
Deciphering the Role of p53 and TAp73 in Neuroblastoma: From Pathogenesis to Treatment

 , ,
, ,  ,
,  and
and 
Simple Summary
Abstract
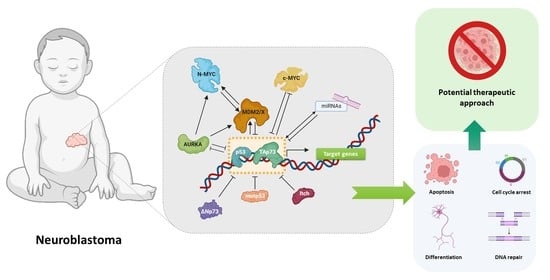
1. Introduction
2. The p53 Family Proteins: p53 and TAp73
2.1. p53 and TAp73 Interaction with MDM2 and MDMX
2.2. p53 and TAp73 Interaction with Mutant p53
2.3. p53 and TAp73 Interaction with ΔNp73
2.4. Proteasomal-Dependent Degradation of TAp73 by Itch
2.5. p53 and TAp73 Interaction with AURKA
3. The N-MYC and p53/TAp73 Interplay
4. Crosstalk between miRNAs and p53/TAp73

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | Target | Function | References |
|---|---|---|---|
| Upregulated OncomiRs | |||
| miR-15a | RECK | Induces migration and invasion | [197] |
| miR-21 | PTEN, PDCD4, FOXO3A | Induces proliferation and invasion | [198] |
| miR-23a | CDH1 | Induces migration and invasion | [199] |
| miR-221 | NLK | Induces proliferation and cell cycle progression | [200] |
| miR-380-5p | TP53 | Increases proliferation and self-renewal | [201] |
| miR-558 | HPSE | Induces proliferation, invasion, metastasis, and angiogenesis | [202] |
| miR-1303 | GSK3β, SFRP1 | Induces proliferation | [203] |
| miR-3934-5p | TP53INP1 | Inhibits apoptosis and promotes viability | [204] |
| Downregulated TSmiRs | |||
| Let-7 | MYCN | Induces differentiation | [161,165] |
| miR-9 | MMP-14, TP73 | Inhibits invasion, metastasis, and angiogenesis | [205] |
| miR-15a/b | MYCN | Reduces proliferation, migration, and invasion | [206] |
| miR-16 | MYCN | Reduces proliferation, migration, and invasion | [206] |
| miR-34a | MYCN, E2F3, BCL2 | Induces cell cycle arrest and apoptosis; reduces angiogenesis | [207,208] |
| miR-192 | DICER1 | Inhibits proliferation and migration | [193,209] |
| miR-203 | KHDRBS1 | Inhibits invasion, proliferation, and migration | [210] |
| miR-338-3p | PREX2a | Inhibits proliferation and survival; induces cell cycle arrest | [211] |
| miR-1247 | ZNF346 | Inhibits proliferation; induces cell-cycle arrest and cell death | [212] |
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Wang, Y.; Zhang, L.; Zhao, W.; Dai, X.; Yang, Y.-G.; Zhang, X. Targeting bromodomain and extra-terminal proteins to inhibit neuroblastoma tumorigenesis through regulating MYCN. Front. Cell Dev. Biol. 2022, 10, 1021820. [Google Scholar] [CrossRef] [PubMed]
- Newman, E.A.; Abdessalam, S.; Aldrink, J.H.; Austin, M.; Heaton, T.E.; Bruny, J.; Ehrlich, P.; Dasgupta, R.; Baertschiger, R.M.; Lautz, T.B.; et al. Update on neuroblastoma. J. Pediatr. Surg. 2019, 54, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Kameneva, P.; Artemov, A.V.; Kastriti, M.E.; Faure, L.; Olsen, T.K.; Otte, J.; Erickson, A.; Semsch, B.; Andersson, E.R.; Ratz, M.; et al. Single-cell transcriptomics of human embryos identifies multiple sympathoblast lineages with potential implications for neuroblastoma origin. Nat. Genet. 2021, 53, 694–706. [Google Scholar] [CrossRef]
- Jansky, S.; Sharma, A.K.; Körber, V.; Quintero, A.; Toprak, U.H.; Wecht, E.M.; Gartlgruber, M.; Greco, A.; Chomsky, E.; Grünewald, T.G.P.; et al. Single-cell transcriptomic analyses provide insights into the developmental origins of neuroblastoma. Nat. Genet. 2021, 53, 683–693. [Google Scholar] [CrossRef]
- Kastriti, M.E.; Faure, L.; Von Ahsen, D.; Bouderlique, T.G.; Boström, J.; Solovieva, T.; Jackson, C.; Bronner, M.; Meijer, D.; Hadjab, S.; et al. Schwann cell precursors represent a neural crest-like state with biased multipotency. EMBO J. 2022, 41, e108780. [Google Scholar] [CrossRef]
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Prim. 2016, 2, 16078. [Google Scholar] [CrossRef]
- Lundberg, K.I.; Treis, D.; Johnsen, J.I. Neuroblastoma Heterogeneity, Plasticity, and Emerging Therapies. Curr. Oncol. Rep. 2022, 24, 1053–1062. [Google Scholar] [CrossRef]
- Nicolai, S.; Pieraccioli, M.; Peschiaroli, A.; Melino, G.; Raschellà, G. Neuroblastoma: Oncogenic mechanisms and therapeutic exploitation of necroptosis. Cell Death Dis. 2015, 6, e2010. [Google Scholar] [CrossRef]
- Nakagawara, A.; Li, Y.; Izumi, H.; Muramori, K.; Inada, H.; Nishi, M. Neuroblastoma. Jpn. J. Clin. Oncol. 2018, 48, 214–241. [Google Scholar] [CrossRef]
- Park, J.R.; Eggert, A.; Caron, H. Neuroblastoma: Biology, Prognosis, and Treatment. Hematol. Oncol. Clin. N. Am. 2010, 24, 65–86. [Google Scholar] [CrossRef] [PubMed]
- Bresler, S.C.; Weiser, D.A.; Huwe, P.J.; Park, J.H.; Krytska, K.; Ryles, H.; Laudenslager, M.; Rappaport, E.F.; Wood, A.C.; McGrady, P.W.; et al. ALK Mutations Confer Differential Oncogenic Activation and Sensitivity to ALK Inhibition Therapy in Neuroblastoma. Cancer Cell 2014, 26, 682–694. [Google Scholar] [CrossRef]
- Mossé, Y.P.; Laudenslager, M.; Longo, L.; Cole, K.A.; Wood, A.; Attiyeh, E.F.; Laquaglia, M.J.; Sennett, R.; Lynch, J.E.; Perri, P.; et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008, 455, 930–935. [Google Scholar] [CrossRef]
- Janoueix-Lerosey, I.; Lequin, D.; Brugières, L.; Ribeiro, A.; De Pontual, L.; Combaret, V.; Raynal, V.; Puisieux, A.; Schleiermacher, G.; Pierron, G.; et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 2008, 455, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Thiele, C.J. Molecular Genetics of Neuroblastoma. In Diagnostic and Therapeutic Nuclear Medicine for Neuroendocrine Tumors; Pacak, K., Taïeb, D., Eds.; Humana Press: Totowaa, NJ, USA, 2017; pp. 83–125. ISBN 978-3-319-46038-3. [Google Scholar]
- Kameneva, P.; Kastriti, M.E.; Adameyko, I. Neuronal lineages derived from the nerve-associated Schwann cell precursors. Cell. Mol. Life Sci. 2021, 78, 513–529. [Google Scholar] [CrossRef] [PubMed]
- Mosse, Y.P.; Laudenslager, M.; Khazi, D.; Carlisle, A.J.; Winter, C.L.; Rappaport, E.; Maris, J.M. Germline PHOX2B Mutation in Hereditary Neuroblastoma. Am. J. Hum. Genet. 2004, 75, 727–730. [Google Scholar] [CrossRef]
- Trochet, D.; Bourdeaut, F.; Janoueix-Lerosey, I.; Deville, A.; De Pontual, L.; Schleiermacher, G.; Coze, C.; Philip, N.; Frébourg, T.; Munnich, A.; et al. Germline Mutations of the Paired-Like Homeobox 2B (PHOX2B) Gene in Neuroblastoma. Am. J. Hum. Genet. 2004, 74, 761–764. [Google Scholar] [CrossRef]
- Valentijn, L.J.; Koster, J.; Zwijnenburg, D.A.; Hasselt, N.E.; Van Sluis, P.; Volckmann, R.; Van Noesel, M.M.; George, R.E.; Tytgat, G.A.M.; Molenaar, J.J.; et al. TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat. Genet. 2015, 47, 1411–1414. [Google Scholar] [CrossRef]
- Brodeur, G.M.; Minturn, J.E.; Ho, R.; Simpson, A.M.; Iyer, R.; Varela, C.R.; Light, J.E.; Kolla, V.; Evans, A.E. Trk receptor expression and inhibition in neuroblastomas. Clin. Cancer Res. 2009, 15, 3244–3250. [Google Scholar] [CrossRef]
- Carén, H.; Kryh, H.; Nethander, M.; Sjöberg, R.M.; Träger, C.; Nilsson, S.; Abrahamsson, J.; Kogner, P.; Martinsson, T. High-risk neuroblastoma tumors with 11q-deletion display a poor prognostic, chromosome instability phenotype with later onset. Proc. Natl. Acad. Sci. USA 2010, 107, 4323–4328. [Google Scholar] [CrossRef]
- J Ribelles, A.; Barberá, S.; Yáñez, Y.; Gargallo, P.; Segura, V.; Juan, B.; Noguera, R.; Piqueras, M.; Fornés-Ferrer, V.; de Mora, J.F.; et al. Clinical Features of Neuroblastoma With 11q Deletion: An Increase in Relapse Probabilities In Localized And 4S Stages. Sci. Rep. 2019, 9, 13806. [Google Scholar] [CrossRef] [PubMed]
- Hogarty, M.D.; Liu, X.; Guo, C.; Thompson, P.M.; Weiss, M.J.; White, P.S.; Sulman, E.P.; Brodeur, G.M.; Maris, J.M. Identification of a 1-megabase consensus region of deletion at 1p36.3 in Primary neuroblastomas. Med. Pediatr. Oncol. 2000, 35, 512–515. [Google Scholar] [CrossRef]
- Ichimiya, S.; Nimura, Y.; Kageyama, H.; Takada, N.; Sunahara, M.; Shishikura, T.; Nakamura, Y.; Sakiyama, S.; Seki, N.; Ohira, M.; et al. P73 At Chromosome 1P36.3 Is Lost in Advanced Stage Neuroblastoma But Its Mutation Is Infrequent. Oncogene 1999, 18, 1061–1066. [Google Scholar] [CrossRef] [PubMed]
- Collavin, L.; Lunardi, A.; Del Sal, G. P53-family proteins and their regulators: Hubs and spokes in tumor suppression. Cell Death Differ. 2010, 17, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Ramos, H.; Raimundo, L.; Saraiva, L. p73: From the p53 shadow to a major pharmacological target in anticancer therapy. Pharmacol. Res. 2020, 162, 105245. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Teo, C.R.; Sabapathy, K. P53-related transcription targets of TAp73 in cancer cells—Bona fide or distorted reality? Int. J. Mol. Sci. 2020, 21, 1346. [Google Scholar] [CrossRef]
- Yang, A.; Schweitzer, R.; Sun, D.; Kaghad, M.; Walker, N.; Bronson, R.T.; Tabin, C.; Sharpe, A.; Caput, D.; Crum, C.; et al. P63 Is Essential for Regenerative Proliferation in Limb, Craniofacial and Epithelial Development. Nature 1999, 398, 714–718. [Google Scholar] [CrossRef]
- Yang, A.; Walker, N.; Bronson, R.; Kaghad, M.; Oosterwegel, M.; Bonnin, J.; Vagner, C.; Bonnet, H.; Dikkesk, P.; Sharpe, A.; et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature 2000, 404, 99–103. [Google Scholar] [CrossRef]
- Wei, J.; Zaika, E.; Zaika, A. P53 family: Role of protein isoforms in human cancer. J. Nucleic Acids 2012, 2012, 687359. [Google Scholar] [CrossRef]
- Dötsch, V.; Bernassola, F.; Coutandin, D.; Candi, E.; Melino, G. P63 and P73, the Ancestors of P53. Cold Spring Harb. Perspect. Biol. 2010, 2, a004887. [Google Scholar] [CrossRef]
- Stiewe, T. The p53 family in differentiation and tumorigenesis. Nat. Rev. Cancer 2007, 7, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lane, D.P. P53 in Health and Disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Machado-Silva, A.; Perrier, S.; Bourdon, J.C. P53 family members in cancer diagnosis and treatment. Semin. Cancer Biol. 2010, 20, 57–62. [Google Scholar] [CrossRef]
- Ichimiya, S.; Nakagawara, A.; Sakuma, Y.; Kimura, S.; Ikeda, T.; Satoh, M.; Takahashi, N.; Sato, N.; Mori, M. p73: Structure and function. Pathol. Int. 2000, 50, 589–593. [Google Scholar] [CrossRef] [PubMed]
- De Laurenzi, V.; Raschellá, G.; Barcaroli, D.; Annicchiarico-Petruzzelli, M.; Ranalli, M.; Catani, M.V.; Tanno, B.; Costanzo, A.; Levrero, M.; Melino, G. Induction of neuronal differentiation by p73 in a neuroblastoma cell line. J. Biol. Chem. 2000, 275, 15226–15231. [Google Scholar] [CrossRef]
- Wagner, L.M.; Danks, M.K. New therapeutic targets for the treatment of high-risk neuroblastoma. J. Cell. Biochem. 2009, 107, 46–57. [Google Scholar] [CrossRef]
- Vogan, K.; Bernstein, M.; Leclerc, J.M.; Brisson, L.; Brossard, J.; Brodeur, G.M.; Pelletier, J.; Gros, P. Absence of p53 Gene Mutations in Primary Neuroblastomas. Cancer Res. 1993, 53, 5269–5273. [Google Scholar]
- Zhu, X.; Wimmer, K.; Kuick, R.; Lamb, B.J.; Motyka, S.; Jasty, R.; Castle, V.P.; Hanash, S.M. N-myc modulates expression of p73 in neuroblastoma. Neoplasia 2002, 4, 432–439. [Google Scholar] [CrossRef][Green Version]
- Ikawa, S.; Nakagawara, A.; Ikawa, Y. p53 family genes: Structural comparison, expression and mutation. Cell Death Differ. 1999, 6, 1154–1161. [Google Scholar] [CrossRef]
- Inomistova, M.V.; Svergun, N.M.; Khranovska, N.M.; Skachkova, O.V.; Gorbach, O.I.; Klymnyuk, G.I. Prognostic significance of MDM2 gene expression in childhood neuroblastoma. Exp. Oncol. 2015, 37, 111–115. [Google Scholar] [CrossRef]
- Berberich, S.J. Mdm2 and MdmX involvement in human cancer. In Sub-Cellular Biochemistry; Springer: Dordrecht, The Netherlands, 2014; Volume 85, pp. 263–280. ISBN 9789401792110. [Google Scholar]
- Corvi, R.; Savelyeva, L.; Breit, S.; Wenzel, A.; Handgretinger, R.; Barak, J.; Oren, M.; Amler, L.; Schwab, M. Non-syntenic amplification of MDM2 and MYCN in human neuroblastoma. Oncogene 1995, 10, 1081–1086. [Google Scholar] [PubMed]
- Rayburn, E.; Zhang, R.; He, J.; Wang, H. MDM2 and human malignancies: Expression, clinical pathology, prognostic markers, and implications for chemotherapy. Curr. Cancer Drug Targets 2005, 5, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Zafar, A.; Wang, W.; Liu, G.; Xian, W.; McKeon, F.; Zhou, J.; Zhang, R. Targeting the p53-MDM2 pathway for neuroblastoma therapy: Rays of hope. Cancer Lett. 2021, 496, 16–29. [Google Scholar] [CrossRef]
- Wade, M.; Li, Y.C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Karni-Schmidt, O.; Lokshin, M.; Prives, C. The Roles of MDM2 and MDMX in Cancer. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 617–644. [Google Scholar] [CrossRef]
- Bálint, E.; Bates, S.; Vousden, K.H. Mdm2 binds p73α without targeting degradation. Oncogene 1999, 18, 3923–3929. [Google Scholar] [CrossRef] [PubMed]
- Dobbelstein, M.; Wienzek, S.; König, C.; Roth, J. Inactivation of the p53-homologue p73 by the mdm2-oncoprotein. Oncogene 1999, 18, 2101–2106. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Leng, R.P. MDM2 mediates p73 ubiquitination: A new molecular mechanism for suppression of p73 function. Oncotarget 2015, 6, 21479–21492. [Google Scholar] [CrossRef]
- Gu, J.; Nie, L.; Wiederschain, D.; Yuan, Z.-M. Identification of p53 Sequence Elements That Are Required for MDM2-Mediated Nuclear Export. Mol. Cell. Biol. 2001, 21, 8533–8546. [Google Scholar] [CrossRef]
- Shi, Y.; Takenobu, H.; Kurata, K.; Yamaguchi, Y.; Yanagisawa, R.; Ohira, M.; Koike, K.; Nakagawara, A.; Jiang, L.L.; Kamijo, T. HDM2 impairs Noxa transcription and affects apoptotic cell death in a p53/p73-dependent manner in neuroblastoma. Eur. J. Cancer 2010, 46, 2324–2334. [Google Scholar] [CrossRef]
- Barbieri, E.; Mehta, P.; Chen, Z.; Zhang, L.; Slack, A.; Berg, S.; Shohet, J.M. MDM2 inhibition sensitizes neuroblastoma to chemotherapy-induced apoptotic cell death. Mol. Cancer Ther. 2006, 5, 2358–2365. [Google Scholar] [CrossRef] [PubMed]
- Van Maerken, T.; Speleman, F.; Vermeulen, J.; Lambertz, I.; De Clercq, S.; De Smet, E.; Yigit, N.; Coppens, V.; Philippé, J.; De Paepe, A.; et al. Small-molecule MDM2 antagonists as a new therapy concept for neuroblastoma. Cancer Res. 2006, 66, 9646–9655. [Google Scholar] [CrossRef] [PubMed]
- Gamble, L.D.; Kees, U.R.; Tweddle, D.A.; Lunec, J. MYCN sensitizes neuroblastoma to the MDM2-p53 antagonists Nutlin-3 and MI-63. Oncogene 2012, 31, 752–763. [Google Scholar] [CrossRef]
- Kung, C.P.; Weber, J.D. It’s Getting Complicated—A Fresh Look at p53-MDM2-ARF Triangle in Tumorigenesis and Cancer Therapy. Front. Cell Dev. Biol. 2022, 10, 818744. [Google Scholar] [CrossRef] [PubMed]
- Van Maerken, T.; Vandesompele, J.; Rihani, A.; De Paepe, A.; Speleman, F. Escape from p53-mediated tumor surveillance in neuroblastoma: Switching off the p14ARF-MDM2-p53 axis. Cell Death Differ. 2009, 16, 1563–1572. [Google Scholar] [CrossRef]
- Carr-Wilkinson, J.; O’Toole, K.; Wood, K.M.; Challen, C.C.; Baker, A.G.; Board, J.R.; Evans, L.; Cole, M.; Cheung, N.K.V.; Boos, J.; et al. High frequency of p53/MDM2/p14ARF pathway abnormalities in relapsed neuroblastoma. Clin. Cancer Res. 2010, 16, 1108–1118. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.S.; Ramos, H.; Inga, A.; Sousa, E.; Saraiva, L. Structural and drug targeting insights on mutant p53. Cancers 2021, 13, 3344. [Google Scholar] [CrossRef]
- Muller, P.A.J.; Vousden, K.H. P53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef]
- Bargonetti, J.; Reynisdottir, I.; Friedman, P.N.; Prives, C. Site-specific binding of wild-type p53 to cellular DNA is inhibited by SV40 T antigen and mutant p53. Genes Dev. 1992, 6, 1886–1898. [Google Scholar] [CrossRef][Green Version]
- Kern, S.E.; Pietenpol, J.A.; Thiagalingam, S.; Seymour, A.; Kinzler, K.W.; Vogelstein, B. Oncogenic forms of p53 inhibit p53-regulated gene expression. Science 1992, 256, 827–830. [Google Scholar] [CrossRef]
- Gaiddon, C.; Lokshin, M.; Ahn, J.; Zhang, T.; Prives, C. A Subset of Tumor-Derived Mutant Forms of p53 Down-Regulate p63 and p73 through a Direct Interaction with the p53 Core Domain. Mol. Cell. Biol. 2001, 21, 1874–1887. [Google Scholar] [CrossRef] [PubMed]
- Di Agostino, S.; Cortese, G.; Monti, O.; Dell’Orso, S.; Sacchi, A.; Eisenstein, M.; Citro, G.; Strano, S.; Blandino, G. The disruption of the protein complex mutantp53/p73 increases selectively the response of tumor cells to anticancer drugs. Cell Cycle 2008, 7, 3440–3447. [Google Scholar] [CrossRef] [PubMed]
- Schulz-Heddergott, R.; Moll, U.M. Gain-of-function (GOF) mutant p53 as actionable therapeutic target. Cancers 2018, 10, 188. [Google Scholar] [CrossRef] [PubMed]
- Di Agostino, S. The impact of mutant p53 in the non-coding RNA world. Biomolecules 2020, 10, 472. [Google Scholar] [CrossRef] [PubMed]
- Dong, P.; Karaayvaz, M.; Jia, N.; Kaneuchi, M.; Hamada, J.; Watari, H.; Sudo, S.; Ju, J.; Sakuragi, N. Mutant p53 gain-of-function induces epithelial-mesenchymal transition through modulation of the miR-130b-ZEB1 axis. Oncogene 2013, 32, 3286–3295. [Google Scholar] [CrossRef]
- Neilsen, P.M.; Noll, J.E.; Mattiske, S.; Bracken, C.P.; Gregory, P.A.; Schulz, R.B.; Lim, S.P.; Kumar, R.; Suetani, R.J.; Goodall, G.J.; et al. Mutant p53 drives invasion in breast tumors through up-regulation of miR-155. Oncogene 2013, 32, 2992–3000. [Google Scholar] [CrossRef]
- Marin, M.C.; Jost, C.A.; Brooks, L.A.; Irwin, M.S.; O’Nions, J.; Tidy, J.A.; James, N.; McGregor, J.M.; Harwood, C.A.; Yulug, I.G.; et al. A common polymorphism acts as an intragenic modifier of mutant p53 behaviour. Nat. Genet. 2000, 25, 47–54. [Google Scholar] [CrossRef]
- Bergamaschi, D.; Gasco, M.; Hiller, L.; Sullivan, A.; Syed, N.; Trigiante, G.; Yulug, I.; Merlano, M.; Numico, G.; Comino, A.; et al. P53 Polymorphism Influences Response in Cancer Chemotherapy Via Modulation of P73-Dependent Apoptosis. Cancer Cell 2003, 3, 387–402. [Google Scholar] [CrossRef]
- Irwin, M.S.; Kondo, K.; Marin, M.C.; Cheng, L.S.; Hahn, W.C.; Kaelin, W.G. Chemosensitivity linked to p73 function. Cancer Cell 2003, 3, 403–410. [Google Scholar] [CrossRef]
- Hosoi, G.; Hara, J.; Okamura, T.; Osugi, Y.; Fukuzawa, M.; Okada, A.; Tawa, A. Low frequency of the p53 gene mutations in neuroblastoma. Cancer 1994, 73, 3087–3093. [Google Scholar] [CrossRef]
- Davidoff, A.M.; Pence, J.C.; Shorter, N.A.; Iglehart, J.D.; Marks, J.R. Expression of p53 in human neuroblastoma- and neuroepithelioma-derived cell lines. Oncogene 1992, 7, 127–133. [Google Scholar] [PubMed]
- Keshelava, N.; Zuo, J.J.; Sitara Waidyaratne, N.; Triche, T.J.; Patrick Reynolds, C. p53 Mutations and loss of p53 function confer multidrug resistance in neuroblastoma. Med. Pediatr. Oncol. 2000, 35, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Keshelava, N.; Zuo, J.J.; Chen, P.; Waidyaratne, S.N.; Luna, M.C.; Gomer, C.J.; Triche, T.J.; Reynolds, C.P.; Keshelava, N.; Zuo, J.J.; et al. Loss of p53 function confers high-level multidrug resistance in neuroblastoma cell lines. Cancer Res. 2001, 61, 6185–6193. [Google Scholar]
- Imamura, J.; Bartram, C.R.; Berthold, F.; Harms, D.; Nakamura, H.; Koeffler, H.P. Mutation of the p53 Gene in Neuroblastoma and Its Relationship with N-myc Amplification. Cancer Res. 1993, 53, 4053–4058. [Google Scholar] [PubMed]
- Manhani, R.; Cristofani, L.M.; Filho, V.O.; Bendit, I. Concomitant p53 mutation and MYCN amplification if neuroblastoma. Med. Pediatr. Oncol. 1997, 29, 206–207. [Google Scholar] [CrossRef]
- Tweddle, D.A.; Malcolm, A.J.; Bown, N.; Pearson, A.D.J.; Lunec, J. Evidence for the Development of p53 Mutations after Cytotoxic Therapy in a Neuroblastoma Cell Line. Cancer Res. 2001, 61, 8–13. [Google Scholar] [PubMed]
- Maris, J.M.; Matthay, K.K. Molecular biology of neuroblastoma. J. Clin. Oncol. 1999, 17, 2264–2279. [Google Scholar] [CrossRef]
- Kravchenko, J.E.; Ilyinskaya, G.V.; Komarov, P.G.; Agapova, L.S.; Kochetkov, D.V.; Strom, E.; Frolova, E.I.; Kovriga, I.; Gudkov, A.V.; Feinstein, E.; et al. Small-molecule RETRA suppresses mutant p53-bearing cancer cells through a p73-dependent salvage pathway. Proc. Natl. Acad. Sci. USA. 2008, 105, 6302–6307. [Google Scholar] [CrossRef]
- Muller, P.A.J.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef]
- Gomes, S.; Raimundo, L.; Soares, J.; Loureiro, J.B.; Leão, M.; Ramos, H.; Monteiro, M.N.; Lemos, A.; Moreira, J.; Pinto, M.; et al. New inhibitor of the TAp73 interaction with MDM2 and mutant p53 with promising antitumor activity against neuroblastoma. Cancer Lett. 2019, 446, 90–102. [Google Scholar] [CrossRef]
- Tomasini, R.; Tsuchihara, K.; Wilhelm, M.; Fujitani, M.; Rufini, A.; Cheung, C.C.; Khan, F.; Itie-Youten, A.; Wakeham, A.; Tsao, M.S.; et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev. 2008, 22, 2677–2691. [Google Scholar] [CrossRef] [PubMed]
- Casciano, I.; Mazzocco, K.; Boni, L.; Pagnan, G.; Banelli, B.; Allemanni, G.; Ponzoni, M.; Tonini, G.P.; Romani, M. Expression of ΔNp73 is a molecular marker for adverse outcome in neuroblastoma patients. Cell Death Differ. 2002, 9, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Wolter, J.; Angelini, P.; Irwin, M. p53 family: Therapeutic targets in neuroblastoma. Futur. Oncol. 2010, 6, 429–444. [Google Scholar] [CrossRef]
- Romani, M.; Tonini, G.P.; Banelli, B.; Allemanni, G.; Mazzocco, K.; Scaruffi, P.; Boni, L.; Ponzoni, M.; Pagnan, G.; Raffaghello, L.; et al. Biological and clinical role of p73 in neuroblastoma. Cancer Lett. 2003, 197, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Rufini, A.; Agostini, M.; Grespi, F.; Tomasini, R.; Sayan, B.S.; Niklison-Chirou, M.V.; Conforti, F.; Velletri, T.; Mastino, A.; Mak, T.W.; et al. P73 in cancer. Genes Cancer 2011, 2, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Delgado, B.; Melendez, B.; Cuadros, M.; Garcia, M.J.; Nomdedeu, J.; Rivas, C.; Fernandez-Piqueras, J.; Benítez, J. Frequent inactivation of the p73 gene by abnormal methylation or LOH in Non-Hodgkin’s Lymphomas. Int. J. Cancer 2002, 102, 15–19. [Google Scholar] [CrossRef]
- Banelli, B.; Casciano, I.; Romani, M. Methylation-independent silencing of the p73 gene in neuroblastoma. Oncogene 2000, 19, 4553–4556. [Google Scholar] [CrossRef]
- Pozniak, C.D.; Radinovic, S.; Yang, A.; McKeon, F.; Kaplan, D.R.; Miller, F.D. An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science 2000, 289, 304–306. [Google Scholar] [CrossRef]
- Wilhelm, M.T.; Rufini, A.; Wetzel, M.K.; Tsuchihara, K.; Inoue, S.; Tomasini, R.; Itie-Youten, A.; Wakeham, A.; Arsenian-Henriksson, M.; Melino, G.; et al. Isoform-specific p73 knockout mice reveal a novel role for ΔNp73 in the DNA damage response pathway. Genes Dev. 2010, 24, 549–560. [Google Scholar] [CrossRef]
- Casciano, I.; Banelli, B.; Croce, M.; Allemanni, G.; Ferrini, S.; Tonini, G.P.; Ponzoni, M.; Romani, M. Role of methylation in the control of ΔNp73 expression in neuroblastoma. Cell Death Differ. 2002, 9, 343–345. [Google Scholar] [CrossRef]
- Melino, G.; Gallagher, E.; Aqeilan, R.I.; Knight, R.; Peschiaroli, A.; Rossi, M.; Scialpi, F.; Malatesta, M.; Zocchi, L.; Browne, G.; et al. Itch: A HECT-type E3 ligase regulating immunity, skin and cancer. Cell Death Differ. 2008, 15, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Bernassola, F.; Karin, M.; Ciechanover, A.; Melino, G. The HECT Family of E3 Ubiquitin Ligases: Multiple Players in Cancer Development. Cancer Cell 2008, 14, 10–21. [Google Scholar] [CrossRef]
- Hansen, T.M.; Rossi, M.; Roperch, J.P.; Ansell, K.; Simpson, K.; Taylor, D.; Mathon, N.; Knight, R.A.; Melino, G. Itch inhibition regulates chemosensitivity in vitro. Biochem. Biophys. Res. Commun. 2007, 361, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Bongiorno-Borbone, L.; Giacobbe, A.; Compagnone, M.; Eramo, A.; De Maria, R.; Peschiaroli, A.; Melino, G. Anti-tumoral effect of desmethylclomipramine in lung cancer stem cells. Oncotarget 2015, 6, 16926–16938. [Google Scholar] [CrossRef] [PubMed]
- de la Fuente, M.; Jones, M.C.; Santander-Ortega, M.J.; Mirenska, A.; Marimuthu, P.; Uchegbu, I.; Schätzlein, A. A nano-enabled cancer-specific ITCH RNAi chemotherapy booster for pancreatic cancer. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 369–377. [Google Scholar] [CrossRef]
- Meng, J.; Tagalakis, A.D.; Hart, S.L. Silencing E3 Ubiqutin ligase ITCH as a potential therapy to enhance chemotherapy efficacy in p53 mutant neuroblastoma cells. Sci. Rep. 2020, 10, 1046. [Google Scholar] [CrossRef]
- Yin, Q.; Wyatt, C.J.; Han, T.; Smalley, K.S.M.; Wan, L. ITCH as a potential therapeutic target in human cancers. Semin. Cancer Biol. 2020, 67, 117–130. [Google Scholar] [CrossRef]
- Chaudhary, N.; Maddika, S. WWP2-WWP1 Ubiquitin Ligase Complex Coordinated by PPM1G Maintains the Balance between Cellular p73 and ΔNp73 Levels. Mol. Cell. Biol. 2014, 34, 3754–3764. [Google Scholar] [CrossRef]
- Peschiaroli, A.; Scialpi, F.; Bernassola, F.; Pagano, M.; Melino, G. The F-box protein FBXO45 promotes the proteasome-dependent degradation of p73. Oncogene 2009, 28, 3157–3166. [Google Scholar] [CrossRef]
- Sayan, B.S.; Yang, A.L.; Conforti, F.; Tucci, P.; Piro, M.C.; Browne, G.J.; Agostini, M.; Bernardini, S.; Knight, R.A.; Mak, T.W.; et al. Differential control of TAp73 and ΔNp73 protein stability by the ring finger ubiquitin ligase PIR2. Proc. Natl. Acad. Sci. USA 2010, 107, 12877–12882. [Google Scholar] [CrossRef]
- Rossi, M.; De Laurenzi, V.; Munarriz, E.; Green, D.R.; Liu, Y.C.; Vousden, K.H.; Cesareni, G.; Melino, G. The ubiquitin-protein ligase Itch regulates p73 stability. EMBO J. 2005, 24, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Rotblat, B.; Ansell, K.; Amelio, I.; Caraglia, M.; Misso, G.; Bernassola, F.; Cavasotto, C.N.; Knight, R.A.; Ciechanover, A.; et al. High throughput screening for inhibitors of the HECT ubiquitin E3 ligase ITCH identifies antidepressant drugs as regulators of autophagy. Cell Death Dis. 2014, 5, e1203. [Google Scholar] [CrossRef] [PubMed]
- Gillman, P.K. Tricyclic antidepressant pharmacology and therapeutic drug interactions updated. Br. J. Pharmacol. 2007, 151, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Mou, P.K.; Yang, E.J.; Shi, C.; Ren, G.; Tao, S.; Shim, J.S. Aurora kinase A, a synthetic lethal target for precision cancer medicine. Exp. Mol. Med. 2021, 53, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ding, L.; Zhou, Q.; Fen, L.; Cao, Y.; Sun, J.; Zhou, X.; Liu, A. Silencing of AURKA augments the antitumor efficacy of the AURKA inhibitor MLN8237 on neuroblastoma cells. Cancer Cell Int. 2020, 20, 9. [Google Scholar] [CrossRef]
- Goldenson, B.; Crispino, J.D. The aurora kinases in cell cycle and leukemia. Oncogene 2015, 34, 537–545. [Google Scholar] [CrossRef]
- Nikonova, A.S.; Astsaturov, I.; Serebriiskii, I.G.; Dunbrack, R.L.; Golemis, E.A. Aurora A kinase (AURKA) in normal and pathological cell division. Cell. Mol. Life Sci. 2013, 70, 661–687. [Google Scholar] [CrossRef]
- Sasai, K.; Treekitkarnmongkol, W.; Kai, K.; Katayama, H.; Sen, S. Functional significance of Aurora kinases-p53 protein family interactions in cancer. Front. Oncol. 2016, 6, 247. [Google Scholar] [CrossRef]
- Marumoto, T.; Hirota, T.; Morisaki, T.; Kunitoku, N.; Zhang, D.; Ichikawa, Y.; Sasayama, T.; Kuninaka, S.; Mimori, T.; Tamaki, N.; et al. Roles of aurora-A kinase in mitotic entry and G2 checkpoint in mammalian cells. Genes Cells 2002, 7, 1173–1182. [Google Scholar] [CrossRef]
- Shao, S.; Wang, Y.; Jin, S.; Song, Y.; Wang, X.; Fan, W.; Zhao, Z.; Fu, M.; Tong, T.; Dong, L.; et al. Gadd45a interacts with aurora-A and inhibits its kinase activity. J. Biol. Chem. 2006, 281, 28943–28950. [Google Scholar] [CrossRef]
- Mao, J.H.; Perez-Iosada, J.; Wu, D.; DelRosario, R.; Tsunematsu, R.; Nakayama, K.I.; Brown, K.; Bryson, S.; Balmain, A. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature 2004, 432, 775–779. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Yang, T.Y.; Yu, C.T.R.; Phan, L.; Ivan, C.; Sood, A.K.; Hsu, S.L.; Lee, M.H. p53 negatively regulates Aurora A via both transcriptional and posttranslational regulation. Cell Cycle 2012, 11, 3433–3442. [Google Scholar] [CrossRef] [PubMed]
- Teng, C.L.; Hsieh, Y.C.; Phan, L.; Shin, J.; Gully, C.; Velazquez-Torres, G.; Skerl, S.; Yeung, S.C.J.; Hsu, S.L.; Lee, M.H. FBXW7 is involved in Aurora B degradation. Cell Cycle 2012, 11, 4059–4068. [Google Scholar] [CrossRef]
- Kwon, Y.W.; Kim, I.J.; Wu, D.; Lu, J.; Stock, W.A.; Liu, Y.; Huang, Y.; Kang, H.C.; DelRosario, R.; Jen, K.Y.; et al. Pten regulates Aurora-A and cooperates with Fbxw7 in modulating radiation-induced tumor development. Mol. Cancer Res. 2012, 10, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.H.; Wu, D.; Perez-Losada, J.; Jiang, T.; Li, Q.; Neve, R.M.; Gray, J.W.; Cai, W.W.; Balmain, A. Crosstalk between Aurora-A and p53: Frequent Deletion or Downregulation of Aurora-A in Tumors from p53 Null Mice. Cancer Cell 2007, 11, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Otto, T.; Horn, S.; Brockmann, M.; Eilers, U.; Schüttrumpf, L.; Popov, N.; Kenney, A.M.; Schulte, J.H.; Beijersbergen, R.; Christiansen, H.; et al. Stabilization of N-Myc Is a Critical Function of Aurora A in Human Neuroblastoma. Cancer Cell 2009, 15, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Brockmann, M.; Poon, E.; Berry, T.; Carstensen, A.; Deubzer, H.E.; Rycak, L.; Jamin, Y.; Thway, K.; Robinson, S.P.; Roels, F.; et al. Small Molecule Inhibitors of Aurora-A Induce Proteasomal Degradation of N-Myc in Childhood Neuroblastoma. Cancer Cell 2013, 24, 75–89. [Google Scholar] [CrossRef]
- Katayama, H.; Sasai, K.; Kawai, H.; Yuan, Z.M.; Bondaruk, J.; Suzuki, F.; Fujii, S.; Arlinghaus, R.B.; Czerniak, B.A.; Sen, S. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat. Genet. 2004, 36, 55–62. [Google Scholar] [CrossRef]
- Liu, Q.; Kaneko, S.; Yang, L.; Feldman, R.I.; Nicosia, S.V.; Chen, J.; Cheng, J.Q. Aurora-A abrogation of p53 DNA binding and transactivation activity by phosphorylation of serine 215. J. Biol. Chem. 2004, 279, 52175–52182. [Google Scholar] [CrossRef]
- Dar, A.A.; Belkhiri, A.; Ecsedy, J.; Zaika, A.; El-Rifai, W. Aurora kinase A inhibition leads to p73-dependent apoptosis in p53-deficient cancer cells. Cancer Res. 2008, 68, 8998–9004. [Google Scholar] [CrossRef]
- Katayama, H.; Wang, J.; Treekitkarnmongkol, W.; Kawai, H.; Sasai, K.; Zhang, H.; Wang, H.; Adams, H.P.; Jiang, S.; Chakraborty, S.N.; et al. Aurora Kinase-A Inactivates DNA Damage-Induced Apoptosis and Spindle Assembly Checkpoint Response Functions of p73. Cancer Cell 2012, 21, 196–211. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Ponte, J.F.; Levy, M.; Papageorgis, P.; Cook, N.M.; Ozturk, S.; Lambert, A.W.; Thiagalingam, A.; Abdolmaleky, H.M.; Sullivan, B.A.; et al. hBub1 negatively regulates p53 mediated early cell death upon mitotic checkpoint activation. Cancer Biol. Ther. 2009, 8, 636–644. [Google Scholar] [CrossRef]
- Fujiwara, T.; Bandi, M.; Nitta, M.; Ivanova, E.V.; Bronson, R.T.; Pellman, D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005, 437, 1043–1047. [Google Scholar] [CrossRef]
- Margolis, R.L.; Lohez, O.D.; Andreassen, P.R. G1 tetraploidy checkpoint and the suppression of tumorigenesis. J. Cell. Biochem. 2003, 88, 673–683. [Google Scholar] [CrossRef]
- Andreassen, P.R.; Lohez, O.D.; Lacroix, F.B.; Margolis, R.L. Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1. Mol. Biol. Cell 2001, 12, 1315–1328. [Google Scholar] [CrossRef]
- Tomasini, R.; Tsuchihara, K.; Tsuda, C.; Lau, S.K.; Wilhelm, M.; Ruffini, A.; Tsao, M.S.; Iovanna, J.L.; Jurisicova, A.; Melino, G.; et al. TAp73 regulates the spindle assembly checkpoint by modulating BubR1 activity. Proc. Natl. Acad. Sci. USA 2009, 106, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Vernole, P.; Neale, M.H.; Barcaroli, D.; Munarriz, E.; Knight, R.A.; Tomasini, R.; Mak, T.W.; Melino, G.; De Laurenzi, V. TAp73α binds the kinetochore proteins Bub1 and Bub3 resulting in polyploidy. Cell Cycle 2009, 8, 421–429. [Google Scholar] [CrossRef]
- Marrazzo, E.; Marchini, S.; Tavecchio, M.; Alberio, T.; Previdi, S.; Erba, E.; Rotter, V.; Broggini, M. The expression of the ΔNp73β isoform of p73 leads to tetraploidy. Eur. J. Cancer 2009, 45, 443–453. [Google Scholar] [CrossRef]
- Yi, J.S.; Sias-Garcia, O.; Nasholm, N.; Hu, X.; Iniguez, A.B.; Hall, M.D.; Davis, M.; Guha, R.; Moreno-Smith, M.; Barbieri, E.; et al. The synergy of BET inhibitors with aurora A kinase inhibitors in MYCN-amplified neuroblastoma is heightened with functional TP53. Neoplasia 2021, 23, 624–633. [Google Scholar] [CrossRef]
- Nguyen, R.; Wang, H.; Sun, M.; Lee, D.G.; Peng, J.; Thiele, C.J. Combining selinexor with alisertib to target the p53 pathway in neuroblastoma. Neoplasia 2022, 26, 100776. [Google Scholar] [CrossRef]
- Schwab, M.; Ellison, J.; Busch, M. Enhanced expression of the human gene N-myc consequent to amplification of DNA may contribute to malignant progression of neuroblastoma. Proc. Natl. Acad. Sci. USA 1984, 81, 4940–4944. [Google Scholar] [CrossRef] [PubMed]
- Kohl, N.E.; Kanda, N.; Schreck, R.R.; Bruns, G.; Latt, S.A.; Gilbert, F.; Alt, F.W. Transposition and amplification of oncogene-related sequences in human neuroblastomas. Cell 1983, 35, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Higashi, M.; Sakai, K.; Fumino, S.; Aoi, S.; Furukawa, T.; Tajiri, T. The roles played by the MYCN, Trk, and ALK genes in neuroblastoma and neural development. Surg. Today 2019, 49, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Wakamatsu, Y.; Watanabe, Y.; Nakamura, H.; Kondoh, H. Regulation of the neural crest cell fate by N-myc: Promotion of ventral migration and neuronal differentiation. Development 1997, 124, 1953–1962. [Google Scholar] [CrossRef]
- Dominguez-Sola, D.; Ying, C.Y.; Grandori, C.; Ruggiero, L.; Chen, B.; Li, M.; Galloway, D.A.; Gu, W.; Gautier, J.; Dalla-Favera, R. Non-transcriptional control of DNA replication by c-Myc. Nature 2007, 448, 445–451. [Google Scholar] [CrossRef]
- Shachaf, C.M.; Kopelman, A.M.; Arvanitis, C.; Karlsson, Å.; Beer, S.; Mandl, S.; Bachmann, M.H.; Borowsky, A.D.; Ruebner, B.; Cardiff, R.D.; et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 2004, 431, 1112–1117. [Google Scholar] [CrossRef]
- Jain, M.; Arvanitis, C.; Chu, K.; Dewey, W.; Leonhardt, E.; Trinh, M.; Sundberg, C.D.; Bishop, J.M.; Felsher, D.W. Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science 2002, 297, 102–104. [Google Scholar] [CrossRef]
- Kubota, Y.; Kim, S.H.; Iguchi-Ariga, S.M.M.; Ariga, H. Transrepression of the N-MYC expression by C-MYC protein. Biochem. Biophys. Res. Commun. 1989, 162, 991–997. [Google Scholar] [CrossRef]
- Levy, N.; Yonish-Rouach, E.; Oren, M.; Kimchi, A. Complementation by wild-type p53 of interleukin-6 effects on M1 cells: Induction of cell cycle exit and cooperativity with c-myc suppression. Mol. Cell. Biol. 1993, 13, 7942–7952. [Google Scholar] [CrossRef]
- Ho, J.S.L.; Ma, W.; Mao, D.Y.L.; Benchimol, S. p53-Dependent Transcriptional Repression of c-myc Is Required for G1 Cell Cycle Arrest. Mol. Cell. Biol. 2005, 25, 7423–7431. [Google Scholar] [CrossRef]
- Feng, Y.C.; Liu, X.Y.; Teng, L.; Ji, Q.; Wu, Y.; Li, J.M.; Gao, W.; Zhang, Y.Y.; La, T.; Tabatabaee, H.; et al. c-Myc inactivation of p53 through the pan-cancer lncRNA MILIP drives cancer pathogenesis. Nat. Commun. 2020, 11, 4980. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.I.; Ozaki, T.; Nakagawa, T.; Miyazaki, K.; Takahashi, M.; Hosoda, M.; Hayashi, S.; Todo, S.; Nakagawara, A. Physical interaction of p73 with c-Myc and MM1, a c-Myc-binding protein, and modulation of the p73 function. J. Biol. Chem. 2002, 277, 15113–15123. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Tweddle, D.A. p53, SKP2, and DKK3 as MYCN Target Genes and Their Potential Therapeutic Significance. Front. Oncol. 2012, 2, 173. [Google Scholar] [CrossRef]
- Zindy, F.; Eischen, C.M.; Randle, D.H.; Kamijo, T.; Cleveland, J.L.; Sherr, C.J.; Roussel, M.F. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 1998, 12, 2424–2433. [Google Scholar] [CrossRef] [PubMed]
- Otte, J.; Dyberg, C.; Pepich, A.; Johnsen, J.I. MYCN Function in Neuroblastoma Development. Front. Oncol. 2021, 10, 624079. [Google Scholar] [CrossRef]
- Chen, J.; Guan, Z. Function of Oncogene Mycn in Adult Neurogenesis and Oligodendrogenesis. Mol. Neurobiol. 2022, 59, 77–92. [Google Scholar] [CrossRef]
- Knoepfler, P.S.; Cheng, P.F.; Eisenman, R.N. N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev. 2002, 16, 2699–2712. [Google Scholar] [CrossRef]
- Alam, G.; Cui, H.; Shi, H.; Yang, L.; Ding, J.; Mao, L.; Maltese, W.A.; Ding, H.F. MYCN promotes the expansion of Phox2B-positive neuronal progenitors to drive neuroblastoma development. Am. J. Pathol. 2009, 175, 856–866. [Google Scholar] [CrossRef]
- Kapeli, K.; Hurlin, P.J. Differential Regulation of N-Myc and c-Myc Synthesis, Degradation, and Transcriptional Activity by the Ras/Mitogen-activated Protein Kinase Pathway. J. Biol. Chem. 2011, 286, 38498–38508. [Google Scholar] [CrossRef]
- Maris, J.M. Recent Advances in Neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef]
- Huang, M.; Weiss, W.A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 2013, 3, a014415. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M.; Alitalo, K.; Klempnauer, K.H.; Varmus, H.E.; Bishop, J.M.; Gilbert, F.; Brodeur, G.; Goldstein, M.; Trent, J. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature 1983, 305, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Seeger, R.C.; Brodeur, G.M.; Sather, H.; Dalton, A.; Siegel, S.E.; Wong, K.Y.; Hammond, D. Association of multiple copies of the N-MYC oncogene with rapid progression of neuroblastomas. N. Engl. J. Med. 1985, 313, 1111–1116. [Google Scholar] [CrossRef]
- Brodeur, G.; Seeger, R.; Schwab, M.; Varmus, H.; Bishop, J. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 1984, 224, 1121–1124. [Google Scholar] [CrossRef] [PubMed]
- Weiss, W.A.; Aldape, K.; Mohapatra, G.; Feuerstein, B.G.; Bishop, J.M.; Hooper, G.W. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997, 16, 2985–2995. [Google Scholar] [CrossRef]
- Althoff, K.; Beckers, A.; Bell, E.; Nortmeyer, M.; Thor, T.; Sprüssel, A.; Lindner, S.; De Preter, K.; Florin, A.; Heukamp, L.C. A Cre-conditional MYCN -driven neuroblastoma mouse model as an improved tool for preclinical studies. Oncogene 2015, 34, 3357–3368. [Google Scholar] [CrossRef] [PubMed]
- Olsen, R.R.; Otero, J.H.; Wallace, K.; Finkelstein, D.; Rehg, J.E.; Yin, Z.; Wang, Y.; Freeman, K.W. MYCN induces neuroblastoma in primary neural crest cells. Oncogene 2017, 36, 5075–5082. [Google Scholar] [CrossRef]
- Zhu, S.; Lee, J.; Guo, F.; Shin, J.; Perez-atayde, A.R.; Kutok, J.L.; Rodig, S.J.; Neuberg, D.S.; Helman, D.; Feng, H.; et al. Article Activated ALK Collaborates with MYCN in Neuroblastoma Pathogenesis. Cancer Cell 2012, 21, 362–373. [Google Scholar] [CrossRef]
- Powers, J.T.; Tsanov, K.M.; Pearson, D.S.; Roels, F.; Spina, C.S.; Ebright, R.; Seligson, M.; De Soysa, Y.; Cahan, P.; Theißen, J.; et al. Multiple mechanisms disrupt the let-7 microRNA family in neuroblastoma. Nature 2016, 535, 246–251. [Google Scholar] [CrossRef]
- Viswanathan, S.R.; Daley, G.Q.; Gregory, R.I. Selective Blockade of MicroRNA Processing by Lin28. Science 2008, 320, 97–100. [Google Scholar] [CrossRef]
- Rickman, D.S.; Schulte, J.H.; Eilers, M. The Expanding World of N-MYC–Driven Tumors. Cancer Discov. 2018, 8, 150–163. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Duong, C.V.; Pape, M.; Geissen, M.; Burbach, G.; Deller, T.; Parlato, R.; Schütz, G.; Development, H.R.; Duong, C.V.; et al. Target-dependent specification of the neurotransmitter phenotype: Cholinergic differentiation of sympathetic neurons is mediated in vivo by gp130 signaling. Development 2006, 133, 141–150. [Google Scholar] [CrossRef]
- Molenaar, J.J.; Domingo-Fernández, R.; Ebus, M.E.; Lindner, S.; Koster, J.; Drabek, K.; Mestdagh, P.; Van Sluis, P.; Valentijn, L.J.; Van Nes, J.; et al. LIN28B induces neuroblastoma and enhances MYCN levels via let-7 suppression. Nat. Genet. 2012, 44, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.H.; Robinton, D.A.; Seligson, M.T.; Wu, L.; Li, L.; Rakheja, D.; Comerford, S.A.; Ramezani, S.; Sun, X.; Parikh, M.S.; et al. Article Lin28b Is Sufficient to Drive Liver Cancer and Necessary for Its Maintenance in Murine Models. Cancer Cell 2014, 26, 248–261. [Google Scholar] [CrossRef] [PubMed]
- Urbach, A.; Yermalovich, A.; Zhang, J.; Spina, C.S.; Zhu, H.; Perez-atayde, A.R.; Shukrun, R.; Charlton, J.; Sebire, N.; Mifsud, W.; et al. Lin28 sustains early renal progenitors and induces Wilms tumor. Genes Dev. 2014, 28, 971–982. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Zhang, H.; He, J.; Li, J.; Huang, M.; Zhou, M. MDM2 regulates MYCN mRNA stabilization and translation in human neuroblastoma cells. Oncogene 2012, 31, 1342–1353. [Google Scholar] [CrossRef]
- He, J.; Gu, L.; Zhang, H.; Zhou, M. Crosstalk between MYCN and MDM2-p53 signal pathways regulates tumor cell growth and apoptosis in neuroblastoma. Cell Cycle 2011, 10, 2994–3002. [Google Scholar] [CrossRef]
- Slack, A.; Chen, Z.; Tonelli, R.; Pule, M.; Hunt, L.; Pession, A.; Shohet, J.M. The p53 regulatory gene MDM2 is a direct transcriptional target of MYCN in neuroblastoma. Proc. Natl. Acad. Sci. USA 2005, 102, 731–736. [Google Scholar] [CrossRef]
- Agarwal, S.; Milazzo, G.; Rajapakshe, K.; Bernardi, R.; Chen, Z.; Barberi, E.; Koster, J.; Perini, G.; Coarfa, C.; Shohet, J.M. MYCN acts as a direct co-regulator of p53 in MYCN amplified neuroblastoma. Oncotarget 2018, 9, 20323–20338. [Google Scholar] [CrossRef]
- Horvilleur, E.; Bauer, M.; Goldschneider, D.; Mergui, X.; De la motte, A.; Bénard, J.; Douc-rasy, S.; Cappellen, D. p73α isoforms drive opposite transcriptional and post-transcriptional regulation of MYCN expression in neuroblastoma cells. Nucleic Acids Res. 2008, 36, 4222–4232. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, S.S.; Clarke, S.; Veschi, V.; Thiele, C.J. Targeting MYCN in Pediatric and Adult Cancers. Front. Oncol. 2021, 10, 623679. [Google Scholar] [CrossRef] [PubMed]
- Clausen, D.M.; Guo, J.; Parise, R.A.; Beumer, J.H.; Egorin, M.J.; Lazo, J.S.; Prochownik, E.V.; Eiseman, J.L. In vitro cytotoxicity and in vivo efficacy, pharmacokinetics, and metabolism of 10074-G5, a novel small-molecule inhibitor of c-Myc/Max dimerization. J. Pharmacol. Exp. Ther. 2010, 335, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, J.I.; Dyberg, C.; Fransson, S.; Wickström, M. Molecular mechanisms and therapeutic targets in neuroblastoma. Pharmacol. Res. 2018, 131, 164–176. [Google Scholar] [CrossRef] [PubMed]
- DuBois, S.G.; Marachelian, A.; Fox, E.; Kudgus, R.A.; Reid, J.M.; Groshen, S.; Malvar, J.; Bagatell, R.; Wagner, L.; Maris, J.M.; et al. Phase I study of the Aurora A kinase inhibitor Alisertib in combination with irinotecan and temozolomide for patients with relapsed or refractory neuroblastoma: A NANT (new approaches to neuroblastoma therapy) trial. J. Clin. Oncol. 2016, 34, 1368–1375. [Google Scholar] [CrossRef]
- Zafar, A.; Wang, W.; Liu, G.; Wang, X.; Xian, W.; McKeon, F.; Foster, J.; Zhou, J.; Zhang, R. Molecular targeting therapies for neuroblastoma: Progress and challenges. Med. Res. Rev. 2021, 41, 961–1021. [Google Scholar] [CrossRef]
- Smith, J.R.; Moreno, L.; Heaton, S.P.; Chesler, L.; Pearson, A.D.J.; Garrett, M.D. Novel pharmacodynamic biomarkers for MYCN protein and PI3K/AKT/mTOR pathway signaling in children with neuroblastoma. Mol. Oncol. 2016, 10, 538–552. [Google Scholar] [CrossRef]
- Hassan, B.; Akcakanat, A.; Holder, A.M.; Meric-Bernstam, F. Targeting the PI3-kinase/Akt/mTOR Signaling Pathway. Surg. Oncol. Clin. N. Am. 2013, 22, 641–664. [Google Scholar] [CrossRef]
- Saliminejad, K.; Khorram Khorshid, H.R.; Soleymani Fard, S.; Ghaffari, S.H. An overview of microRNAs: Biology, functions, therapeutics, and analysis methods. J. Cell. Physiol. 2019, 234, 5451–5465. [Google Scholar] [CrossRef]
- Ambros, V.; Bartel, B.; Bartel, D.P.; Burge, C.B.; Carrington, J.C.; Chen, X.; Dreyfuss, G.; Eddy, S.R.; Griffiths-jones, S.M.; Marshall, M.; et al. A uniform system for microRNA annotation. RNA 2003, 9, 277–279. [Google Scholar] [CrossRef]
- Li, M.; Li, J.; Ding, X.; He, M.; Cheng, S.Y. MicroRNA and cancer. AAPS J. 2010, 12, 309–317. [Google Scholar] [CrossRef]
- Bhaskaran, M.; Mohan, M. MicroRNAs: History, Biogenesis, and Their Evolving Role in Animal Development and Disease. Vet. Pathol. 2014, 51, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006, 34, 140–144. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Saini, H.K.; Van Dongen, S.; Enright, A.J. miRBase: Tools for microRNA genomics. Nucleic Acids Res. 2008, 36, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Syeda, Z.A.; Langden, S.S.S.; Munkhzul, C.; Lee, M.; Song, S.J. Regulatory mechanism of microrna expression in cancer. Int. J. Mol. Sci. 2020, 21, 1723. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.J.; Alemany-cosme, E.; Goñi, S.; Bandres, E.; Palanca-Ballester, C.; Sandoval, J. Epigenetic regulation of microRNAs in cancer: Shortening the distance from bench to bedside. Int. J. Mol. Sci. 2021, 22, 7350. [Google Scholar] [CrossRef]
- Logotheti, S.; Marquardt, S.; Putzer, B.M. p73-Governed miRNA Networks: Translating Bioinformatics Approaches to Therapeutic Solutions for Cancer Metastasis. In Computational Biology of Non-Coding RNA: Methods and Protocols; Lai, X., Gupta, S.K., Vera, J., Eds.; Humana Press: New York, NY, USA, 2019; Volume 1912, pp. 33–52. ISBN 9781493989829. [Google Scholar]
- Liu, J.; Zhang, C.; Zhao, Y.; Feng, Z. MicroRNA Control of p53. J. Cell. Biochem. 2017, 118, 7–14. [Google Scholar] [CrossRef]
- Veeraraghavan, V.P.; Jayaraman, S.; Rengasamy, G.; Mony, U.; Ganapathy, D.M.; Geetha, R.V.; Sekar, D. Deciphering the role of micrornas in neuroblastoma. Molecules 2022, 27, 99. [Google Scholar] [CrossRef]
- Galardi, A.; Colletti, M.; Businaro, P.; Quintarelli, C.; Locatelli, F.; Di Giannatale, A. MicroRNAs in Neuroblastoma: Biomarkers with Therapeutic Potential. Curr. Med. Chem. 2017, 25, 584–600. [Google Scholar] [CrossRef]
- Tivnan, A.; Orr, W.S.; Gubala, V.; Nooney, R.; Williams, D.E.; McDonagh, C.; Prenter, S.; Harvey, H.; Domingo-Fernández, R.; Bray, I.M.; et al. Inhibition of neuroblastoma tumor growth by targeted delivery of microRNA-34a using anti-disialoganglioside GD2 coated nanoparticles. PLoS ONE 2012, 7, e38129. [Google Scholar] [CrossRef]
- Boominathan, L. The Tumor Suppressors p53, p63, and p73 Are Regulators of MicroRNA Processing Complex. PLoS ONE 2010, 5, e10615. [Google Scholar] [CrossRef]
- Boominathan, L. Tumor suppressors p53, p63, and p73 inhibit migrating cancer stem cells by increasing the expression of stem cell suppressing miRNAs. Cell 2010, 1, 1–20. [Google Scholar] [CrossRef]
- Madrigal, T.; Hernández-Monge, J.; Herrera, L.A.; González-De la Rosa, C.H.; Domínguez-Gómez, G.; Candelaria, M.; Luna-Maldonado, F.; Calderón González, K.G.; Díaz-Chávez, J. Regulation of miRNAs Expression by Mutant p53 Gain of Function in Cancer. Front. Cell Dev. Biol. 2021, 9, 695723. [Google Scholar] [CrossRef]
- Li, X.L.; Jones, M.F.; Subramanian, M.; Lal, A. Mutant p53 exerts oncogenic effects through microRNAs and their target gene networks. FEBS Lett. 2014, 588, 2610–2615. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Buhe, B.; Hongtime, L.; Chuanmin, Y.; Xiwei, H.; Hong, Z.; Lulu, H.; Qian, D.; Renjie, W. MicroRNA-15a promotes neuroblastoma migration by targeting reversion-inducing cysteine-rich protein with Kazal motifs (RECK) and regulating matrix metalloproteinase-9 expression. FEBS J. 2013, 280, 855–866. [Google Scholar] [CrossRef]
- Wang, Z.; Yao, W.; Li, K.; Zheng, N.; Zheng, C.; Zhao, X.; Zheng, S. Reduction of miR-21 induces SK-N-SH cell apoptosis and inhibits proliferation via PTEN/PDCD4. Oncol. Lett. 2017, 13, 4727–4733. [Google Scholar] [CrossRef]
- Cheng, L.; Yang, T.; Kuang, Y.; Kong, B.; Yu, S.; Shu, H.; Zhou, H.; Gu, J. MicroRNA-23a promotes neuroblastoma cell metastasis by targeting CDH1. Oncol. Lett. 2014, 7, 839–845. [Google Scholar] [CrossRef]
- He, X.Y.; Tan, Z.L.; Mou, Q.; Liu, F.J.; Liu, S.; Yu, C.W.; Zhu, J.; Lv, L.Y.; Zhang, J.; Wang, S.; et al. microRNA-221 enhances MYCN via targeting nemo-like kinase and functions as an oncogene related to poor prognosis in neuroblastoma. Clin. Cancer Res. 2017, 23, 2905–2918. [Google Scholar] [CrossRef]
- Swarbrick, A.; Woods, S.L.; Shaw, A.; Phua, Y.; Nguyen, A.; Chanthery, Y.; Lim, L.; Lesley, J.; Judson, R.L.; Huskey, N.; et al. miR-380-5p represses p53 to control cellular survival and is associated with poor outcome in MYCN amplified neuroblastoma. Nat. Med. 2011, 16, 1134–1140. [Google Scholar] [CrossRef]
- Qu, H.; Zheng, L.; Pu, J.; Mei, H.; Xiang, X.; Zhao, X.; Li, D.; Li, S.; Mao, L.; Huang, K.; et al. miRNA-558 promotes tumorigenesis and aggressiveness of neuroblastoma cells through activating the transcription of heparanase. Hum. Mol. Genet. 2015, 24, 2539–2551. [Google Scholar] [CrossRef]
- Li, Z.; Xu, Z.; Xie, Q.; Gao, W.; Xie, J.; Zhou, L. miR-1303 promotes the proliferation of neuroblastoma cell SH-SY5Y by targeting GSK3β and SFRP1. Biomed. Pharmacother. 2016, 83, 508–513. [Google Scholar] [CrossRef]
- Ye, W.; Liang, F.; Ying, C.; Zhang, M.; Feng, D.; Jiang, X. Downregulation of microRNA-3934-5p induces apoptosis and inhibits the proliferation of neuroblastoma cells by targeting TP53INP1. Exp. Ther. Med. 2019, 18, 3729–3736. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Qi, M.; Li, S.; Qi, T.; Mei, H.; Huang, K.; Zheng, L.; Tong, Q. MicroRNA-9 targets matrix metalloproteinase 14 to inhibit invasion, metastasis, and angiogenesis of neuroblastoma cells. Mol. Cancer Ther. 2012, 11, 1454–1466. [Google Scholar] [CrossRef] [PubMed]
- Chava, S.; Reynolds, C.P.; Pathania, A.S.; Gorantla, S.; Poluektova, L.Y.; Couldter, D.W.; Gupta, S.C.; Pandey, M.K.; Challagundia, K.B. miR-15a-5p, miR-15b-5p, and miR-16-5p inhibit tumor progression by directly targeting MYCN in neuroblastoma. Mol. Oncol. 2019, 14, 180–196. [Google Scholar] [CrossRef]
- De Antonellis, P.; Carotenuto, M.; Vandenbussche, J.; De Vita, G.; Ferrucci, V.; Medaglia, C.; Boffa, I.; Galiero, A.; Di Somma, S.; Magliulo, D.; et al. Early targets of miR-34a in neuroblastoma. Mol. Cell. Proteom. 2014, 13, 2114–2131. [Google Scholar] [CrossRef] [PubMed]
- Agostini, M.; Tucci, P.; Killick, R.; Candi, E.; Sayan, B.S.; Di Val Cervo, P.R.; Nicoterad, P.; McKeon, F.; Knight, R.A.; Mak, T.W.; et al. Neuronal differentiation by TAp73 is mediated by microRNA-34a regulation of synaptic protein targets. Proc. Natl. Acad. Sci. USA 2011, 108, 21093–21098. [Google Scholar] [CrossRef]
- Feinberg-Gorenshtein, G.; Guedj, A.; Shichrur, K.; Jeison, M.; Luria, D.; Kodman, Y.; Ash, S.; Feinmesser, M.; Edry, L.; Shomron, N.; et al. miR-192 directly binds and regulates Dicer1 expression in neuroblastoma. PLoS ONE 2013, 8, e78713. [Google Scholar] [CrossRef]
- Zhao, D.; Tian, Y.; Li, P.; Wang, L.; Xiao, A.; Zhang, M.; Shi, T. MicroRNA-203 inhibits the malignant progression of neuroblastoma by targeting Sam68. Mol. Med. Rep. 2015, 12, 5554–5560. [Google Scholar] [CrossRef]
- Chen, X.; Pan, M.; Han, L.; Lu, H.; Hao, X.; Dong, Q. miR-338-3p suppresses neuroblastoma proliferation invasion and migration through targeting PREX2a. FEBS Lett. 2013, 587, 3729–3737. [Google Scholar] [CrossRef]
- Wu, T.; Lin, Y.; Xie, Z. MicroRNA-1247 inhibits cell proliferation by directly targeting ZNF346 in childhood neuroblastoma. Biol. Res. 2018, 51, 13. [Google Scholar] [CrossRef]
- Afanasyeva, E.A.; Mestdagh, P.; Kumps, C.; Vandesompele, J.; Ehemann, V.; Theissen, J.; Fischer, M.; Zapatka, M.; Brors, B.; Savelyeva, L.; et al. MicroRNA miR-885-5p targets CDK2 and MCM5, activates p53 and inhibits proliferation and survival. Cell Death Differ. 2011, 18, 974–984. [Google Scholar] [CrossRef]
- Guglielmi, L.; Cinnella, C.; Nardella, M.; Maresca, G.; Valentini, A.; Mercanti, D.; Felsani, A.; D’Agnano, I. MYCN gene expression is required for the onset of the differentiation programme in neuroblastoma cells. Cell Death Dis. 2014, 5, e1081. [Google Scholar] [CrossRef] [PubMed]
- Slabáková, E.; Culig, Z.; Remšík, J.; Souček, K. Alternative mechanisms of MIR-34a regulation in cancer. Cell Death Dis. 2017, 8, e3100. [Google Scholar] [CrossRef]
- Rihani, A.; Van Goethem, A.; Ongenaert, M.; De Brouwer, S.; Volders, P.J.; Agarwal, S.; De Preter, K.; Mestdagh, P.; Shohet, J.; Speleman, F.; et al. Genome wide expression profiling of p53 regulated miRNAs in neuroblastoma. Sci. Rep. 2015, 5, 9027. [Google Scholar] [CrossRef]
- Le, M.T.N.; Teh, C.; Shyh-Chang, N.; Xie, H.; Zhou, B.; Korzh, V.; Lodish, H.F.; Lim, B. MicroRNA-125b is a novel negative regulator of p53. Genes Dev. 2009, 23, 862–876. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Zeng, S.; Dai, X.; Wu, J.; Ma, W. MiR-504 promotes tumour growth and metastasis in human osteosarcoma by targeting TP53INP1. Oncol. Rep. 2017, 38, 2993–3000. [Google Scholar] [CrossRef] [PubMed]
- Irwin, M.S.; Naranjo, A.; Zhang, F.F.; Cohn, S.L.; London, W.B.; Gastier-Foster, J.M.; Ramirez, N.C.; Pfau, R.; Reshmi, S.; Wagner, E.; et al. Revised Neuroblastoma Risk Classification System: A Report From the Children’s Oncology Group. J. Clin. Oncol. 2021, 39, 3229–3241. [Google Scholar] [CrossRef]



| Clinical Approach | Drugs Tested | Study Phase | Clinical Trials.gov Identifier or Ref. |
|---|---|---|---|
| Treatment with a MAPK inhibitor for relapsed or high-risk NB with activation mutations | Selumetinib sulfate | Phase 2 | NCT03213691 |
| Treatment with a ALK inhibitor for NB with ALK mutations | Crizotinib | Phase 1 | NCT01121588 |
| Combination therapy of ALK inhibitor (crizotinib) with chemotherapeutics | Crizotinib + dexrazoxane hydrochloride + topotecan hydrochloride + cyclophosphamide + doxorubicin + vincristine sulfate | Phase 1 | NCT01606878 |
| Combination therapy of ALK inhibitor (lorlatinib) with/without other chemotherapeutics | Lorlatinib + cyclophosphamide + topotecan | Phase 1 | NCT03107988 |
| Combination therapy of ALK inhibitor (ceritinib) with CDK 4/6 inhibitor (ribociclib) | Ceritinib + ribociclib | Phase 1 | NCT02780128 |
| Therapy with PI3K/mTOR inhibitor in relapsed or high-risk NB with PI3K/mTOR mutations | Samotolisib | Phase 2 | NCT03213678 |
| Treatment of NB with PI3K/mTOR inhibitor | SF1126 | Phase 1 | NCT02337309 |
| Combination therapy of mTOR inhibitor (rapamycin) with multi-kinase inhibitor (dasatinib) with other chemotherapeutics | Rapamycin + dasatinib + irinotecan + temozolomide | Phase 2 | NCT01467986 |
| Combination therapy of mTOR inhibitor (temsirolimus) with perifosine | Temsirolimus + perifosine | Phase 1 | NCT01049841 |
| Combination therapy of AURKA inhibitor (alisertib) with chemotherapeutic agents | Alisertib + irinotecan + temozolomide | Phase 1/2 | NCT01601535 |
| Combination therapy of AURKA inhibitor (LY3295668 Erbumine) with/without other chemotherapeutics | LY3295668 Erbumine + topotecan + cyclophosphamide | Phase 1 | NCT04106219 |
| Combination therapy of MDM2 inhibitor (idasanutlin) with/without other chemotherapeutics or venetoclax | Idasanutlin + chemotherapy (cyclophosphamide/topotecan/fludarabine/cytarabine) or venetoclax | Phase 1/2 | NCT04029688 |
| Combination therapy of HDAC inhibitor (vorinostat) with 13-cis-retinoic acid (isotretinoin) | Vorinostat + isotretinoin | Phase 1 | NCT01208454 |
| Combination therapy of HDAC inhibitor (vorinostat) with bortezomib | Vorinostat + bortezomib | Phase 1 | NCT01132911 |
| Combination therapy of HDAC inhibitor (vorinostat) with 131I-MIBG in resistant or relapsed NB | Vorinostat + 131I-MIBG | Phase 1 | NCT01019850 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almeida, J.; Mota, I.; Skoda, J.; Sousa, E.; Cidade, H.; Saraiva, L. Deciphering the Role of p53 and TAp73 in Neuroblastoma: From Pathogenesis to Treatment. Cancers 2022, 14, 6212. https://doi.org/10.3390/cancers14246212
Almeida J, Mota I, Skoda J, Sousa E, Cidade H, Saraiva L. Deciphering the Role of p53 and TAp73 in Neuroblastoma: From Pathogenesis to Treatment. Cancers. 2022; 14(24):6212. https://doi.org/10.3390/cancers14246212
Chicago/Turabian StyleAlmeida, Joana, Inês Mota, Jan Skoda, Emília Sousa, Honorina Cidade, and Lucília Saraiva. 2022. "Deciphering the Role of p53 and TAp73 in Neuroblastoma: From Pathogenesis to Treatment" Cancers 14, no. 24: 6212. https://doi.org/10.3390/cancers14246212
APA StyleAlmeida, J., Mota, I., Skoda, J., Sousa, E., Cidade, H., & Saraiva, L. (2022). Deciphering the Role of p53 and TAp73 in Neuroblastoma: From Pathogenesis to Treatment. Cancers, 14(24), 6212. https://doi.org/10.3390/cancers14246212