
Pancreatic Cancer Organoids in the Field of Precision Medicine: A Review of Literature and Experience on Drug Sensitivity Testing with Multiple Readouts and Synergy Scoring

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
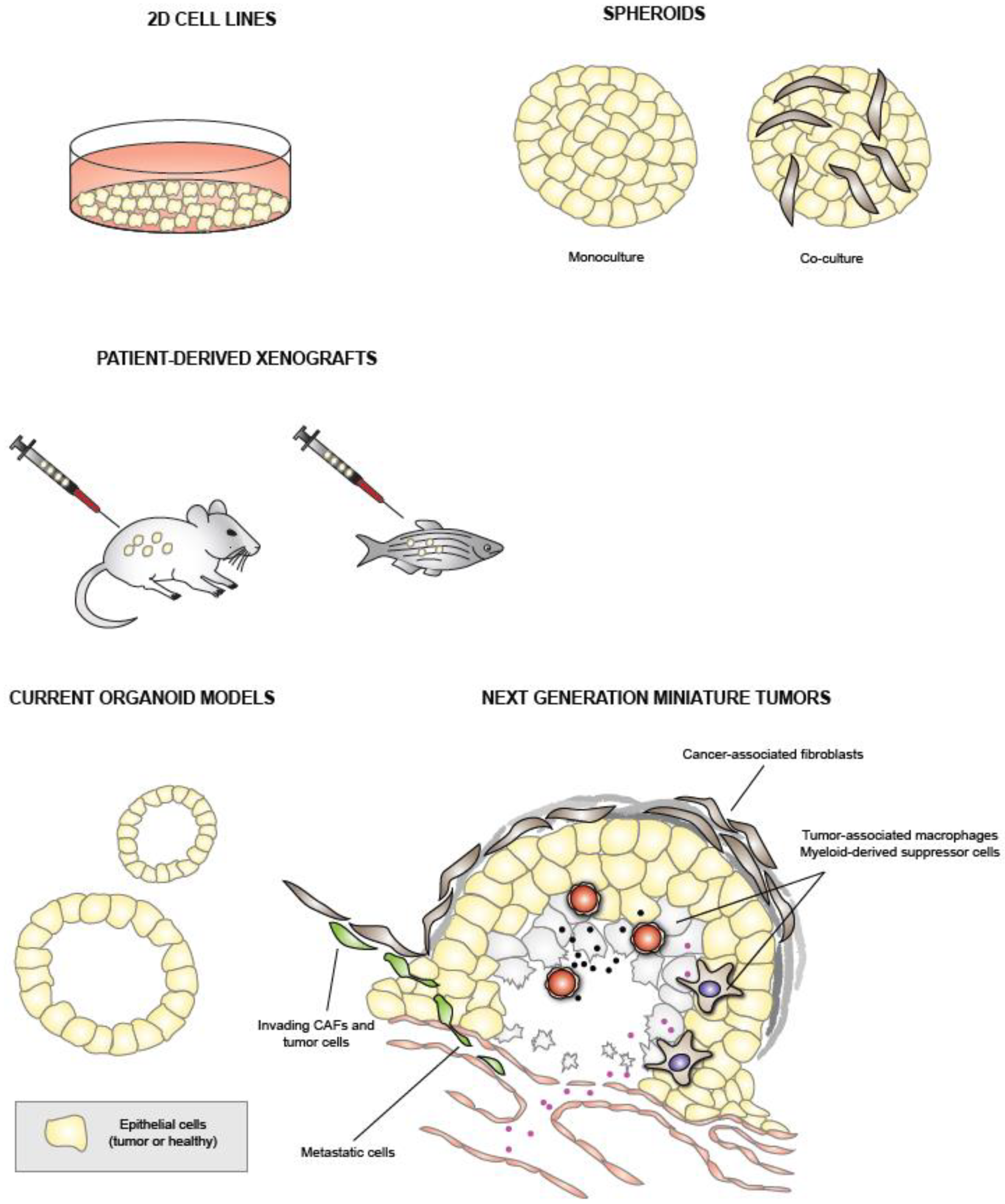
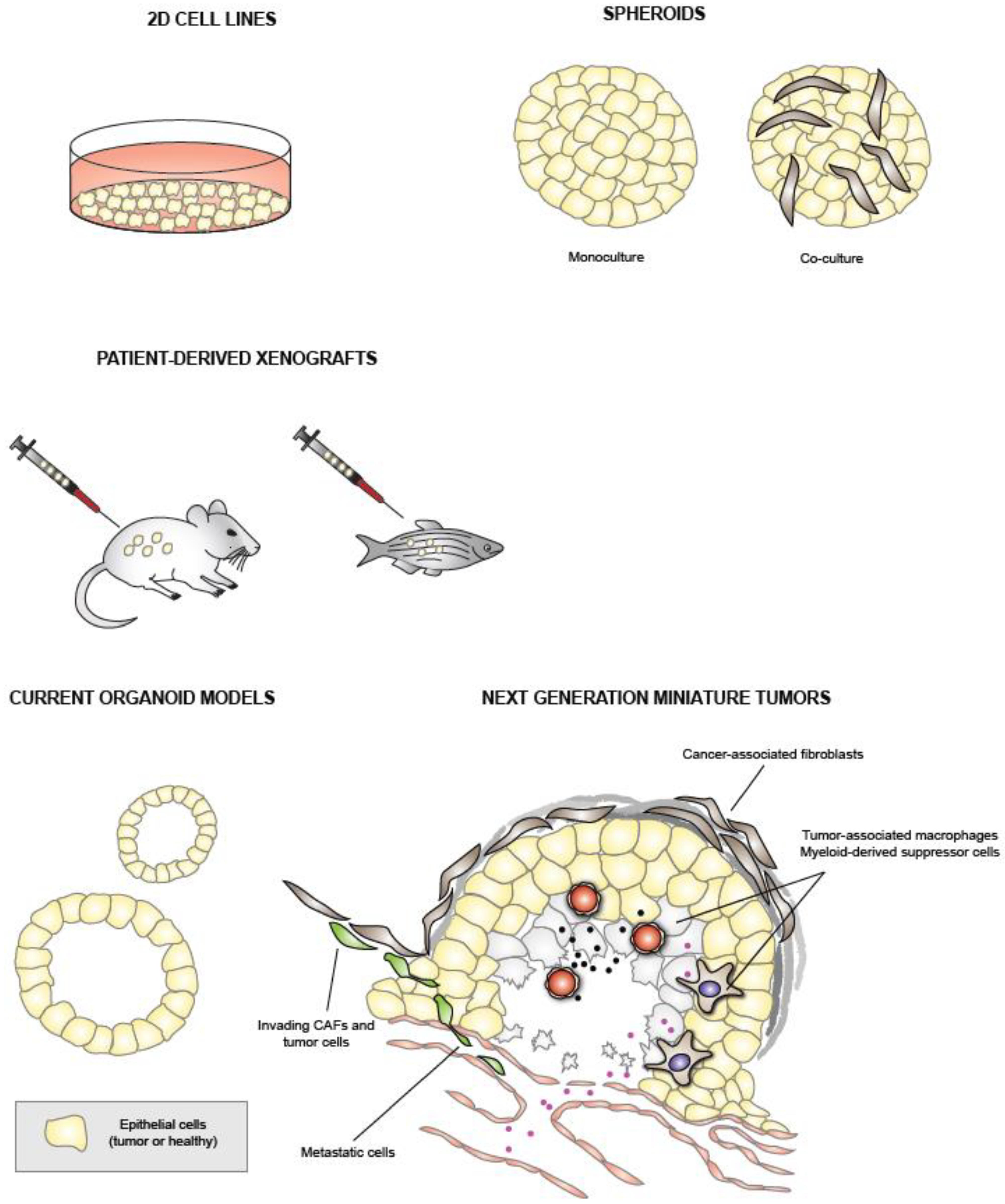
2. Traditional PDAC Models
2.1. PDAC Cell Lines
2.2. Patient-Derived Xenografts
2.3. Genetically Engineered Mouse Models
3. Pancreatic Cancer Organoids
3.1. Organoid Technology
3.2. Organoid Culture Conditions and Success Rates
4. Organoid Applications
4.1. Drug Sensitivity and Resistance Testing and Personalized Medicine
4.2. Neoplasia Modeling
4.3. Co-Cultures
4.4. Organoid Biobanks
5. A Single-Institute Experience: Organoids as a Drug Sensitivity Testing Platform for Pancreatic Cancer
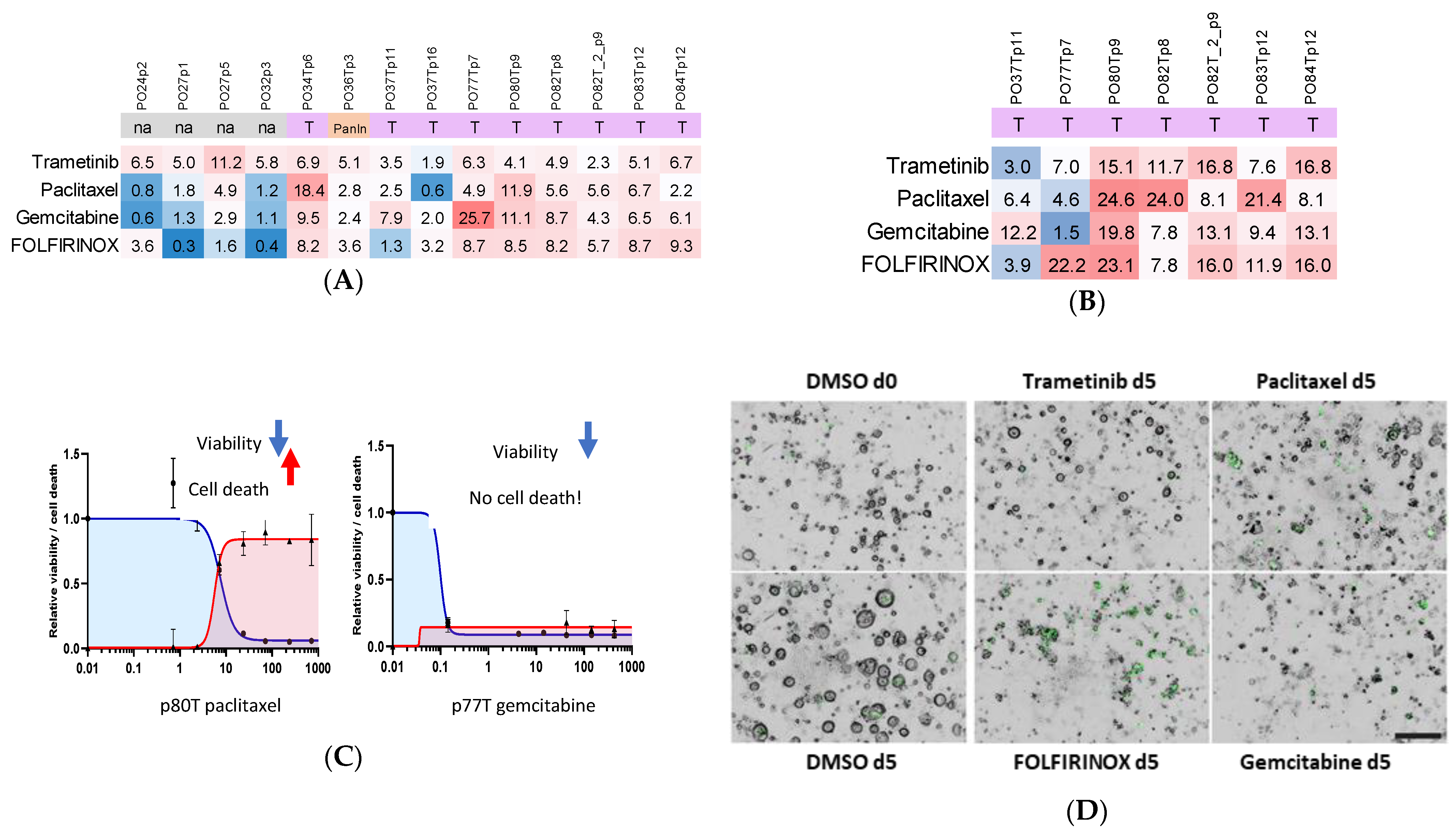
5.1. Organoid Culture, Drug Administration and Response Quantification
5.2. Organoid Viability Responses Show Both Individual and Shared Patterns
5.3. Measuring Organoid Cell Death Together with Viability Important for Future Clinical Correlation Studies
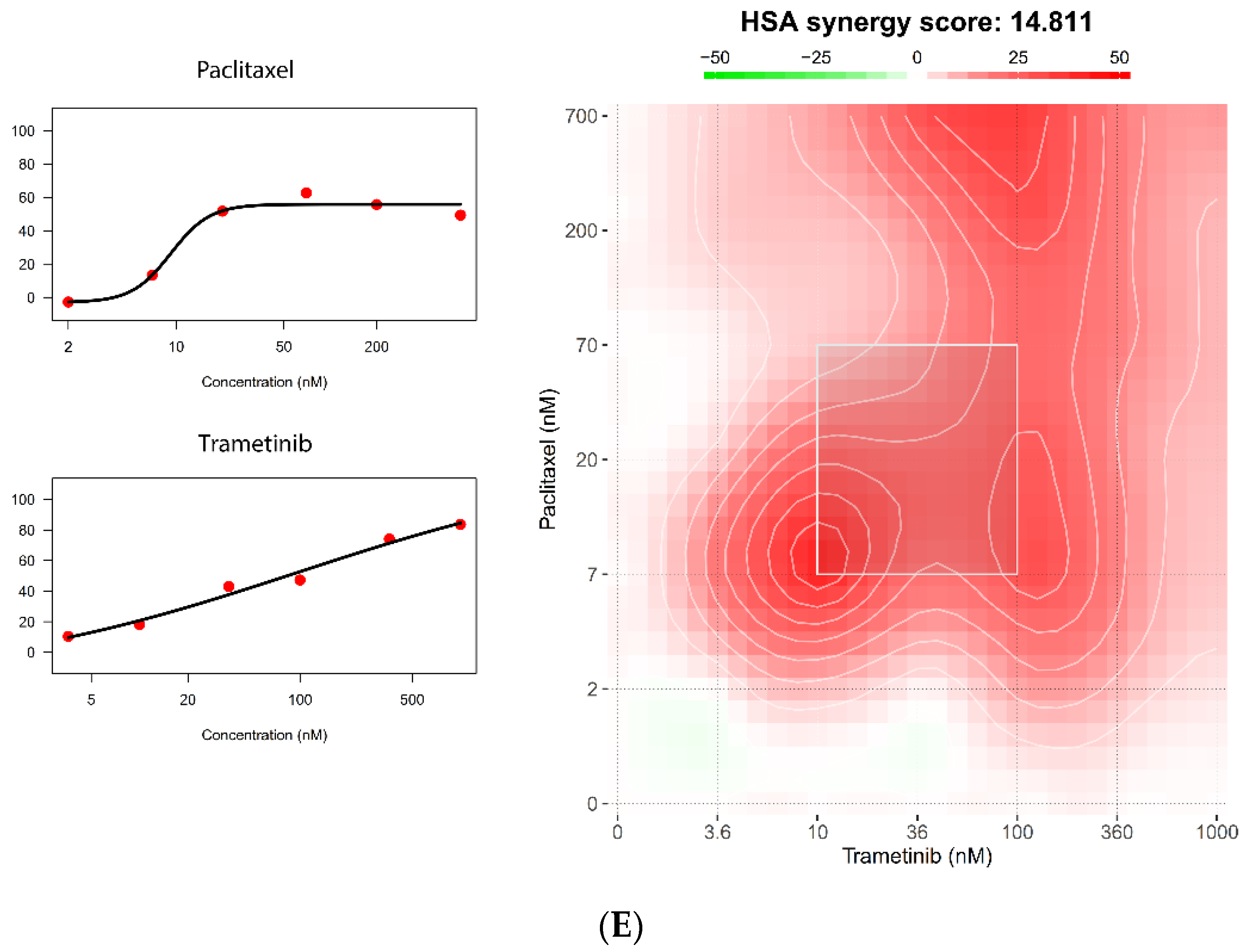
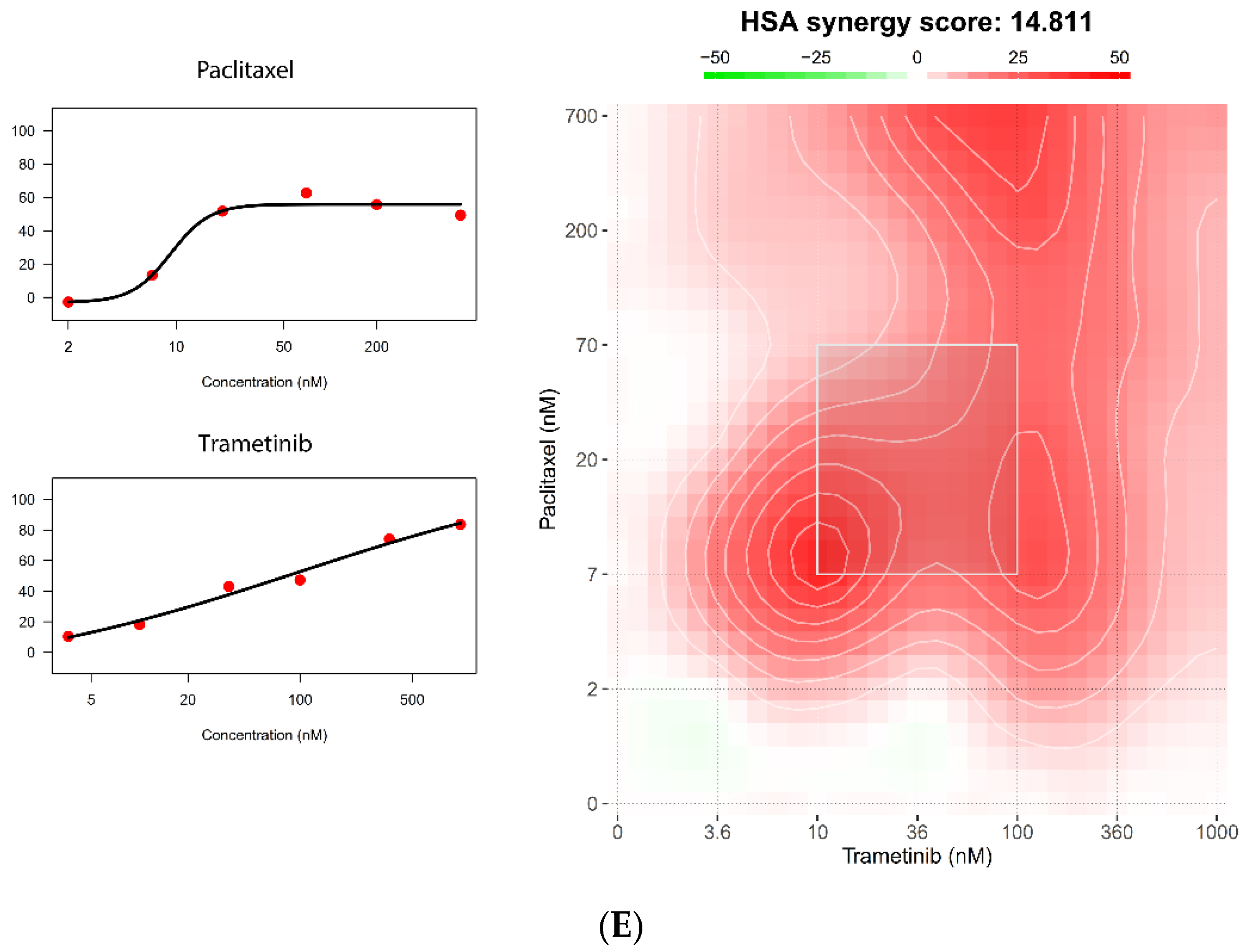
5.4. The 384-Well Organoid Profiling Platform Is Useful for Drug Combination Studies
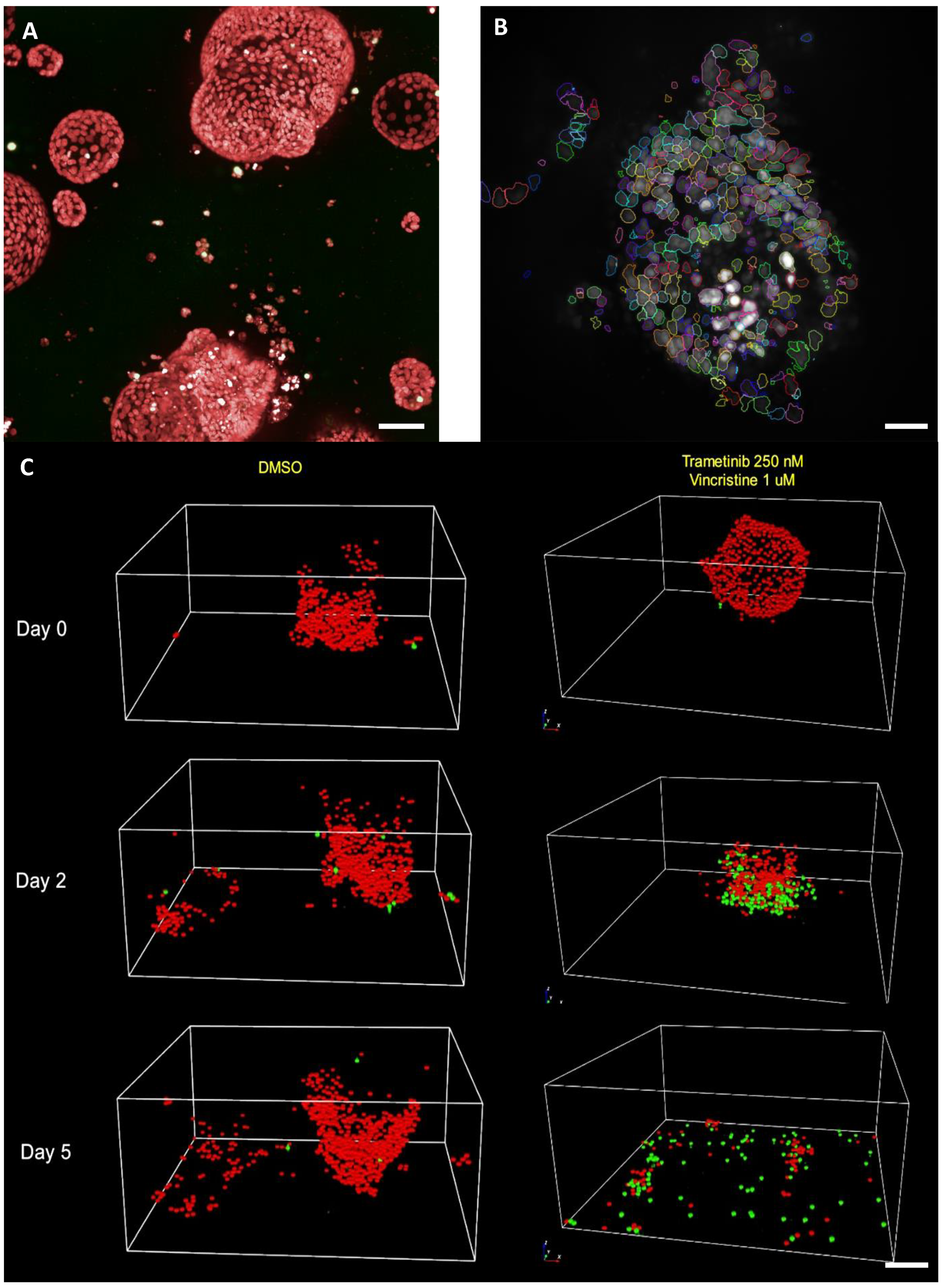
5.5. Single Cell Organoid Drug Sensitivity and Resistance Testing Reveals Single Surviving Tumor Cells
6. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Aaltonen, P.; Carpen, O.; Mustonen, H.; Puolakkainen, P.; Haglund, C.; Peltola, K.; Seppänen, H. Long-term nationwide trends in the treatment of and outcomes among pancreatic cancer patients. Eur. J. Surg. Oncol. 2021. [Google Scholar] [CrossRef]
- Versteijne, E.; Suker, M.; Groothuis, K.; Akkermans-Vogelaar, J.M.; Besselink, M.G.; Bonsing, B.A.; Buijsen, J.; Busch, O.R.; Creemers, G.M.; van Dam, R.M.; et al. Preoperative Chemoradiotherapy Versus Immediate Surgery for Resectable and Borderline Resectable Pancreatic Cancer: Results of the Dutch Randomized Phase III PREOPANC Trial. J. Clin. Oncol. 2020, 38, 1763–1773. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. NEJM 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; De La Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. NEJM 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [Green Version]
- Pusceddu, S.; Ghidini, M.; Torchio, M.; Corti, F.; Tomasello, G.; Niger, M.; Prinzi, N.; Nichetti, F.; Coinu, A.; Di Bartolomeo, M.; et al. Comparative Effectiveness of Gemcitabine plus Nab-Paclitaxel and FOLFIRINOX in the First-Line Setting of Metastatic Pancreatic Cancer: A Systematic Review and Meta-Analysis. Cancers 2019, 11, 484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perri, G.; Prakash, L.; Qiao, W.; Varadhachary, G.R.; Wolff, R.; Fogelman, D.; Overman, M.; Pant, S.; Javle, M.; Koay, E.J.; et al. Response and Survival Associated With First-line FOLFIRINOX vs Gemcitabine and nab-Paclitaxel Chemotherapy for Localized Pancreatic Ductal Adenocarcinoma. JAMA Surgery 2020, 155, 832. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Pajic, M.; Patch, A.-M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cicenas, J.; Kvederaviciute, K.; Meskinyte, I.; Meskinyte-Kausiliene, E.; Skeberdyte, A.; Cicenas, J. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 Mutations in Pancreatic Cancer. Cancers 2017, 9, 42. [Google Scholar] [CrossRef] [Green Version]
- Pishvaian, M.J.; Blais, E.M.; Brody, J.R.; Lyons, E.; DeArbeloa, P.; Hendifar, A.; Mikhail, S.; Chung, V.; Sahai, V.; Sohal, D.P.S.; et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: A retrospective analysis of the Know Your Tumor registry trial. Lancet. Oncol. 2020, 21, 508–518. [Google Scholar] [CrossRef]
- Wattenberg, M.M.; Asch, D.; Yu, S.; O’Dwyer, P.J.; Domchek, S.M.; Nathanson, K.L.; Rosen, M.A.; Beatty, G.L.; Siegelman, E.S.; Reiss, K.A. Platinum response characteristics of patients with pancreatic ductal adenocarcinoma and a germline BRCA1, BRCA2 or PALB2 mutation. Br. J. Cancer 2020, 122, 333–339. [Google Scholar] [CrossRef]
- Torres, C.; Grippo, P.J. Pancreatic cancer subtypes: A roadmap for precision medicine. Ann. Med. 2018, 50, 277–287. [Google Scholar] [CrossRef]
- Aung, K.L.; Fischer, S.E.; Denroche, R.E.; Jang, G.H.; Dodd, A.; Creighton, S.; Southwood, B.; Liang, S.B.; Chadwick, D.; Zhang, A.; et al. Genomics-Driven Precision Medicine for Advanced Pancreatic Cancer: Early Results from the COMPASS Trial. Clin. Cancer Res. 2018, 24, 1344–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.C.; Walker, D. A predictive test for the selection of cancer chemotherapeutic agents for the treatment of human cancer. J. Surg. Oncol. 1975, 7, 381–393. [Google Scholar] [CrossRef]
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Chio, I.I.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [Green Version]
- Dobrynin, Y.V. Establishment and characteristics of cell strains from some epithelial tumors of human origin. J. Natl. Cancer Inst. 1963, 31, 1173–1195. [Google Scholar]
- Gadaleta, E.; Cutts, R.J.; Kelly, G.P.; Crnogorac-Jurcevic, T.; Kocher, H.M.; Lemoine, N.R.; Chelala, C. A global insight into a cancer transcriptional space using pancreatic data: Importance, findings and flaws. Nucleic Acids Res. 2011, 39, 7900–7907. [Google Scholar] [CrossRef]
- Deer, E.L.; González-Hernández, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and Genotype of Pancreatic Cancer Cell Lines. Pancreas 2010, 39, 425–435. [Google Scholar] [CrossRef] [Green Version]
- Longati, P.; Jia, X.; Eimer, J.; Wagman, A.; Witt, M.-R.; Rehnmark, S.; Verbeke, C.; Toftgård, R.; Löhr, M.; Heuchel, R.L. 3D pancreatic carcinoma spheroids induce a matrix-rich, chemoresistant phenotype offering a better model for drug testing. BMC Cancer 2013, 13, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, H.J.; Oh, M.S.; Lee, D.W.; Kuh, H.J. Multiplex quantitative analysis of stroma-mediated cancer cell invasion, matrix remodeling, and drug response in a 3D co-culture model of pancreatic tumor spheroids and stellate cells. J. Exp. Clin. Cancer Res. 2019, 38, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazzari, G.; Nicolas, V.; Matsusaki, M.; Akashi, M.; Couvreur, P.; Mura, S. Multicellular spheroid based on a triple co-culture: A novel 3D model to mimic pancreatic tumor complexity. Acta Biomater. 2018, 78, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Di Modugno, F.; Colosi, C.; Trono, P.; Antonacci, G.; Ruocco, G.; Nistico, P. 3D models in the new era of immune oncology: Focus on T cells, CAF and ECM. J. Exp. Clin. Cancer Res. 2019, 38, 117. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo, M.; Bruckheimer, E.; Rajeshkumar, N.V.; Garrido-Laguna, I.; De Oliveira, E.; Rubio-Viqueira, B.; Strawn, S.; Wick, M.J.; Martell, J.; Sidransky, D. A Pilot Clinical Study of Treatment Guided by Personalized Tumorgrafts in Patients with Advanced Cancer. Mol. Cancer Ther. 2011, 10, 1311–1316. [Google Scholar] [CrossRef] [Green Version]
- Rubio-Viqueira, B.; Jimeno, A.; Cusatis, G.; Zhang, X.; Iacobuzio-Donahue, C.; Karikari, C.; Shi, C.; Danenberg, K.; Danenberg, P.V.; Kuramochi, H.; et al. An In vivo Platform for Translational Drug Development in Pancreatic Cancer. Clin. Cancer Res. 2006, 12, 4652–4661. [Google Scholar] [CrossRef] [Green Version]
- Ben-David, U.; Ha, G.; Tseng, Y.Y.; Greenwald, N.F.; Oh, C.; Shih, J.; McFarland, J.M.; Wong, B.; Boehm, J.S.; Beroukhim, R.; et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat. Genet. 2017, 49, 1567–1575. [Google Scholar] [CrossRef] [Green Version]
- Barriuso, J.; Nagaraju, R.; Hurlstone, A. Zebrafish: A new companion for translational research in oncology. Clin. Cancer Res. 2015, 21, 969–975. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.Q.; Zhai, J.; Li, C.Y.; Tan, A.M.; Wei, P.; Shen, L.Z.; He, M.F. Patient-derived xenograft in zebrafish embryos: A new platform for translational research in gastric cancer. J. Exp. Clin. Cancer Res. 2017, 36, 160. [Google Scholar] [CrossRef] [Green Version]
- Al-Samadi, A.; Tuomainen, K.; Kivimaki, A.; Salem, A.; Al-Kubati, S.; Hyytiainen, A.; Parikka, M.; Mesimaki, K.; Wilkman, T.; Makitie, A.; et al. PCR-based zebrafish model for personalised medicine in head and neck cancer. J. Transl. Med. 2019, 17, 235. [Google Scholar] [CrossRef]
- Di Franco, G.; Usai, A.; Funel, N.; Palmeri, M.; Montesanti, I.E.R.; Bianchini, M.; Gianardi, D.; Furbetta, N.; Guadagni, S.; Vasile, E.; et al. Use of zebrafish embryos as avatar of patients with pancreatic cancer: A new xenotransplantation model towards personalized medicine. World J. Gastroenterol. 2020, 26, 2792–2809. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.W.; Komar, C.A.; Bengsch, F.; Graham, K.; Beatty, G.L. Genetically Engineered Mouse Models of Pancreatic Cancer: The KPC Model (LSL-Kras(G12D/+); LSL-Trp53(R172H/+); Pdx-1-Cre), Its Variants, and Their Application in Immuno-oncology Drug Discovery. Curr. Protoc. Pharmacol. 2016, 73, 143911–143920. [Google Scholar] [CrossRef] [Green Version]
- Evans, R.A.; Diamond, M.S.; Rech, A.J.; Chao, T.; Richardson, M.W.; Lin, J.H.; Bajor, D.L.; Byrne, K.T.; Stanger, B.Z.; Riley, J.L.; et al. Lack of immunoediting in murine pancreatic cancer reversed with neoantigen. JCI Insight 2016, 1, e88328. [Google Scholar] [CrossRef]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef]
- Huch, M.; Bonfanti, P.; Boj, S.F.; Sato, T.; Loomans, C.J.M.; Van De Wetering, M.; Sojoodi, M.; Li, V.S.W.; Schuijers, J.; Gracanin, A.; et al. Unlimited in vitro expansion of adult bi-potent pancreas progenitors through the Lgr5/R-spondin axis. EMBO J. 2013, 32, 2708–2721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.J.; Van Es, J.H.; Van Den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term Expansion of Epithelial Organoids from Human Colon, Adenoma, Adenocarcinoma, and Barrett’s Epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef]
- van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [Green Version]
- Broutier, L.; Andersson-Rolf, A.; Hindley, C.J.; Boj, S.F.; Clevers, H.; Koo, B.-K.; Huch, M. Culture and establishment of self-renewing human and mouse adult liver and pancreas 3D organoids and their genetic manipulation. Nat. Protoc. 2016, 11, 1724–1743. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Holtzinger, A.; Jagan, I.; Begora, M.; Lohse, I.; Ngai, N.; Nostro, C.; Wang, R.; Muthuswamy, L.B.; Crawford, H.C.; et al. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat. Med. 2015, 21, 1364–1371. [Google Scholar] [CrossRef] [PubMed]
- Walsh, A.J.; Castellanos, J.A.; Nagathihalli, N.S.; Merchant, N.B.; Skala, M.C. Optical Imaging of Drug-Induced Metabolism Changes in Murine and Human Pancreatic Cancer Organoids Reveals Heterogeneous Drug Response. Pancreas 2016, 45, 863–869. [Google Scholar] [CrossRef]
- Seino, T.; Kawasaki, S.; Shimokawa, M.; Tamagawa, H.; Toshimitsu, K.; Fujii, M.; Ohta, Y.; Matano, M.; Nanki, K.; Kawasaki, K.; et al. Human Pancreatic Tumor Organoids Reveal Loss of Stem Cell Niche Factor Dependence during Disease Progression. Cell Stem Cell 2018, 22, 454–467.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, L.A.; Tiriac, H.; Tuveson, D.A. Generation and Culture of Human Pancreatic Ductal Adenocarcinoma Organoids from Resected Tumor Specimens; Humana Press: New York, NY, USA, 2019; pp. 97–115. [Google Scholar] [CrossRef]
- Li, X.; Nadauld, L.; Ootani, A.; Corney, D.C.; Pai, R.K.; Gevaert, O.; Cantrell, M.A.; Rack, P.G.; Neal, J.T.; Chan, C.W.M.; et al. Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nat. Med. 2014, 20, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Mihara, E.; Hirai, H.; Yamamoto, H.; Tamura-Kawakami, K.; Matano, M.; Kikuchi, A.; Sato, T.; Takagi, J. Active and water-soluble form of lipidated Wnt protein is maintained by a serum glycoprotein afamin/alpha-albumin. eLife 2016, 5, e11621. [Google Scholar] [CrossRef]
- Tuysuz, N.; van Bloois, L.; van den Brink, S.; Begthel, H.; Verstegen, M.M.; Cruz, L.J.; Hui, L.; van der Laan, L.J.; de Jonge, J.; Vries, R.; et al. Lipid-mediated Wnt protein stabilization enables serum-free culture of human organ stem cells. Nat. Commun. 2017, 8, 14578. [Google Scholar] [CrossRef]
- Sackett, S.D.; Tremmel, D.M.; Ma, F.; Feeney, A.K.; Maguire, R.M.; Brown, M.E.; Zhou, Y.; Li, X.; O’Brien, C.; Li, L.; et al. Extracellular matrix scaffold and hydrogel derived from decellularized and delipidized human pancreas. Sci. Rep. 2018, 8, 10452. [Google Scholar] [CrossRef]
- Broguiere, N.; Isenmann, L.; Hirt, C.; Ringel, T.; Placzek, S.; Cavalli, E.; Ringnalda, F.; Villiger, L.; Zullig, R.; Lehmann, R.; et al. Growth of Epithelial Organoids in a Defined Hydrogel. Adv. Mater. 2018, 30, e1801621. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulos, N.; Prior, N.; Angres, B.; Mastrogiovanni, G.; Cagan, A.; Harrison, D.; Hindley, C.J.; Arnes-Benito, R.; Liau, S.-S.; Curd, A.; et al. Long-term expansion, genomic stability and in vivo safety of adult human pancreas organoids. BMC Dev. Biol. 2020, 20, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Tiriac, H.; Belleau, P.; Engle, D.D.; Plenker, D.; Deschenes, A.; Somerville, T.D.D.; Froeling, F.E.M.; Burkhart, R.A.; Denroche, R.E.; Jang, G.H.; et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov. 2018, 8, 1112–1129. [Google Scholar] [CrossRef] [Green Version]
- Tiriac, H.; Bucobo, J.C.; Tzimas, D.; Grewel, S.; Lacomb, J.F.; Rowehl, L.M.; Nagula, S.; Wu, M.; Kim, J.; Sasson, A.; et al. Successful creation of pancreatic cancer organoids by means of EUS-guided fine-needle biopsy sampling for personalized cancer treatment. Gastrointest. Endosc. 2018, 87, 1474–1480. [Google Scholar] [CrossRef] [PubMed]
- Seppälä, T.T.; Zimmerman, J.W.; Sereni, E.; Plenker, D.; Suri, R.; Rozich, N.; Blair, A.; Thomas, D.L.; Teinor, J.; Javed, A.; et al. Patient-derived Organoid Pharmacotyping is a Clinically Tractable Strategy for Precision Medicine in Pancreatic Cancer. Ann. Surg. 2020, 272, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Driehuis, E.; Van Hoeck, A.; Moore, K.; Kolders, S.; Francies, H.E.; Gulersonmez, M.C.; Stigter, E.C.A.; Burgering, B.; Geurts, V.; Gracanin, A.; et al. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc. Natl. Acad. Sci. USA 2019, 116, 26580–26590. [Google Scholar] [CrossRef]
- Grossman, J.E.; Muthuswamy, L.; Huang, L.; Akshinthala, D.; Perea, S.; Gonzalez, R.S.; Tsai, L.L.; Cohen, J.; Bockorny, B.; Bullock, A.J.; et al. Organoid Sensitivity Correlates with Therapeutic Response in Patients with Pancreatic Cancer. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Hou, S.; Tiriac, H.; Sridharan, B.P.; Scampavia, L.; Madoux, F.; Seldin, J.; Souza, G.R.; Watson, D.; Tuveson, D.; Spicer, T.P. Advanced Development of Primary Pancreatic Organoid Tumor Models for High-Throughput Phenotypic Drug Screening. SLAS Discov. 2018, 23, 574–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghavan, S.; Winter, P.S.; Navia, A.W.; Williams, H.L.; DenAdel, A.; Lowder, K.E.; Galvez-Reyes, J.; Kalekar, R.L.; Mulugeta, N.; Kapner, K.S.; et al. Microenvironment drives cell state, plasticity, and drug response in pancreatic cancer. Cell 2021, 184, 6119–6137.e26. [Google Scholar] [CrossRef]
- Porter, R.L.; Magnus, N.K.C.; Thapar, V.; Morris, R.; Szabolcs, A.; Neyaz, A.; Kulkarni, A.S.; Tai, E.; Chougule, A.; Hillis, A.; et al. Epithelial to mesenchymal plasticity and differential response to therapies in pancreatic ductal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2019, 116, 26835–26845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osuna de la Pena, D.; Trabulo, S.M.D.; Collin, E.; Liu, Y.; Sharma, S.; Tatari, M.; Behrens, D.; Erkan, M.; Lawlor, R.T.; Scarpa, A.; et al. Bioengineered 3D models of human pancreatic cancer recapitulate in vivo tumour biology. Nat. Commun. 2021, 12, 5623. [Google Scholar] [CrossRef]
- Zhang, Z.; Shiratsuchi, H.; Lin, J.; Chen, G.; Reddy, R.M.; Azizi, E.; Fouladdel, S.; Chang, A.C.; Lin, L.; Jiang, H.; et al. Expansion of CTCs from early stage lung cancer patients using a microfluidic co-culture model. Oncotarget 2014, 5, 12383–12397. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnoletti, J.P.; Fanaian, N.; Reza, J.; Sause, R.; Almodovar, A.J.; Srivastava, M.; Patel, S.; Veldhuis, P.P.; Griffith, E.; Shao, Y.P.; et al. Pancreatic and bile duct cancer circulating tumor cells (CTC) form immune-resistant multi-cell type clusters in the portal venous circulation. Cancer Biol. Ther. 2018, 19, 887–897. [Google Scholar] [CrossRef] [Green Version]
- Sharick, J.T.; Walsh, C.M.; Sprackling, C.M.; Pasch, C.A.; Pham, D.L.; Esbona, K.; Choudhary, A.; Garcia-Valera, R.; Burkard, M.E.; McGregor, S.M.; et al. Metabolic Heterogeneity in Patient Tumor-Derived Organoids by Primary Site and Drug Treatment. Front. Oncol. 2020, 10, 553. [Google Scholar] [CrossRef] [PubMed]
- Farshadi, E.A.; Chang, J.; Sampadi, B.; Doukas, M.; Van’t Land, F.; van der Sijde, F.; Vietsch, E.E.; Pothof, J.; Koerkamp, B.G.; van Eijck, C.H.J. Organoids Derived from Neoadjuvant FOLFIRINOX Patients Recapitulate Therapy Resistance in Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2021, 27, 6602–6612. [Google Scholar] [CrossRef]
- Wensink, G.E.; Elias, S.G.; Mullenders, J.; Koopman, M.; Boj, S.F.; Kranenburg, O.W.; Roodhart, J.M.L. Patient-derived organoids as a predictive biomarker for treatment response in cancer patients. NPJ Precis. Oncol. 2021, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, G.; Mato-Berciano, A.; Pascual-Sabater, S.; Rovira-Rigau, M.; Cuatrecasas, M.; Fondevila, C.; Sanchez-Cabus, S.; Begthel, H.; Boj, S.F.; Clevers, H.; et al. Patient-derived pancreatic tumour organoids identify therapeutic responses to oncolytic adenoviruses. EBioMedicine 2020, 56, 102786. [Google Scholar] [CrossRef] [PubMed]
- Miyabayashi, K.; Baker, L.A.; Deschenes, A.; Traub, B.; Caligiuri, G.; Plenker, D.; Alagesan, B.; Belleau, P.; Li, S.; Kendall, J.; et al. Intraductal Transplantation Models of Human Pancreatic Ductal Adenocarcinoma Reveal Progressive Transition of Molecular Subtypes. Cancer Discov. 2020, 10, 1566–1589. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Huang, C.; Cui Zhou, D.; Hu, Y.; Lih, T.M.; Savage, S.R.; Krug, K.; Clark, D.J.; Schnaubelt, M.; Chen, L.; et al. Proteogenomic characterization of pancreatic ductal adenocarcinoma. Cell 2021, 184, 5031–5052.e26. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [Green Version]
- Garrido-Laguna, I.; Uson, M.; Rajeshkumar, N.V.; Tan, A.C.; de Oliveira, E.; Karikari, C.; Villaroel, M.C.; Salomon, A.; Taylor, G.; Sharma, R.; et al. Tumor engraftment in nude mice and enrichment in stroma- related gene pathways predict poor survival and resistance to gemcitabine in patients with pancreatic cancer. Clin. Cancer Res. 2011, 17, 5793–5800. [Google Scholar] [CrossRef] [Green Version]
- Grunwald, B.T.; Devisme, A.; Andrieux, G.; Vyas, F.; Aliar, K.; McCloskey, C.W.; Macklin, A.; Jang, G.H.; Denroche, R.; Romero, J.M.; et al. Spatially confined sub-tumor microenvironments in pancreatic cancer. Cell 2021, 184, 5577–5592.e18. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liang, Y.; Xu, H.; Zhang, X.; Mao, T.; Cui, J.; Yao, J.; Wang, Y.; Jiao, F.; Xiao, X.; et al. Single-cell analysis of pancreatic ductal adenocarcinoma identifies a novel fibroblast subtype associated with poor prognosis but better immunotherapy response. Cell Discov. 2021, 7, 36. [Google Scholar] [CrossRef]
- Dominguez, C.X.; Muller, S.; Keerthivasan, S.; Koeppen, H.; Hung, J.; Gierke, S.; Breart, B.; Foreman, O.; Bainbridge, T.W.; Castiglioni, A.; et al. Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15(+) Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov. 2020, 10, 232–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krieger, T.G.; Le Blanc, S.; Jabs, J.; Ten, F.W.; Ishaque, N.; Jechow, K.; Debnath, O.; Leonhardt, C.S.; Giri, A.; Eils, R.; et al. Single-cell analysis of patient-derived PDAC organoids reveals cell state heterogeneity and a conserved developmental hierarchy. Nat. Commun. 2021, 12, 5826. [Google Scholar] [CrossRef]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [Green Version]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutton, C.; Heider, F.; Blanco-Gomez, A.; Banyard, A.; Kononov, A.; Zhang, X.; Karim, S.; Paulus-Hock, V.; Watt, D.; Steele, N.; et al. Single-cell analysis defines a pancreatic fibroblast lineage that supports anti-tumor immunity. Cancer Cell 2021, 39, 1227–1244.e20. [Google Scholar] [CrossRef]
- Finnberg, N.K.; Gokare, P.; Lev, A.; Grivennikov, S.I.; MacFarlane, A.W.t.; Campbell, K.S.; Winters, R.M.; Kaputa, K.; Farma, J.M.; Abbas, A.E.; et al. Application of 3D tumoroid systems to define immune and cytotoxic therapeutic responses based on tumoroid and tissue slice culture molecular signatures. Oncotarget 2017, 8, 66747–66757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988.e16. [Google Scholar] [CrossRef] [Green Version]
- Tsai, S.; McOlash, L.; Palen, K.; Johnson, B.; Duris, C.; Yang, Q.; Dwinell, M.B.; Hunt, B.; Evans, D.B.; Gershan, J.; et al. Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer 2018, 18, 335. [Google Scholar] [CrossRef]
- Holokai, L.; Chakrabarti, J.; Lundy, J.; Croagh, D.; Adhikary, P.; Richards, S.S.; Woodson, C.; Steele, N.; Kuester, R.; Scott, A.; et al. Murine- and Human-Derived Autologous Organoid/Immune Cell Co-Cultures as Pre-Clinical Models of Pancreatic Ductal Adenocarcinoma. Cancers 2020, 12, 3816. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.I.; Jang, S.I.; Hong, J.; Kim, C.H.; Kwon, S.S.; Park, J.S.; Lim, J.B. Cancer-initiating cells in human pancreatic cancer organoids are maintained by interactions with endothelial cells. Cancer Lett. 2021, 498, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Koikawa, K.; Ohuchida, K.; Ando, Y.; Kibe, S.; Nakayama, H.; Takesue, S.; Endo, S.; Abe, T.; Okumura, T.; Iwamoto, C.; et al. Basement membrane destruction by pancreatic stellate cells leads to local invasion in pancreatic ductal adenocarcinoma. Cancer Lett. 2018, 425, 65–77. [Google Scholar] [CrossRef]
- Gendoo, D.M.A.; Denroche, R.E.; Zhang, A.; Radulovich, N.; Jang, G.H.; Lemire, M.; Fischer, S.; Chadwick, D.; Lungu, I.M.; Ibrahimov, E.; et al. Whole genomes define concordance of matched primary, xenograft, and organoid models of pancreas cancer. PLoS Comput. Biol. 2019, 15, e1006596. [Google Scholar] [CrossRef] [Green Version]
- Pemovska, T.; Johnson, E.; Kontro, M.; Repasky, G.A.; Chen, J.; Wells, P.; Cronin, C.N.; McTigue, M.; Kallioniemi, O.; Porkka, K.; et al. Axitinib effectively inhibits BCR-ABL1(T315I) with a distinct binding conformation. Nature 2015, 519, 102–105. [Google Scholar] [CrossRef]
- Pemovska, T.; Kontro, M.; Yadav, B.; Edgren, H.; Eldfors, S.; Szwajda, A.; Almusa, H.; Bespalov, M.M.; Ellonen, P.; Elonen, E.; et al. Individualized systems medicine strategy to tailor treatments for patients with chemorefractory acute myeloid leukemia. Cancer Discov. 2013, 3, 1416–1429. [Google Scholar] [CrossRef] [Green Version]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 2.0: Visual analytics of multi-drug combination synergies. Nucleic Acids. Res. 2020, 48, W488–W493. [Google Scholar] [CrossRef]
- Awasthi, N.; Kronenberger, D.; Stefaniak, A.; Hassan, M.S.; von Holzen, U.; Schwarz, M.A.; Schwarz, R.E. Dual inhibition of the PI3K and MAPK pathways enhances nab-paclitaxel/gemcitabine chemotherapy response in preclinical models of pancreatic cancer. Cancer Lett. 2019, 459, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Chung, V.; McDonough, S.; Philip, P.A.; Cardin, D.; Wang-Gillam, A.; Hui, L.; Tejani, M.A.; Seery, T.E.; Dy, I.A.; Al Baghdadi, T.; et al. Effect of Selumetinib and MK-2206 vs. Oxaliplatin and Fluorouracil in Patients with Metastatic Pancreatic Cancer after Prior Therapy: SWOG S1115 Study Randomized Clinical Trial. JAMA Oncol. 2017, 3, 516–522. [Google Scholar] [CrossRef]
- Kun, E.; Tsang, Y.T.M.; Ng, C.W.; Gershenson, D.M.; Wong, K.K. MEK inhibitor resistance mechanisms and recent developments in combination trials. Cancer Treat Rev. 2021, 92, 102137. [Google Scholar] [CrossRef]
- Vena, F.; Li Causi, E.; Rodriguez-Justo, M.; Goodstal, S.; Hagemann, T.; Hartley, J.A.; Hochhauser, D. The MEK1/2 Inhibitor Pimasertib Enhances Gemcitabine Efficacy in Pancreatic Cancer Models by Altering Ribonucleotide Reductase Subunit-1 (RRM1). Clin. Cancer Res. 2015, 21, 5563–5577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, K.; Bilal, F.; Bernado Morales, C.; Salcedo, M.T.; Macarulla, T.; Masso-Valles, D.; Mohan, V.; Vivancos, A.; Carreras, M.J.; Serres, X.; et al. Pancreatic cancer heterogeneity and response to Mek inhibition. Oncogene 2017, 36, 5639–5647. [Google Scholar] [CrossRef]
- Garrido-Laguna, I.; Hidalgo, M. Pancreatic cancer: From state-of-the-art treatments to promising novel therapies. Nat. Rev. Clin. Oncol. 2015, 12, 319–334. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Hidalgo, M.; Canon, J.L.; Macarulla, T.; Bazin, I.; Poddubskaya, E.; Manojlovic, N.; Radenkovic, D.; Verslype, C.; Raymond, E.; et al. Phase I/II trial of pimasertib plus gemcitabine in patients with metastatic pancreatic cancer. Int. J. Cancer 2018, 143, 2053–2064. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.R.; Somer, B.G.; Park, J.O.; Li, C.P.; Scheulen, M.E.; Kasubhai, S.M.; Oh, D.Y.; Liu, Y.; Redhu, S.; Steplewski, K.; et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur. J. Cancer 2014, 50, 2072–2081. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Brosnan, J.A.; Blackford, A.L.; Sur, S.; Hruban, R.H.; Kinzler, K.W.; Vogelstein, B.; Maitra, A.; Diaz, L.A., Jr.; Iacobuzio-Donahue, C.A.; et al. Genetically defined subsets of human pancreatic cancer show unique in vitro chemosensitivity. Clin. Cancer. Res. 2012, 18, 6519–6530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Julkunen, H.; Cichonska, A.; Gautam, P.; Szedmak, S.; Douat, J.; Pahikkala, T.; Aittokallio, T.; Rousu, J. Leveraging multi-way interactions for systematic prediction of pre-clinical drug combination effects. Nat. Commun. 2020, 11, 6136. [Google Scholar] [CrossRef] [PubMed]
- Hayes, T.K.; Neel, N.F.; Hu, C.; Gautam, P.; Chenard, M.; Long, B.; Aziz, M.; Kassner, M.; Bryant, K.L.; Pierobon, M.; et al. Long-Term ERK Inhibition in KRAS-Mutant Pancreatic Cancer Is Associated with MYC Degradation and Senescence-like Growth Suppression. Cancer Cell 2016, 29, 75–89. [Google Scholar] [CrossRef] [Green Version]
- Hadj Bachir, E.; Poiraud, C.; Paget, S.; Stoup, N.; El Moghrabi, S.; Duchêne, B.; Jouy, N.; Bongiovanni, A.; Tardivel, M.; Weiswald, L.B.; et al. A new pancreatic adenocarcinoma-derived organoid model of acquired chemoresistance to FOLFIRINOX: First insight of the underlying mechanisms. Biol. Cell 2022, 114, 32–55. [Google Scholar] [CrossRef]
- Peng, J.; Sun, B.F.; Chen, C.Y.; Zhou, J.Y.; Chen, Y.S.; Chen, H.; Liu, L.; Huang, D.; Jiang, J.; Cui, G.S.; et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019, 29, 725–738. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | Mutations | Drug Screen | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | Surgical Site | Diagnosis | TNM | Grade | KRAS | TP53 | Others | Final ID | Passage | Readout | ||||
| Tissue | Organoid | Codon | Tissue | Organoid | CTG | CTX | ||||||||
| PO24 | Tumor | PDAC | pT3N1 | G2 | na | na | na | na | na | na | PO24 | p2 | 1 | 0 |
| PO27 | Tumor | PDAC | pT2N1 | G2 | na | na | na | na | na | na | PO27 | p1 | 1 | 0 |
| p5 | 1 | 0 | ||||||||||||
| PO32 | Tumor | PDAC | pT3N1 | G2-3 | na | na | na | na | na | na | PO32 | p3 | 1 | 0 |
| PO34 | Tumor | PDAC | pT2N0 | G2 | 46.8 | 47 | G12D | 100 | 100 | CDKN2A-/-, SMAD4-/- | PO34T | p6 | 1 | 0 |
| PO36 | Tumor | PanIN2 | na | na | na | 48 | G13D | 0 | 0 | 0 | PO36PanIN | p3 | 1 | 0 |
| PO37 | Tumor | PDAC | pT3N2 | G2 | 0 | 100 | G12D | 0 | 100 | CDKN2A-/- | PO37T | p11 | 1 | 1 |
| p16 | 1 | 0 | ||||||||||||
| PO77 | Tumor | PDAC | pT3N1 | G3 | 48.7 | 64.1 | G12D | 38.6 | 97.7 | 0 | PO77T | p7 | 1 | 1 |
| PO80 | Tumor | PDAC | pT2N1 | G2 | na | 66.3 | G12D | na | 100 | 0 | PO80T | p9 | 1 | 1 |
| PO82 | Tumor | PDAC | pT2N1 | G2 | na | 58 | G12R | na | 99 | CDKN2A-/- | PO82T | p8 | 1 | 1 |
| Adjacent | G2 | na | 72 | G12R | na | 98 | 0 | PO82T_2 | p9 | 1 | 1 | |||
| PO83 | Tumor | PDAC | pT3N2R1 | G2 | na | 69 | G12D | na | 100 | 0 | PO83T | p12 | 1 | 1 |
| PO84 | Tumor | PDAC | pT2N1 | G2 | na | 48 | G12D | na | na | 0 | PO84T | p12 | 1 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mäkinen, L.; Vähä-Koskela, M.; Juusola, M.; Mustonen, H.; Wennerberg, K.; Hagström, J.; Puolakkainen, P.; Seppänen, H. Pancreatic Cancer Organoids in the Field of Precision Medicine: A Review of Literature and Experience on Drug Sensitivity Testing with Multiple Readouts and Synergy Scoring. Cancers 2022, 14, 525. https://doi.org/10.3390/cancers14030525
Mäkinen L, Vähä-Koskela M, Juusola M, Mustonen H, Wennerberg K, Hagström J, Puolakkainen P, Seppänen H. Pancreatic Cancer Organoids in the Field of Precision Medicine: A Review of Literature and Experience on Drug Sensitivity Testing with Multiple Readouts and Synergy Scoring. Cancers. 2022; 14(3):525. https://doi.org/10.3390/cancers14030525
Chicago/Turabian StyleMäkinen, Lotta, Markus Vähä-Koskela, Matilda Juusola, Harri Mustonen, Krister Wennerberg, Jaana Hagström, Pauli Puolakkainen, and Hanna Seppänen. 2022. "Pancreatic Cancer Organoids in the Field of Precision Medicine: A Review of Literature and Experience on Drug Sensitivity Testing with Multiple Readouts and Synergy Scoring" Cancers 14, no. 3: 525. https://doi.org/10.3390/cancers14030525
APA StyleMäkinen, L., Vähä-Koskela, M., Juusola, M., Mustonen, H., Wennerberg, K., Hagström, J., Puolakkainen, P., & Seppänen, H. (2022). Pancreatic Cancer Organoids in the Field of Precision Medicine: A Review of Literature and Experience on Drug Sensitivity Testing with Multiple Readouts and Synergy Scoring. Cancers, 14(3), 525. https://doi.org/10.3390/cancers14030525