
Primaquine as a Candidate for HHV-8-Associated Primary Effusion Lymphoma and Kaposi’s Sarcoma Treatment

 , ,
, ,  , ,
, ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Reagents
2.3. Cell Viability Assay
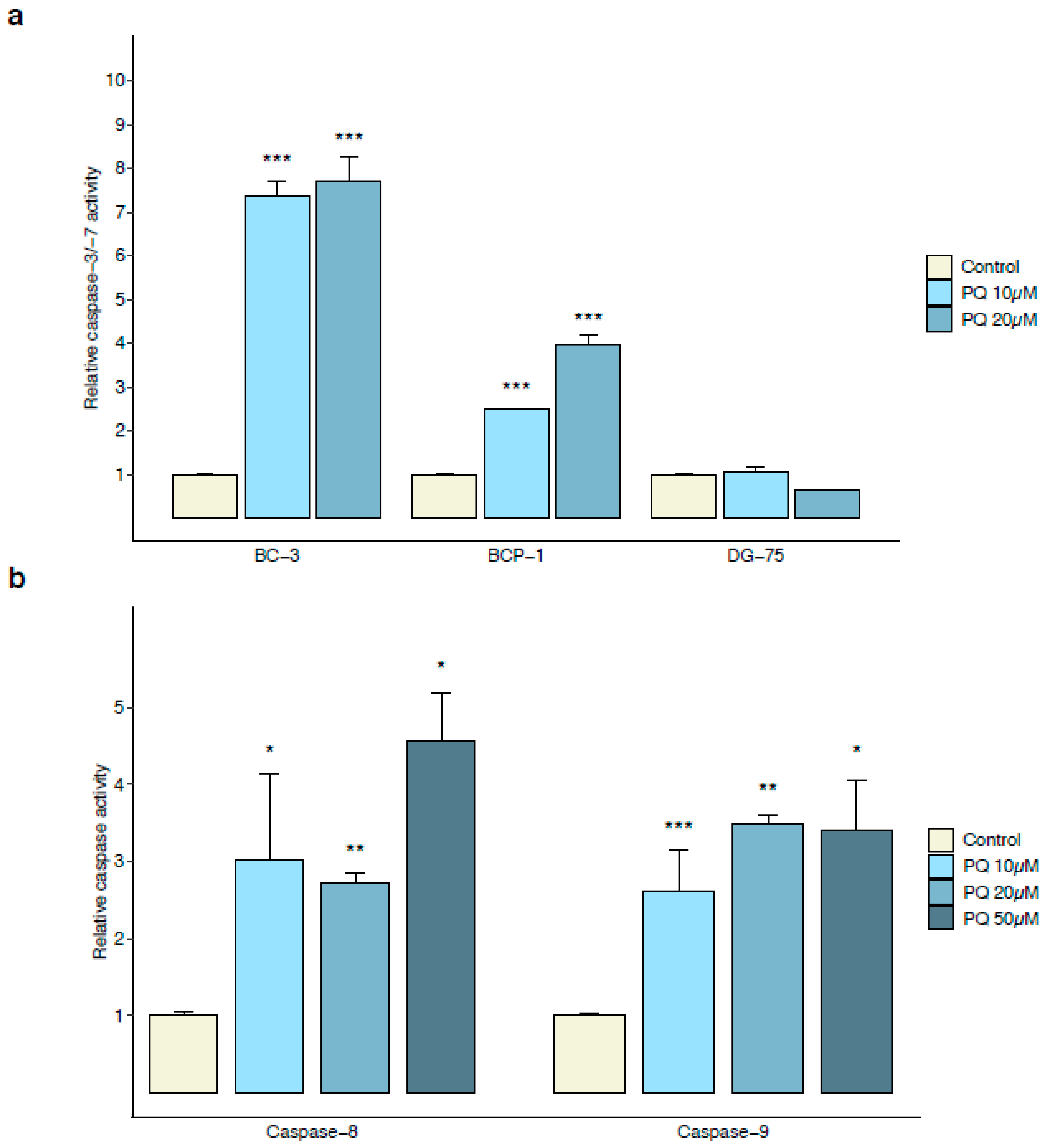
2.4. Caspases-3/-7, -8, and -9 Assays
2.5. Caspase-4 Assay
2.6. Phenotypic Screening Assay
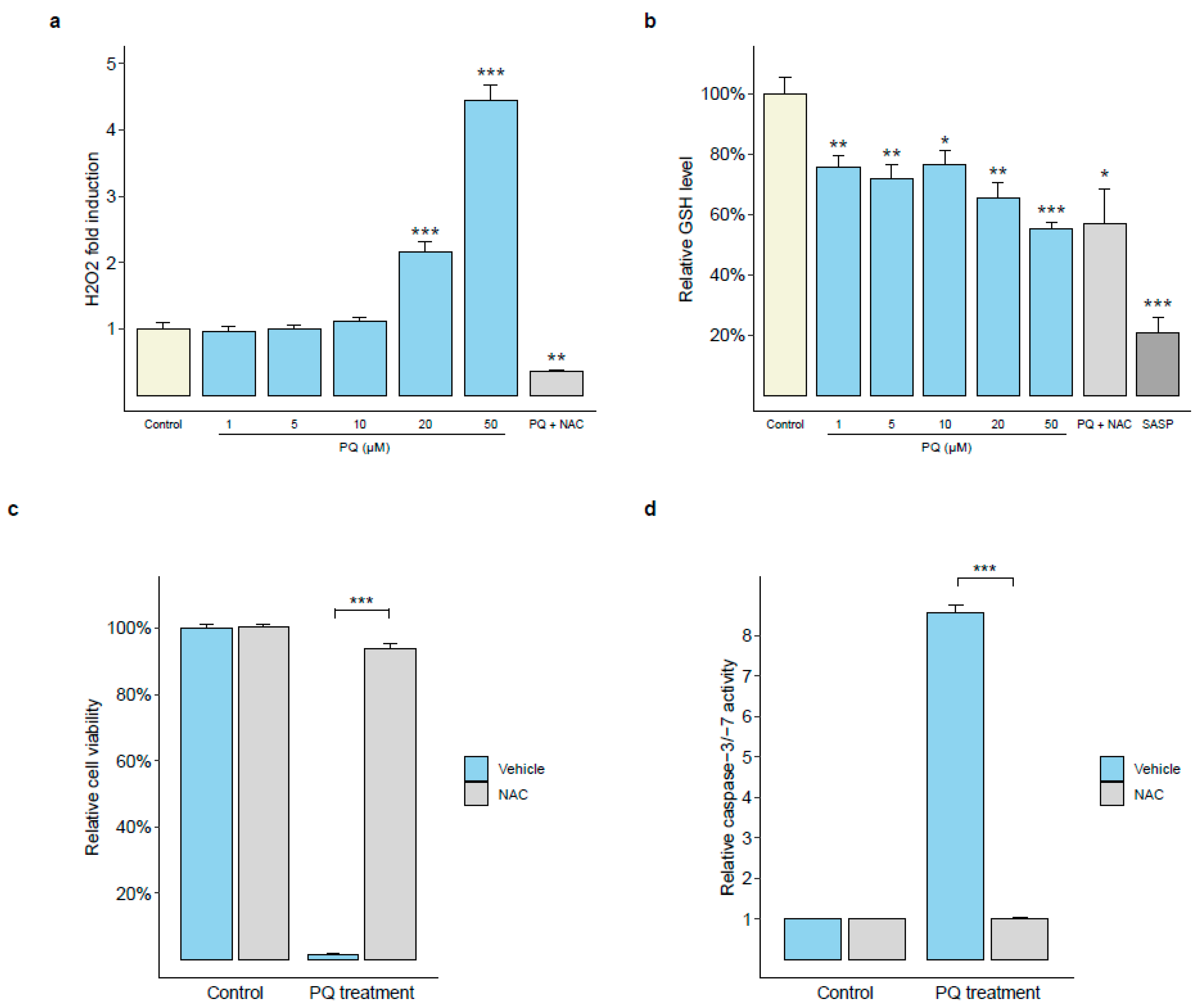
2.7. Measurement of Intracellular Hydrogen Peroxide (H2O2) and Reduced Glutathione (GSH) Productions
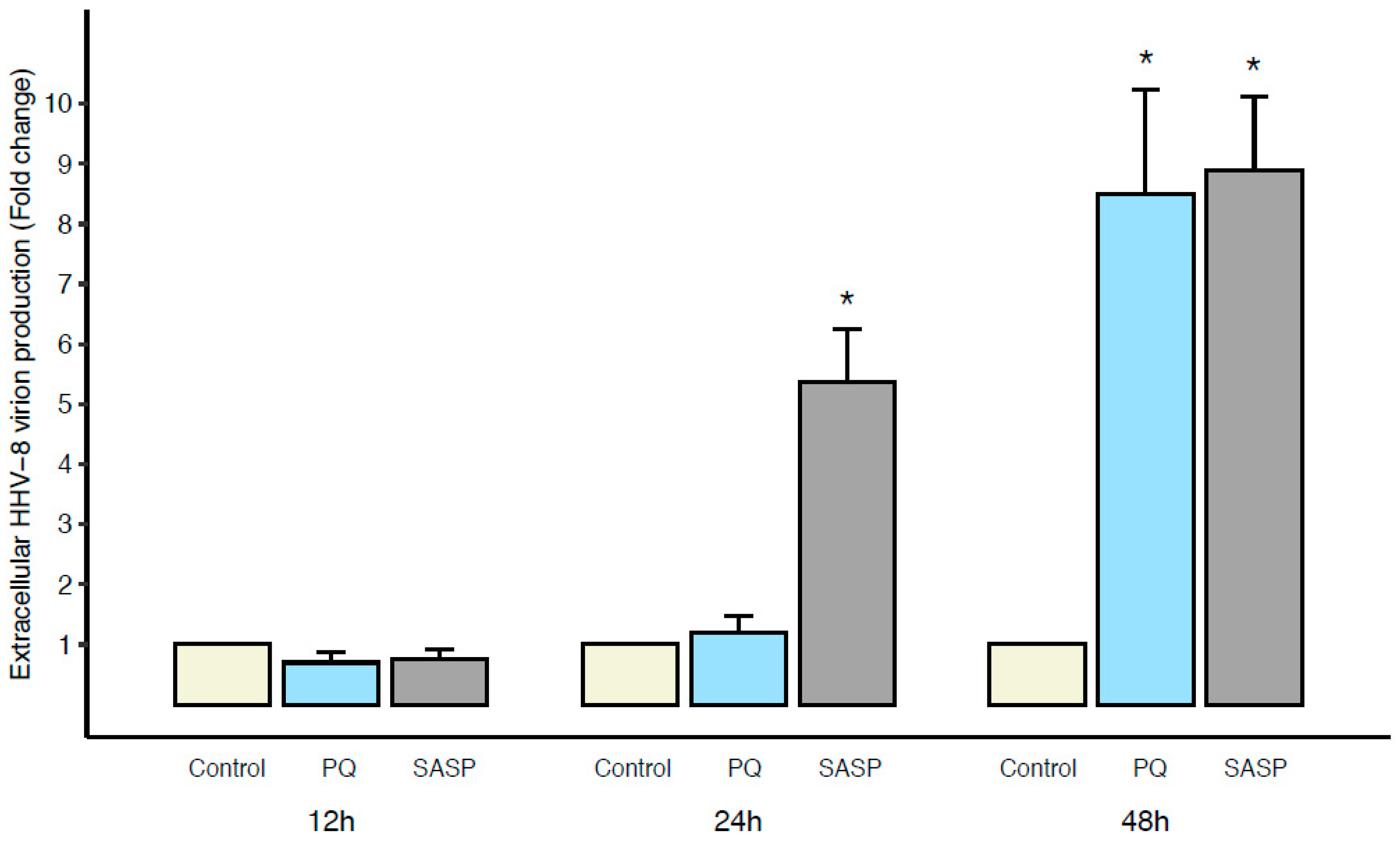
2.8. Measurement of Extracellular HHV-8 Virus Production by Real-Time PCR
2.9. Transcriptome Profiling by RNA-Seq
2.10. qRT-PCR
2.11. PEL Xenograft Murine Model
2.12. Proof of Concept Clinical Trial
2.13. Statistics
3. Results
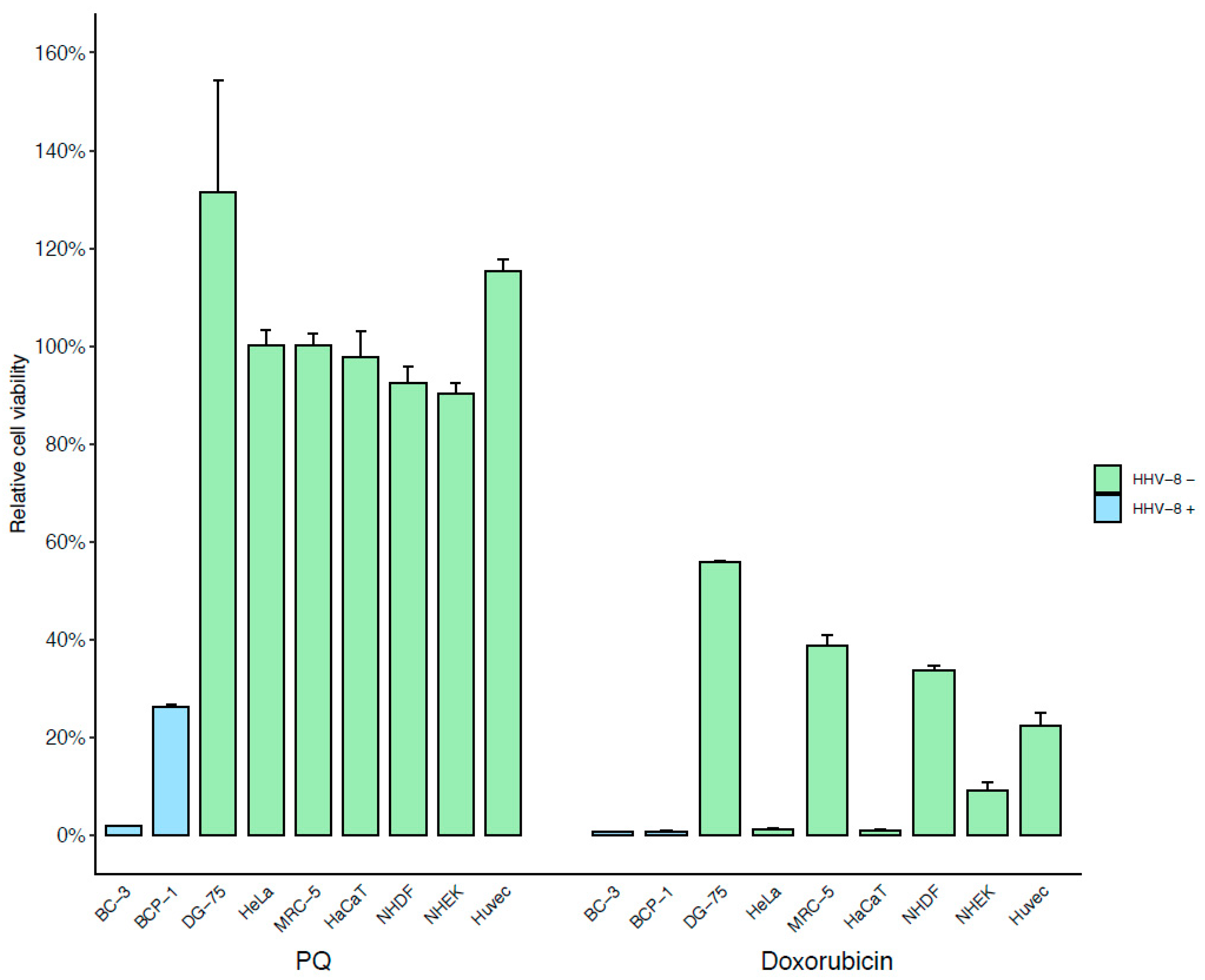
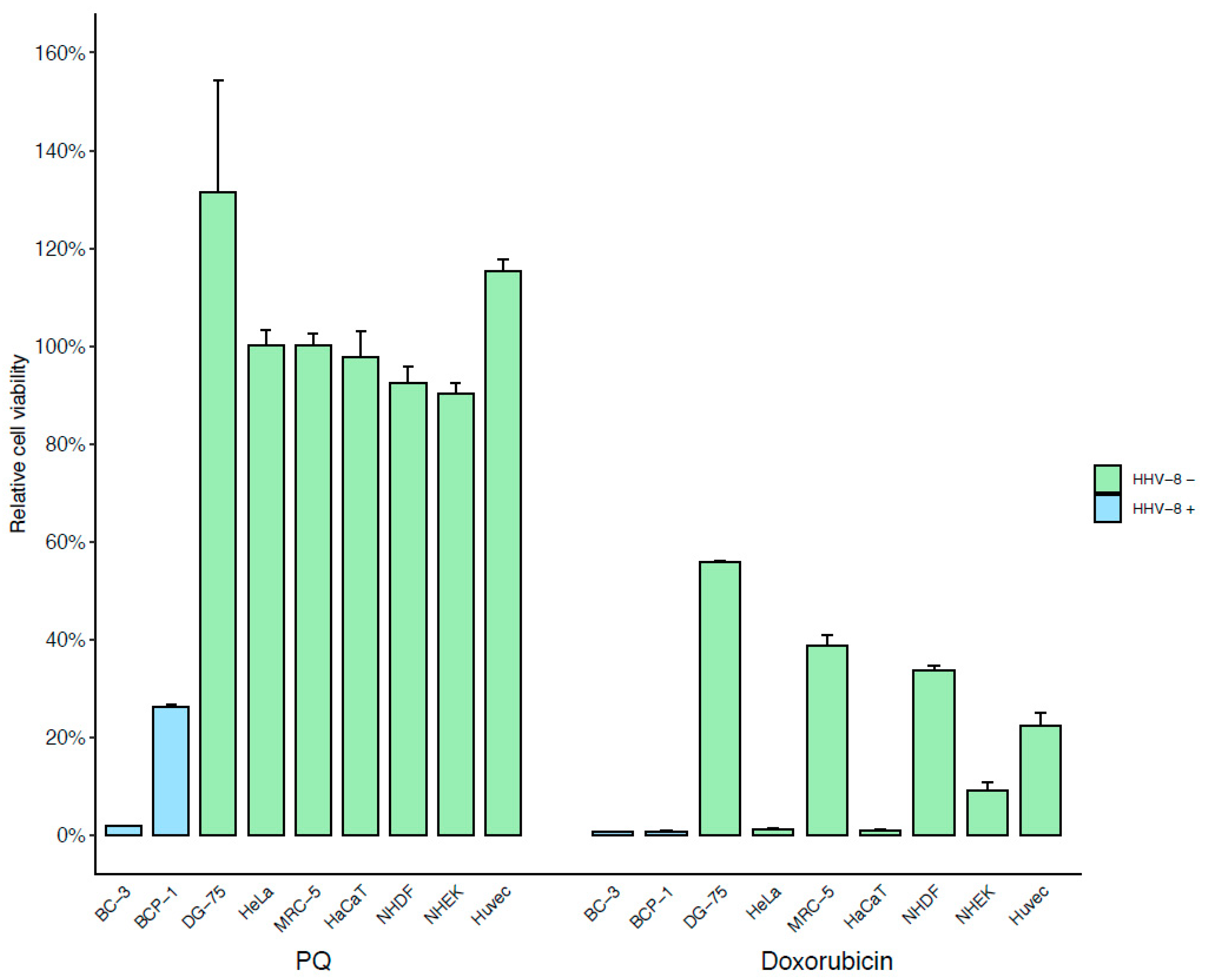
3.1. Identification of a New Drug Specifically Active on PEL Cells Using Phenotypic Screening Assay
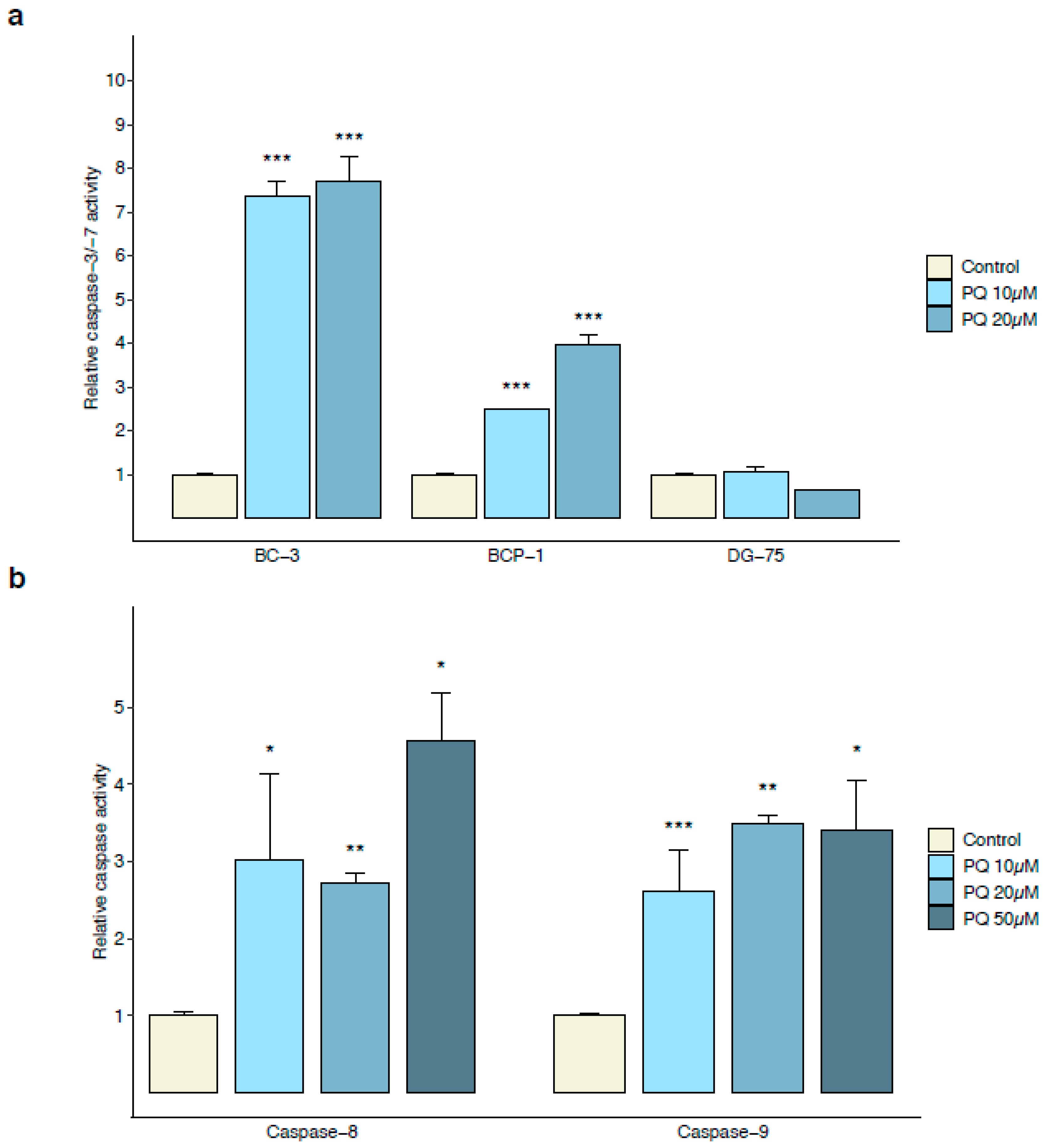
3.2. Primaquine Treatment Induces Oxidative Stress-Mediated Apoptosis in PEL Cells
3.3. Early Primaquine-Induced Apoptosis in PEL Cells Is Not Associated with Extracellular HHV-8 Virion Production
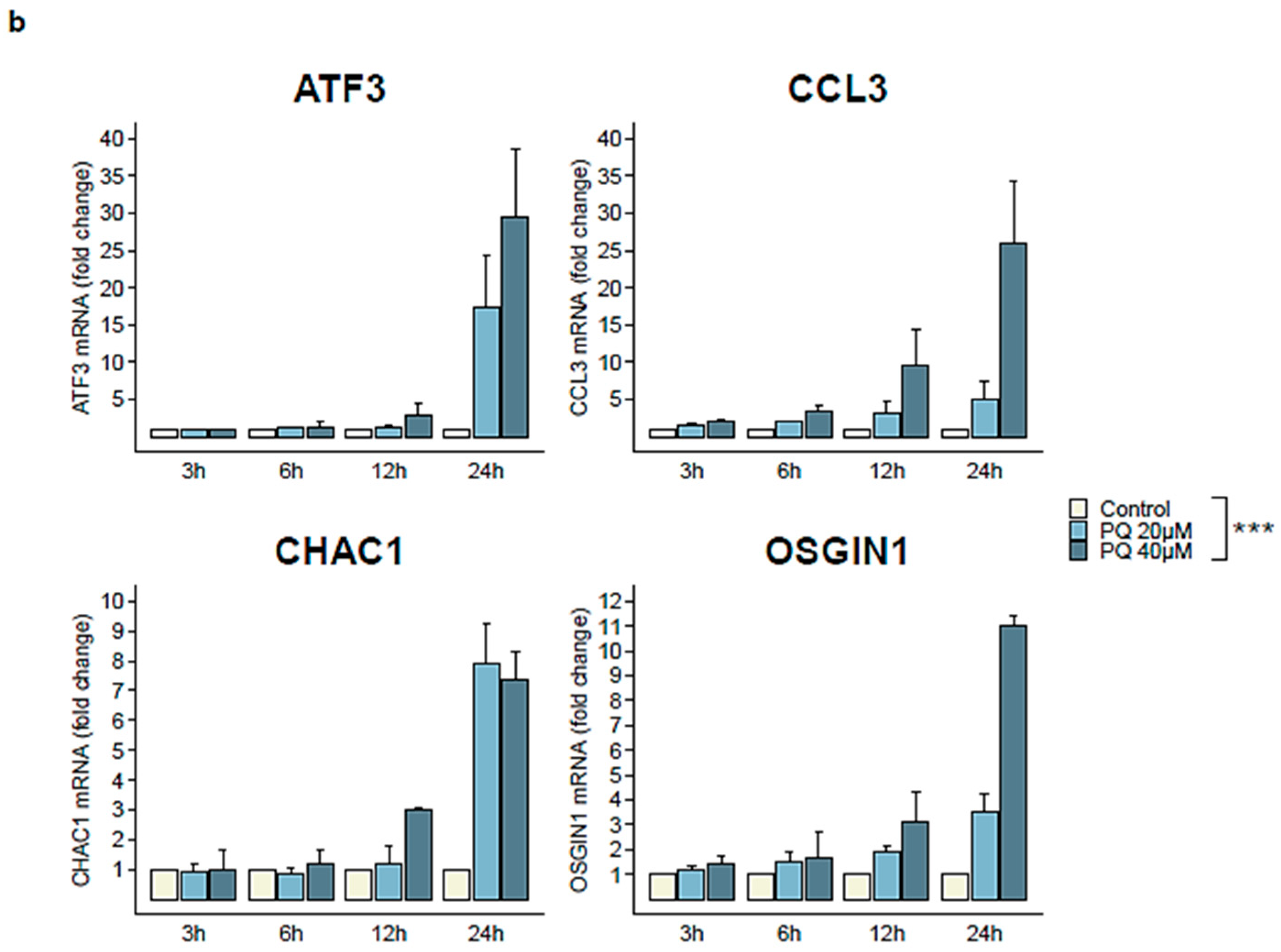
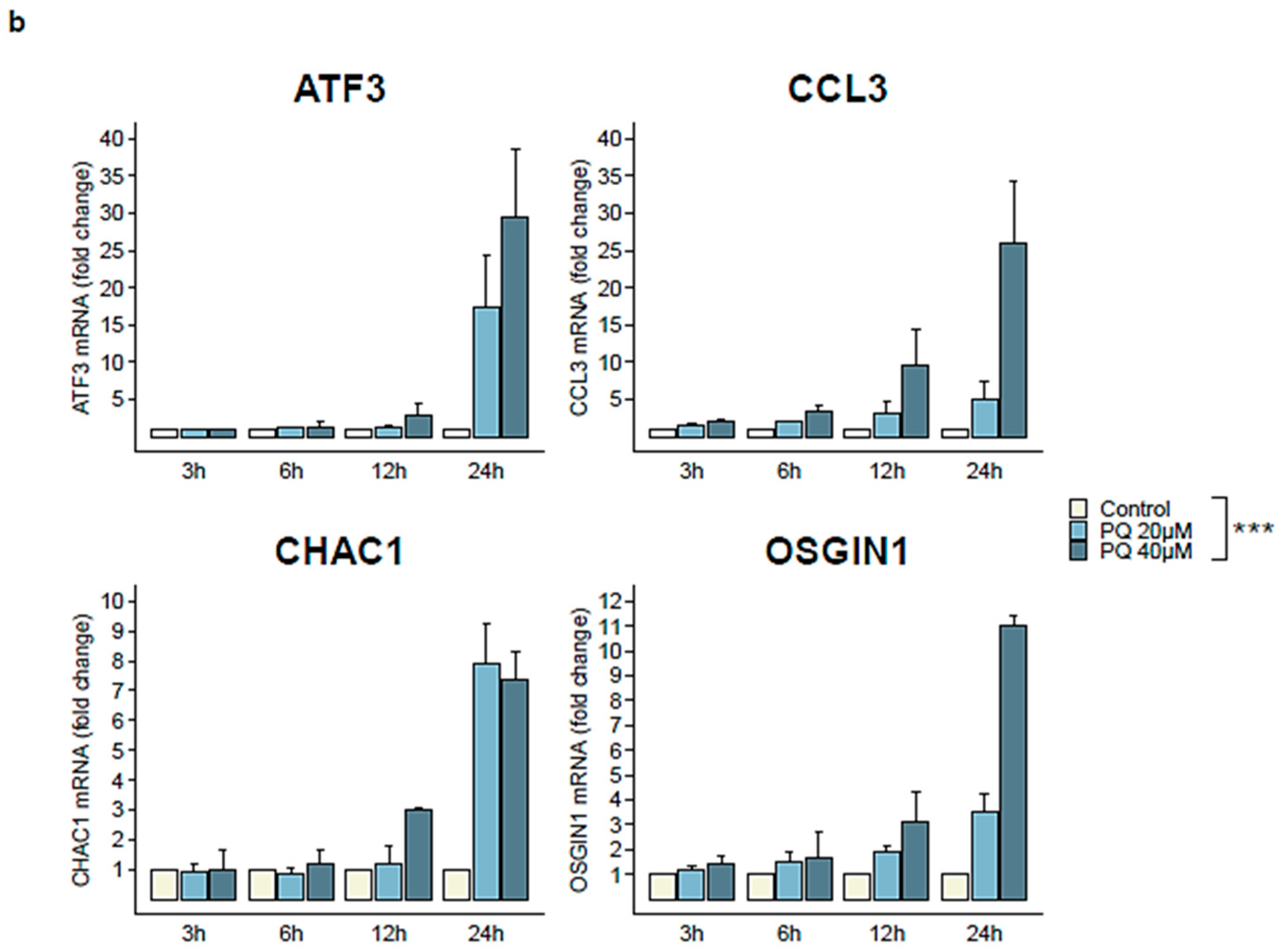
3.4. Four Cellular Genes Involved in Apoptotic Process Are Up-Regulated during Primaquine Treatment of HHV-8-Infected Cells
3.5. Primaquine Treatment Induces ER Stress-Mediated Apoptosis through Transcriptional Expression of CHOP and Activation of Caspase-4
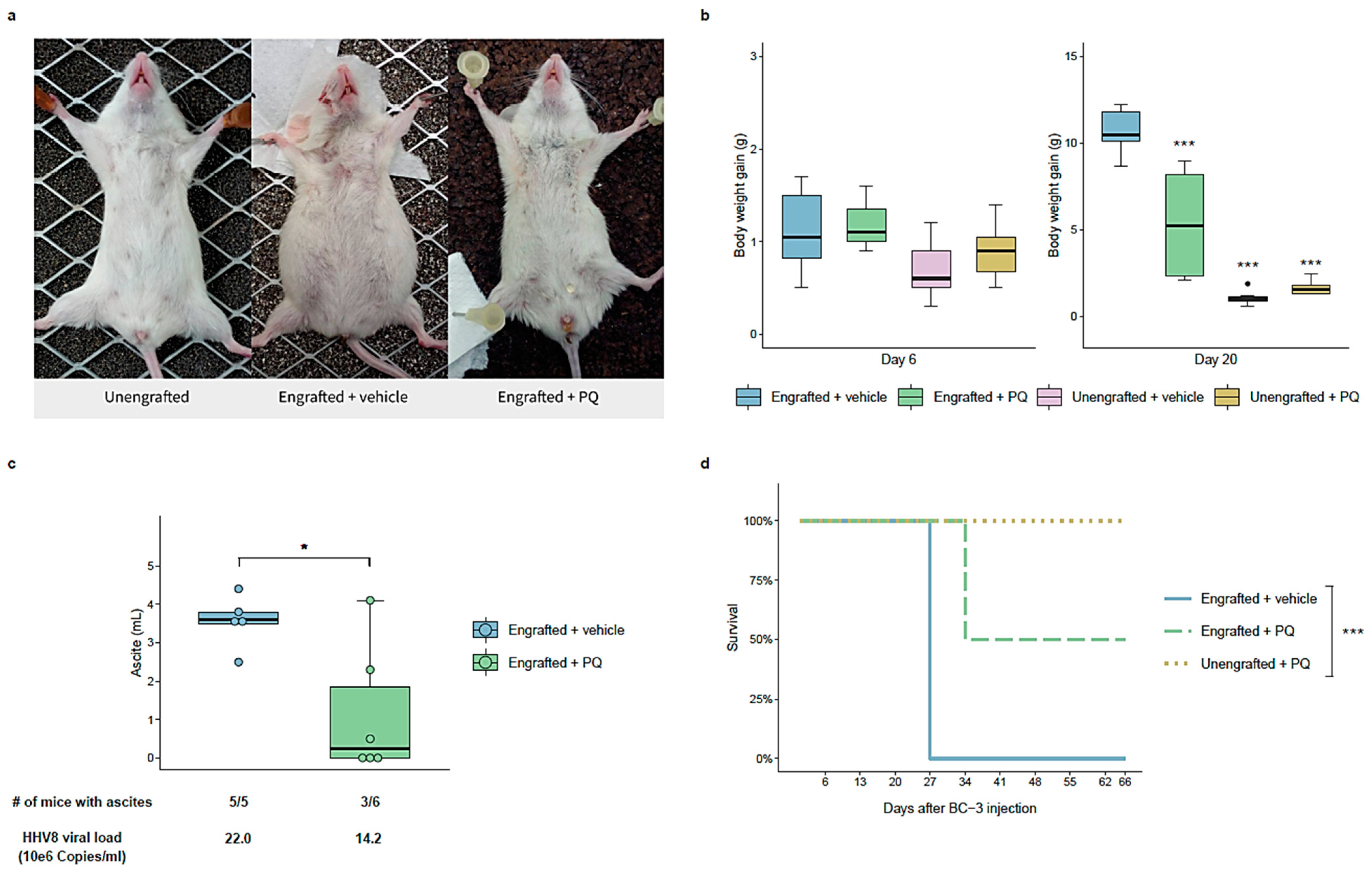
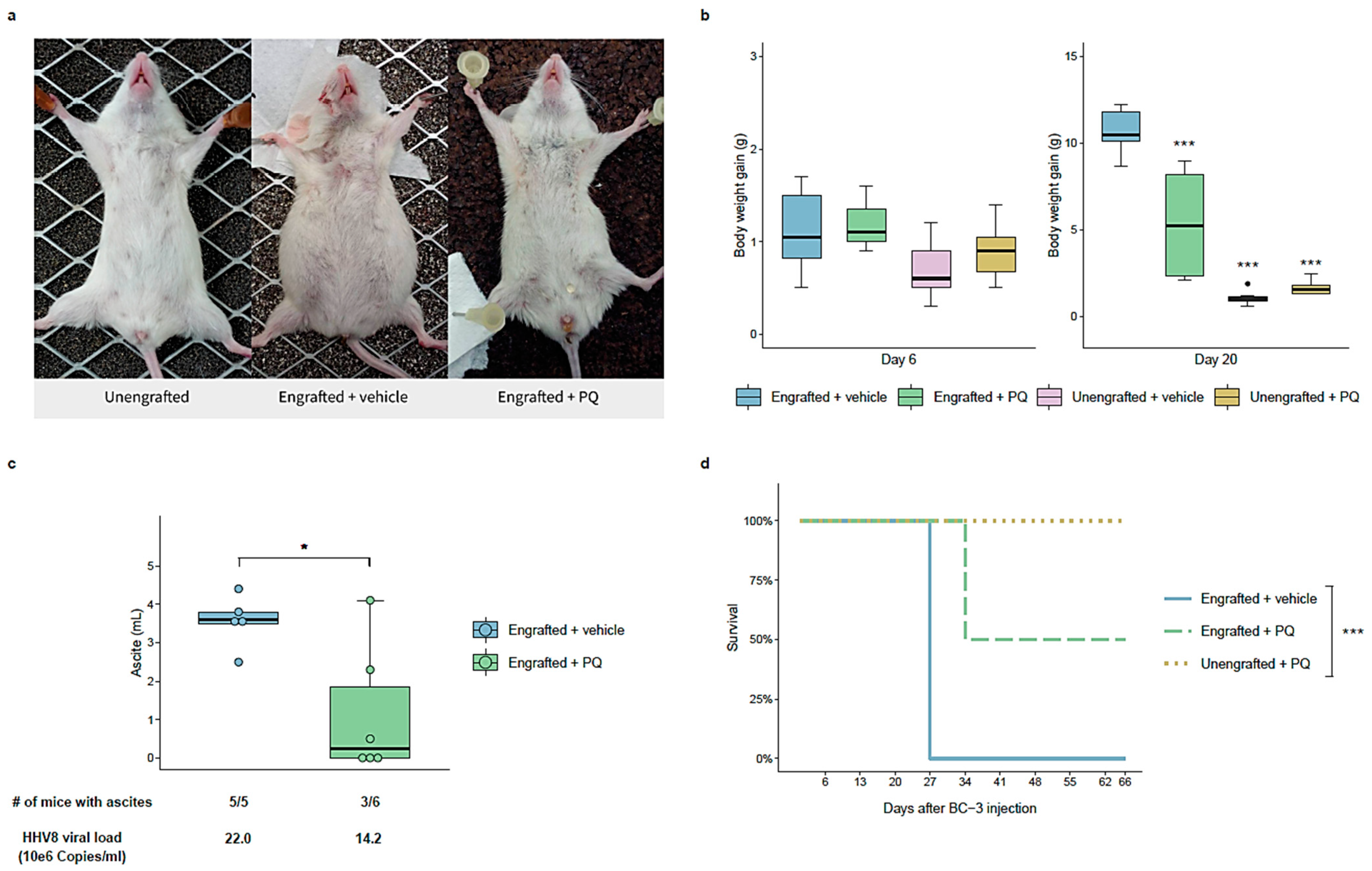
3.6. Primaquine Treatment Reduces PEL Tumor Growth and Improves Survival in a NOD/SCID Mice Xenografted with a PEL
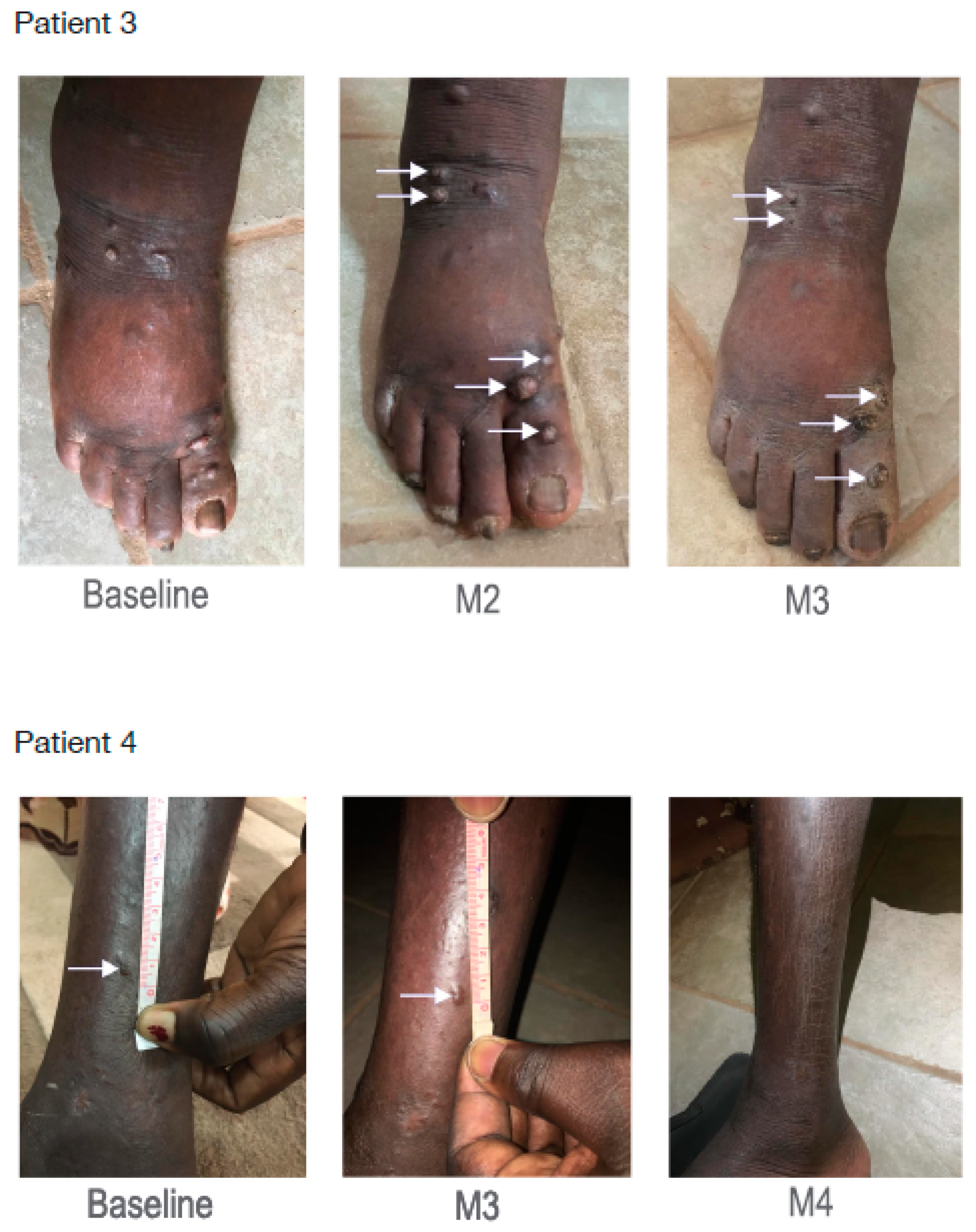
3.7. Primaquine Treatment Decreases Kaposi’s Sarcoma-Related Lesions and Lymphedema in Humans
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kaplan, L.D. Human herpesvirus-8: Kaposi sarcoma, multicentric Castleman disease, and primary effusion lymphoma. Hematology 2013, 2013, 103–108. [Google Scholar] [CrossRef]
- Shimada, K.; Hayakawa, F.; Kiyoi, H. Biology and management of primary effusion lymphoma. Blood 2018, 132, 1879–1888. [Google Scholar] [CrossRef] [Green Version]
- Goncalves, P.H.; Ziegelbauer, J.; Uldrick, T.S.; Yarchoan, R. Kaposi-Sarcoma Herpesvirus Associated Cancers and Related Diseases. Curr. Opin. HIV AIDS 2017, 12, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E.; Damania, B.; Krown, S.E.; Martin, J.; Bower, M.; Whitby, D. Kaposi sarcoma. Nat. Rev. Dis. Primer 2019, 5, 9. [Google Scholar]
- Dai, L.; Cao, Y.; Chen, Y.; Parsons, C.; Qin, Z. Targeting xCT, a cystine-glutamate transporter induces apoptosis and tumor regression for KSHV/HIV-associated lymphoma. J. Hematol. Oncol. 2014, 7, 30. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Majerciak, V.; Zheng, Z.-M.; Lan, K. Towards Better Understanding of KSHV Life Cycle: From Transcription and Posttranscriptional Regulations to Pathogenesis. Virol. Sin. 2019, 34, 135–161. [Google Scholar]
- Cancian, L.; Hansen, A.; Boshoff, C. Cellular origin of Kaposi’s sarcoma and Kaposi’s sarcoma-associated herpesvirus-induced cell reprogramming. Trends Cell Biol. 2013, 23, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Purushothaman, P.; Dabral, P.; Gupta, N.; Sarkar, R.; Verma, S.C. KSHV Genome Replication and Maintenance. Front. Microbiol. 2016, 7, 54. [Google Scholar]
- Bruce, A.G.; Barcy, S.; DiMaio, T.; Gan, E.; Garrigues, H.J.; Lagunoff, M.; Rose, T.M. Quantitative Analysis of the KSHV Transcriptome Following Primary Infection of Blood and Lymphatic Endothelial Cells. Pathogens 2017, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuno, T.; Kariya, R.; Yano, S.; Marino-Koga, S.; Taura, M.; Suico, M.A.; Shimauchi, Y.; Matsuyama, S.; Okamoto, Y.; Shuto, T.; et al. Diethyldithiocarbamate induces apoptosis in HHV-8-infected primary effusion lymphoma cells via inhibition of the NF-κB pathway. Int. J. Oncol. 2012, 40, 1071–1078. [Google Scholar]
- Lallemand, F.; Desire, N.; Rozenbaum, W.; Nicolas, J.-C.; Marechal, V. Quantitative Analysis of Human Herpesvirus 8 Viral Load Using a Real-Time PCR Assay. J. Clin. Microbiol. 2000, 38, 1404–1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Rochford, R.; Ohrt, C.; Baresel, P.C.; Campo, B.; Sampath, A.; Magill, A.J.; Tekwani, B.L.; Walker, L.A. Humanized mouse model of glucose 6-phosphate dehydrogenase deficiency for in vivo assessment of hemolytic toxicity. Proc. Natl. Acad. Sci. USA 2013, 110, 17486–17491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdes, A.; Epelboin, L.; Mosnier, E.; Walter, G.; Vesin, G.; Abboud, P.; Melzani, A.; Blanchet, D.; Blaise, N.; Nacher, M.; et al. Primaquine 30 mg/day versus 15 mg/day during 14 days for the prevention of Plasmodium vivax relapses in adults in French Guiana: A historical comparison. Malar. J. 2018, 17, 237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariggiò, G.; Koch, S.; Schulz, T.F. Kaposi sarcoma herpesvirus pathogenesis. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160275. [Google Scholar]
- Li, X.; Feng, J.; Sun, R. Oxidative Stress Induces Reactivation of Kaposi’s Sarcoma-Associated Herpesvirus and Death of Primary Effusion Lymphoma Cells. J. Virol. 2011, 85, 715–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, L.; Cao, Y.; Chen, Y.; Kaleeba, J.A.R.; Zabaleta, J.; Qin, Z. Genomic analysis of xCT-mediated regulatory network: Identification of novel targets against AIDS-associated lymphoma. Oncotarget 2015, 13, 12710. [Google Scholar] [CrossRef] [Green Version]
- Oslowski, C.M.; Urano, F. Measuring ER Stress and the Unfolded Protein Response Using Mammalian Tissue Culture System. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2011; pp. 71–92. [Google Scholar]
- Merlot, A.M.; Shafie, N.H.; Yu, Y.; Richardson, V.; Jansson, P.J.; Sahni, S.; Lane, D.J.R.; Kovacevic, Z.; Kalinowski, D.S.; Richardson, D.R. Mechanism of the induction of endoplasmic reticulum stress by the anti-cancer agent, di-2-pyridylketone 4,4-dimethyl-3-thiosemicarbazone (Dp44mT): Activation of PERK/eIF2α, IRE1α, ATF6 and calmodulin kinase. Biochem. Pharmacol. 2016, 109, 27–47. [Google Scholar] [PubMed]
- Mahdi, A.A.; Rizvi, S.H.M.; Parveen, A. Role of Endoplasmic Reticulum Stress and Unfolded Protein Responses in Health and Diseases. Indian J. Clin. Biochem. 2016, 31, 127–137. [Google Scholar]
- Shigemi, Z.; Manabe, K.; Hara, N.; Baba, Y.; Hosokawa, K.; Kagawa, H.; Watanabe, T.; Fujimuro, M. Methylseleninic acid and sodium selenite induce severe ER stress and subsequent apoptosis through UPR activation in PEL cells. Chem. Biol. Interact. 2017, 266, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urra, H.; Dufey, E.; Lisbona, F.; Rojas-Rivera, D.; Hetz, C. When ER stress reaches a dead end. Biochim. Biophys. Acta BBA Mol. Cell Res. 2013, 1833, 3507–3517. [Google Scholar]
- Shigemi, Z.; Baba, Y.; Hara, N.; Matsuhiro, J.; Kagawa, H.; Watanabe, T.; Fujimuro, M. Effects of ER stress on unfolded protein responses, cell survival, and viral replication in primary effusion lymphoma. Biochem. Biophys. Res. Commun. 2016, 469, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Bottero, V.; Sadagopan, S.; Johnson, K.E.; Dutta, S.; Veettil, M.V.; Chandran, B. Kaposi’s Sarcoma-Associated Herpesvirus-Positive Primary Effusion Lymphoma Tumor Formation in NOD/SCID Mice Is Inhibited by Neomycin and Neamine Blocking Angiogenin’s Nuclear Translocation. J. Virol. 2013, 87, 11806–11820. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Rochford, R.; Toomey, L.; Harrington, W.; Feuer, G. Inhibition of HHV-8/KSHV infected primary effusion lymphomas in NOD/SCID mice by azidothymidine and interferon-α. Leuk. Res. 2005, 29, 545–555. [Google Scholar] [CrossRef]
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [PubMed] [Green Version]
- Schieber, M. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [PubMed] [Green Version]
- Ganesan, S.; Chaurasiya, N.D.; Sahu, R.; Walker, L.A.; Tekwani, B.L. Understanding the mechanisms for metabolism-linked hemolytic toxicity of primaquine against glucose 6-phosphate dehydrogenase deficient human erythrocytes: Evaluation of eryptotic pathway. Toxicology 2012, 294, 54–60. [Google Scholar] [CrossRef]
- Duparc, S.; Beutler, E. Glucose-6-Phosphate Dehydrogenase Deficiency and Antimalarial Drug Development. Am. J. Trop. Med. Hyg. 2007, 77, 779–789. [Google Scholar]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta BBA Mol. Cell Res. 2013, 1833, 3460–3470. [Google Scholar]
- Hitomi, J.; Katayama, T.; Eguchi, Y.; Kudo, T.; Taniguchi, M.; Koyama, Y.; Manabe, T.; Yamagishi, S.; Bando, Y.; Imaizumi, K.; et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Aβ-induced cell death. J. Cell Biol. 2004, 165, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Baba, Y.; Shigemi, Z.; Hara, N.; Moriguchi, M.; Ikeda, M.; Watanabe, T.; Fujimuro, M. Arctigenin induces the apoptosis of primary effusion lymphoma cells under conditions of glucose deprivation. Int. J. Oncol. 2017, 52, 505–517. [Google Scholar] [CrossRef]
- Alam, M.M.; Kariya, R.; Kawaguchi, A.; Matsuda, K.; Kudo, E.; Okada, S. Inhibition of autophagy by chloroquine induces apoptosis in primary effusion lymphoma in vitro and in vivo through induction of endoplasmic reticulum stress. Apoptosis 2016, 21, 1191–1201. [Google Scholar] [CrossRef] [PubMed]
- Crawford, R.R.; Prescott, E.T.; Sylvester, C.F.; Higdon, A.N.; Shan, J.; Kilberg, M.S.; Mungrue, I.N. Human CHAC1 Protein Degrades Glutathione, and mRNA Induction Is Regulated by the Transcription Factors ATF4 and ATF3 and a Bipartite ATF/CRE Regulatory Element. J. Biol. Chem. 2015, 290, 15878–15891. [Google Scholar] [CrossRef] [Green Version]
- Mungrue, I.N.; Pagnon, J.; Kohannim, O.; Gargalovic, P.S.; Lusis, A.J. CHAC1/MGC4504 Is a Novel Proapoptotic Component of the Unfolded Protein Response, Downstream of the ATF4-ATF3-CHOP Cascade. J. Immunol. 2009, 182, 466–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purushothaman, P.; Thakker, S.; Verma, S.C. Transcriptome Analysis of Kaposi’s Sarcoma-Associated Herpesvirus during De Novo Primary Infection of Human B and Endothelial Cells. J. Virol. 2015, 89, 3093–3111. [Google Scholar] [CrossRef] [Green Version]
- Baird, K. Origins and implications of neglect of G6PD deficiency and primaquine toxicity in Plasmodium vivax malaria. Pathog. Glob. Health 2015, 109, 93–106. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | BC-3 | BCP-1 | DG-75 | HaCaT | HeLa P4 | MRC-5 | NHDF | NHEK | HUVEC |
|---|---|---|---|---|---|---|---|---|---|
| PQ (µM) | 4.93 | 5.33 | 80.51 | 47.37 | 90.2 | 89.02 | 98.05 | 40.74 | 331.1 |
| Gene | Type | logFC | Status in PQ-Treated Group |
|---|---|---|---|
| ATF3 | Cellular | 1.57 | Up-regulated |
| CCL3 | Cellular | 3.18 | Up-regulated |
| CHAC1 | Cellular | 2.07 | Up-regulated |
| OSGIN1 | Cellular | 1.74 | Up-regulated |
| v-FLIP | Viral (latent) | −0.15 | Not regulated |
| v-Cyclin | Viral (latent) | −0.2 | Not regulated |
| LANA | Viral (latent) | 0.22 | Not regulated |
| RTA | Viral (lytic) | 0.1 | Not regulated |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gothland, A.; Leducq, V.; Grange, P.; Faye, O.; Beauvais Remigereau, L.; Sayon, S.; Désiré, N.; Jary, A.; Laplantine, E.; Maiga, A.I.; et al. Primaquine as a Candidate for HHV-8-Associated Primary Effusion Lymphoma and Kaposi’s Sarcoma Treatment. Cancers 2022, 14, 543. https://doi.org/10.3390/cancers14030543
Gothland A, Leducq V, Grange P, Faye O, Beauvais Remigereau L, Sayon S, Désiré N, Jary A, Laplantine E, Maiga AI, et al. Primaquine as a Candidate for HHV-8-Associated Primary Effusion Lymphoma and Kaposi’s Sarcoma Treatment. Cancers. 2022; 14(3):543. https://doi.org/10.3390/cancers14030543
Chicago/Turabian StyleGothland, Adélie, Valentin Leducq, Philippe Grange, Ousmane Faye, Laurianne Beauvais Remigereau, Sophie Sayon, Nathalie Désiré, Aude Jary, Emmanuel Laplantine, Almoustapha Issiaka Maiga, and et al. 2022. "Primaquine as a Candidate for HHV-8-Associated Primary Effusion Lymphoma and Kaposi’s Sarcoma Treatment" Cancers 14, no. 3: 543. https://doi.org/10.3390/cancers14030543
APA StyleGothland, A., Leducq, V., Grange, P., Faye, O., Beauvais Remigereau, L., Sayon, S., Désiré, N., Jary, A., Laplantine, E., Maiga, A. I., Dupin, N., Marcelin, A.-G., & Calvez, V. (2022). Primaquine as a Candidate for HHV-8-Associated Primary Effusion Lymphoma and Kaposi’s Sarcoma Treatment. Cancers, 14(3), 543. https://doi.org/10.3390/cancers14030543