Res. 2011, 71, 3110–3120. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.S.; Jones, R.E.; Ransom, R.C.; Longaker, M.T.; Norton, J.A. The evolving relationship of wound healing and tumor stroma. JCI Insight 2018, 3, e99911. [Google Scholar] [CrossRef]
- Evan, G.I.; Hah, N.; Littlewood, T.D.; Sodir, N.M.; Campos, T.; Downes, M.; Evans, R.M. Re-engineering the Pancreas Tumor Microenvironment: A "Regenerative Program" Hacked. Clin. Cancer Res. 2017, 23, 1647–1655. [Google Scholar] [CrossRef]
- Xiao, Q.; Zhou, D.; Rucki, A.A.; Williams, J.; Zhou, J.; Mo, G.; Murphy, A.; Fujiwara, K.; Kleponis, J.; Salman, B.; et al. Cancer-Associated Fibroblasts in Pancreatic Cancer Are Reprogrammed by Tumor-Induced Alterations in Genomic DNA Methylation. Cancer Res. 2016, 76, 5395–5404. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Kapoor, A.; Zhang, Q.; Li, J.; Wu, C.J.; Li, J.; Lan, Z.; Tang, M.; Ma, X.; Ackroyd, J.J.; et al. Tumor Microenvironment Remodeling Enables Bypass of Oncogenic KRAS Dependency in Pancreatic Cancer. Cancer Discov. 2020, 10, 1058–1077. [Google Scholar] [CrossRef]
- Borgoni, S.; Iannello, A.; Cutrupi, S.; Allavena, P.; D’Incalci, M.; Novelli, F.; Cappello, P. Depletion of tumor-associated macrophages switches the epigenetic profile of pancreatic cancer infiltrating T cells and restores their anti-tumor phenotype. Oncoimmunology 2018, 7, e1393596. [Google Scholar] [CrossRef] [PubMed]
- Morrison, A.H.; Byrne, K.T.; Vonderheide, R.H. Immunotherapy and Prevention of Pancreatic Cancer. Trends Cancer 2018, 4, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Balli, D.; Rech, A.J.; Stanger, B.Z.; Vonderheide, R.H. Immune Cytolytic Activity Stratifies Molecular Subsets of Human Pancreatic Cancer. Clin. Cancer Res. 2017, 23, 3129–3138. [Google Scholar] [CrossRef] [PubMed]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018, 362. [Google Scholar] [CrossRef]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Markosyan, N.; Li, J.; Sun, Y.H.; Richman, L.P.; Lin, J.H.; Yan, F.; Quinones, L.; Sela, Y.; Yamazoe, T.; Gordon, N.; et al. Tumor cell-intrinsic EPHA2 suppresses anti-tumor immunity by regulating PTGS2 (COX-2). J. Clin. Investig. 2019, 129, 3594–3609. [Google Scholar] [CrossRef]
- Sheng, W.; LaFleur, M.W.; Nguyen, T.H.; Chen, S.; Chakravarthy, A.; Conway, J.R.; Li, Y.; Chen, H.; Yang, H.; Hsu, P.H.; et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell 2018, 174, 549–563.e19. [Google Scholar] [CrossRef]
- Li, J.; Yuan, S.; Norgard, R.J.; Yan, F.; Yamazoe, T.; Blanco, A.; Stanger, B.Z. Tumor Cell-Intrinsic USP22 Suppresses Antitumor Immunity in Pancreatic Cancer. Cancer Immunol. Res. 2020, 8, 282–291. [Google Scholar] [CrossRef]
- Wang, S.S.; Xu, J.; Ji, K.Y.; Hwang, C.I. Epigenetic Alterations in Pancreatic Cancer Metastasis. Biomolecules 2021, 11, 1082. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; Fu, B.; Kamiyama, M.; Hruban, R.H.; Eshleman, J.R.; Nowak, M.A.; et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010, 467, 1114–1117. [Google Scholar] [CrossRef] [PubMed]
- Dhara, S.; Chhangawala, S.; Chintalapudi, H.; Askan, G.; Aveson, V.; Massa, A.L.; Zhang, L.; Torres, D.; Makohon-Moore, A.P.; Lecomte, N.; et al. Pancreatic cancer prognosis is predicted by an ATAC-array technology for assessing chromatin accessibility. Nat. Commun. 2021, 12, 3044. [Google Scholar] [CrossRef]
- Fritsche, P.; Seidler, B.; Schuler, S.; Schnieke, A.; Gottlicher, M.; Schmid, R.M.; Saur, D.; Schneider, G. HDAC2 mediates therapeutic resistance of pancreatic cancer cells via the BH3-only protein NOXA. Gut 2009, 58, 1399–1409. [Google Scholar] [CrossRef]
- Lehmann, A.; Denkert, C.; Budczies, J.; Buckendahl, A.C.; Darb-Esfahani, S.; Noske, A.; Muller, B.M.; Bahra, M.; Neuhaus, P.; Dietel, M.; et al. High class I HDAC activity and expression are associated with RelA/p65 activation in pancreatic cancer in vitro and in vivo. BMC Cancer 2009, 9, 395. [Google Scholar] [CrossRef]
- Krauss, L.; Urban, B.C.; Hastreiter, S.; Schneider, C.; Wenzel, P.; Hassan, Z.; Wirth, M.; Lankes, K.; Terrasi, A.; Klement, C.; et al. HDAC2 facilitates pancreatic cancer metastasis. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Bararia, A.; Dey, S.; Gulati, S.; Ghatak, S.; Ghosh, S.; Banerjee, S.; Sikdar, N. Differential methylation landscape of pancreatic ductal adenocarcinoma and its precancerous lesions. Hepatobiliary Pancreat. Dis. Int. 2020, 19, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S.; Raimondo, M.; Taylor, W.R.; Yab, T.C.; Berger, C.K.; Dukek, B.A.; Cao, X.; Foote, P.H.; Wu, C.W.; Devens, M.E.; et al. Methylated DNA in Pancreatic Juice Distinguishes Patients With Pancreatic Cancer From Controls. Clin. Gastroenterol. Hepatol. 2020, 18, 676–683.e3. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, S.; Kitamoto, S.; Higashi, M.; Goto, Y.; Hara, T.; Ikebe, D.; Yamaguchi, T.; Arisaka, Y.; Niihara, T.; Nishimata, H.; et al. Diagnosis of pancreatic neoplasms using a novel method of DNA methylation analysis of mucin expression in pancreatic juice. PLoS ONE 2014, 9, e93760. [Google Scholar] [CrossRef]
- Yokoyama, S.; Hamada, T.; Higashi, M.; Matsuo, K.; Maemura, K.; Kurahara, H.; Horinouchi, M.; Hiraki, T.; Sugimoto, T.; Akahane, T.; et al. Predicted Prognosis of Patients with Pancreatic Cancer by Machine Learning. Clin. Cancer Res. 2020, 26, 2411–2421. [Google Scholar] [CrossRef]
- Brancaccio, M.; Natale, F.; Falco, G.; Angrisano, T. Cell-Free DNA Methylation: The New Frontiers of Pancreatic Cancer Biomarkers’ Discovery. Genes 2019, 11, 14. [Google Scholar] [CrossRef]
- Cao, F.; Wei, A.; Hu, X.; He, Y.; Zhang, J.; Xia, L.; Tu, K.; Yuan, J.; Guo, Z.; Liu, H.; et al. Integrated epigenetic biomarkers in circulating cell-free DNA as a robust classifier for pancreatic cancer. Clin. Epigenet. 2020, 12, 112. [Google Scholar] [CrossRef]
- Sole, C.; Arnaiz, E.; Manterola, L.; Otaegui, D.; Lawrie, C.H. The circulating transcriptome as a source of cancer liquid biopsy biomarkers. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2019; Volume 58, pp. 100–108. [Google Scholar] [CrossRef]
- Zhao, F.; Wei, C.; Cui, M.Y.; Xia, Q.Q.; Wang, S.B.; Zhang, Y. Prognostic value of microRNAs in pancreatic cancer: A meta-analysis. Aging 2020, 12, 9380–9404. [Google Scholar] [CrossRef] [PubMed]
- Daoud, A.Z.; Mulholland, E.J.; Cole, G.; McCarthy, H.O. MicroRNAs in Pancreatic Cancer: Biomarkers, prognostic, and therapeutic modulators. BMC Cancer 2019, 19, 1130. [Google Scholar] [CrossRef] [PubMed]
- Aita, A.; Millino, C.; Sperti, C.; Pacchioni, B.; Plebani, M.; De Pitta, C.; Basso, D. Serum miRNA Profiling for Early PDAC Diagnosis and Prognosis: A Retrospective Study. Biomedicines 2021, 9, 845. [Google Scholar] [CrossRef]
- Yoshizawa, N.; Sugimoto, K.; Tameda, M.; Inagaki, Y.; Ikejiri, M.; Inoue, H.; Usui, M.; Ito, M.; Takei, Y. miR-3940-5p/miR-8069 ratio in urine exosomes is a novel diagnostic biomarker for pancreatic ductal adenocarcinoma. Oncol. Lett. 2020, 19, 2677–2684. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Taniue, K.; Ono, Y.; Fujiya, M.; Mizukami, Y.; Okumura, T. Long Non-Coding RNAs in Epithelial-Mesenchymal Transition of Pancreatic Cancer. Front. Mol. Biosci. 2021, 8, 717890. [Google Scholar] [CrossRef]
- Gong, R.; Jiang, Y. Non-coding RNAs in Pancreatic Ductal Adenocarcinoma. Front. Oncol. 2020, 10, 309. [Google Scholar] [CrossRef]
- Pandya, G.; Kirtonia, A.; Sethi, G.; Pandey, A.K.; Garg, M. The implication of long non-coding RNAs in the diagnosis, pathogenesis and drug resistance of pancreatic ductal adenocarcinoma and their possible therapeutic potential. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188423. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Hu, M.; Zhou, L.; Ling, S.; Li, Y.; Kong, B.; Huang, P. Long non-coding RNA HOTAIR promotes cancer cell energy metabolism in pancreatic adenocarcinoma by upregulating hexokinase-2. Oncol. Lett. 2019, 18, 2212–2219. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Shen, M.; Zhou, M.; Shi, X.; He, R.; Yin, T.; Wang, M.; Guo, X.; Qin, R. Long noncoding RNA LINC01111 suppresses pancreatic cancer aggressiveness by regulating DUSP1 expression via microRNA-3924. Cell Death Dis. 2019, 10, 883. [Google Scholar] [CrossRef] [PubMed]
- Limb, C.; Liu, D.S.K.; Veno, M.T.; Rees, E.; Krell, J.; Bagwan, I.N.; Giovannetti, E.; Pandha, H.; Strobel, O.; Rockall, T.A.; et al. The Role of Circular RNAs in Pancreatic Ductal Adenocarcinoma and Biliary-Tract Cancers. Cancers 2020, 12, 3250. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zhou, Q.; Su, D.; Luo, Y.; Fu, Z.; Huang, L.; Li, Z.; Jiang, D.; Kong, Y.; Li, Z.; et al. Circular RNA circBFAR promotes the progression of pancreatic ductal adenocarcinoma via the miR-34b-5p/MET/Akt axis. Mol. Cancer 2020, 19, 83. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, J.; Yu, S.; Wang, Z.; He, X.; Su, Y.; Guo, T.; Sheng, H.; Chen, J.; Zheng, Q.; et al. Extracellular Vesicles Long RNA Sequencing Reveals Abundant mRNA, circRNA, and lncRNA in Human Blood as Potential Biomarkers for Cancer Diagnosis. Clin. Chem. 2019, 65, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Li, Y.; Liao, Z.; Wang, Z.; Wang, Z.; Li, Y.; Qian, L.; Zhao, J.; Zong, H.; Kang, B.; et al. Plasma extracellular vesicle long RNA profiling identifies a diagnostic signature for the detection of pancreatic ductal adenocarcinoma. Gut 2020, 69, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Hessmann, E.; Johnsen, S.A.; Siveke, J.T.; Ellenrieder, V. Epigenetic treatment of pancreatic cancer: Is there a therapeutic perspective on the horizon? Gut 2017, 66, 168–179. [Google Scholar] [CrossRef]
- Mohammad, H.P.; Barbash, O.; Creasy, C.L. Targeting epigenetic modifications in cancer therapy: Erasing the roadmap to cancer. Nat. Med. 2019, 25, 403–418. [Google Scholar] [CrossRef]
- Chan, E.; Arlinghaus, L.R.; Cardin, D.B.; Goff, L.; Berlin, J.D.; Parikh, A.; Abramson, R.G.; Yankeelov, T.E.; Hiebert, S.; Merchant, N.; et al. Phase I trial of vorinostat added to chemoradiation with capecitabine in pancreatic cancer. Radiother. Oncol. 2016, 119, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Ohtani, H.; Chakravarthy, A.; De Carvalho, D.D. Epigenetic therapy in immune-oncology. Nat. Rev. Cancer 2019, 19, 151–161. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Nahar, S.; Nakagawa, A.; Fernandez-Barrena, M.G.; Mertz, J.A.; Bryant, B.M.; Adams, C.E.; Mino-Kenudson, M.; Von Alt, K.N.; Chang, K.; et al. Regulation of GLI Underlies a Role for BET Bromodomains in Pancreatic Cancer Growth and the Tumor Microenvironment. Clin. Cancer Res. 2016, 22, 4259–4270. [Google Scholar] [CrossRef]
- Smith, S.G.; Zhou, M.M. The Bromodomain: A New Target in Emerging Epigenetic Medicine. ACS Chem. Biol. 2016, 11, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Kloesch, B.; Ionasz, V.; Paliwal, S.; Hruschka, N.; Martinez de Villarreal, J.; Ollinger, R.; Mueller, S.; Dienes, H.P.; Schindl, M.; Gruber, E.S.; et al. A GATA6-centred gene regulatory network involving HNFs and DeltaNp63 controls plasticity and immune escape in pancreatic cancer. Gut 2021. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.; Steuber, B.; Kopp, W.; Kari, V.; Urbach, L.; Wang, X.; Kuffer, S.; Bohnenberger, H.; Spyropoulou, D.; Zhang, Z.; et al. EZH2 Regulates Pancreatic Cancer Subtype Identity and Tumor Progression via Transcriptional Repression of GATA6. Cancer Res. 2020, 80, 4620–4632. [Google Scholar] [CrossRef] [PubMed]
- Eyres, M.; Lanfredini, S.; Xu, H.; Burns, A.; Blake, A.; Willenbrock, F.; Goldin, R.; Hughes, D.; Hughes, S.; Thapa, A.; et al. TET2 Drives 5hmc Marking of GATA6 and Epigenetically Defines Pancreatic Ductal Adenocarcinoma Transcriptional Subtypes. Gastroenterology 2021, 161, 653–668.e16. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, A.; Arimondo, P.B.; Rots, M.G.; Jeronimo, C.; Berdasco, M. The timeline of epigenetic drug discovery: From reality to dreams. Clin. Epigenet. 2019, 11, 174. [Google Scholar] [CrossRef] [PubMed]
- Richards, D.A.; Boehm, K.A.; Waterhouse, D.M.; Wagener, D.J.; Krishnamurthi, S.S.; Rosemurgy, A.; Grove, W.; Macdonald, K.; Gulyas, S.; Clark, M.; et al. Gemcitabine plus CI-994 offers no advantage over gemcitabine alone in the treatment of patients with advanced pancreatic cancer: Results of a phase II randomized, double-blind, placebo-controlled, multicenter study. Ann. Oncol. 2006, 17, 1096–1102. [Google Scholar] [CrossRef]
- Mross, K.; Dittrich, C.; Aulitzky, W.E.; Strumberg, D.; Schutte, J.; Schmid, R.M.; Hollerbach, S.; Merger, M.; Munzert, G.; Fleischer, F.; et al. A randomised phase II trial of the Polo-like kinase inhibitor BI 2536 in chemo-naive patients with unresectable exocrine adenocarcinoma of the pancreas - a study within the Central European Society Anticancer Drug Research (CESAR) collaborative network. Br. J. Cancer 2012, 107, 280–286. [Google Scholar] [CrossRef]
- Hauge, A.; Rofstad, E.K. Antifibrotic therapy to normalize the tumor microenvironment. J. Transl. Med. 2020, 18, 207. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, R.; Musteanu, M.; Garcia-Garcia, E.; Lopez-Casas, P.P.; Megias, D.; Guerra, C.; Munoz, M.; Quijano, Y.; Cubillo, A.; Rodriguez-Pascual, J.; et al. Stromal disrupting effects of nab-paclitaxel in pancreatic cancer. Br. J. Cancer 2013, 109, 926–933. [Google Scholar] [CrossRef]
- Hessmann, E.; Buchholz, S.M.; Demir, I.E.; Singh, S.K.; Gress, T.M.; Ellenrieder, V.; Neesse, A. Microenvironmental Determinants of Pancreatic Cancer. Physiol. Rev. 2020, 100, 1707–1751. [Google Scholar] [CrossRef]
- Mazur, P.K.; Herner, A.; Mello, S.S.; Wirth, M.; Hausmann, S.; Sanchez-Rivera, F.J.; Lofgren, S.M.; Kuschma, T.; Hahn, S.A.; Vangala, D.; et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat. Med. 2015, 21, 1163–1171. [Google Scholar] [CrossRef]
- Nguyen, A.H.; Elliott, I.A.; Wu, N.; Matsumura, C.; Vogelauer, M.; Attar, N.; Dann, A.; Ghukasyan, R.; Toste, P.A.; Patel, S.G.; et al. Histone deacetylase inhibitors provoke a tumor supportive phenotype in pancreatic cancer associated fibroblasts. Oncotarget 2017, 8, 19074–19088. [Google Scholar] [CrossRef] [PubMed]
- Shakya, R.; Gonda, T.; Quante, M.; Salas, M.; Kim, S.; Brooks, J.; Hirsch, S.; Davies, J.; Cullo, A.; Olive, K.; et al. Hypomethylating therapy in an aggressive stroma-rich model of pancreatic carcinoma. Cancer Res. 2013, 73, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Kohi, S.; Sato, N.; Cheng, X.B.; Koga, A.; Higure, A.; Hirata, K. A novel epigenetic mechanism regulating hyaluronan production in pancreatic cancer cells. Clin. Exp. Metastasis 2016, 33, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Espinet, E.; Gu, Z.; Imbusch, C.D.; Giese, N.A.; Büscher, M.; Safavi, M.; Weisenburger, S.; Klein, C.; Vogel, V.; Falcone, M.; et al. Aggressive PDACs Show Hypomethylation of Repetitive Elements and the Execution of an Intrin.sic IFN Program Linked to a Ductal Cell of Origin. Cancer Discov. 2021, 11, 638–659. [Google Scholar] [CrossRef] [PubMed]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- Kubli, S.P.; Berger, T.; Araujo, D.V.; Siu, L.L.; Mak, T.W. Beyond immune checkpoint blockade: Emerging immunological strategies. Nat. Rev. Drug Discov. 2021, 20, 899–919. [Google Scholar] [CrossRef]
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M.; et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165. [Google Scholar] [CrossRef]
- Sen, D.R.; Kaminski, J.; Barnitz, R.A.; Kurachi, M.; Gerdemann, U.; Yates, K.B.; Tsao, H.W.; Godec, J.; LaFleur, M.W.; Brown, F.D.; et al. The epigenetic landscape of T cell exhaustion. Science 2016, 354, 1165–1169. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Topper, M.J.; Vaz, M.; Marrone, K.A.; Brahmer, J.R.; Baylin, S.B. The emerging role of epigenetic therapeutics in immuno-oncology. Nat. Rev. Clin. Oncol. 2020, 17, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.I.; Shia, J.; Stadler, Z.K.; Varghese, A.M.; Capanu, M.; Salo-Mullen, E.; Lowery, M.A.; Diaz, L.A., Jr.; Mandelker, D.; Yu, K.H.; et al. Evaluating Mismatch Repair Deficiency in Pancreatic Adenocarcinoma: Challenges and Recommendations. Clin. Cancer Res. 2018, 24, 1326–1336. [Google Scholar] [CrossRef] [PubMed]
- Ebelt, N.D.; Zuniga, E.; Johnson, B.L.; Diamond, D.J.; Manuel, E.R. 5-Azacytidine Potentiates Anti-tumor Immunity in a Model of Pancreatic Ductal Adenocarcinoma. Front. Immunol. 2020, 11, 538. [Google Scholar] [CrossRef] [PubMed]
- Weber, R.; Fleming, V.; Hu, X.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front. Immunol. 2018, 9, 1310. [Google Scholar] [CrossRef] [PubMed]
- Christmas, B.J.; Rafie, C.I.; Hopkins, A.C.; Scott, B.A.; Ma, H.S.; Cruz, K.A.; Woolman, S.; Armstrong, T.D.; Connolly, R.M.; Azad, N.A.; et al. Entinostat Converts Immune-Resistant Breast and Pancreatic Cancers into Checkpoint-Responsive Tumors by Reprogramming Tumor-Infiltrating MDSCs. Cancer Immunol. Res. 2018, 6, 1561–1577. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Drug(s) | Combination Agent(s) | Phase of Study | Status | NCT Number |
|---|---|---|---|---|
| Panobinostat Vorinostat | Various antineoplastic drugs | Phase 1 | Recruiting | NCT03878524 |
| Tazemetostat | Durvalumab | Phase 2 | Recruiting | NCT04705818 |
| Romidepsin Azacitidine | Gemcitabine Durvalumab Lenalidomide nab-Paclitaxel | Phase 1/2 | Recruiting | NCT04257448 |
| Azacitidine | Chemotherapy after progression | Phase 2 | Active, not recruiting | NCT01845805 |
| Vorinostat | Gemcitabine Sorafenib | Phase 1 | Active, not recruiting | NCT02349867 |
| Azacitidine | Pembrolizumab | Phase 2 | Active, not recruiting | NCT03264404 |
| Entinostat | Nivolumab | Phase 2 | Completed | NCT03250273 |
| Decitabine | Tetrahydrouridine | Phase 1 | Completed | NCT02847000 |
| Entinostat | Phase 1 | Completed | NCT00020579 | |
| Vorinostat | Capecitabine | Phase 1 | Completed | NCT00983268 |
| Azacitidine | nab-Paclitaxel Carboplatin | Phase 1 | Completed | NCT01478685 |
| Vorinostat | NPI-0052 (marizomib) | Phase 1 | Completed | NCT00667082 |
| Panobinostat | Bortezomib | Phase 2 | Terminated (funding not available) | NCT01056601 |
| Entinostat | FOLFOX regimen | Phase 1 | Withdrawn (lack of funding) | NCT03760614 |
| Entinostat Molibresib | Phase 1 | Withdrawn (protocol moved to disapproved) | NCT03925428 | |
| Vorinostat | Phase 1/2 | Terminated (slow accrual) | NCT00831493 | |
| Vorinostat | 5-FU | Phase 1/2 | Terminated (funding withdrawn) | NCT00948688 |
| Azatacidine | Gemcitabine | Phase 1 | Terminated (miscellaneous reasons) | NCT01167816 |
| Epigenetic targets | ||||
| Azacitidine | Hypomethylates DNA by inhibition of DNA methyltransferase (DNMTi), halting cell division. | |||
| Decitabane | ||||
| Tazemetostat | Lysine histone methyltransferase inhibitor (HMTi), selective inhibition of EZH2. | |||
| Molibresib | Molibresib is a bromodomain and extra-terminal motif inhibitor (BETi), downregulating transcription of oncogenes. | |||
| Vorinostat | Histone deacetylase inhibitors (HDACi) that induce growth arrest, differentiation, autophagy, and apoptosis in tumor cells. | |||
| Entinostat | ||||
| Panobinostat | ||||
| Romidepsin | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roalsø, M.T.T.; Hald, Ø.H.; Alexeeva, M.; Søreide, K. Emerging Role of Epigenetic Alterations as Biomarkers and Novel Targets for Treatments in Pancreatic Ductal Adenocarcinoma. Cancers 2022, 14, 546. https://doi.org/10.3390/cancers14030546
Roalsø MTT, Hald ØH, Alexeeva M, Søreide K. Emerging Role of Epigenetic Alterations as Biomarkers and Novel Targets for Treatments in Pancreatic Ductal Adenocarcinoma. Cancers. 2022; 14(3):546. https://doi.org/10.3390/cancers14030546
Chicago/Turabian StyleRoalsø, Marcus T. T., Øyvind H. Hald, Marina Alexeeva, and Kjetil Søreide. 2022. "Emerging Role of Epigenetic Alterations as Biomarkers and Novel Targets for Treatments in Pancreatic Ductal Adenocarcinoma" Cancers 14, no. 3: 546. https://doi.org/10.3390/cancers14030546
APA StyleRoalsø, M. T. T., Hald, Ø. H., Alexeeva, M., & Søreide, K. (2022). Emerging Role of Epigenetic Alterations as Biomarkers and Novel Targets for Treatments in Pancreatic Ductal Adenocarcinoma. Cancers, 14(3), 546. https://doi.org/10.3390/cancers14030546