

Targeting Mutant p53 for Cancer Treatment: Moving Closer to Clinical Use?

 , ,
, ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Mutant p53: A Highly Attractive Target for Cancer Treatment
2.1. High Prevalence in Cancer
2.2. Reactivation of Mutant p53 Can Potentially Induce Both Cell Intrinsic and Extrinsic Anti-Tumor Effects
2.3. Inhibition of Mutant p53 Function May Enhance Response to Standard Therapies
2.4. Mutations in p53 Tend to Be Clonal
2.5. Mutant p53 Proteins Accumulates in Malignant Cells
3. Difficulties in Targeting Mutant p53
3.1. Multiplicity of Mutations with Different Structures and Functions
3.2. Most Mutant Forms of p53 Lack a Suitable Pocket for High-Affinity Binding of Low Molecular Weight Compounds
3.3. Location of Mutant p53 in Tumor Cells Renders It Largely Inaccessible for Certain Types of Drugs
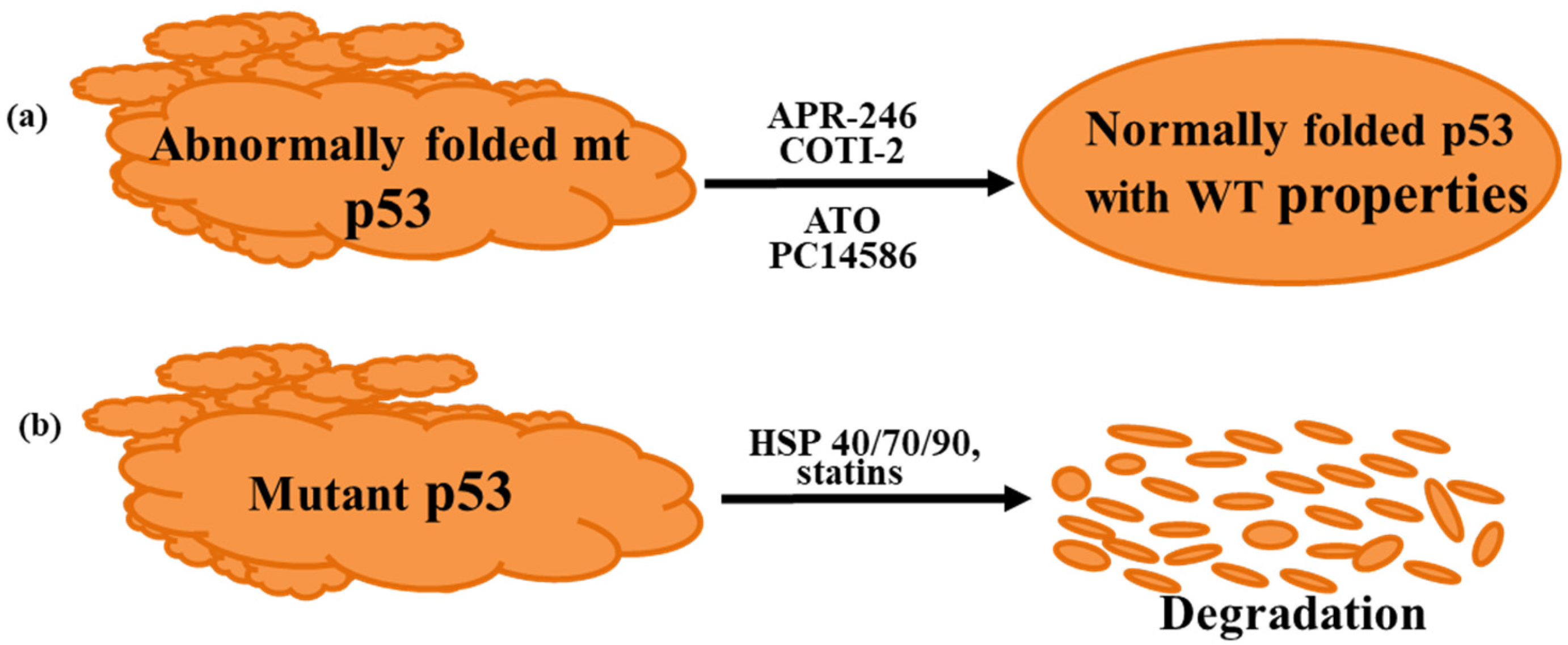
4. Mutant p53 Reactivating Drugs Undergoing Clinical Trials
4.1. Eprenetapopt/APR-246
4.2. COTI-2
4.3. Arsenic Trioxide
4.4. PC14586
5. Mutant p53 Degrading Drugs Undergoing Clinical Trials
6. Exploiting Mutant p53 for Vaccination
7. Other Strategies for Targeting Mutant p53
8. Targeting Mutant p53 Looking to the Future
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hayes, D.F. HER2 and breast cancer—A phenomenal success story. N. Engl. J. Med. 2019, 381, 1284–1286. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Gu, T.; Lee, M.H.; Dong, Z. Challenge and countermeasures for EGFR targeted therapy in non-small cell lung cancer. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188645. [Google Scholar] [CrossRef] [PubMed]
- Giugliano, F.; Crimini, E.; Tarantino, P.; Zagami, P.; Uliano, J.; Corti, C.; Trapani, D.; Curigliano, G.; Ascierto, P.A. First line treatment of BRAF mutated advanced melanoma: Does one size fit all? Cancer Treat. Rev. 2021, 99, 102253. [Google Scholar] [CrossRef] [PubMed]
- Dolgin, E. The most popular genes in the human genome. Nature 2017, 551, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. Spontaneous and inherited TP53 genetic alterations. Oncogene 2021, 40, 5975–5983. [Google Scholar] [CrossRef] [PubMed]
- Priestley, P.; Baber, J.; Lolkema, M.P.; Steeghs, N.; de Bruijn, E.; Shale, C.; Duyvesteyn, K.; Haidari, S.; van Hoeck, A.; Onstenk, W.; et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 2019, 575, 210–216. [Google Scholar] [CrossRef]
- Duffy, M.J.; Synnott, N.C.; O’Grady, S.; Crown, J. Targeting p53 for the treatment of cancer. Semin. Cancer Biol. 2022, 79, 58–67. [Google Scholar] [CrossRef]
- Alexandrova, E.M.; Moll, U.M. Depleting stabilized GOF mutant p53 proteins by inhibiting molecular folding chaperones: A new promise in cancer therapy. Cell Death Differ. 2017, 24, 3–5. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Song, Y.; Li, L.; Ou, Y.; Gao, Z.; Li, E.; Li, X.; Zhang, W.; Wang, J.; Xu, L.; Zhou, Y.; et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature 2014, 509, 91–95. [Google Scholar] [CrossRef]
- Peifer, M.; Fernández-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 2012, 44, 1104–1110. [Google Scholar] [CrossRef]
- Pilley, S.; Rodriguez, T.A.; Vousden, K.H. Mutant p53 in cell-cell interactions. Genes Dev. 2021, 35, 433–448. [Google Scholar] [CrossRef]
- Levine, A.J. P53 and the immune response: 40 years of exploration-a plan for the future. Int. J. Mol. Sci. 2020, 21, 541. [Google Scholar] [CrossRef]
- Agupitan, A.D.; Neeson, P.; Williams, S.; Howitt, J.; Haupt, S.; Haupt, Y. P53: A guardian of immunity becomes its saboteur through mutation. Int. J. Mol. Sci. 2020, 21, 3452. [Google Scholar] [CrossRef]
- Hientz, K.; Mohr, A.; Bhakta-Guha, D.; Efferth, T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget 2017, 8, 8921–8946. [Google Scholar] [CrossRef]
- Wallace-Brodeur, R.R.; Lowe, S.W. Clinical implications of p53 mutations. Cell Mol. Life Sci. 1999, 55, 64–75. [Google Scholar] [CrossRef]
- Fransson, Å.; Glaessgen, D.; Alfredsson, J.; Wiman, K.G.; Bajalica-Lagercrantz, S.; Mohell, N. Strong synergy with APR-246 and DNA-damaging drugs in primary cancer cells from patients with TP53 mutant high-grade serous ovarian cancer. J. Ovarian Res. 2016, 9, 27. [Google Scholar] [CrossRef]
- Ceder, S.; Eriksson, S.E.; Liang, Y.Y.; Cheteh, E.H.; Zhang, S.M.; Fujihara, K.M.; Bianchi, J.; Bykov, V.J.N.; Abrahmsen, L.; Clemons, N.J.; et al. Mutant p53-reactivating compound APR-246 synergizes with asparaginase in inducing growth suppression in acute lymphoblastic leukemia cells. Cell Death Dis. 2021, 12, 709. [Google Scholar] [CrossRef]
- Synnott, N.C.; Murray, A.; McGowan, P.M.; Kiely, M.; Kiely, P.A.; O’Donovan, N.; O’Connor, D.P.; Gallagher, W.M.; Crown, J.; Duffy, M.J. Mutant p53: A novel target for the treatment of patients with triple-negative breast cancer? Int. J. Cancer 2017, 140, 234–246. [Google Scholar] [CrossRef]
- Maslah, N.; Salomao, N.; Drevon, L.; Verger, E.; Partouche, N.; Ly, P.; Aubin, P.; Naoui, N.; Schlageter, M.H.; Bally, C.; et al. Synergistic effects of PRIMA-1Met (APR-246) and 5-azacitidine in TP53-mutated myelodysplastic syndromes and acute myeloid leukemia. Haematologica 2020, 105, 1539–1551. [Google Scholar] [CrossRef]
- Sampath, J.; Sun, D.; Kidd, V.J.; Grenet, J.; Gandhi, A.; Shapiro, L.H.; Wang, Q.; Zambetti, G.P.; Schuetz, J.D. Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1. J. Biol. Chem. 2001, 276, 39359–39367. [Google Scholar] [CrossRef]
- Gerstung, M.; Jolly, C.; Leshchiner, I.; Dentro, S.C.; Gonzalez, S.; Rosebrock, D.; Mitchell, T.J.; Rubanova, Y.; Anur, P.; Yu, K.; et al. The evolutionary history of 2658 cancers. Nature 2020, 578, 122–128. [Google Scholar] [CrossRef]
- Terzian, T.; Suh, Y.A.; Iwakuma, T.; Post, S.M.; Neumann, M.; Lang, G.A.; Van Pelt, C.S.; Lozano, G. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev. 2008, 22, 1337–1344. [Google Scholar] [CrossRef]
- D’Orazi, G.; Cirone, M. Mutant p53 and cellular stress pathways: A criminal alliance that promotes cancer progression. Cancers 2019, 11, 614. [Google Scholar] [CrossRef]
- Oren, M.; Tal, P.; Rotter, V. Targeting mutant p53 for cancer therapy. Aging 2016, 8, 1159–1160. [Google Scholar] [CrossRef][Green Version]
- Sabapathy, K.; Lane, D.P. Therapeutic targeting of p53: All mutants are equal, but some mutants are more equal than others. Nat. Rev. Clin. Oncol. 2018, 15, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. The many faces of p53: Something for everyone. J. Mol. Cell Biol. 2019, 11, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. TP53 variations in human cancers: New lessons from the IARC TP53 database and genomics data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. The p53 pathway: Origins, inactivation in cancer, and emerging therapeutic approaches. Annu. Rev. Biochem. 2016, 85, 375–404. [Google Scholar] [CrossRef]
- Hernández Borrero, L.J.; El-Deiry, W.S. Tumor suppressor p53: Biology, signaling pathways, and therapeutic targeting. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188556. [Google Scholar] [CrossRef]
- Moxley, A.H.; Reisman, D. Context is key: Understanding the regulation, functional control, and activities of the p53 tumour suppressor. Cell Biochem. Funct. 2021, 39, 235–247. [Google Scholar] [CrossRef]
- McCann, J.J.; Vasilevskaya, I.A.; McNair, C.; Gallagher, P.; Neupane, N.P.; de Leeuw, R.; Shafi, A.A.; Dylgjeri, E.; Mandigo, A.C.; Schiewer, M.J.; et al. Mutant p53 elicits context-dependent pro-tumorigenic phenotypes. Oncogene 2022, 41, 444–458. [Google Scholar] [CrossRef]
- Kadosh, E.; Snir-Alkalay, I.; Venkatachalam, A.; May, S.; Lasry, A.; Elyada, E.; Zinger, A.; Shaham, M.; Vaalani, G.; Mernberger, M.; et al. The gut microbiome switches mutant p53 from tumour-suppressive to oncogenic. Nature 2020, 586, 133–138. [Google Scholar] [CrossRef]
- Nguyen, T.A.; Menendez, D.; Resnick, M.A.; Anderson, C.W. Mutant TP53 posttranslational modifications: Challenges and opportunities. Hum. Mutat. 2014, 35, 738–755. [Google Scholar] [CrossRef]
- Bauer, M.R.; Jones, R.N.; Tareque, R.K.; Springett, B.; Dingler, F.A.; Verduci, L.; Patel, K.J.; Fersht, A.R.; Joerger, A.C.; Spencer, J. A structure-guided molecular chaperone approach for restoring the transcriptional activity of the p53 cancer mutant Y220C. Future Med. Chem. 2019, 11, 2491–2504. [Google Scholar] [CrossRef]
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.B.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef]
- Zhang, Q.; Bykov, V.J.N.; Wiman, K.G.; Zawacka-Pankau, J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 2018, 9, 439, Correction: Zhang, Q.; Bykov, V.J.N.; Wiman, K.G.; Zawacka-Pankau, J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 2019, 10, 769. [Google Scholar] [CrossRef]
- Wassman, C.D.; Baronio, R.; Demir, Ö.; Wallentine, B.D.; Chen, C.K.; Hall, L.V.; Salehi, F.; Lin, D.W.; Chung, B.P.; Hatfield, G.W.; et al. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nat. Commun. 2013, 4, 1407. [Google Scholar] [CrossRef]
- Degtjarik, O.; Golovenko, D.; Diskin-Posner, Y.; Abrahmsén, L.; Rozenberg, H.; Shakked, Z. Structural basis of reactivation of oncogenic p53 mutants by a small molecule: Methylene quinuclidinone (MQ). Nat. Commun. 2021, 12, 7057. [Google Scholar] [CrossRef] [PubMed]
- Hsiue, E.H.; Wright, K.M.; Douglass, J.; Hwang, M.S.; Mog, B.J.; Pearlman, A.H.; Paul, S.; DiNapoli, S.R.; Konig, M.F.; Wang, Q.; et al. Targeting a neoantigen derived from a common TP53 mutation. Science 2021, 371, eabc8697. [Google Scholar] [CrossRef] [PubMed]
- Chasov, V.; Zaripov, M.; Mirgayazova, R.; Khadiullina, R.; Zmievskaya, E.; Ganeeva, I.; Valiullina, A.; Rizvanov, A.; Bulatov, E. Promising new tools for targeting p53 mutant cancers: Humoral and cell-based immunotherapies. Front. Immunol. 2021, 12, 707734. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, S.E.; Ceder, S.; Bykov, V.J.N.; Wiman, K.G. p53 as a hub in cellular redox regulation and therapeutic target in cancer. J. Mol. Cell Biol. 2019, 11, 330–341. [Google Scholar] [CrossRef] [PubMed]
- Perdrix, A.; Najem, A.; Saussez, S.; Awada, A.; Journe, F.; Ghanem, G.; Krayem, M. PRIMA-1 and PRIMA-1Met (APR-246): From mutant/wild type p53 reactivation to unexpected mechanisms underlying their potent anti-tumor effect in combinatorial therapies. Cancers 2017, 9, 172. [Google Scholar] [CrossRef] [PubMed]
- Tessoulin, B.; Descamps, G.; Moreau, P.; Maïga, S.; Lodé, L.; Godon, C.; Marionneau-Lambot, S.; Oullier, T.; Le Gouill, S.; Amiot, M.; et al. PRIMA-1Met induces myeloma cell death independent of p53 by impairing the GSH/ROS balance. Blood J. Am. Soc. Hematol. 2014, 124, 1626–1636. [Google Scholar] [CrossRef]
- Ogiwara, H.; Takahashi, K.; Sasaki, M.; Kuroda, T.; Yoshida, H.; Watanabe, R.; Maruyama, A.; Makinoshima, H.; Chiwaki, F.; Sasaki, H.; et al. Targeting the vulnerability of glutathione metabolism in ARID1a-deficient cancers. Cancer Cell 2019, 35, 177–190.e8. [Google Scholar] [CrossRef]
- Peng, X.; Zhang, M.Q.; Conserva, F.; Hosny, G.; Selivanova, G.; Bykov, V.J.; Arnér, E.S.; Wiman, K.G. APR-246/PRIMA-1MET inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell Death Dis. 2013, 4, e881. [Google Scholar] [CrossRef]
- Haffo, L.; Lu, J.; Bykov, V.J.N.; Martin, S.S.; Ren, X.; Coppo, L.; Wiman, K.G.; Holmgren, A. Inhibition of the glutaredoxin and thioredoxin systems and ribonucleotide reductase by mutant p53-targeting compound APR-246. Sci. Rep. 2018, 8, 12671. [Google Scholar] [CrossRef]
- Cheung, E.C.; Vousden, K.H. The role of ROS in tumour development and progression. Nat. Rev. Cancer 2022, 22, 280–297. [Google Scholar] [CrossRef]
- Zhang, S.; Zhou, L.; El-Deiry, W.S. Small-Molecule NSC59984 Induces Mutant p53 Degradation through a ROS-ERK2-MDM2 Axis in Cancer Cells. Mol. Cancer Res. 2022, 20, 622–636. [Google Scholar] [CrossRef]
- Lehmann, S.; Bykov, V.J.; Ali, D.; Andrén, O.; Cherif, H.; Tidefelt, U.; Uggla, B.; Yachnin, J.; Juliusson, G.; Moshfegh, A.; et al. Targeting p53 in vivo: A first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J. Clin. Oncol. 2012, 30, 3633–3639. [Google Scholar] [CrossRef]
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.; McGraw, K.L.; et al. Eprenetapopt (APR-246) and azacitidine in TP53-mutant myelodysplastic syndromes. J. Clin. Oncol. 2021, 39, 1584–1594. [Google Scholar] [CrossRef]
- Cluzeau, T.; Sebert, M.; Rahmé, R.; Cuzzubbo, S.; Lehmann-Che, J.; Madelaine, I.; Peterlin, P.; Bève, B.; Attalah, H.; Chermat, F.; et al. Eprenetapopt plus azacitidine in TP53-mutated myelodysplastic syndromes and acute myeloid leukemia: A phase II study by the Groupe Francophone des Myélodysplasies (GFM). J. Clin. Oncol. 2021, 39, 1575–1583. [Google Scholar] [CrossRef]
- Salim, K.Y.; Maleki Vareki, S.; Danter, W.R.; Koropatnick, J. COTI-2, a novel small molecule that is active against multiple human cancer cell lines in vitro and in vivo. Oncotarget 2016, 7, 41363–41379. [Google Scholar] [CrossRef]
- Synnott, N.C.; O’Connell, D.; Crown, J.; Duffy, M.J. COTI-2 reactivates mutant p53 and inhibits growth of triple-negative breast cancer cells. Breast Cancer Res. Treat. 2020, 179, 47–56. [Google Scholar] [CrossRef]
- Lindemann, A.; Patel, A.A.; Silver, N.L.; Tang, L.; Liu, Z.; Wang, L.; Tanaka, N.; Rao, X.; Takahashi, H.; Maduka, N.K.; et al. COTI-2, a novel thiosemicarbazone derivative, exhibits antitumor activity in HNCC through p53-dependent and -independent mechanisms. Clin. Cancer Res. 2019, 25, 5650–5662. [Google Scholar] [CrossRef]
- Coronel, L.; Häckes, D.; Schwab, K.; Riege, K.; Hoffmann, S.; Fischer, M. p53-mediated AKT and mTOR inhibition requires RFX7 and DDIT4 and depends on nutrient abundance. Oncogene 2022, 41, 1063–1069. [Google Scholar] [CrossRef]
- Feng, Z.; Hu, W.; de Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: Stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007, 67, 3043–3053. [Google Scholar] [CrossRef]
- Alzahrani, A.S. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin. Cancer Biol. 2019, 59, 125–132. [Google Scholar] [CrossRef]
- Westin, S.N.; Nieves-Neira, W.; Lynam, C.; Salim, K.Y.; Silva, A.D.; Ho, R.; Mills, G.B.; Coleman, R.L.; Janku, F.; Matei, D. Safety and early efficacy signals for COTI-2, an orally available small molecule targeting p53, in a phase I trial of recurrent gynecologic cancer. Cancer Res. 2018, 78, CT033. [Google Scholar] [CrossRef]
- Chen, S.; Wu, J.L.; Liang, Y.; Tang, Y.G.; Song, H.X.; Wu, L.L.; Xing, Y.F.; Yan, N.; Li, Y.T.; Wang, Z.Y.; et al. Arsenic trioxide rescues structural p53 mutations through a cryptic allosteric site. Cancer Cell 2021, 39, 225–239.e8. [Google Scholar] [CrossRef]
- Yan, W.; Zhang, Y.; Zhang, J.; Liu, S.; Cho, S.J.; Chen, X. Mutant p53 protein is targeted by arsenic for degradation and plays a role in arsenic-mediated growth suppression. J. Biol. Chem. 2011, 286, 17478–17486. [Google Scholar] [CrossRef]
- Autore, F.; Chiusolo, P.; Sorà, F.; Giammarco, S.; Laurenti, L.; Innocenti, I.; Metafuni, E.; Piccirillo, N.; Pagano, L.; Sica, S. Efficacy and tolerability of first line arsenic trioxide in combination with all-trans retinoic acid in patients with acute promyelocytic leukemia: Real life experience. Front. Oncol. 2021, 11, 614721. [Google Scholar] [CrossRef]
- Dumble, M.; Xu, L.; Dominique, R.; Liu, B.; Yang, H.; McBrayer, M.K.; Thomas, D.; Fahr, B.; Li, H.; Huang, K.S.; et al. Abstract LB006: PC14586: The First Orally Bioavailable Small Molecule Reactivator of Y220C Mutant p53 in Clinical Development. In Proceedings of the American Association for Cancer Research Annual Meeting 2021, Washington, DC, USA, 10–15 April 2021; AACR: Philadelphia, PA, USA, 2021; (Suppl. 13). [Google Scholar]
- Liu, X.; Wilcken, R.; Joerger, A.C.; Chuckowree, I.S.; Amin, J.; Spencer, J.; Fersht, A.R. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res. 2013, 41, 6034–6044. [Google Scholar] [CrossRef]
- Puzio-Kuter, A.M.; Mulligan, C.; Russo, B.; Wiebesiek, A.; Xu, L.; Yang, H.; Vu, B.; Dumble, M. Small Molecule Reactivators of Y220C Mutant p53 Modulate Tumor Infiltrating Leukocytes and Synergize with Immune Checkpoint Inhibitors. In Proceedings of the 113th Annual Meeting of the American Association for Cancer Research, New Orleans, LA, USA, 8–13 April 2021; AACR: Philadelphia, PA, USA, 2022. Abstract nr 1295/5.. [Google Scholar]
- Dumbrava, E.E.; Johnson, M.L.; Tolcher, A.W.; Shapiro, G.; Thompson, J.A.; El-Khoueiry, A.B.; Vandross, A.L.; Kummar, S.; Parikh, A.R.; Munster, P.N.; et al. First-in-human study of PC14586, a small molecule structural corrector of Y220C mutant p53, in patients with advanced solid tumors harboring a TP53 Y220C mutation. J. Clin. Oncol. 2022, 40 (Suppl. 16), 3003. [Google Scholar] [CrossRef]
- Howell, A.; Bergh, J. Insights into the place of fulvestrant for the treatment of advanced endocrine responsive breast cancer. J. Clin. Oncol. 2010, 28, 4548–4550. [Google Scholar] [CrossRef]
- Charliński, G.; Vesole, D.H.; Jurczyszyn, A. rapid progress in the use of immunomodulatory drugs and cereblon E3 Ligase modulators in the treatment of multiple myeloma. Cancers 2021, 13, 4666. [Google Scholar] [CrossRef]
- Alexandrova, E.M.; Yallowitz, A.R.; Li, D.; Xu, S.; Schulz, R.; Proia, D.A.; Lozano, G.; Dobbelstein, M.; Moll, U.M. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature 2015, 523, 352–356. [Google Scholar] [CrossRef]
- Chatterjee, S.; Burns, T.F. Targeting heat shock proteins in cancer: A promising therapeutic approach. Int. J. Mol. Sci. 2017, 18, 1978. [Google Scholar] [CrossRef]
- Schulz-Heddergott, R.; Moll, U.M. Gain-of-function (GOF) mutant p53 as actionable therapeutic target. Cancers 2018, 10, 188. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Marchenko, N.D.; Schulz, R.; Fischer, V.; Velasco-Hernandez, T.; Talos, F.; Moll, U.M. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol. Cancer Res. 2011, 9, 577–588. [Google Scholar] [CrossRef]
- Jhaveri, K.; Ochiana, S.O.; Dunphy, M.P.; Gerecitano, J.F.; Corben, A.D.; Peter, R.I.; Janjigian, Y.Y.; Gomes-DaGama, E.M.; Koren, J.; Modi, S.; et al. Heat shock protein 90 inhibitors in the treatment of cancer: Current status and future directions. Expert Opin. Investig. Drugs 2014, 23, 611–628. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.; Goss, G.; Rosell, R.; Schmid-Bindert, G.; Zaric, B.; Andric, Z.; Bondarenko, I.; Komov, D.; Ceric, T.; Khuri, F.; et al. A randomized phase II study of ganetespib, a heat shock protein 90 inhibitor, in combination with docetaxel in second-line therapy of advanced non-small cell lung cancer (GALAXY-1). Ann. Oncol. 2015, 26, 1741–1748. [Google Scholar] [CrossRef] [PubMed]
- Goyal, L.; Chaudhary, S.P.; Kwak, E.L.; Abrams, T.A.; Carpenter, A.N.; Wolpin, B.M.; Wadlow, R.C.; Allen, J.N.; Heist, R.; McCleary, N.J.; et al. A phase 2 clinical trial of the heat shock protein 90 (HSP 90) inhibitor ganetespib in patients with refractory advanced esophagogastric cancer. Invest. N. Drugs 2020, 38, 1533–1539. [Google Scholar] [CrossRef]
- Pillai, R.N.; Fennell, D.A.; Kovcin, V.; Ciuleanu, T.E.; Ramlau, R.; Kowalski, D.; Schenker, M.; Yalcin, I.; Teofilovici, F.; Vukovic, V.M.; et al. Randomized Phase III study of ganetespib, a heat shock protein 90 inhibitor, with docetaxel versus docetaxel in advanced non-small-cell lung cancer (GALAXY-2). J. Clin. Oncol. 2020, 38, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Ranjan, A.; Iyer, S.V.; Padhye, S.; Weir, S.J.; Roy, A.; Iwakuma, T. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell Biol. 2016, 18, 1233–1243. [Google Scholar] [CrossRef]
- Ingallina, E.; Sorrentino, G.; Bertolio, R.; Lisek, K.; Zannini, A.; Azzolin, L.; Severino, L.U.; Scaini, D.; Mano, M.; Mantovani, F.; et al. Mechanical cues control mutant p53 stability through a mevalonate-RhoA axis. Nat. Cell Biol. 2018, 20, 28–35. [Google Scholar] [CrossRef]
- Chou, C.W.; Lin, C.H.; Hsiao, T.H.; Lo, C.C.; Hsieh, C.Y.; Huang, C.C.; Sher, Y.P. Therapeutic effects of statins against lung adenocarcinoma via p53 mutant-mediated apoptosis. Sci. Rep. 2019, 9, 20403. [Google Scholar] [CrossRef]
- Xu, D.; Tong, X.; Sun, L.; Li, H.; Jones, R.D.; Liao, J.; Yang, G.Y. Inhibition of mutant Kras and p53-driven pancreatic carcinogenesis by atorvastatin: Mainly via targeting of the farnesylated DNAJA1 in chaperoning mutant p53. Mol. Carcinog. 2019, 58, 2052–2064. [Google Scholar] [CrossRef]
- Longo, J.; van Leeuwen, J.E.; Elbaz, M.; Branchard, E.; Penn, L.Z. Statins as anticancer agents in the era of precision medicine. Clin. Cancer Res. 2020, 26, 5791–5800. [Google Scholar] [CrossRef]
- O’Grady, S.; Crown, J.; Duffy, M.J. Statins inhibit proliferation and induce apoptosis in triple-negative breast cancer. Med. Oncol. 2022, in press. [Google Scholar] [CrossRef]
- Parrales, A.; Thoenen, E.; Iwakuma, T. The interplay between mutant p53 and the mevalonate pathway. Cell Death Differ. 2018, 25, 460–470. [Google Scholar] [CrossRef]
- Deluche, E.; Antoine, A.; Bachelot, T.; Lardy-Cleaud, A.; Dieras, V.; Brain, E.; Debled, M.; Jacot, W.; Mouret-Reynier, M.A.; Goncalves, A.; et al. Contemporary outcomes of metastatic breast cancer among 22,000 women from the multicentre ESME cohort 2008–2016. Eur. J. Cancer 2020, 129, 60–70. [Google Scholar] [CrossRef]
- Matusewicz, L.; Meissner, J.; Toporkiewicz, M.; Sikorski, A.F. The effect of statins on cancer cells—Review. Tumor Biol. 2015, 36, 4889–4904. [Google Scholar] [CrossRef]
- Lv, H.; Shi, D.; Fei, M.; Chen, Y.; Xie, F.; Wang, Z.; Wang, Y.; Hu, P. Association between statin use and prognosis of breast cancer: A meta-analysis of cohort studies. Front. Oncol. 2020, 10, 556243. [Google Scholar] [CrossRef]
- Ahmadi, M.; Amiri, S.; Pecic, S.; Machaj, F.; Rosik, J.; Łos, M.J.; Alizadeh, J.; Mahdian, R.; da Silva Rosa, S.C.; Schaafsma, D.; et al. Pleiotropic effects of statins: A focus on cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165968. [Google Scholar] [CrossRef]
- Trotta, F.; Avena, P.; Chimento, A.; Rago, V.; De Luca, A.; Sculco, S.; Nocito, M.C.; Malivindi, R.; Fallo, F.; Pezzani, R.; et al. Statins reduce intratumor cholesterol affecting adrenocortical cancer growth. Mol. Cancer Ther. 2020, 19, 1909–1921. [Google Scholar] [CrossRef]
- Wang, Y.; You, S.; Su, S.; Yeon, A.; Lo, E.M.; Kim, S.; Mohler, J.L.; Freeman, M.R.; Kim, H.L. Cholesterol-lowering intervention decreases mTOR complex 2 signaling and enhances antitumor immunity. Clin. Cancer Res. 2022, 28, 414–424. [Google Scholar] [CrossRef]
- Zhou, S.; Fan, C.; Zeng, Z.; Young, K.H.; Li, Y. Clinical and immunological effects of p53-targeting vaccines. Front. Cell Dev. Biol. 2021, 9, 762796. [Google Scholar] [CrossRef]
- Roth, J.; Dittmer, D.; Rea, D.; Tartaglia, J.; Paoletti, E.; Levine, A.J. p53 as a target for cancer vaccines: Recombinant canarypox virus vectors expressing p53 protect mice against lethal tumor cell challenge. Proc. Natl. Acad. Sci. USA 1996, 93, 4781–4786. [Google Scholar] [CrossRef]
- Morse, M.A.; Gwin, W.R.; Mitchell, D.A. Vaccine therapies for cancer: Then and now. Target Oncol. 2021, 16, 121–152. [Google Scholar] [CrossRef]
- Li, Y.; Guo, W.; Li, X.; Zhang, J.; Sun, M.; Tang, Z.; Ran, W.; Yang, K.; Huang, G.; Li, L. Expert consensus on the clinical application of recombinant adenovirus human p53 for head and neck cancers. Int. J. Oral. Sci. 2021, 13, 38. [Google Scholar] [CrossRef]
- Hwang, L.A.; Phang, B.H.; Liew, O.W.; Iqbal, J.; Koh, X.H.; Koh, X.Y.; Othman, R.; Xue, Y.; Richards, A.M.; Lane, D.P.; et al. Monoclonal antibodies against specific p53 hotspot mutants as potential tools for precision medicine. Cell Rep. 2018, 22, 299–312. [Google Scholar] [CrossRef]
- Ubby, I.; Krueger, C.; Rosato, R.; Qian, W.; Chang, J.; Sabapathy, K. Cancer therapeutic targeting using mutant-p53-specific siRNAs. Oncogene 2019, 38, 3415–3427. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Cao, J.; Topatana, W.; Juengpanich, S.; Li, S.; Zhang, B.; Shen, J.; Cai, L.; Cai, X.; Chen, M. Targeting mutant p53 for cancer therapy: Direct and indirect strategies. J. Hematol. Oncol. 2021, 14, 157. [Google Scholar] [CrossRef]
- Hu, W.; Feng, Z. Hypothermia is a potential new therapy for a subset of tumors with mutant p53. Cancer Res. 2021, 81, 3762–3763. [Google Scholar] [CrossRef] [PubMed]
- Isobe, Y.; Okumura, M.; McGregor, L.M.; Brittain, S.M.; Jones, M.D.; Liang, X.; White, R.; Forrester, W.; McKenna, J.M.; Tallarico, J.A.; et al. Manumycin polyketides act as molecular glues between UBR7 and P53. Nat. Chem. Biol. 2020, 16, 1189–1198. [Google Scholar] [CrossRef] [PubMed]
- Chira, S.; Gulei, D.; Hajitou, A.; Berindan-Neagoe, I. Restoring the p53 ‘guardian’ phenotype in p53-deficient tumor cells with CRISPR/Cas9. Trends Biotechnol. 2018, 36, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Sayed, S.; Sidorova, O.A.; Hennig, A.; Augsburg, M.; Cortés Vesga, C.P.; Abohawya, M.; Schmitt, L.T.; Sürün, D.; Stange, D.E.; Mircetic, J.; et al. Efficient Correction of Oncogenic KRAS and TP53 Mutations through CRISPR Base Editing. Cancer Res. 2022, 82, 3002–3015. [Google Scholar] [CrossRef]


{kind=link}
| Multiplicity of mutations with different structures and functions |
| Absence of a readily identifiable pocket suitable for binding of drugs * |
| Absence of enzyme activity which might be blocked by catalytic inhibitors |
| Predominantly nuclear localization prevents access by standard antibodies |
| Strategy | Example of Drug | Refs. |
|---|---|---|
| Reactivation to WT form | Eprenetapopt, COTI-2, arsenic trioxide, PC14586 | [8,44,46,57,58,63,66] |
| Degradation of mt 53 | Ganetespib, statins | [73,80,81,82,83] |
| Vaccines targeting mt p53 | p53-SLP, p53MVA | [93,94] |
| Gene therapy | Gendicine * | [96] |
| P53 mt-specific antibodies | anti-R248Q antibody | [97] |
| P53 mt-specific siRNAs | [98] | |
| T-cell receptor mimic antibodies | H2-scDb | [42] |
| Inhibiting genes exhibiting synthetic lethality with mt p53 | WEE1, ATR, CHK1 | [99] |
| Induction of therapeutic hypothermia via temperature sensitive mt forms of p53 | CHA | [100] |
| Molecular glues/PROTACS | Manumycin polyketides | [101] |
| CRISPR/Cas9 | [102,103] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duffy, M.J.; Tang, M.; Rajaram, S.; O’Grady, S.; Crown, J. Targeting Mutant p53 for Cancer Treatment: Moving Closer to Clinical Use? Cancers 2022, 14, 4499. https://doi.org/10.3390/cancers14184499
Duffy MJ, Tang M, Rajaram S, O’Grady S, Crown J. Targeting Mutant p53 for Cancer Treatment: Moving Closer to Clinical Use? Cancers. 2022; 14(18):4499. https://doi.org/10.3390/cancers14184499
Chicago/Turabian StyleDuffy, Michael J., Minhong Tang, Subhasree Rajaram, Shane O’Grady, and John Crown. 2022. "Targeting Mutant p53 for Cancer Treatment: Moving Closer to Clinical Use?" Cancers 14, no. 18: 4499. https://doi.org/10.3390/cancers14184499
APA StyleDuffy, M. J., Tang, M., Rajaram, S., O’Grady, S., & Crown, J. (2022). Targeting Mutant p53 for Cancer Treatment: Moving Closer to Clinical Use? Cancers, 14(18), 4499. https://doi.org/10.3390/cancers14184499