
Mechanisms of Drug Resistance in Ovarian Cancer and Associated Gene Targets

Abstract
:Simple Summary
Abstract
1. Introduction
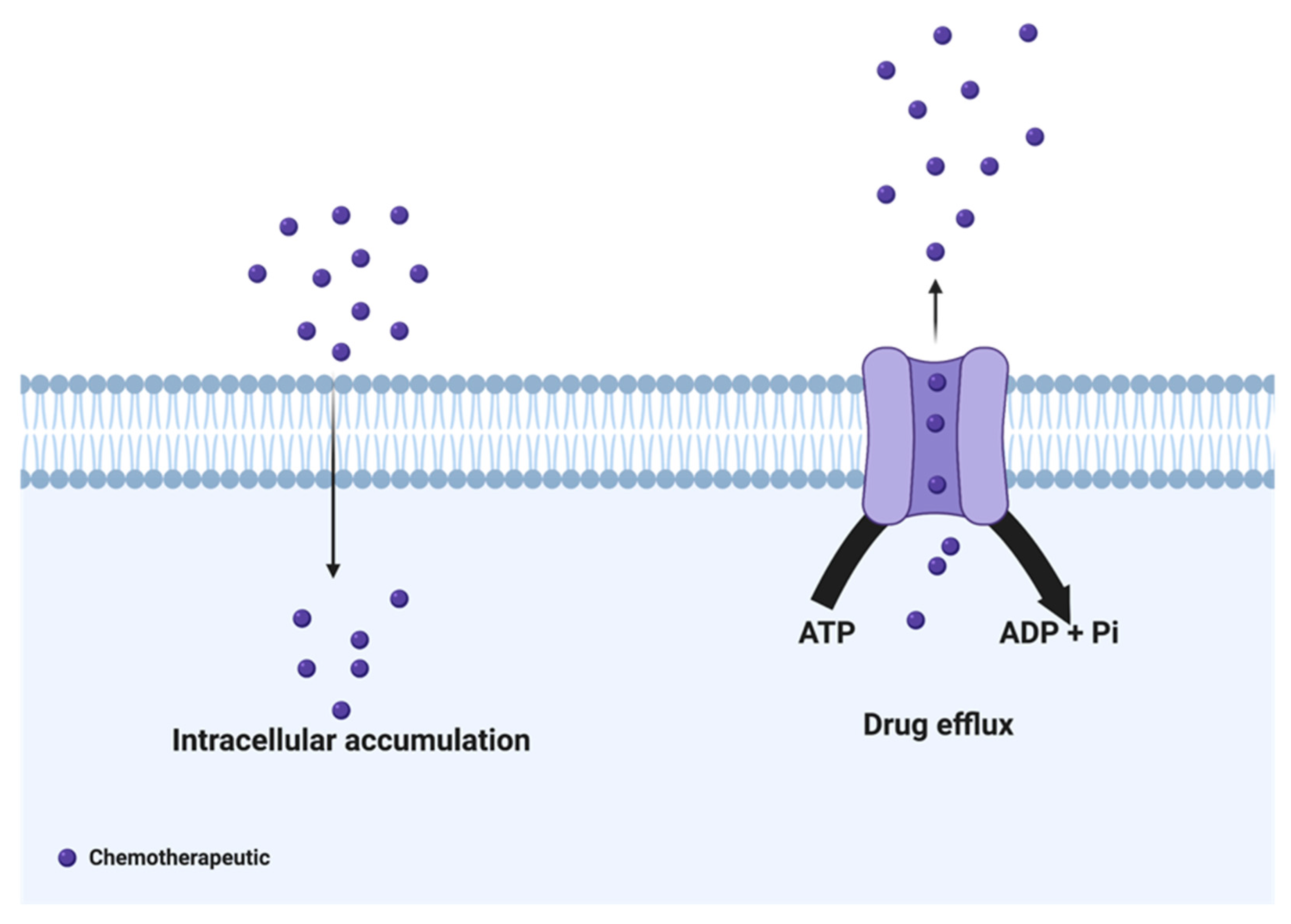
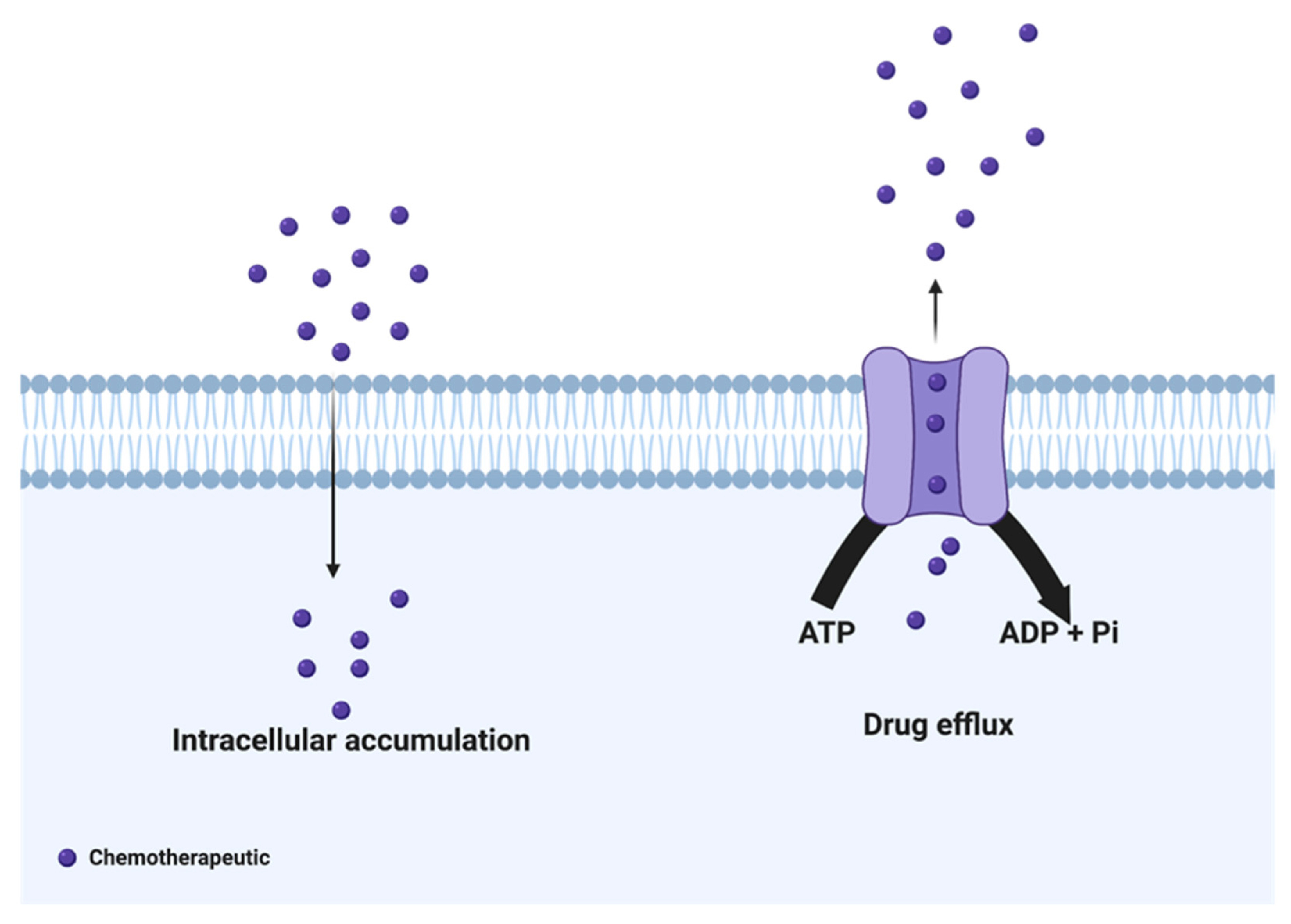
2. Drug Efflux Proteins
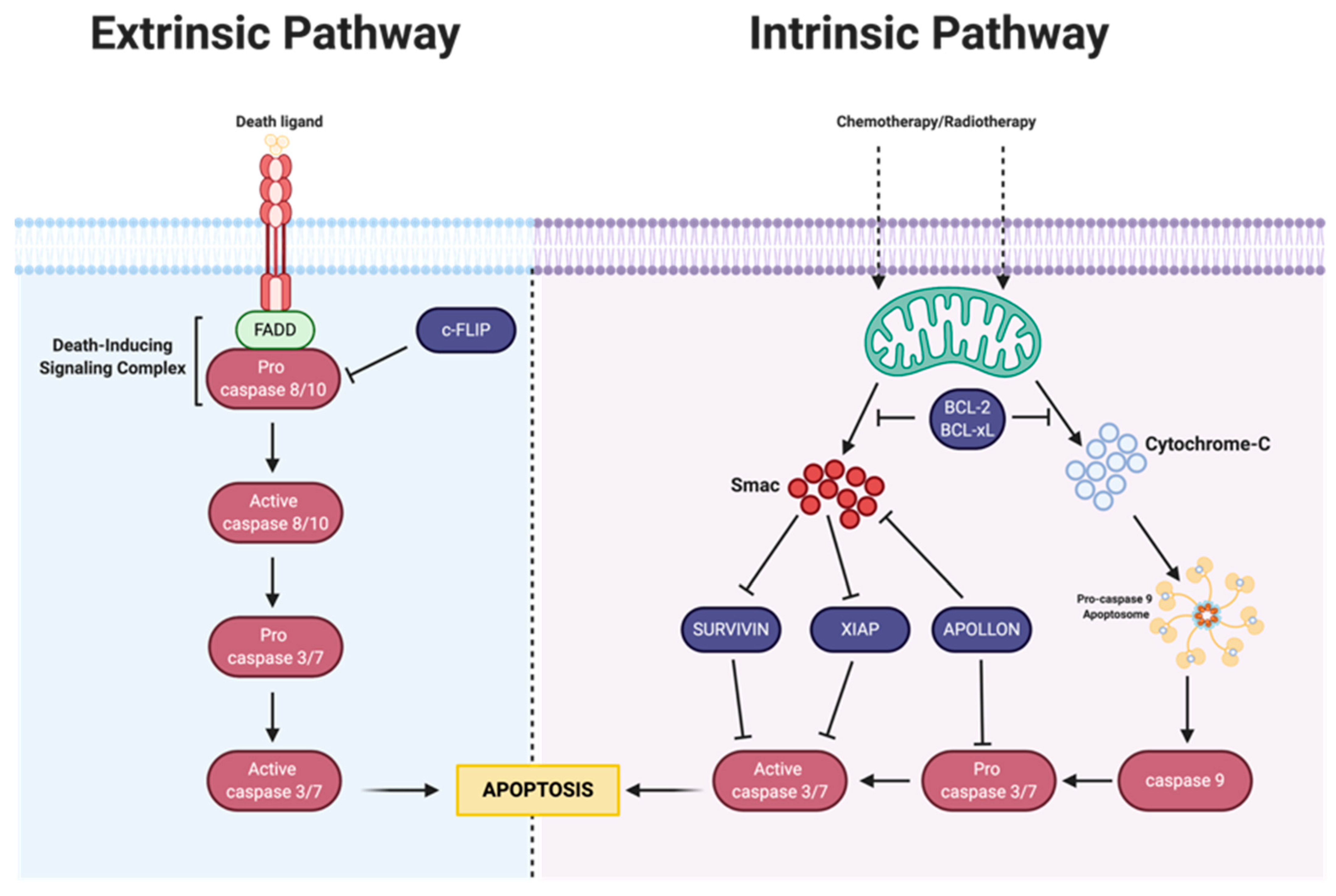
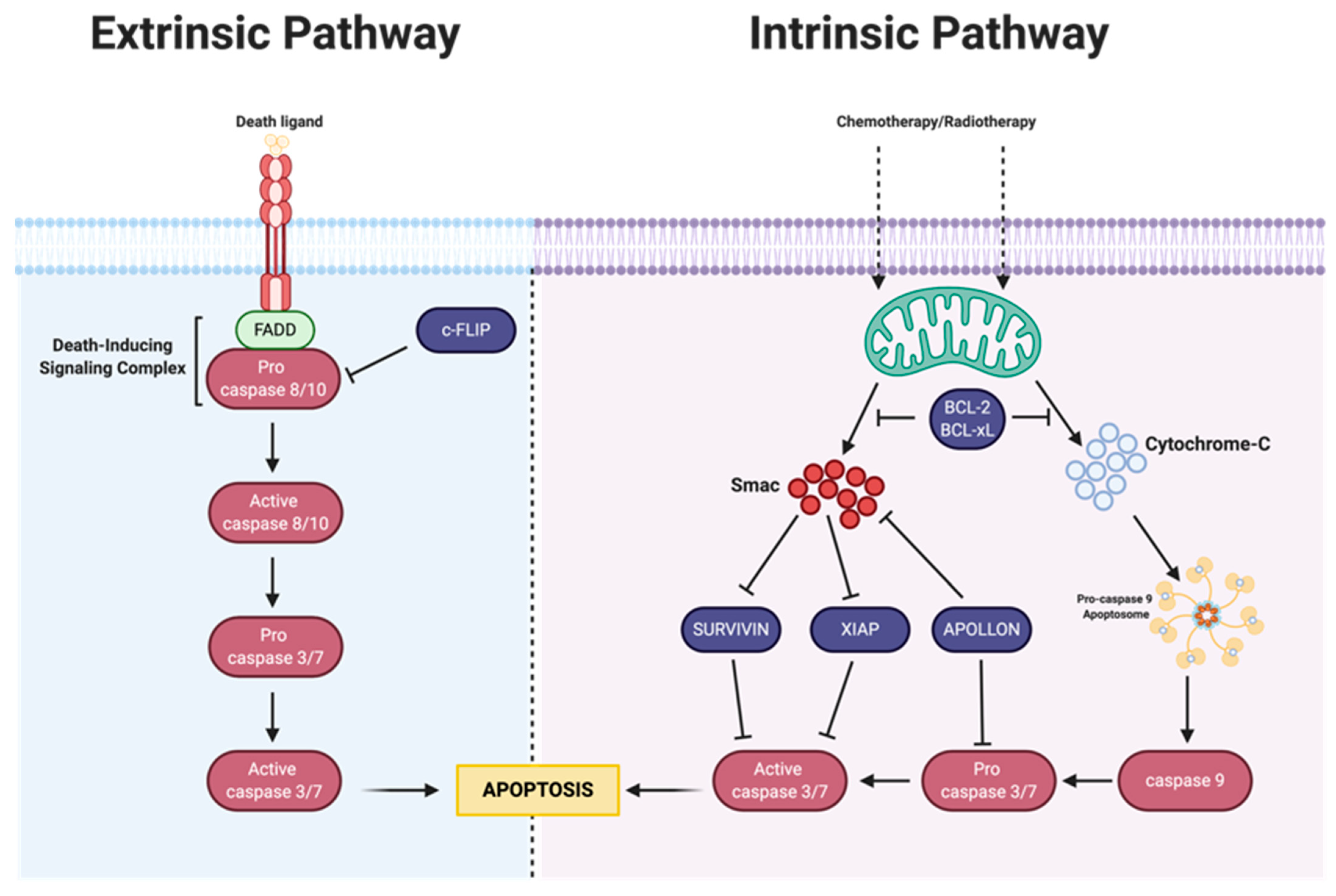
3. Apoptosis
3.1. Intrinsic Apoptosis
3.1.1. Bcl-2 Family
3.1.2. IAP Family
3.2. Extrinsic Pathway
MAPK-Activation Death Domain Protein and cFLIP
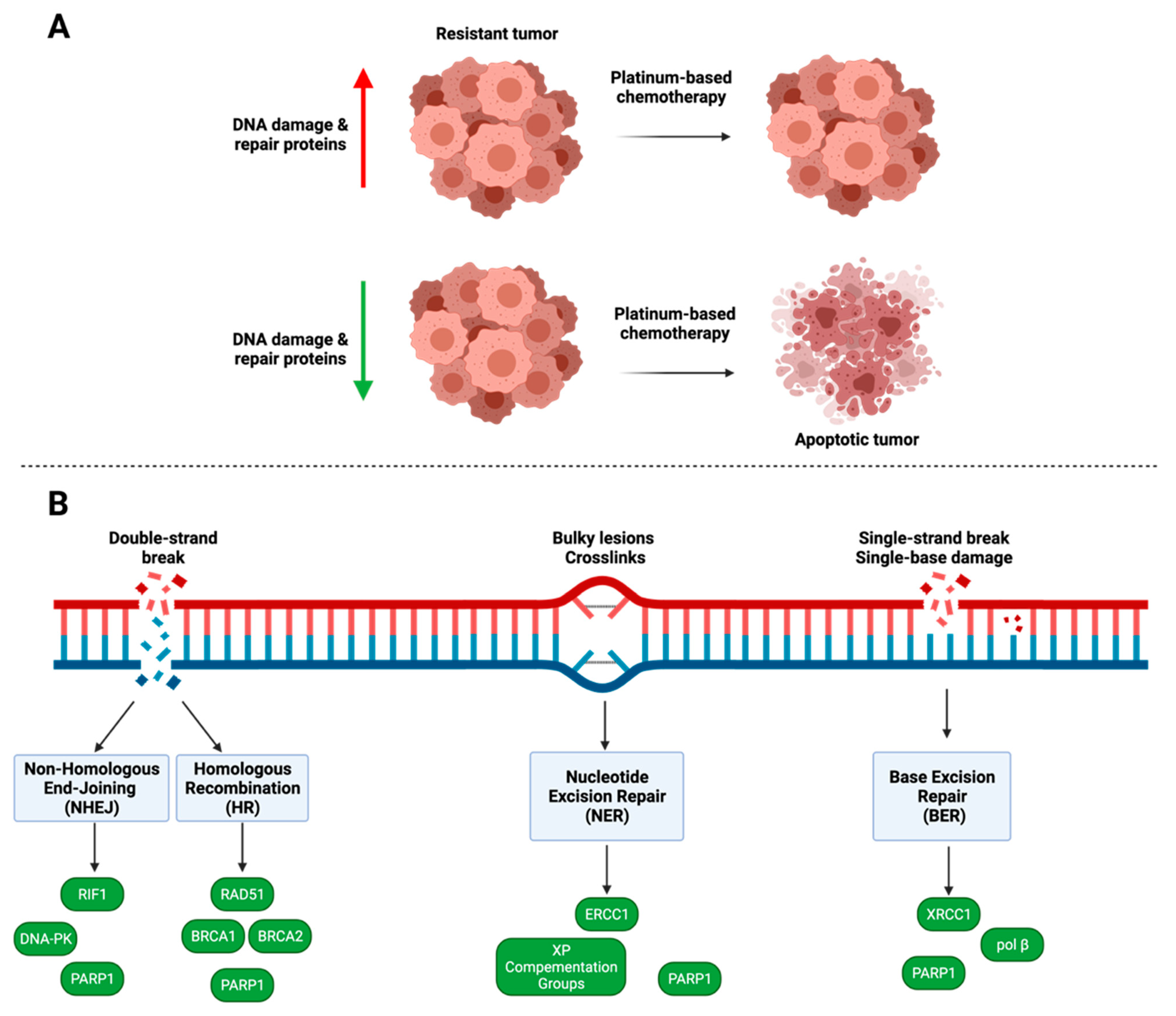
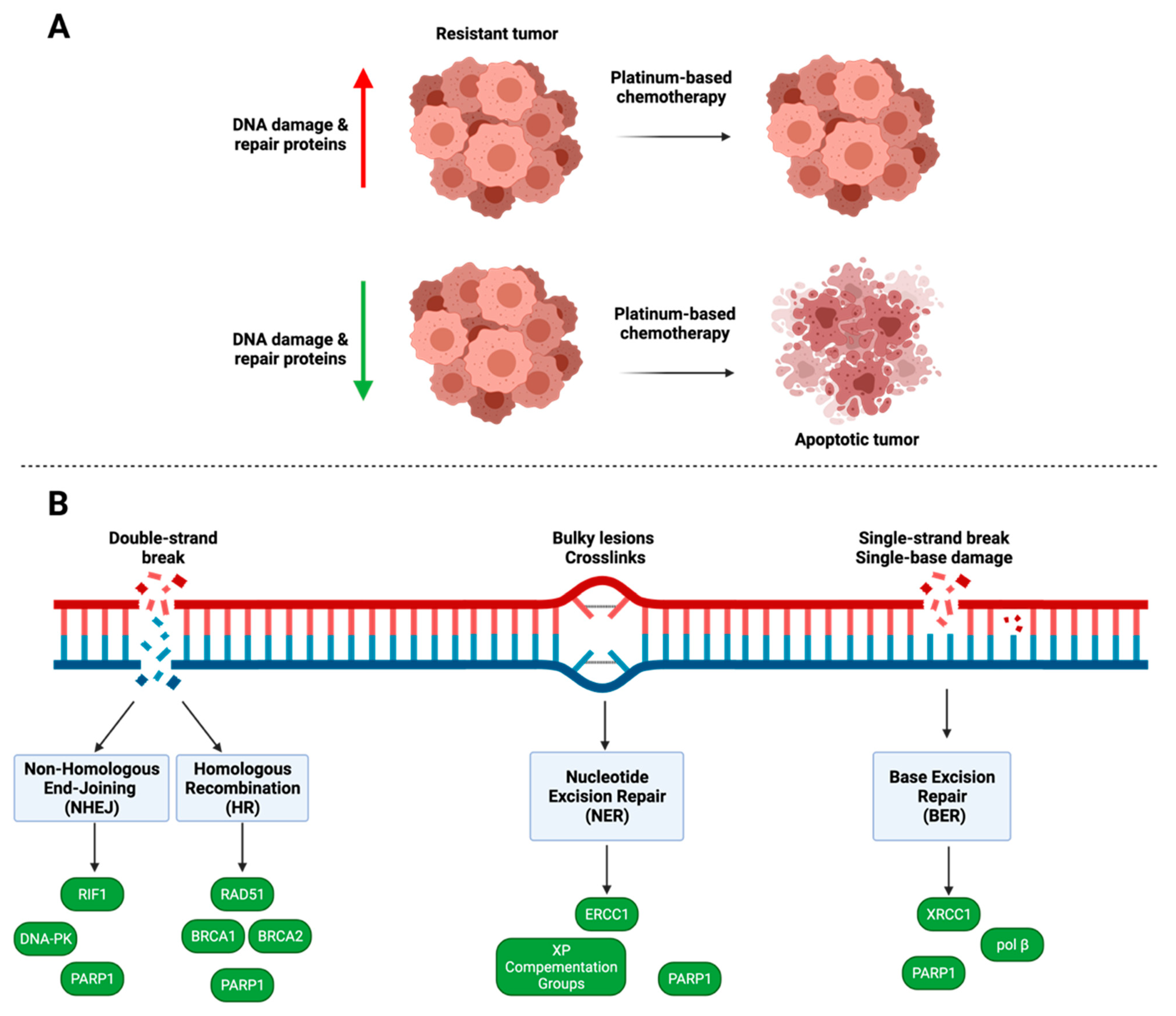
4. DNA Damage and Repair
4.1. PARP1
4.2. Homologous Recombination
4.3. Nucleotide Excision Repair
4.4. Non-Homologous End Joining
4.5. Base Excision Repair
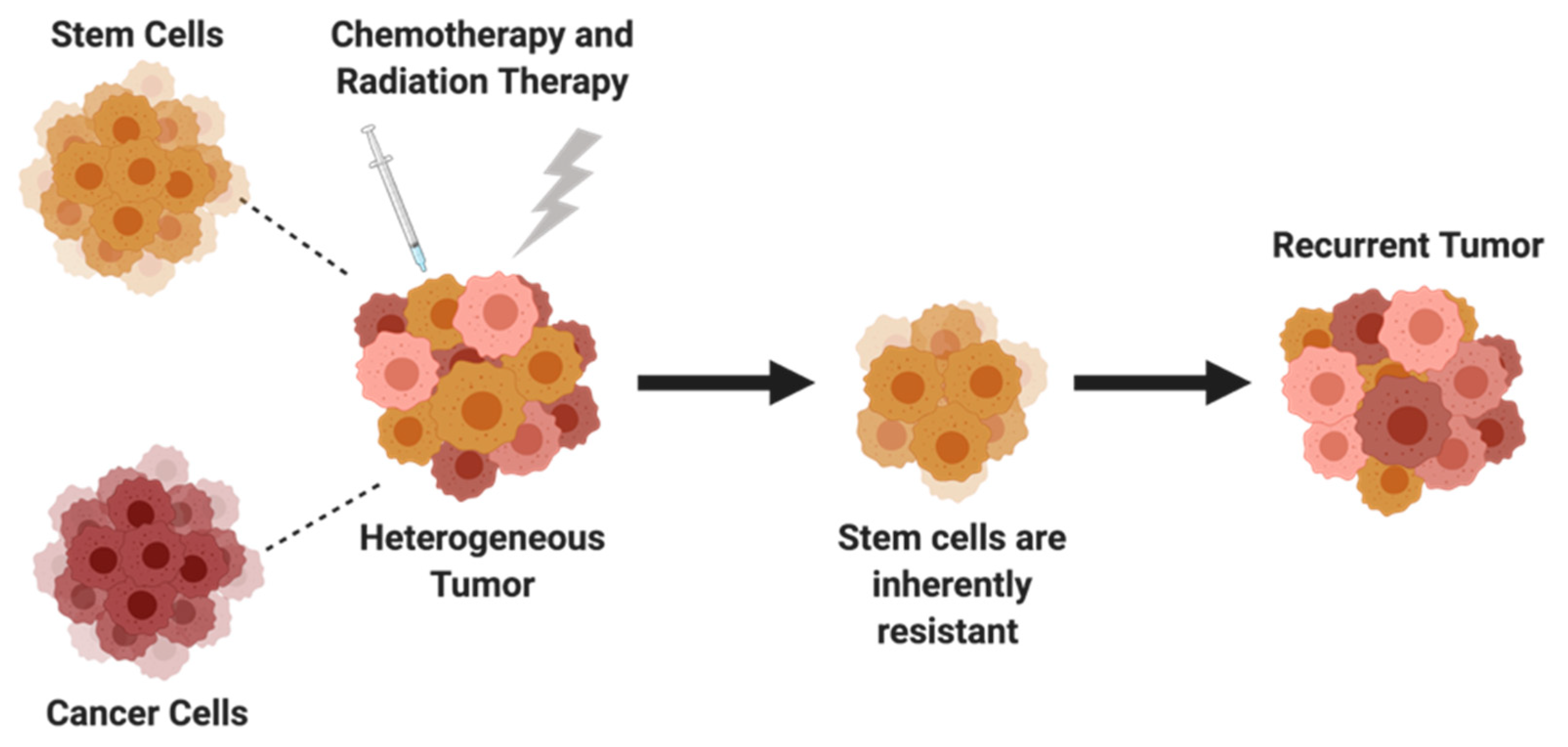
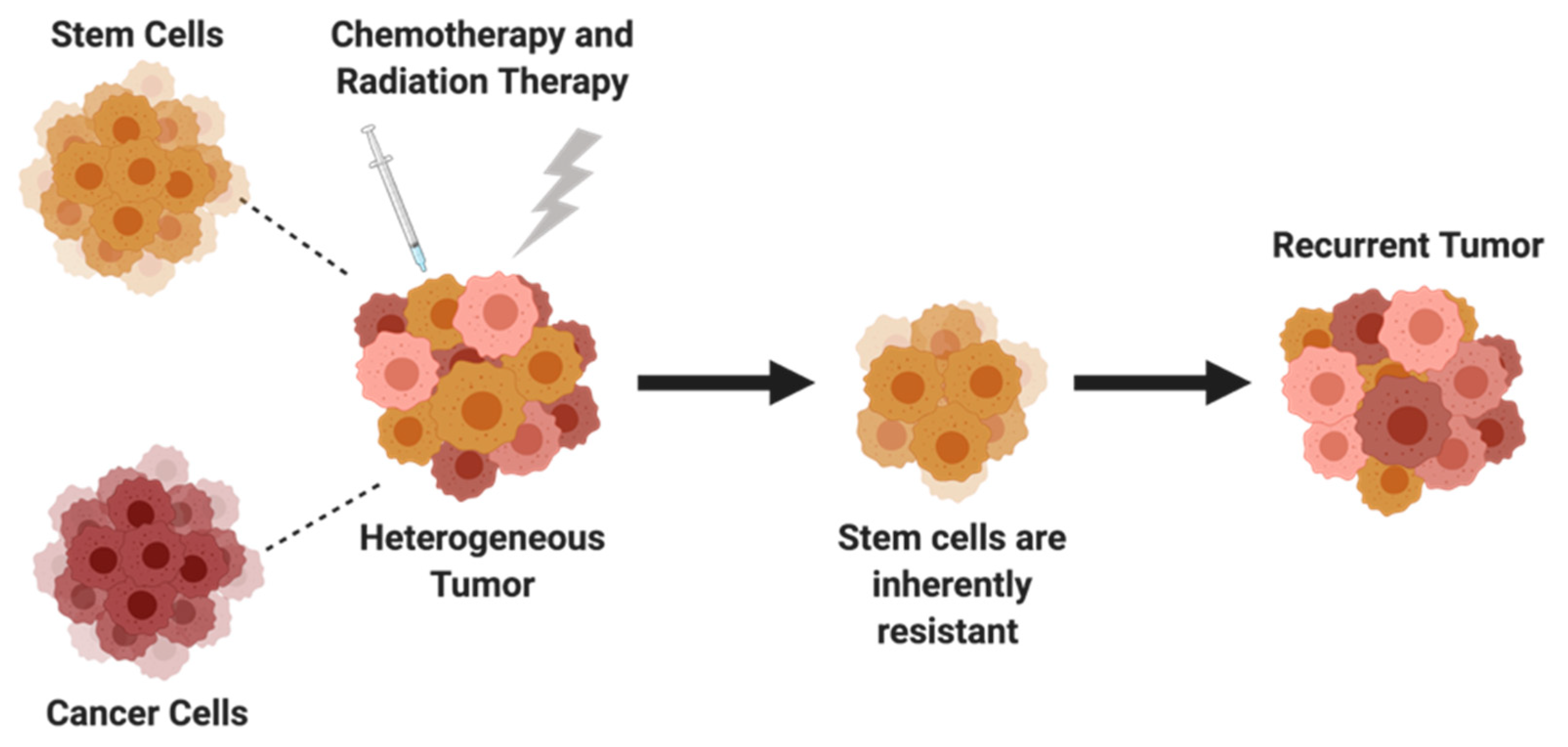
5. Cancer Stem Cells
5.1. SOX2, OCT4, NANOG
5.2. JAK/STAT Pathway
6. Clinical Relevance and Future Directions
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chandra, A.; Pius, C.; Nabeel, M.; Nair, M.; Vishwanatha, J.K.; Ahmad, S.; Basha, R. Ovarian Cancer: Current Status and Strategies for Improving Therapeutic Outcomes. Cancer Med. 2019, 8, 7018–7031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, C.; Ralyea, C.; Lockwood, S. Ovarian Cancer: An Integrated Review. In Seminars Oncology Nursing; WB Saunders: Philadelphia, PA, USA, 2019; Volume 35, pp. 151–156. [Google Scholar]
- American Cancer Society Key Statistics for Ovarian Cancer. 2022. Available online: https://www.cancer.org/cancer/ovarian-cancer/about/key-statistics.html (accessed on 16 December 2022).
- Ovarian Cancer—Cancer Stat Facts. National Cancer Institute: Surveillance, Epidemiology, and End Results Program. 2022. Available online: https://seer.cancer.gov/statfacts/html/ovary.html (accessed on 30 November 2022).
- Cortez, A.J.; Tudrej, P.; Kujawa, K.A.; Lisowska, K.M. Advances in Ovarian Cancer Therapy. Cancer Chemother. Pharmacol. 2018, 81, 17–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brasseur, K.; Gévry, N.; Asselin, E. Chemoresistance and Targeted Therapies in Ovarian and Endometrial Cancers. Oncotarget 2017, 8, 4008–4042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornelison, R.; Llaneza, D.C.; Landen, C.N. Emerging Therapeutics to Overcome Chemoresistance in Epithelial Ovarian Cancer: A Mini-Review. Int. J. Mol. Sci. 2017, 18, 2171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer Drug Resistance: An Evolving Paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Gatti, L.; Zunino, F. Overview of Tumor Cell Chemoresistance Mechanisms. Methods Mol. Med. 2005, 111, 127–148. [Google Scholar] [CrossRef]
- Nikolaou, M.; Pavlopoulou, A.; Georgakilas, A.G.; Kyrodimos, E. The Challenge of Drug Resistance in Cancer Treatment: A Current Overview. Clin. Exp. Metastasis 2018, 35, 309–318. [Google Scholar] [CrossRef]
- Dean, M.; Allikmets, R. Complete Characterization of the Human ABC Gene Family. J. Bioenerg. Biomembr. 2001, 33, 475–479. [Google Scholar] [CrossRef]
- Dean, M.; Hamon, Y.; Chimini, G. The Human ATP-Binding Cassette (ABC) Transporter Superfamily. J. Lipid Res. 2001, 42, 1007–1017. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the Role of ABC Transporters in Multidrug-Resistant Cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Lage, H. MDR1/P-Glycoprotein (ABCB1) as Target for RNA Interference-Mediated Reversal of Multidrug Resistance. Curr. Drug Targets 2006, 7, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Sadée, W. Membrane Transporters and Channels in Chemoresistance and -Sensitivity of Tumor Cells. Cancer Lett. 2006, 239, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Freimund, A.E.; Beach, J.A.; Christie, E.L.; Bowtell, D.D.L. Mechanisms of Drug Resistance in High-Grade Serous Ovarian Cancer. Hematol. Oncol. Clin. North Am. 2018, 32, 983–996. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.; Shen, J.; Shi, H.; Hornicek, F.J.; Kan, Q.; Duan, Z. Novel Mechanisms and Approaches to Overcome Multidrug Resistance in the Treatment of Ovarian Cancer. Biochim. Biophys. Acta Rev. Cancer 2016, 1866, 266–275. [Google Scholar] [CrossRef]
- Choi, C.H. ABC Transporters as Multidrug Resistance Mechanisms and the Development of Chemosensitizers for Their Reversal. Cancer Cell Int. 2005, 5, 30. [Google Scholar] [CrossRef] [Green Version]
- Penson, R.T.; Oliva, E.; Skates, S.J.; Glyptis, T.; Fuller, A.F.; Goodman, A.; Seiden, M.V. Expression of Multidrug Resistance-1 Protein Inversely Correlates with Paclitaxel Response and Survival in Ovarian Cancer Patients: A Study in Serial Samples. Gynecol. Oncol. 2004, 93, 98–106. [Google Scholar] [CrossRef]
- Hille, S.; Rein, D.T.; Riffelmann, M.; Neumann, R.; Sartorius, J.; Pfützner, A.; Kurbacher, C.M.; Schöndorf, T.; Breidenbach, M. Anticancer Drugs Induce Mdr1 Gene Expression in Recurrent Ovarian Cancer. Anticancer Drugs 2006, 17, 1041–1044. [Google Scholar] [CrossRef]
- Masanek, U.; Stammler, G.; Volm, M. Messenger RNA Expression of Resistance Proteins and Related Factors in Human Ovarian Carcinoma Cell Lines Resistant to Doxorubicin, Taxol and Cisplatin. Anticancer Drugs 1997, 8, 189–198. [Google Scholar] [CrossRef]
- Pan, J.; Mendes, L.P.; Yao, M.; Filipczak, N.; Garai, S.; Thakur, G.A.; Sarisozen, C.; Torchilin, V.P. Polyamidoamine Dendrimers-Based Nanomedicine for Combination Therapy with SiRNA and Chemotherapeutics to Overcome Multidrug Resistance. Eur. J. Pharm. Biopharm. 2019, 136, 18–28. [Google Scholar] [CrossRef]
- Yang, X.; Lyer, A.K.; Singh, A.; Choy, E.; Hornicek, F.J.; Amiji, M.M.; Duan, Z. MDR1 SiRNA Loaded Hyaluronic Acid-Based CD44 Targeted Nanoparticle Systems Circumvent Paclitaxel Resistance in Ovarian Cancer. Sci. Rep. 2015, 5, 8509. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, M.F.; Mao, Q.; Holzenburg, A.; Ford, R.C.; Deeley, R.G.; Cole, S.P.C. The Structure of the Multidrug Resistance Protein 1 (MRP1/ABCC1): Crystallization and Single-Particle Analysis. J. Biol. Chem. 2001, 276, 16076–16082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakos, É.; Homolya, L. Portrait of Multifaceted Transporter, the Multidrug Resistance-Associated Protein 1 (MRP1/ABCC1). Pflug. Arch. Eur. J. Physiol. 2007, 453, 621–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almquist, K.C.; Loe, D.W.; Hipfner, D.R.; Mackie, J.E.; Cole, S.P.C.; Deeley, R.G. Characterization of the Mr 190,000 Multidrug Resistance Protein (MRP) in Drug-Selected and Transfected Human Tumor Cells. Cancer Res. 1995, 55, 102–110. [Google Scholar] [PubMed]
- Cole, S.P.C.; Bhardwaj, G.; Gerlach, J.H.; Mackie, J.E.; Grant, C.E.; Almquist, K.C.; Stewart, A.J.; Kurz, E.U.; Duncan, A.M.V.; Deeley, R.G. Overexpression of a Transporter Gene in a Multidrug-Resistant Human Lung Cancer Cell Line. Science 1992, 258, 1650–1654. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Zhao, J.; Zhang, Y.; Mu, P.; Wang, X. Expression Levels of MRP1, GST-π, and GSK3Β in Ovarian Cancer and the Relationship with Drug Resistance and Prognosis of Patients. Oncol. Lett. 2019, 18, 22–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kavallaris, M.; Leary, J.A.; Barrett, J.A.; Friedlander, M.L. MDR1 and Multidrug Resistance-Associated Protein (MRP) Gene Expression in Epithelial Ovarian Tumors. Cancer Lett. 1996, 102, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Ohishi, Y.; Oda, Y.; Uchiumi, T.; Kobayashi, H.; Hirakawa, T.; Miyamoto, S.; Kinukawa, N.; Nakano, H.; Kuwano, M.; Tsuneyoshi, M. ATP-Binding Cassette Superfamily Transporter Gene Expression in Human Primary Ovarian Carcinoma. Clin. Cancer Res. 2002, 8, 3767–3775. [Google Scholar]
- Ehrlichova, M.; Mohelnikova-Duchonova, B.; Hrdy, J.; Brynychova, V.; Mrhalova, M.; Kodet, R.; Rob, L.; Pluta, M.; Gut, I.; Soucek, P.; et al. The Association of Taxane Resistance Genes with the Clinical Course of Ovarian Carcinoma. Genomics 2013, 102, 96–101. [Google Scholar] [CrossRef] [Green Version]
- Tong, W.Y.; Alnakhli, M.; Bhardwaj, R.; Apostolou, S.; Sinha, S.; Fraser, C.; Kuchel, T.; Kuss, B.; Voelcker, N.H. Delivery of SiRNA in Vitro and in Vivo Using PEI-Capped Porous Silicon Nanoparticles to Silence MRP1 and Inhibit Proliferation in Glioblastoma. J. Nanobiotechnol. 2018, 16, 38. [Google Scholar] [CrossRef]
- Tivnan, A.; Zakaria, Z.; O’Leary, C.; Kögel, D.; Pokorny, J.L.; Sarkaria, J.N.; Prehn, J.H.M. Inhibition of Multidrug Resistance Protein 1 (MRP1) Improves Chemotherapy Drug Response in Primary and Recurrent Glioblastoma Multiforme. Front. Neurosci. 2015, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Shao, S.L.; Cui, T.T.; Zhao, W.; Zhang, W.W.; Xie, Z.L.; Wang, C.H.; Jia, H.S.; Liu, Q. RNAi-Based Knockdown of Multidrug Resistance-Associated Protein 1 Is Sufficient to Reverse Multidrug Resistance of Human Lung Cells. Asian Pac. J. Cancer Prev. 2014, 15, 10597–10601. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Kong, Y.; Wu, Y.; Gao, Q.; Fu, J.; Sun, X.; Geng, Q. CAC1 Knockdown Reverses Drug Resistance through the Downregulation of P-Gp and MRP-1 Expression in Colorectal Cancer. PLoS ONE 2019, 14, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gana, C.C.; Hanssen, K.M.; Yu, D.M.T.; Flemming, C.L.; Wheatley, M.S.; Conseil, G.; Cole, S.P.C.; Norris, M.D.; Haber, M.; Fletcher, J.I. MRP1 Modulators Synergize with Buthionine Sulfoximine to Exploit Collateral Sensitivity and Selectively Kill MRP1-Expressing Cancer Cells. Biochem. Pharmacol. 2019, 168, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.H.; Jonker, J.W. Mammalian Drug Efflux Transporters of the ATP Binding Cassette (ABC) Family: An Overview. Adv. Drug Deliv. Rev. 2003, 55, 3–29. [Google Scholar] [CrossRef]
- Natarajan, K.; Xie, Y.; Baer, M.R.; Ross, D.D. Role of Breast Cancer Resistance Protein (BCRP/ABCG2) in Cancer Drug Resistance. Biochem. Pharmacol. 2012, 83, 1084–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Austin Doyle, L.; Yang, W.; Abruzzo, L.V.; Krogmann, T.; Gao, Y.; Rishi, A.K.; Ross, D.D. A Multidrug Resistance Transporter from Human MCF-7 Breast Cancer Cells. Proc. Natl. Acad. Sci. USA 1998, 95, 15665–15670. [Google Scholar] [CrossRef] [Green Version]
- Messenger, P.; Expression, R.N.A.; Ross, D.D.; Yang, W.; Abruzzo, L.V.; Dalton, W.S.; Lage, H.; Dietel, M.; Greenberger, L.; Cole, S.P.C.; et al. Atypical Multidrug Resistance: Breast Cancer Resistance. J. Natl. Cancer Inst. 1999, 91, 429–433. [Google Scholar]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug Resistance in Cancer: Role of ATP-Dependent Transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Zahreddine, H.; Borden, K.L.B. Mechanisms and Insights into Drug Resistance in Cancer. Front. Pharmacol. 2013, 4, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Maliepaard, M.; Van Gastelen, A.; De Jong, L.A.; Pluim, D.; Van Waardenburg, R.C.A.M.; Ruevekamp-Helmers, M.C.; Floot, B.G.J.; Schellens, J.H.M. Advances in Brief Overexpression of the BCRP/MXR/ABCP Gene in a Topotecan-Selected Ovarian. Cancer Res. 1999, 59, 4559–4563. [Google Scholar]
- Januchowski, R.; Sterzyńska, K.; Zaorska, K.; Sosińska, P.; Klejewski, A.; Brązert, M.; Nowicki, M.; Zabel, M. Analysis of MDR Genes Expression and Cross-Resistance in Eight Drug Resistant Ovarian Cancer Cell Lines. J. Ovarian Res. 2016, 9, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, L.; Pospichalova, V.; Huang, Z.; Murphy, S.K.; Payne, S.; Wang, F.; Kennedy, M.; Cianciolo, G.J.; Bryja, V.; Pizzo, S.V.; et al. Ascites Increases Expression/Function of Multidrug Resistance Proteins in Ovarian Cancer Cells. PLoS ONE 2015, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ricci, J.W.; Lovato, D.M.; Severns, V.; Sklar, L.A.; Richard, S. Novel ABCG2 Antagonists Reverse Topotecan-Mediated Chemotherapeutic Resistance in Ovarian Carcinoma Xenografts. Mol. Cancer Ther. 2017, 15, 2853–2862. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, S.H.; Earnshaw, W.C. Induction of Apoptosis by Cancer Chemotherapy. Exp. Cell Res. 2000, 256, 42–49. [Google Scholar] [CrossRef]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as Anticancer Mechanism: Function and Dysfunction of Its Modulators and Targeted Therapeutic Strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef] [Green Version]
- Binju, M.; Amaya-Padilla, M.A.; Wan, G.; Gunosewoyo, H.; Rahmanto, Y.S.; Yu, Y. Therapeutic Inducers of Apoptosis in Ovarian Cancer. Cancers 2019, 11, 1786. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448–458. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Yuan, J. Caspases in Apoptosis and Beyond. Oncogene 2008, 27, 6194–6206. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Hernandez, L.; Annunziata, C.M. Caspase 8 Expression May Determine the Survival of Women with Ovarian Cancer. Cell Death Dis. 2016, 7, e2045. [Google Scholar] [CrossRef] [Green Version]
- Cory, S.; Adams, J.M. The BCL2 Family: Regulators of the Cellular Life-or-Death Switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef]
- Maji, S.; Panda, S.; Samal, S.K.; Shriwas, O.; Rath, R.; Pellecchia, M.; Emdad, L.; Das, S.K.; Fisher, P.B.; Dash, R. Bcl-2 Antiapoptotic Family Proteins and Chemoresistance in Cancer, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2018; Volume 137. [Google Scholar]
- Villedieu, M.; Louis, M.H.; Dutoit, S.; Brotin, E.; Lincet, H.; Duigou, F.; Staedel, C.; Gauduchon, P.; Poulain, L. Absence of Bcl-XL down-Regulation in Response to Cisplatin Is Associated with Chemoresistance in Ovarian Carcinoma Cells. Gynecol. Oncol. 2007, 105, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Brotin, E.; Meryet-Figuière, M.; Simonin, K.; Duval, R.E.; Villedieu, M.; Leroy-Dudal, J.; Saison-Behmoaras, E.; Gauduchon, P.; Denoyelle, C.; Poulain, L. Bcl-XL and MCL-1 Constitute Pertinent Targets in Ovarian Carcinoma and Their Concomitant Inhibition Is Sufficient to Induce Apoptosis. Int. J. Cancer 2010, 126, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.; Lucas, P.C.; Griffith, K.A.; Choi, M.; Fogoros, S.; Hu, Y.Y.; Liu, J.R. Expression of Bcl-XL in Ovarian Carcinoma Is Associated with Chemoresistance and Recurrent Disease. Gynecol. Oncol. 2005, 96, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Mano, Y.; Kikuchi, Y.; Yamamoto, K.; Kita, T.; Hirata, J.; Tode, T.; Ishii, K.; Nagata, I. Bcl-2 as a Predictor of Chemosensitivity and Prognosis in Primary Epithelial Ovarian Cancer. Eur. J. Cancer 1999, 35, 1214–1219. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, S.; Sun, Y.; Zhang, D.; Zhao, Z.; Liu, L. Reversing Platinum Resistance in Ovarian Cancer Multicellular Spheroids by Targeting Bcl-2. Onco. Targets. Ther. 2019, 12, 897–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.R.; Fletcher, B.; Page, C.; Hu, C.; Nunez, G.; Baker, V. Bcl-x(L) Is Expressed in Ovarian Carcinoma and Modulates Chemotherapy- Induced Apoptosis. Gynecol. Oncol. 1998, 70, 398–403. [Google Scholar] [CrossRef]
- Igney, F.H.; Krammer, P.H. Death and Anti-Death: Tumour Resistance to Apoptosis. Nat. Rev. Cancer 2002, 2, 277–288. [Google Scholar] [CrossRef]
- Fulda, S. Targeting Extrinsic Apoptosis in Cancer: Challenges and Opportunities. Semin. Cell Dev. Biol. 2015, 39, 20–25. [Google Scholar] [CrossRef]
- Kim, R. Recent Advances in Understanding the Cell Death Pathways Activated by Anticancer Therapy. Cancer 2005, 103, 1551–1560. [Google Scholar] [CrossRef]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a Mitochondrial Protein That Promotes Cytochrome c-Dependent Caspase Activation by Eliminating IAP Inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Al-Alem, L.F.; Baker, A.T.; Pandya, U.M.; Eisenhauer, E.L.; Rueda, B.R. Understand and Targeting Apoptotic Pathways in Ovarian Cancer. Cancers 2019, 11, 1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, M.; Watari, H.; Abualmaaty, A.; Ohba, Y.; Sakuragi, N. Apoptosis and Molecular Targeting Therapy in Cancer. Biomed. Res. Int. 2014, 2014, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.Q.; Jiang, X.; Fraser, M.; Li, M.; Dan, H.C.; Sun, M.; Tsang, B.K. Role of X-Linked Inhibitor of Apoptosis Protein in Chemoresistance in Ovarian Cancer: Possible Involvement of the Phosphoinositide-3 Kinase/Akt Pathway. Drug Resist. Updat. 2002, 5, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Sapi, E.; Alvero, A.B.; Chen, W.; O’Malley, D.; Hao, X.Y.; Dwipoyono, B.; Garg, M.; Kamsteeg, M.; Rutherford, T.; Mor, G. Resistance of Ovarian Carcinoma Cells to Docetaxel Is XIAP Dependent and Reversible by Phenoxodiol. Oncol. Res. 2004, 14, 567–578. [Google Scholar] [CrossRef]
- Asselin, E.; Mills, G.B.; Tsang, B.K. XIAP Regulates Akt Activity and Caspase-3-Dependent Cleavage during Cisplatin-Induced Apoptosis in Human Ovarian Epithelial Cancer Cells. Cancer Res. 2001, 61, 1862–1868. [Google Scholar] [PubMed]
- Zhang, Y.; Huang, F.; Luo, Q.; Wu, X.; Liu, Z.; Chen, H.; Huang, Y. Inhibition of XIAP Increases Carboplatin Sensitivity in Ovarian Cancer. Onco. Targets. Ther. 2018, 11, 8751–8759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, M.; Takano, M.; Iwaya, K.; Shinomiya, N.; Kato, M.; Aoyama, T.; Sasaki, N.; Goto, T.; Suzuki, A.; Hitrata, J.; et al. X-Chromosome-Linked Inhibitor of Apoptosis as a Key Factor for Chemoresistance in Clear Cell Carcinoma of the Ovary. Br. J. Cancer 2014, 110, 2881–2886. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.J.; Chen, B.L.; Xin, X.Y. XIAP Gene Downregulation by Small Interfering RNA Inhibits Proliferation, Induces Apoptosis, and Reverses the Cisplatin Resistance of Ovarian Carcinoma. Eur. J. Obstet. Gynecol. Reprod. Biol. 2009, 146, 222–226. [Google Scholar] [CrossRef]
- Cossu, F.; Milani, M.; Mastrangelo, E.; Lecis, D. Targeting the BIR Domains of Inhibitor of Apoptosis (IAP) Proteins in Cancer Treatment. Comput. Struct. Biotechnol. J. 2019, 17, 142–150. [Google Scholar] [CrossRef]
- Cho, H.J.; Kim, H.R.; Park, Y.S.; Kim, Y.H.; Kim, D.K.; Park, S. Il Prognostic Value of Survivin Expression in Stage III Non-Small Cell Lung Cancer Patients Treated with Platinum-Based Therapy. Surg. Oncol. 2015, 24, 329–334. [Google Scholar] [CrossRef]
- Ambrosini, G.; Adida, C.; Altieri, D.C. A Novel Anti-Apoptosis Gene, Survivin, Expressed in Cancer and Lymphoma. Nat. Med. 1997, 3, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Ai, Z.; Yin, L.; Zhou, X.; Zhu, Y.; Zhu, D.; Yu, Y.; Feng, Y. Inhibition of Survivin Reduces Cell Proliferation and Induces Apoptosis in Human Endometrial Cancer. Cancer 2006, 107, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Welm, A.; Bishop, J.M. Cell Division and Cell Survival in the Absence of Survivin. Proc. Natl. Acad. Sci. USA 2004, 101, 15100–15105. [Google Scholar] [CrossRef] [PubMed]
- Altieri, D.C. Validating Survivin as a Cancer Therapeutic Target. Nat. Rev. Cancer 2003, 3, 46–54. [Google Scholar] [CrossRef]
- Mita, A.C.; Mita, M.M.; Nawrocki, S.T.; Giles, F.J. Survivin: Key Regulator of Mitosis and Apoptosis and Novel Target for Cancer Therapeutics. Clin. Cancer Res. 2008, 14, 5000–5005. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Ackermann, E.J.; Bennett, C.F.; Rothermel, A.L.; Plescia, J.; Tognin, S.; Villa, A.; Marchisio, P.C.; Altieri, D.C. Pleiotropic Cell-Division Defects and Apoptosis Induced by Interference with Survivin Function. Nat. Cell Biol. 1999, 1, 461–466. [Google Scholar] [CrossRef]
- Carvalho, A.; Carmena, M.; Sambade, C.; Earnshaw, W.C.; Wheatley, S.P. Survivin Is Required for Stable Checkpoint Activation in Taxol-Treated HeLa Cells. J. Cell Sci. 2003, 116, 2987–2998. [Google Scholar] [CrossRef] [Green Version]
- Skoufias, D.A.; Mollinari, C.; Lacroix, F.B.; Margolis, R.L. Human Survivin Is a Kinetochore-Associated Passenger Protein. J. Cell Biol. 2000, 151, 1575–1581. [Google Scholar] [CrossRef] [Green Version]
- Fortugno, P.; Wall, N.R.; Giodini, A.; O’Connor, D.S.; Plescia, J.; Padgett, K.M.; Tognin, S.; Marchisio, P.C.; Altieri, D.C. Survivin Exists in Immunochemically Distinct Subcellular Pools and Is Involved in Spindle Microtubule Function. J. Cell Sci. 2002, 115, 575–585. [Google Scholar] [CrossRef]
- Li, F.; Ambrosini, G.; Chu, E.Y.; Plescia, J.; Tognin, S.; Marchisio, P.C.; Altieri, D.C. Control of Apoptosis and Mitotic Spindle Checkpoint by Survivin. Nature 1998, 396, 580–884. [Google Scholar] [CrossRef]
- Zaffaroni, N.; Pennati, M.; Colella, G.; Perego, P.; Supino, R.; Gatti, L.; Pilotti, S.; Zunino, F.; Daidone, M.G. Expression of the Anti-Apoptotic Gene Survivin Correlates with Taxol Resistance in Human Ovarian Cancer. Cell. Mol. Life Sci. 2002, 59, 1406–1412. [Google Scholar] [CrossRef] [PubMed]
- Kar, R.; Palanichamy, J.K.; Banerjee, A.; Chattopadhyay, P.; Jain, S.K.; Singh, N. Survivin SiRNA Increases Sensitivity of Primary Cultures of Ovarian Cancer Cells to Paclitaxel. Clin. Transl. Oncol. 2015, 17, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Chandele, A.; Prasad, V.; Jagtap, J.C.; Shukla, R.; Shastry, P.R. Upregulation of Survivin in G2/M Cells and Inhibition of Caspase 9 Activity Enhances Resistance in Staurosporine-Induced Apoptosis. Neoplasia 2004, 6, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Sung, B.J.; Cho, Y.S.; Kim, H.J.; Ha, N.C.; Hwang, J.I.; Chung, C.W.; Jung, Y.K.; Oh, B.H. An Anti-Apoptotic Protein Human Survivin Is a Direct Inhibitor of Caspase-3 and -7. Biochemistry 2001, 40, 1117–1123. [Google Scholar] [CrossRef]
- Banks, D.P.; Plescia, J.; Altieri, D.C.; Haven, N.; Chen, J.; Rosenberg, S.H.; Zhang, H.; Ng, S.; Laboratories, A.; Park, A. Survivin Does Not Inhibit Caspase-3 Activity. Blood J. Am. Soc. Hematol. 2000, 96, 4002–4003. [Google Scholar]
- Conway, E.M.; Pollefeyt, S.; Cornelissen, J.; DeBaere, I.; Steiner-Mosonyi, M.; Ong, K.; Baens, M.; Collen, D.; Schuh, A.C. Three Differentially Expressed Survivin CDNA Variants Encode Proteins with Distinct Antiapoptotic Functions. Blood 2000, 95, 1435–1442. [Google Scholar] [CrossRef]
- Song, Z.; Yao, X.; Wu, M. Direct Interaction between Survivin and Smac/DIABLO Is Essential for the Anti-Apoptotic Activity of Survivin during Taxol-Induced Apoptosis. J. Biol. Chem. 2003, 278, 23130–23140. [Google Scholar] [CrossRef] [Green Version]
- Blanc-Brude, O.P.; Mesri, M.; Wall, N.R.; Plescia, J.; Dohi, T.; Altieri, D.C. Therapeutic Targeting of the Survivin Pathway in Cancer: Initiation of Mitochondrial Apoptosis and Suppression of Tumor-Associated Angiogenesis. Clin. Cancer Res. 2003, 9, 2683–2692. [Google Scholar]
- Cohen, C.; Lohmann, C.M.; Cotsonis, G.; Lawson, D.; Santoianni, R. Survivin Expression in Ovarian Carcinoma: Correlation with Apoptotic Markers and Prognosis. Mod. Pathol. 2003, 16, 574–583. [Google Scholar] [CrossRef] [Green Version]
- Sui, L.; Dong, Y.; Ohno, M.; Watanabe, Y.; Sugimoto, K.; Tokuda, M. Survivin Expression and Its Correlation with Cell Proliferation and Prognosis in Epithelial Ovarian Tumors. Int. J. Oncol. 2002, 21, 315–320. [Google Scholar] [CrossRef]
- Gąsowska-Bodnar, A.; Bodnar, L.; Dąbek, A.; Cichowicz, M.; Jerzak, M.; Cierniak, S.; Kozłowski, W.; Baranowski, W. Survivin Expression as a Prognostic Factor in Patients with Epithelial Ovarian Cancer or Primary Peritoneal Cancer Treated with Neoadjuvant Chemotherapy. Int. J. Gynecol. Cancer 2014, 24, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Felisiak-Golabek, A.; Rembiszewska, A.; Rzepecka, I.K.; Szafron, L.; Madry, R.; Murawska, M.; Napiorkowski, T.; Sobiczewski, P.; Osuch, B.; Kupryjanczyk, J. Nuclear Survivin Expression Is a Positive Prognostic Factor in Taxane-Platinum-Treated Ovarian Cancer Patients. J. Ovarian Res. 2011, 4, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Naito, M.; Hori, S.; Mashima, T.; Yamori, T.; Tsuruo, T. A Human IAP-Family Gene, Apollon, Expressed in Human Brain Cancer Cells. Biochem. Biophys. Res. Commun. 1999, 264, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Hauser, H.P.; Bardroff, M.; Pyrowolakis, G.; Jentsch, S. A Giant Ubiquitin-Conjugating Enzyme Related to IAP Apoptosis Inhibitors. J. Cell Biol. 1998, 141, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Sekine, K.; Kawabata, A.; Nakamura, H.; Ishioka, T.; Ohata, H.; Katayama, R.; Hashimoto, C.; Zhang, X.; Noda, T.; et al. Apollon Ubiquitinates SMAC and Caspase-9, and Has an Essential Cytoprotection Function. Nat. Cell Biol. 2004, 6, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.B.; Goldberg, A.L. The Membrane-Associated Inhibitor of Apoptosis Protein, BRUCE/Apollon, Antagonizes Both the Precursor and Mature Forms of Smac and Caspase-9. J. Biol. Chem. 2005, 280, 174–182. [Google Scholar] [CrossRef] [Green Version]
- Low, C.G.; Luk, I.S.U.; Lin, D.; Fazli, L.; Yang, K.; Xu, Y.; Gleave, M.; Gout, P.W.; Wang, Y. BIRC6 Protein, an Inhibitor of Apoptosis: Role in Survival of Human Prostate Cancer Cells. PLoS ONE 2013, 8, e55837. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Lin, D.; Low, C.; Vucic, E.A.; English, J.C.; Yee, J.; Murray, N.; Lam, W.L.; Ling, V.; Lam, S.; et al. Elevated Expression of Birc6 Protein in Non-Small-Cell Lung Cancers Is Associated with Cancer Recurrence and Chemoresistance. J. Thorac. Oncol. 2013, 8, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Hu, T.; Weng, S.; Tang, W.; Xe, R.; Chen, S.; Cai, G.; Cai, Y.; Shen, X.; Zhang, S.; Dong, L. Overexpression of BIRC6 Is a Predictor of Prognosis for Colorectal Cancer. PLoS ONE 2015, 10, 1–18. [Google Scholar] [CrossRef]
- Li, R.; Chen, B.L.; Zhou, Y.W.; Guo, R.W.; Shuai, M.T.; Zeng, J.X.; Leng, A.M. Expression and Clinical Significance of Apollon in Esophageal Squamous Cell Carcinoma. Mol. Med. Rep. 2016, 14, 1933–1940. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Chen, Y.J.; Hou, J.; Wang, Y.Y.; Tang, W.Q.; Shen, X.Z.; Tu, R.Q. Expression and Clinical Significance of BIRC6 in Human Epithelial Ovarian Cancer. Tumor Biol. 2014, 35, 4891–4896. [Google Scholar] [CrossRef] [PubMed]
- Lopergolo, A.; Pennati, M.; Gandellini, P.; Orlotti, N.I.; Poma, P.; Daidone, M.G.; Folini, M.; Zaffaroni, N. Apollon Gene Silencing Induces Apoptosis in Breast Cancer Cells through P53 Stabilisation and Caspase-3 Activation. Br. J. Cancer 2009, 100, 739–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrison, J.B. Knockdown of the Inhibitor of Apoptosis BRUCE Sensitizes Resistant Breast Cancer Cells to Chemotherapeutic Agents. J. Cancer Sci. Ther. 2015, 7, 121–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodmer, J.L.; Holler, N.; Reynard, S.; Vinciguerra, P.; Schneider, P.; Juo, P.; Blenis, J.; Tschopp, J. TRAIL Receptor-2 Signals Apoptosis through FADD and Caspase-8. Nat. Cell Biol. 2000, 2, 241–243. [Google Scholar] [CrossRef] [PubMed]
- Kischkel, F.C.; Lawrence, D.A.; Chuntharapai, A.; Schow, P.; Kim, K.J.; Ashkenazi, A. Apo2L/TRAIL-Dependent Recruitment of Endogenous FADD and Caspase-8 to Death Receptors 4 and 5. Immunity 2000, 12, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Fang, B. Mechanisms of Resistance to TRAIL-Induced Apoptosis in Cancer. Cancer Gene Ther. 2005, 12, 228–237. [Google Scholar] [CrossRef]
- Mulherkar, N.; Ramaswamy, M.; Mordi, D.C.; Prabhakar, B.S. MADD/DENN Splice Variant of the IG20 Gene Is Necessary and Sufficient for Cancer Cell Survival. Oncogene 2006, 25, 6252–6261. [Google Scholar] [CrossRef] [Green Version]
- Mulherkar, N.; Prasad, K.V.; Prabhakar, B.S. MADD/DENN Splice Variant of the IG20 Gene Is a Negative Regulator of Caspase-8 Activation: Knockdown Enhances Trail-Induced Apoptosis of Cancer Cells. J. Biol. Chem. 2007, 282, 11715–11721. [Google Scholar] [CrossRef] [Green Version]
- Al-Zoubi, A.M.; Efimova, E.V.; Kaithamana, S.; Martinez, O.; El-Idrissi, M.E.A.; Dogan, R.E.; Prabhakar, B.S. Contrasting Effects of IG20 and Its Splice Isoforms, MADD and DENN-SV, on Tumor Necrosis Factor α-Induced Apoptosis and Activation of Caspase-8 and -3. J. Biol. Chem. 2001, 276, 47202–47211. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Jayarama, S.; Ganesh, L.; Mordi, D.; Carr, R.; Kanteti, P.; Hay, N.; Prabhakar, B.S. Akt-Phosphorylated Mitogen-Activated Kinase-Activating Death Domain Protein (MADD) Inhibits TRAIL-Induced Apoptosis by Blocking Fas-Associated Death Domain (FADD) Association with Death Receptor 4. J. Biol. Chem. 2010, 285, 22713–22722. [Google Scholar] [CrossRef] [Green Version]
- Saini, S.; Sripada, L.; Tulla, K.; Kumar, P.; Yue, F.; Kunda, N.; Maker, A.V.; Prabhakar, B.S. Loss of MADD Expression Inhibits Cellular Growth and Metastasis in Anaplastic Thyroid Cancer. Cell Death Dis. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, M.; Pilli, T.; Bhattacharya, P.; Pacini, F.; Nikiforov, Y.E.; Kanteti, P.V.; Prabhakar, B.S. Knockdown of IG20 Gene Expression Renders Thyroid Cancer Cells Susceptible to Apoptosis. J. Clin. Endocrinol. Metab. 2009, 94, 1467–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, A.; Li, L.C.; Pilli, T.; Qian, L.; Wiley, E.L.; Setty, S.; Christov, K.; Ganesh, L.; Maker, A.V.; Li, P.; et al. MADD Knock-Down Enhances Doxorubicin and TRAIL Induced Apoptosis in Breast Cancer Cells. PLoS ONE 2013, 8, e0154756. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.; Wei, Y.; Wu, J.; Sun, G.; Guo, Y.; Zhang, Q.; Dong, L. MADD Promotes the Survival of Human Lung Adenocarcinoma Cells by Inhibiting Apoptosis. Oncol. Rep. 2013, 29, 1533–1539. [Google Scholar] [CrossRef] [Green Version]
- Li, L.-C.; Jayaram, S.; Ganesh, L.; Qian, L.; Rotmensch, J.; Maker, A.V.; Prabhakar, B.S. Knockdown of MADD and C-FLIP Overcomes Resistance to TRAIL-Induced Apoptosis in Ovarian Cancer Cells. Am J Obs. Gynecol 2011, 205, 362.e12–362.e25. [Google Scholar] [CrossRef]
- Thome, M.; Schneider, P.; Hofmann, K.; Fickenscher, H.; Meinl, E.; Neipel, F.; Mattmann, C.; Burns, K.; Bodmer, J.L.; Schröter, M.; et al. Viral FLICE-Inhibitory Proteins (FLIPs) Prevent Apoptosis Induced by Death Receptors. Nature 1997, 386, 517–521. [Google Scholar] [CrossRef] [Green Version]
- Irmler, M.; Thome, M.; Hahne, M.; Schneider, P.; Hofmann, K.; Steiner, V.; Bodmer, J.L.; Schröter, M.; Burns, K.; Mattmann, C.; et al. Inhibition of Death Receptor Signals by Cellular FLIP. Nature 1997, 388, 190–195. [Google Scholar] [CrossRef] [Green Version]
- Golks, A.; Brenner, D.; Fritsch, C.; Krammer, P.H.; Lavrik, I.N. C-FLIPR, a New Regulator of Death Receptor-Induced Apoptosis. J. Biol. Chem. 2005, 280, 14507–14513. [Google Scholar] [CrossRef] [Green Version]
- Ram, D.R.; Ilyukha, V.; Volkova, T.; Buzdin, A.; Tai, A.; Smirnova, I.; Poltorak, A. Balance between Short and Long Isoforms of CFLIP Regulates Fas-Mediated Apoptosis in Vivo. Proc. Natl. Acad. Sci. USA 2016, 113, 1606–1611. [Google Scholar] [CrossRef] [Green Version]
- Scaffidi, C.; Schmitz, I.; Krammer, P.H.; Peter, M.E. The Role of C-FLIP in Modulation of CD95-Induced Apoptosis. J. Biol. Chem. 1999, 274, 1541–1548. [Google Scholar] [CrossRef] [Green Version]
- Micheau, O.; Thome, M.; Schneider, P.; Holler, N.; Tschopp, J.; Nicholson, D.W.; Briand, C.; Grütter, M.G. The Long Form of FLIP Is an Activator of Caspase-8 at the Fas Death-Inducing Signaling Complex. J. Biol. Chem. 2002, 277, 45162–45171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, M.A.; Powley, I.R.; Jukes-Jones, R.; Horn, S.; Feoktistova, M.; Fairall, L.; Schwabe, J.W.R.; Leverkus, M.; Cain, K.; MacFarlane, M. Co-Operative and Hierarchical Binding of c-FLIP and Caspase-8: A Unified Model Defines How c-FLIP Isoforms Differentially Control Cell Fate. Mol. Cell 2016, 61, 834–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, E.Y.; Jeong, M.S.; Park, S.Y.; Jang, S.B. Evidence of Complex Formation between FADD and C-FLIP Death Effector Domains for the Death Inducing Signaling Complex. BMB Rep. 2014, 47, 488–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.; Vincenz, C.; Ni, J.; Gentz, R.; Dixit, V.M. I-FLICE, a Novel Inhibitor of Tumor Necrosis Factor Receptor-1- and CD- 95-Induced Apoptosis. J. Biol. Chem. 1997, 272, 17255–17257. [Google Scholar] [CrossRef] [Green Version]
- Krueger, A.; Baumann, S.; Krammer, P.H.; Kirchhoff, S. FLICE-Inhibitory Proteins: Regulators of Regulators of Death. Mol. Cell. Biol. 2001, 21, 8247–8254. [Google Scholar] [CrossRef]
- Hillert, L.K.; Ivanisenko, N.V.; Espe, J.; König, C.; Ivanisenko, V.A.; Kähne, T.; Lavrik, I.N. Long and Short Isoforms of C-FLIP Act as Control Checkpoints of DED Filament Assembly. Oncogene 2020, 39, 1756–1772. [Google Scholar] [CrossRef]
- Yun, H.; Xie, J.; Olumi, A.F.; Ghosh, R.; Kumar, A.P. Activation of AKR1C1/ERβ Induces Apoptosis by Downregulation of c-FLIP in Prostate Cancer Cells: A Prospective Therapeutic Opportunity. Oncotarget 2015, 6, 11600–11613. [Google Scholar] [CrossRef] [Green Version]
- Korkolopoulou, P.; Goudopoulou, A.; Voutsinas, G.; Thomas-Tsagli, E.; Kapralos, P.; Patsouris, E.; Saetta, A.A. C-FLIP Expression in Bladder Urothelial Carcinomas: Its Role in Resistance to Fas-Mediated Apoptosis and Clinicopathologic Correlations. Urology 2004, 63, 1198–1204. [Google Scholar] [CrossRef]
- Ullenhag, G.J.; Mukherjee, A.; Watson, N.F.S.; Al-Attar, A.H.; Scholefield, J.H.; Durrant, L.G. Overexpression of FLIPL Is an Independent Marker of Poor Prognosis in Colorectal Cancer Patients. Clin. Cancer Res. 2007, 13, 5070–5075. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Jin, T.G.; Yang, H.; Dewolf, W.C.; Khosravi-Far, R.; Olumi, A.F. Persistent C-FLIP(L) Expression Is Necessary and Sufficient to Maintain Resistance to Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand-Mediated Apoptosis in Prostate Cancer. Cancer Res. 2004, 64, 7086–7091. [Google Scholar] [CrossRef] [Green Version]
- Bagnoli, M.; Ambrogi, F.; Pilotti, S.; Alberti, P.; Ditto, A.; Barbareschi, M.; Galligioni, E.; Biganzoli, E.; Canevari, S.; Mezzanzanica, D. C-FLIPL Expression Defines Two Ovarian Cancer Patient Subsets and Is a Prognostic Factor of Adverse Outcome. Endocr. Relat. Cancer 2009, 16, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.Y.; Jung, G.A.; Hur, G.C.; Chung, H.Y.; Kim, W.H.; Seol, D.W.; Lee, B.L. Upregulation of FLIPs by Akt, a Possible Inhibition Mechanism of TRAIL-Induced Apoptosis in Human Gastric Cancers. Cancer Sci. 2003, 94, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- El-Gazzar, A.; Wittinger, M.; Perco, P.; Anees, M.; Horvat, R.; Mikulits, W.; Grunt, T.W.; Mayer, B.; Krainer, M. The Role of C-FLIPL in Ovarian Cancer: Chaperoning Tumor Cells from Immunosurveillance and Increasing Their Invasive Potential. Gynecol. Oncol. 2010, 117, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.D.; Yu, J.P.; Liu, J.; Luo, H.S.; Chen, H.X.; Yu, H.G. Overexpression of Cellular FLICE-Inhibitory Protein (FLIP) in Gastric Adenocarcinoma. Clin. Sci. 2004, 106, 397–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chawla-Sarkar, M.; Bae, S.I.; Reu, F.J.; Jacobs, B.S.; Lindner, D.J.; Borden, E.C. Downregulation of Bcl-2, FLIP or IAPs (XIAP and Survivin) by SiRNAs Sensitizes Resistant Melanoma Cells to Apo2L/TRAIL-Induced Apoptosis. Cell Death Differ. 2004, 11, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Sharp, D.A.; Lawrence, D.A.; Ashkenazi, A. Selective Knockdown of the Long Variant of Cellular FLICE Inhibitory Protein Augments Death Receptor-Mediated Caspase-8 Activation and Apoptosis. J. Biol. Chem. 2005, 280, 19401–19409. [Google Scholar] [CrossRef] [Green Version]
- Clarke, P.; Tyler, K.L. Down-Regulation of CFLIP Following Reovirus Infection Sensitizes Human Ovarian Cancer Cells to TRAIL-Induced Apoptosis P. Apoptosis 2007, 12, 211–223. [Google Scholar] [CrossRef]
- Kim, D.Y.; Kim, M.J.; Kim, H.B.; Lee, J.W.; Bae, J.H.; Kim, D.W.; Kang, C.D.; Kim, S.H. Suppression of Multidrug Resistance by Treatment with TRAIL in Human Ovarian and Breast Cancer Cells with High Level of C-Myc. Biochim. Biophys. Acta Mol. Basis Dis. 2011, 1812, 796–805. [Google Scholar] [CrossRef]
- Day, T.W.; Najafi, F.; Wu, C.H.; Safa, A.R. Cellular FLICE-like Inhibitory Protein (c-FLIP): A Novel Target for Taxol-Induced Apoptosis. Biochem. Pharmacol. 2006, 71, 1551–1561. [Google Scholar] [CrossRef]
- Kang, J.; Bu, J.; Hao, Y.; Chen, F. Subtoxic Concentration of Doxorubicin Enhances TRAIL-Induced Apoptosis in Human Prostate Cancer Cell Line LNCaP. Prostate Cancer Prostatic Dis. 2005, 8, 274–279. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.F.; Xiao, C.; Li, H.; Yang, S.J. Resistance to Fas-Mediated Apoptosis in Malignant Tumours Is Rescued by KN-93 and Cisplatin via Downregulation of c-FLIP Expression and Phosphorylation. Clin. Exp. Pharmacol. Physiol. 2007, 34, 1245–1251. [Google Scholar] [CrossRef] [PubMed]
- Damia, G.; Broggini, M. Platinum Resistance in Ovarian Cancer: Role of DNA Repair. Cancers 2019, 11, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirza-Aghazadeh-Attari, M.; Ostadian, C.; Saei, A.A.; Mihanfar, A.; Darband, S.G.; Sadighparvar, S.; Kaviani, M.; Samadi Kafil, H.; Yousefi, B.; Majidinia, M. DNA Damage Response and Repair in Ovarian Cancer: Potential Targets for Therapeutic Strategies. DNA Repair 2019, 80, 59–84. [Google Scholar] [CrossRef] [PubMed]
- Hegan, D.C.; Lu, Y.; Stacheleka, G.C.; Crosbya, M.E.; Bindraa, R.S.; Glazer, P.M. Inhibition of Poly(ADP-Ribose) Polymerase down-Regulates BRCA1 and RAD51 in a Pathway Mediated by E2F4 and P130. Proc. Natl. Acad. Sci. USA 2010, 107, 2201–2206. [Google Scholar] [CrossRef] [Green Version]
- D’Andrea, A.D. Mechanisms of PARP Inhibitor Sensitivity and Resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef]
- Ledermann, J.A.; Drew, Y.; Kristeleit, R.S. Homologous Recombination Deficiency and Ovarian Cancer. Eur. J. Cancer 2016, 60, 49–58. [Google Scholar] [CrossRef]
- Pascal, J.M. The Comings and Goings of PARP-1 in Response to DNA Damage. DNA Repair 2018, 71, 177–182. [Google Scholar] [CrossRef]
- Swisher, E.M.; Sakai, W.; Karlan, B.Y.; Wurz, K.; Urban, N.; Taniguchi, T. Secondary BRCA1 Mutations in BRCA1-Mutated Ovarian Carcinomas with Platinum Resistance. Cancer Res. 2008, 68, 2581–2586. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Liu, Z.-Y.; Wu, N.; Chen, Y.-C.; Cheng, Q.; Wang, J. PARP Inhibitor Resistance: The Underlying Mechanisms and Clinical Implications. Mol. Cancer 2020, 19, 107. [Google Scholar] [CrossRef]
- Zhang, Z.; Dou, X.; Yang, H.; Jia, L.; Qin, K.; Gao, X.; Yang, B.; Zhang, W.; Qin, C.; Zhang, F.; et al. Association of Expression of P53, Livin, ERCC1, BRCA1 and PARP1 in Epithelial Ovarian Cancer Tissue with Drug Resistance and Prognosis. Pathol. Res. Pract. 2020, 216, 152794. [Google Scholar] [CrossRef]
- Liu, Y.; Burness, M.L.; Martin-Trevino, R.; Guy, J.; Bai, S.; Harouaka, R.; Brooks, M.D.; Shang, L.; Fox, A.; Luther, T.K.; et al. RAD51 Mediates Resistance of Cancer Stem Cells to PARP Inhibition in Triple-Negative Breast Cancer. Clin. Cancer Res. 2017, 23, 514–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, M.; Cai, J.; Cai, L.; Jia, J.; Xie, L.; Zhu, Y.; Huang, B.; Jin, D.; Wang, Z. Let-7e Sensitizes Epithelial Ovarian Cancer to Cisplatin through Repressing DNA Double Strand Break Repair. J. Ovarian Res. 2017, 10, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.G.; Sarkaria, J.N.; Kaufmann, S.H. Nonhomologous End Joining Drives Poly(ADP-Ribose) Polymerase (PARP) Inhibitor Lethality in Homologous Recombination-Deficient Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 3406–3411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-B.; Mei, Y.; Tian, Z.-W.; Long, J.; Luo, C.-H.; Zhou, H.-H. Downregulation of RIF1 Enhances Sensitivity to Platinum-Based Chemotherapy in Epithelial Ovarian Cancer (EOC) by Regulating Nucleotide Excision Repair (NER) Pathway. Cell. Physiol. Biochem. 2018, 46, 1971–1984. [Google Scholar] [CrossRef] [PubMed]
- Gee, M.E.; Faraahi, Z.; McCormick, A.; Edmondson, R.J. DNA Damage Repair in Ovarian Cancer: Unlocking the Heterogeneity. J. Ovarian Res. 2018, 11, 1–12. [Google Scholar] [CrossRef]
- Mei, Y.; Peng, C.; Liu, Y.B.; Wang, J.; Zhou, H.H. Silencing RIF1 Decreases Cell Growth, Migration and Increases Cisplatin Sensitivity of Human Cervical Cancer Cells. Oncotarget 2017, 8, 107044–107051. [Google Scholar] [CrossRef] [Green Version]
- Mattarocci, S.; Hafner, L.; Lezaja, A.; Shyian, M.; Shore, D. Rif1: A Conserved Regulator of DNA Replication and Repair Hijacked by Telomeres in Yeasts. Front. Genet. 2016, 7, 45. [Google Scholar] [CrossRef] [Green Version]
- Wise, H.C.; Iyer, G.V.; Moore, K.; Temkin, S.M.; Gordon, S.; Aghajanian, C.; Grisham, R.N. Activity of M3814, an Oral DNA-PK Inhibitor, In Combination with Topoisomerase II Inhibitors in Ovarian Cancer Models. Sci. Rep. 2019, 9, 18882. [Google Scholar] [CrossRef] [Green Version]
- Mohiuddin, I.S.; Kang, M.H. DNA-PK as an Emerging Therapeutic Target in Cancer. Front. Oncol. 2019, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Fatah, T.M.A.; Arora, A.; Moseley, P.; Coveney, C.; Perry, C.; Johnson, K.; Kent, C.; Ball, G.; Chan, S.; Madhusudan, S. ATM, ATR and DNA-PKcs Expressions Correlate to Adverse Clinical Outcomes in Epithelial Ovarian Cancers. BBA Clin. 2014, 2, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Damia, G. Targeting DNA-PK in Cancer. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2020, 821, 111692. [Google Scholar] [CrossRef] [PubMed]
- Dejmek, J.; Dirk Iglehart, J.; Lazaro, J.B. DNA-Dependent Protein Kinase (DNA-PK)-Dependent Cisplatin-Induced Loss of Nucleolar Facilitator of Chromatin Transcription (FACT) and Regulation of Cisplatin Sensitivity by DNA-PK and FACT. Mol. Cancer Res. 2009, 7, 581–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saldivar, J.S.; Wu, X.; Follen, M.; Gershenson, D. Nucleotide Excision Repair Pathway Review I: Implications in Ovarian Cancer and Platinum Sensitivity. In Proceedings of the Gynecologic Oncology, Venice, Italy, 13–15 April 2007; Volume 107. [Google Scholar]
- Li, Q.; Yu, J.J.; Mu, C.; Yunmbam, M.K.; Slavsky, D.; Cross, C.L.; Bostick-Bruton, F. Association between the Level of ERCC-1 Expression and the Repair of Cisplatin-Induced DNA Damage in Human Ovarian Cancer Cells. Anticancer Res. 2000, 20, 645–652. [Google Scholar] [PubMed]
- Arora, S.; Kothandapani, A.; Tillison, K.; Kalman-Maltese, V.; Patrick, S.M. Downregulation of XPF-ERCC1 Enhances Cisplatin Efficacy in Cancer Cells. DNA Repair 2010, 9, 745–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesquita, K.A.; Alabdullah, M.; Griffin, M.; Toss, M.S.; Fatah, T.M.A.A.; Alblihy, A.; Moseley, P.; Chan, S.Y.T.; Rakha, E.A.; Madhusudan, S. ERCC1-XPF Deficiency Is a Predictor of Olaparib Induced Synthetic Lethality and Platinum Sensitivity in Epithelial Ovarian Cancers. Gynecol. Oncol. 2019, 153, 416–424. [Google Scholar] [CrossRef]
- Pulzová, L.B.; Ward, T.A.; Chovanec, M. XPA: DNA Repair Protein of Significant Clinical Importance. Int. J. Mol. Sci. 2020, 21, 2182. [Google Scholar] [CrossRef] [Green Version]
- Tsodikov, O.V.; Ivanov, D.; Orelli, B.; Staresincic, L.; Shoshani, I.; Oberman, R.; Schärer, O.D.; Wagner, G.; Ellenberger, T. Structural Basis for the Recruitment of ERCC1-XPF to Nucleotide Excision Repair Complexes by XPA. EMBO J. 2007, 26, 4768–4776. [Google Scholar] [CrossRef] [Green Version]
- Yu, J. Platinum-Sensitive and Platinum-Resistant Ovarian Cancer Tissues Show Differences in the Relationships between MRNA Levels of P53, ERCC1 and XPA. Int. J. Oncol. 1996, 8, 313–317. [Google Scholar] [CrossRef]
- Amable, L. Cisplatin Resistance and Opportunities for Precision Medicine. Pharmacol. Res. 2016, 106, 27–36. [Google Scholar] [CrossRef]
- Dabholkar, M.; Thornton, K.; Vionnet, J.; Bostick-Bruton, F.; Yu, J.J.; Reed, E. Increased MRNA Levels of Xeroderma Pigmentosum Complementation Group B (XPB) and Cockayne’s Syndrome Complementation Group B (CSB) without Increased MRNA Levels of Multidrug-Resistance Gene (MDR1) or Metallothionein-II (MT-II) in Platinum-Resistant Human. Biochem. Pharmacol. 2000, 60, 1611–1619. [Google Scholar] [CrossRef]
- Rosenberg, E.; Taher, M.M.; Kuemmerle, N.B.; Farnsworth, J.; Valerie, K. A Truncated Human Xeroderma Pigmentosum Complementation Group A Protein Expressed from an Adenovirus Sensitizes Human Tumor Cells to Ultraviolet Light and Cisplatin Cancer Research. Cancer Res. 2001, 61, 764–770. [Google Scholar] [PubMed]
- Faridounnia, M.; Folkers, G.E.; Boelens, R. Function and Interactions of ERCC1-XPF in DNA Damage Response. Molecules 2018, 23, 3205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirschner, K.; Melton, D.W. Multiple Roles of the ERCC1-XPF Endonuclease in DNA Repair and Resistance to Anticancer Drugs. Anticancer Res. 2010, 30, 3223–3232. [Google Scholar]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-Wide and High-Density CRISPR-Cas9 Screens Identify Point Mutations in PARP1 Causing PARP Inhibitor Resistance. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Fatah, T.; Sultana, R.; Abbotts, R.; Hawkes, C.; Seedhouse, C.; Chan, S.; Madhusudan, S. Clinicopathological and Functional Significance of XRCC1 Expression in Ovarian Cancer. Int. J. Cancer 2013, 132, 2778–2786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Xie, Z.; Sun, G.; Yang, P.; Li, J.; Yang, H.; Xiao, S.; Liu, Y.; Qiu, H.; Qin, L.; et al. Reversing Drug Resistance of Cisplatin by Hsp90 Inhibitors in Human Ovarian Cancer Cells. Int. J. Clin. Exp. Med. 2015, 8, 6687–6701. [Google Scholar] [PubMed]
- Boudsocq, F.; Benaim, P.; Canitrot, Y.; Knibiehler, M.; Ausseil, F.; Capp, J.P.; Bieth, A.; Long, C.; David, B.; Shevelev, I.; et al. Modulation of Cellular Response to Cisplatin by a Novel Inhibitor of DNA Polymerase β. Mol. Pharmacol. 2005, 67, 1485–1492. [Google Scholar] [CrossRef] [Green Version]
- Bergoglio, V.; Canitrot, Y.; Hogarth, L.; Minto, L.; Howell, S.B.; Cazaux, C.; Hoffmann, J.S. Enhanced Expression and Activity of DNA Polymerase β in Human Ovarian Tumor Cells: Impact on Sensitivity towards Antitumor Agents. Oncogene 2001, 20, 6181–6187. [Google Scholar] [CrossRef] [Green Version]
- Nemec, A.A.; Abriola, L.; Merkel, J.S.; De Stanchina, E.; DeVeaux, M.; Zelterman, D.; Glazer, P.M.; Sweasy, J.B. DNA Polymerase Beta Germline Variant Confers Cellular Response to Cisplatin Therapy. Mol. Cancer Res. 2017, 15, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Sawant, A.; Floyd, A.M.; Dangeti, M.; Lei, W.; Sobol, R.W.; Patrick, S.M. Differential Role of Base Excision Repair Proteins in Mediating Cisplatin Cytotoxicity. DNA Repair 2017, 51, 46–59. [Google Scholar] [CrossRef] [Green Version]
- Chiruvella, K.K.; Liang, Z.; Wilson, T.E. Repair of Double-Strand Breaks by End Joining. Cold Spring Harb. Perspect. Biol. 2013, 5, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Kondrashova, O.; Topp, M.; Nesic, K.; Lieschke, E.; Ho, G.Y.; Harrell, M.I.; Zapparoli, G.V.; Hadley, A.; Holian, R.; Boehm, E.; et al. Methylation of All BRCA1 Copies Predicts Response to the PARP Inhibitor Rucaparib in Ovarian Carcinoma. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labidi-Galy, S.I.; Olivier, T.; Rodrigues, M.; Ferraioli, D.; Derbel, O.; Bodmer, A.; Petignat, P.; Rak, B.; Chopin, N.; Tredan, O.; et al. Location of Mutation in BRCA2 Gene and Survival in Patients with Ovarian Cancer. Clin. Cancer Res. 2018, 24, 326–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivera, B.; Di Iorio, M.; Frankum, J.; Nadaf, J.; Fahiminiya, S.; Arcand, S.L.; Burk, D.L.; Grapton, D.; Tomiak, E.; Hastings, V.; et al. Functionally Null RAD51D Missense Mutation Associates Strongly with Ovarian Carcinoma. Cancer Res. 2017, 77, 4517–4529. [Google Scholar] [CrossRef] [Green Version]
- Kondrashova, O.; Nguyen, M.; Shield-Artin, K.; Tinker, A.V.; Teng, N.N.H.; Harrell, M.I.; Kuiper, M.J.; Ho, G.Y.; Barker, H.; Jasin, M.; et al. Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated with Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2017, 7, 984–998. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, N.; Abubaker, K.; Findlay, J.K. Ovarian Cancer Stem Cells: Molecular Concepts and Relevance as Therapeutic Targets. Mol. Aspects Med. 2014, 39, 110–125. [Google Scholar] [CrossRef]
- Mihanfar, A.; Aghazadeh Attari, J.; Mohebbi, I.; Majidinia, M.; Kaviani, M.; Yousefi, M.; Yousefi, B. Ovarian Cancer Stem Cell: A Potential Therapeutic Target for Overcoming Multidrug Resistance. J. Cell. Physiol. 2019, 234, 3238–3253. [Google Scholar] [CrossRef]
- Burgos-Ojeda, D.; Rueda, B.R.; Buckanovich, R.J. Ovarian Cancer Stem Cell Markers: Prognostic and Therapeutic Implications. Cancer Lett. 2012, 322, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Motohara, T.; Katabuchi, H. Ovarian Cancer Stemness: Biological and Clinical Implications for Metastasis and Chemotherapy Resistance. Cancers 2019, 11, 907. [Google Scholar] [CrossRef] [Green Version]
- Piva, M.; Domenici, G.; Iriondo, O.; Rábano, M.; Simões, B.M.; Comaills, V.; Barredo, I.; López-Ruiz, J.A.; Zabalza, I.; Kypta, R.; et al. Sox2 Promotes Tamoxifen Resistance in Breast Cancer Cells. EMBO Mol. Med. 2014, 6, 66–79. [Google Scholar] [CrossRef]
- Chen, B.; Zhu, Z.; Li, L.; Ye, W.; Zeng, J.; Gao, J.; Wang, S.; Zhang, L.; Huang, Z. Effect of Overexpression of Oct4 and Sox2 Genes on the Biological and Oncological Characteristics of Gastric Cancer Cells. Onco. Targets. Ther. 2019, 12, 4667–4682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Ji, X.; Chen, J.; Yan, D.; Zhang, Z.; Wang, Q.; Xi, X.; Feng, Y. SOX2 Enhances the Migration and Invasion of Ovarian Cancer Cells via Src Kinase. PLoS ONE 2014, 9, e99594. [Google Scholar] [CrossRef] [PubMed]
- Belotte, J.; Fletcher, N.M.; Alexis, M.; Morris, R.T.; Munkarah, A.R.; Diamond, M.P.; Saed, G.M. Sox2 Gene Amplification Significantly Impacts Overall Survival in Serous Epithelial Ovarian Cancer. Reprod. Sci. 2015, 22, 38–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, F.; Li, Y.; Hu, Y.; Zhou, C.; Hu, Y.; Chen, H. Expression of Sox2 in Human Ovarian Epithelial Carcinoma. J. Cancer Res. Clin. Oncol. 2011, 137, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chang, D.Y.; Mercado-Uribe, I.; Liu, J. Sex-Determining Region Y-Box 2 Expression Predicts Poor Prognosis in Human Ovarian Carcinoma. Hum. Pathol. 2012, 43, 1405–1412. [Google Scholar] [CrossRef] [Green Version]
- Bareiss, P.M.; Paczulla, A.; Wang, H.; Schairer, R.; Wiehr, S.; Kohlhofer, U.; Rothfuss, O.C.; Fischer, A.; Perner, S.; Staebler, A.; et al. SOX2 Expression Associates with Stem Cell State in Human Ovarian Carcinoma. Cancer Res. 2013, 73, 5544–5555. [Google Scholar] [CrossRef]
- Wen, Y.; Hou, Y.; Huang, Z.; Cai, J.; Wang, Z. SOX2 Is Required to Maintain Cancer Stem Cells in Ovarian Cancer. Cancer Sci. 2017, 108, 719–731. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.L.; Mallanna, S.K.; Luo, X.; Rizzino, A. Sox2 Uses Multiple Domains to Associate with Proteins Present in Sox2-Protein Complexes. PLoS ONE 2010, 5, e15486. [Google Scholar] [CrossRef]
- Wang, J.; Rao, S.; Chu, J.; Shen, X.; Levasseur, D.N.; Theunissen, T.W.; Orkin, S.H. A Protein Interaction Network for Pluripotency of Embryonic Stem Cells. Nature 2006, 444, 364–368. [Google Scholar] [CrossRef]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An Embryonic Stem Cell–like Gene Expression Signature in Poorly Differentiated Aggressive Human Tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef]
- Nagata, T.; Shimada, Y.; Sekine, S.; Hori, R.; Matsui, K.; Okumura, T.; Sawada, S.; Fukuoka, J.; Tsukada, K. Prognostic Significance of NANOG and KLF4 for Breast Cancer. Breast Cancer 2014, 21, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Sun, J.; Cai, B.; Xi, X.; Yang, L.; Zhang, Z.; Feng, Y.; Sun, Y. NANOG Regulates Epithelial-Mesenchymal Transition and Chemoresistance through Activation of the STAT3 Pathway in Epithelial Ovarian Cancer. Tumor Biol. 2016, 37, 9671–9680. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Maihle, N.J.; Huang, Y. Pluripotency Factors Lin28 and Oct4 Identify a Sub-Population of Stem Cell-like Cells in Ovarian Cancer. Oncogene 2010, 29, 2153–2159. [Google Scholar] [CrossRef] [Green Version]
- Meng, H.M.; Zheng, P.; Wang, X.Y.; Liu, C.; Sui, H.M.; Wu, S.J.; Zhou, J.; Ding, Y.Q.; Li, J.M. Overexpression of Nanog Predicts Tumor Progression and Poor Prognosis in Colorectal Cancer. Cancer Biol. Ther. 2010, 9, 295–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Balch, C.; Chan, M.W.; Lai, H.-C.; Matei, D.; Schilder, J.M.; Yan, P.S.; Huang, T.H.; Nephew, K.P. Identification and Characterization of Ovarian Cancer-Initiating Cells from Primary Human Tumors. Cancer Res. 2008, 68, 4311–4320. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Nam, E.J.; Kim, S.W.; Kim, S.; Kim, J.H.; Kim, Y.T. Prognostic Impact of the Cancer Stem Cell-Related Marker NANOG in Ovarian Serous Carcinoma. Int. J. Gynecol. Cancer 2012, 22, 1489–1496. [Google Scholar] [CrossRef]
- Lu, Y.; Zhu, H.; Shan, H.; Lu, J.; Chang, X.; Li, X.; Lu, J.; Fan, X.; Zhu, S.; Wang, Y.; et al. Knockdown of Oct4 and Nanog Expression Inhibits the Stemness of Pancreatic Cancer Cells. Cancer Lett. 2013, 340, 113–123. [Google Scholar] [CrossRef]
- Abubaker, K.; Luwor, R.B.; Zhu, H.; McNally, O.; Quinn, M.A.; Burns, C.J.; Thompson, E.W.; Findlay, J.K.; Ahmed, N. Inhibition of the JAK2/STAT3 Pathway in Ovarian Cancer Results in the Loss of Cancer Stem Cell-like Characteristics and a Reduced Tumor Burden. BMC Cancer 2014, 14, 317. [Google Scholar] [CrossRef] [Green Version]
- Abubaker, K.; Luwor, R.B.; Escalona, R.; McNally, O.; Quinn, M.A.; Thompson, E.W.; Findlay, J.K.; Ahmed, N. Targeted Disruption of the JAK2/STAT3 Pathway in Combination with Systemic Administration of Paclitaxel Inhibits the Priming of Ovarian Cancer Stem Cells Leading to a Reduced Tumor Burden. Front. Oncol. 2014, 4, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Quintás-Cardama, A.; Verstovsek, S. Molecular Pathways: JAK/STAT Pathway: Mutations, Inhibitors, and Resistance. Clin. Cancer Res. 2013, 19, 1933–1940. [Google Scholar] [CrossRef] [Green Version]
- Reeves, P.M.; Abbaslou, M.A.; Kools, F.R.W.; Vutipongsatorn, K.; Tong, X.; Gavegnano, C.; Schinazi, R.F.; Poznansky, M.C. Correction: Ruxolitinib Sensitizes Ovarian Cancer to Reduced Dose Taxol, Limits Tumor Growth and Improves Survival in Immune Competent Mice. Oncotarget 2018, 9, 30472. [Google Scholar] [CrossRef] [PubMed]
- Han, E.S.; Wen, W.; Dellinger, T.H.; Wu, J.; Lu, S.A.; Jove, R.; Yim, J.H. Ruxolitinib Synergistically Enhances the Anti-Tumor Activity of Paclitaxel in Human Ovarian Cancer. Oncotarget 2018, 9, 24304–24319. [Google Scholar] [CrossRef] [PubMed]
- Áyen, Á.; Martínez, Y.J.; Marchal, J.A.; Boulaiz, H. Recent Progress in Gene Therapy for Ovarian Cancer. Int. J. Mol. Sci. 2018, 19, 1–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, M.N.; Malhotra, J.; Tarapore, R.S.; Malhotra, U.; Silk, A.W.; Chan, N.; Rodriguez, L.; Aisner, J.; Aiken, R.D.; Mayer, T.; et al. Safety and Enhanced Immunostimulatory Activity of the DRD2 Antagonist ONC201 in Advanced Solid Tumor Patients with Weekly Oral Administration. J. Immunother. Cancer 2019, 7, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, M.N.; Bertino, J.R.; Kaufman, H.L.; Mayer, T.; Moss, R.; Silk, A.; Chan, N.; Malhotra, J.; Rodriguez, L.; Aisner, J.; et al. First-in-Human Clinical Trial of Oral ONC201 in Patients with Refractory Solid Tumors. Clin. Cancer Res. 2017, 23, 4163–4169. [Google Scholar] [CrossRef] [Green Version]
- Brenner, A.J.; Cohen, Y.C.; Breitbart, E.; Bangio, L.; Sarantopoulos, J.; Giles, F.J.; Borden, E.C.; Harats, D.; Triozzi, P.L. Phase i Dose-Escalation Study of VB-111, an Antiangiogenic Virotherapy, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2013, 19, 3996–4007. [Google Scholar] [CrossRef]
- Blagden, S.P.; Hamilton, A.L.; Mileshkin, L.; Wong, S.; Michael, A.; Hall, M.; Goh, J.C.; Lisyanskaya, A.S.; DeSilvio, M.; Frangou, E.; et al. Phase IB Dose Escalation and Expansion Study of Akt Inhibitor Afuresertib with Carboplatin and Paclitaxel in Recurrent Platinum-Resistant Ovarian Cancer. Clin. Cancer Res. 2019, 25, 1472–1478. [Google Scholar] [CrossRef] [Green Version]
- Risnayanti, C.; Jang, Y.S.; Lee, J.; Ahn, H.J. PLGA Nanoparticles Co-Delivering MDR1 and BCL2 SiRNA for Overcoming Resistance of Paclitaxel and Cisplatin in Recurrent or Advanced Ovarian Cancer. Sci. Rep. 2018, 8, 7498. [Google Scholar] [CrossRef] [Green Version]
- Vaghari-Tabari, M.; Hassanpour, P.; Sadeghsoltani, F.; Malakoti, F.; Alemi, F.; Qujeq, D.; Asemi, Z.; Yousefi, B. CRISPR/Cas9 Gene Editing: A New Approach for Overcoming Drug Resistance in Cancer. Cell. Mol. Biol. Lett. 2022, 27, 49. [Google Scholar] [CrossRef]
- Norouzi-Barough, L.; Sarookhani, M.; Salehi, R.; Sharifi, M.; Moghbelinejad, S. CRISPR/Cas9, a New Approach to Successful Knockdown of ABCB1/P-Glycoprotein and Reversal of Chemosensitivity in Human Epithelial Ovarian Cancer Cell Line. Iran. J. Basic Med. Sci. 2018, 21, 181–187. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T Cell Therapy: Current Limitations and Potential Strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Cai, H.; Zhao, L.; Ning, L.; Lang, J. CAR-T Cell Therapy in Ovarian Cancer: From the Bench to the Bedside. Oncotarget 2017, 8, 64607–64621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Saux, O.; Ray-Coquard, I.; Labidi-Galy, S.I. Challenges for Immunotherapy for the Treatment of Platinum Resistant Ovarian Cancer. Semin. Cancer Biol. 2021, 77, 127–143. [Google Scholar] [CrossRef] [PubMed]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T Cells in Solid Tumors: Challenges and Opportunities. Stem Cell Res. Ther. 2021, 12, 81. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DNA Repair Pathway | Overexpressed Protein(s) | References |
|---|---|---|
| Homologous Recombination | RAD51 and RAD51 paralogs; BRCA1; BRCA2; PARP1 | [147,148,149,150,151,152,153,154,155,156] |
| Non-homologous End Joining | RIF1; DNA-PK; PARP1 | [157,158,159,160,161,162,163,164,165,166] |
| Nucleotide Excision Repair | XP groups ERCC1; PARP1 | [159,167,168,169,170,171,172,173,174,175,176,177,178] |
| Base Excision Repair | XRCC1; pol β; PARP1 | [149,159,179,180,181,182,183,184,185] |
| Therapeutic | Target | Drug Type | Phase | Clinical Trial Identifier |
|---|---|---|---|---|
| ABT-263 (Navitoclax) | Bcl-XL, Bcl-2 | Small molecule mimetic | II | NCT02591095 |
| GDC-0068 (Ipatasertib) | AKT1/2/3 | Small molecule inhibitor | II | NCT04561817 |
| Tivozanib | VEGF | Small molecule inhibitor | II | NCT01853644 |
| XL999 | VEGFR PDGFR FGFR, FLT-3, Src | Small molecule inhibitor | II | NCT00277290 |
| MM-121 (seribantumab) | HER3 Pathway | Monoclonal antibody | II | NCT01447706 |
| VB-111 (ofranergene obadenovec) | TNFR1, FAS | Gene therapy (chimeric gene) | III | NCT03398655 |
| ONC201 | Akt/ERK | Small molecule inhibitor | II | NCT04055649 |
| ZN-c3 | Wee1 | Small molecule inhibitor | I | NCT05198804 |
| ACR-368 | CHK1/2 | Small molecule inhibitor | I | NCT05548296 |
| Navicixizumab | DLL4, VEGF | Monoclonal antibody | III | NCT05043402 |
| Bevacizumab | VEGF | Monoclonal antibody | II | NCT05325229 |
| JPI-547 | PARP1/2, TNKS | Small molecule inhibitor | II | NCT05475184 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alatise, K.L.; Gardner, S.; Alexander-Bryant, A. Mechanisms of Drug Resistance in Ovarian Cancer and Associated Gene Targets. Cancers 2022, 14, 6246. https://doi.org/10.3390/cancers14246246
Alatise KL, Gardner S, Alexander-Bryant A. Mechanisms of Drug Resistance in Ovarian Cancer and Associated Gene Targets. Cancers. 2022; 14(24):6246. https://doi.org/10.3390/cancers14246246
Chicago/Turabian StyleAlatise, Kharimat Lora, Samantha Gardner, and Angela Alexander-Bryant. 2022. "Mechanisms of Drug Resistance in Ovarian Cancer and Associated Gene Targets" Cancers 14, no. 24: 6246. https://doi.org/10.3390/cancers14246246
APA StyleAlatise, K. L., Gardner, S., & Alexander-Bryant, A. (2022). Mechanisms of Drug Resistance in Ovarian Cancer and Associated Gene Targets. Cancers, 14(24), 6246. https://doi.org/10.3390/cancers14246246