
The Tumor Microenvironment of Hepatocellular Carcinoma: Untying an Intricate Immunological Network



Abstract
Simple Summary
Abstract
1. Introduction
2. The Role of Inflammation in HCC Occurrence and Progression
3. Liver Immune Privilege
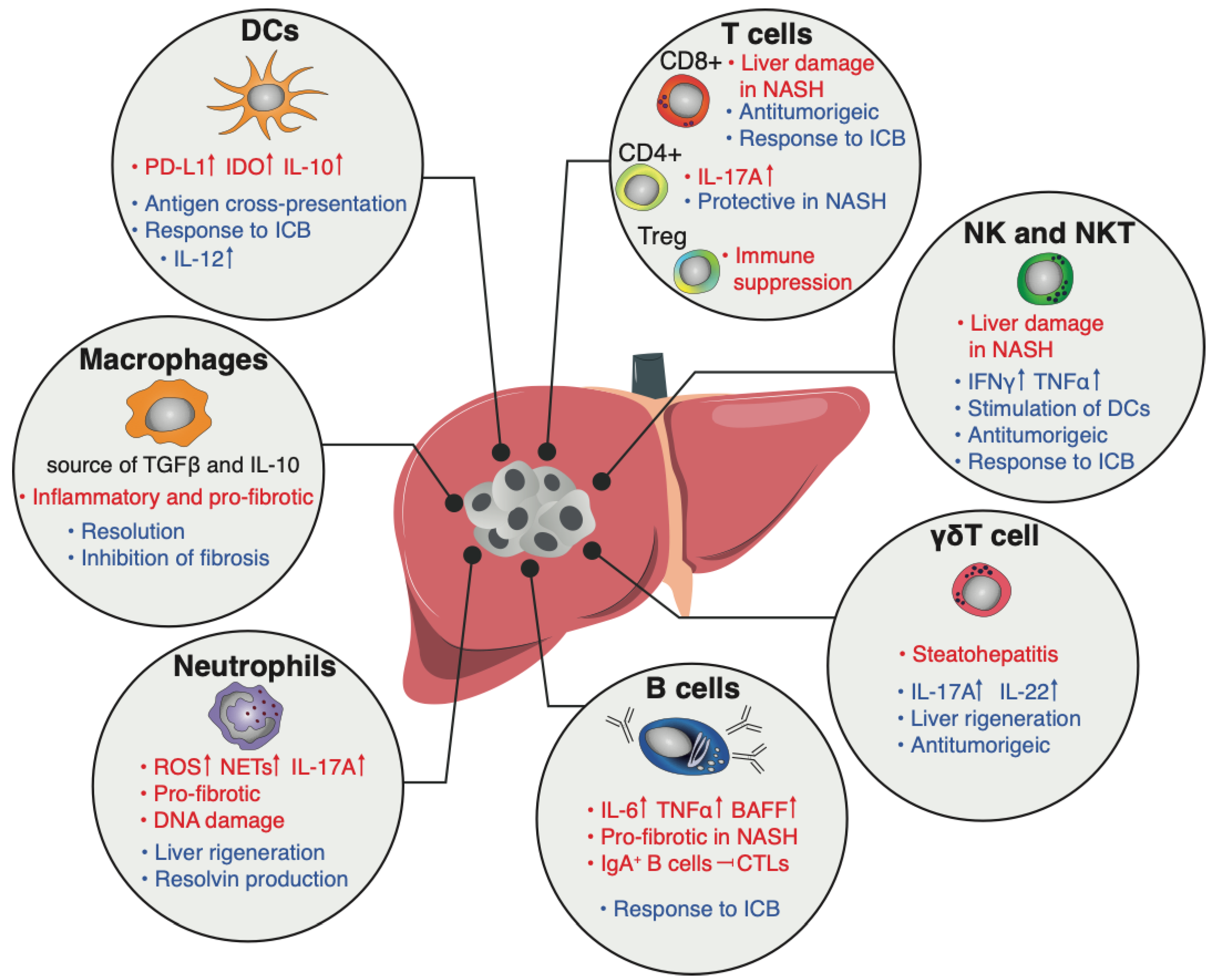
4. The Immune Compartment
4.1. Myeloid Cells
4.1.1. Dendritic Cells
4.1.2. Macrophages and Monocytes
4.1.3. Neutrophils
4.2. Lymphoid Cells
4.2.1. T Cells
4.2.2. B Cells
4.2.3. NK Cells and ILCs
4.2.4. iNKT
4.2.5. γδT Cells
5. Metabolic Syndrome and Liver Cancer
6. Therapy of Liver Cancer
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Jiang, Y.; Que, W.; Zhu, P.; Li, X.K. The Role of Diverse Liver Cells in Liver Transplantation Tolerance. Front. Immunol. 2020, 11, 1203. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Maeda, S. Inflammation- and stress-related signaling pathways in hepatocarcinogenesis. World J. Gastroenterol. 2012, 18, 4071–4081. [Google Scholar] [CrossRef] [PubMed]
- Idalsoaga, F.; Kulkarni, A.V.; Mousa, O.Y.; Arrese, M.; Arab, J.P. Non-alcoholic Fatty Liver Disease and Alcohol-Related Liver Disease: Two Intertwined Entities. Front. Med. 2020, 7, 448. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef]
- Pinato, D.J.; Guerra, N.; Fessas, P.; Murphy, R.; Mineo, T.; Mauri, F.A.; Mukherjee, S.K.; Thursz, M.; Wong, C.N.; Sharma, R.; et al. Immune-based therapies for hepatocellular carcinoma. Oncogene 2020, 39, 3620–3637. [Google Scholar] [CrossRef]
- Greten, T.F.; Sangro, B. Targets for immunotherapy of liver cancer. J. Hepatol. 2017. [Google Scholar] [CrossRef]
- Giraud, J.; Chalopin, D.; Blanc, J.F.; Saleh, M. Hepatocellular Carcinoma Immune Landscape and the Potential of Immunotherapies. Front. Immunol. 2021, 12, 655697. [Google Scholar] [CrossRef]
- Yu, J.; Green, M.D.; Li, S.; Sun, Y.; Journey, S.N.; Choi, J.E.; Rizvi, S.M.; Qin, A.; Waninger, J.J.; Lang, X.; et al. Liver metastasis restrains immunotherapy efficacy via macrophage-mediated T cell elimination. Nat. Med. 2021, 27, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Bonavita, E.; Bromley, C.P.; Jonsson, G.; Pelly, V.S.; Sahoo, S.; Walwyn-Brown, K.; Mensurado, S.; Moeini, A.; Flanagan, E.; Bell, C.R.; et al. Antagonistic Inflammatory Phenotypes Dictate Tumor Fate and Response to Immune Checkpoint Blockade. Immunity 2020, 53, 1215–1229.e1218. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Zelenay, S.; van der Veen, A.G.; Böttcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef]
- Florian, R.; Greten, S.I.G. Inflammation and Cancer:Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar]
- Rothwell, P.M.; Price, J.F.; Fowkes, F.G.; Zanchetti, A.; Roncaglioni, M.C.; Tognoni, G.; Lee, R.; Belch, J.F.; Wilson, M.; Mehta, Z.; et al. Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: Analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet 2012, 379, 1602–1612. [Google Scholar] [CrossRef]
- Ringelhan, M.; Pfister, D.; O’Connor, T.; Pikarsky, E.; Heikenwalder, M. The immunology of hepatocellular carcinoma. Nat. Immunol. 2018, 19, 222–232. [Google Scholar] [CrossRef]
- Schwitalla, S.; Fingerle, A.A.; Cammareri, P.; Nebelsiek, T.; Goktuna, S.I.; Ziegler, P.K.; Canli, O.; Heijmans, J.; Huels, D.J.; Moreaux, G.; et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 2013, 152, 25–38. [Google Scholar] [CrossRef]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Galle, P.R.; Forner, A.; Llovet, J.M.; Mazzaferro, V.; Piscaglia, F.; Raoul, J.L.; Schirmacher, P.; Vilgrain, V. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef]
- Sakurai, T.; He, G.; Matsuzawa, A.; Yu, G.Y.; Maeda, S.; Hardiman, G.; Karin, M. Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell 2008, 14, 156–165. [Google Scholar] [CrossRef]
- Khambu, B.; Huda, N.; Chen, X.; Antoine, D.J.; Li, Y.; Dai, G.; Kohler, U.A.; Zong, W.X.; Waguri, S.; Werner, S.; et al. HMGB1 promotes ductular reaction and tumorigenesis in autophagy-deficient livers. J. Clin. Investig. 2018, 128, 2419–2435. [Google Scholar] [CrossRef]
- Wolf, M.J.; Adili, A.; Piotrowitz, K.; Abdullah, Z.; Boege, Y.; Stemmer, K.; Ringelhan, M.; Simonavicius, N.; Egger, M.; Wohlleber, D.; et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 2014, 26, 549–564. [Google Scholar] [CrossRef]
- Endig, J.; Buitrago-Molina, L.E.; Marhenke, S.; Reisinger, F.; Saborowski, A.; Schutt, J.; Limbourg, F.; Konecke, C.; Schreder, A.; Michael, A.; et al. Dual Role of the Adaptive Immune System in Liver Injury and Hepatocellular Carcinoma Development. Cancer Cell 2016, 30, 308–323. [Google Scholar] [CrossRef]
- Gomes, A.L.; Teijeiro, A.; Buren, S.; Tummala, K.S.; Yilmaz, M.; Waisman, A.; Theurillat, J.P.; Perna, C.; Djouder, N. Metabolic Inflammation-Associated IL-17A Causes Non-alcoholic Steatohepatitis and Hepatocellular Carcinoma. Cancer Cell 2016, 30, 161–175. [Google Scholar] [CrossRef]
- Garnelo, M.; Tan, A.; Her, Z.; Yeong, J.; Lim, C.J.; Chen, J.; Lim, K.H.; Weber, A.; Chow, P.; Chung, A.; et al. Interaction between tumour-infiltrating B cells and T cells controls the progression of hepatocellular carcinoma. Gut 2017, 66, 342–351. [Google Scholar] [CrossRef]
- Ma, C.; Kesarwala, A.H.; Eggert, T.; Medina-Echeverz, J.; Kleiner, D.E.; Jin, P.; Stroncek, D.F.; Terabe, M.; Kapoor, V.; ElGindi, M.; et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature 2016, 531, 253–257. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, H.; Mao, C.; Sun, M.; Dominah, G.; Chen, L.; Zhuang, Z. Fatty acid oxidation contributes to IL-1β secretion in M2 macrophages and promotes macrophage-mediated tumor cell migration. Mol. Immunol. 2018, 94, 27–35. [Google Scholar] [CrossRef]
- Kuang, D.M.; Zhao, Q.; Peng, C.; Xu, J.; Zhang, J.P.; Wu, C.; Zheng, L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J. Exp. Med. 2009, 206, 1327–1337. [Google Scholar] [CrossRef]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef]
- Schouppe, E.; Mommer, C.; Movahedi, K.; Laoui, D.; Morias, Y.; Gysemans, C.; Luyckx, A.; De Baetselier, P.; Van Ginderachter, J.A. Tumor-induced myeloid-derived suppressor cell subsets exert either inhibitory or stimulatory effects on distinct CD8+ T-cell activation events. Eur. J. Immunol. 2013, 43, 2930–2942. [Google Scholar] [CrossRef]
- Tu, S.; Bhagat, G.; Cui, G.; Takaishi, S.; Kurt-Jones, E.A.; Rickman, B.; Betz, K.S.; Penz-Oesterreicher, M.; Bjorkdahl, O.; Fox, J.G.; et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 2008, 14, 408–419. [Google Scholar] [CrossRef]
- Lechner, M.G.; Liebertz, D.J.; Epstein, A.L. Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells. J. Immunol. 2010, 185, 2273–2284. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Kanterman, J.; Ish-Shalom, E.; Elnekave, M.; Horwitz, E.; Baniyash, M. Tumor necrosis factor-alpha blocks differentiation and enhances suppressive activity of immature myeloid cells during chronic inflammation. Immunity 2013, 38, 541–554. [Google Scholar] [CrossRef]
- Condamine, T.; Gabrilovich, D.I. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011, 32, 19–25. [Google Scholar] [CrossRef]
- Chiu, D.K.; Tse, A.P.; Xu, I.M.; Di Cui, J.; Lai, R.K.; Li, L.L.; Koh, H.Y.; Tsang, F.H.; Wei, L.L.; Wong, C.M.; et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat. Commun. 2017, 8, 517. [Google Scholar] [CrossRef]
- Suthen, S.; Lim, C.J.; Nguyen, P.H.D.; Dutertre, C.A.; Lai, H.L.H.; Wasser, M.; Chua, C.; Lim, T.K.H.; Leow, W.Q.; Loh, T.J.; et al. Hypoxia-driven immunosuppression by Treg and type-2 conventional dendritic cells in HCC. Hepatology 2022, 76, 1329–1344. [Google Scholar] [CrossRef]
- Li, X.; Xing, Y.F.; Lei, A.H.; Xiao, Q.; Lin, Z.H.; Hong, Y.F.; Wu, X.-Y.; Zhou, J. Neutrophil count is associated with myeloid derived suppressor cell level and presents prognostic value for hepatocellular carcinoma patients. Oncotarget 2017, 8, 24380. [Google Scholar] [CrossRef]
- Malik, A.; Thanekar, U.; Amarachintha, S.; Mourya, R.; Nalluri, S.; Bondoc, A.; Shivakumar, P. “Complimenting the Complement”: Mechanistic Insights and Opportunities for Therapeutics in Hepatocellular Carcinoma. Front. Oncol. 2020, 10, 627701. [Google Scholar] [CrossRef]
- Bonavita, E.; Gentile, S.; Rubino, M.; Maina, V.; Papait, R.; Kunderfranco, P.; Greco, C.; Feruglio, F.; Molgora, M.; Laface, I.; et al. PTX3 is an extrinsic oncosuppressor regulating complement-dependent inflammation in cancer. Cell 2015, 160, 700–714. [Google Scholar] [CrossRef]
- Calne, R.Y.; Sells, R.A.; Pena, J.R.; Davis, D.R.; Millard, P.R.; Herbertson, B.M.; Binns, R.M.; Davies, D.A. Induction of immunological tolerance by porcine liver allografts. Nature 1969, 223, 472–476. [Google Scholar] [CrossRef]
- Crispe, I.N. Immune tolerance in liver disease. Hepatology 2014, 60, 2109–2117. [Google Scholar] [CrossRef]
- Williams, M.A.; Tyznik, A.J.; Bevan, M.J. Interleukin-2 signals during priming are required for secondary expansion of CD8+ memory T cells. Nature 2006, 441, 890–893. [Google Scholar] [CrossRef]
- Mareike Mueller, H.C.S.; Kersting, N.; Altay, T.; Blum, H.E.; Klenerman, P.; Thimme, R.; Semmo, N. Virus-specific CD4+ T cell responses in chronic HCV infection in blood and liver identified by antigen-specific upregulation of CD154. J. Hepatol. 2010, 52, 800–811. [Google Scholar] [CrossRef]
- Saha, B.; Choudhary, M.C.; Sarin, S.K. Expression of inhibitory markers is increased on effector memory T cells during hepatitis C virus/HIV coinfection as compared to hepatitis C virus or HIV monoinfection. AIDS 2013, 27, 2191–2200. [Google Scholar] [CrossRef]
- Wedemeyer, H.; He, X.S.; Nascimbeni, M.; Davis, A.R.; Greenberg, H.B.; Hoofnagle, J.H.; Liang, T.J.; Alter, H.; Rehermann, B. Impaired effector function of hepatitis C virus-specific CD8+ T cells in chronic hepatitis C virus infection. J. Immunol. 2002, 169, 3447–3458. [Google Scholar] [CrossRef]
- Pillarisetty, V.G.; Shah, A.B.; Miller, G.; Bleier, J.I.; DeMatteo, R.P. Liver dendritic cells are less immunogenic than spleen dendritic cells because of differences in subtype composition. J. Immunol. 2004, 172, 1009–1017. [Google Scholar] [CrossRef]
- Abe, M.; Tokita, D.; Raimondi, G.; Thomson, A.W. Endotoxin modulates the capacity of CpG-activated liver myeloid DC to direct Th1-type responses. Eur. J. Immunol. 2006, 36, 2483–2493. [Google Scholar] [CrossRef]
- Matta, B.M.; Raimondi, G.; Rosborough, B.R.; Sumpter, T.L.; Thomson, A.W. IL-27 production and STAT3-dependent upregulation of B7-H1 mediate immune regulatory functions of liver plasmacytoid dendritic cells. J. Immunol. 2012, 188, 5227–5237. [Google Scholar] [CrossRef]
- Liu, H.; Bakthavatsalam, R.; Meng, Z.; Li, Z.; Li, W.; Perkins, J.D.; Reyes, J. PD-L1 signal on liver dendritic cells is critical for Foxp3(+)CD4(+)CD25(+) Treg and liver tolerance induction in mice. Transpl. Proc. 2013, 45, 1853–1855. [Google Scholar] [CrossRef] [PubMed]
- Jurgens, B.; Hainz, U.; Fuchs, D.; Felzmann, T.; Heitger, A. Interferon-gamma-triggered indoleamine 2,3-dioxygenase competence in human monocyte-derived dendritic cells induces regulatory activity in allogeneic T cells. Blood 2009, 114, 3235–3243. [Google Scholar] [CrossRef]
- Zhang, M.; Xu, S.; Han, Y.; Cao, X. Apoptotic cells attenuate fulminant hepatitis by priming Kupffer cells to produce interleukin-10 through membrane-bound TGF-beta. Hepatology 2011, 53, 306–316. [Google Scholar] [CrossRef]
- Yan, M.L.; Wang, Y.D.; Tian, Y.F.; Lai, Z.D.; Yan, L.N. Inhibition of allogeneic T-cell response by Kupffer cells expressing indoleamine 2,3-dioxygenase. World J. Gastroenterol. 2010, 16, 636–640. [Google Scholar] [CrossRef]
- You, Q.; Cheng, L.; Kedl, R.M.; Ju, C. Mechanism of T cell tolerance induction by murine hepatic Kupffer cells. Hepatology 2008, 48, 978–990. [Google Scholar] [CrossRef]
- Kruse, N.; Neumann, K.; Schrage, A.; Derkow, K.; Schott, E.; Erben, U.; Kuhl, A.; Loddenkemper, C.; Zeitz, M.; Hamann, A.; et al. Priming of CD4+ T cells by liver sinusoidal endothelial cells induces CD25low forkhead box protein 3- regulatory T cells suppressing autoimmune hepatitis. Hepatology 2009, 50, 1904–1913. [Google Scholar] [CrossRef] [PubMed]
- Charles, R.; Chou, H.S.; Wang, L.; Fung, J.J.; Lu, L.; Qian, S. Human hepatic stellate cells inhibit T-cell response through B7-H1 pathway. Transplantation 2013, 96, 17–24. [Google Scholar] [CrossRef]
- Hochst, B.; Schildberg, F.A.; Sauerborn, P.; Gabel, Y.A.; Gevensleben, H.; Goltz, D.; Heukamp, L.C.; Turler, A.; Ballmaier, M.; Gieseke, F.; et al. Activated human hepatic stellate cells induce myeloid derived suppressor cells from peripheral blood monocytes in a CD44-dependent fashion. J. Hepatol. 2013, 59, 528–535. [Google Scholar] [CrossRef]
- Thomson, A.W.; Knolle, P.A. Antigen-presenting cell function in the tolerogenic liver environment. Nat. Rev. Immunol. 2010, 10, 753–766. [Google Scholar] [CrossRef]
- Bamboat, Z.M.; Stableford, J.A.; Plitas, G.; Burt, B.M.; Nguyen, H.M.; Welles, A.P.; Gonen, M.; Young, J.W.; DeMatteo, R.P. Human Liver Dendritic Cells Promote T Cell Hyporesponsiveness. J. Immunol. 2009, 182, 1901–1911. [Google Scholar] [CrossRef]
- Chen, Y.; Jiang, G.; Yang, H.-R.; Gu, X.; Wang, L.; Hsieh, C.-C.; Chou, H.-S.; Fung, J.J.; Qian, S.; Lu, L. Distinct response of liver myeloid dendritic cells to endotoxin is mediated by IL-27. J. Hepatol. 2009, 51, 510–519. [Google Scholar] [CrossRef]
- Munn, D.H.; Sharma, M.D.; Mellor, A.L. Ligation of B7-1/B7-2 by Human CD4+ T Cells Triggers Indoleamine 2,3-Dioxygenase Activity in Dendritic Cells. J. Immunol. 2004, 172, 4100–4110. [Google Scholar] [CrossRef]
- Castellaneta, A.; Sumpter, T.L.; Chen, L.; Tokita, D.; Thomson, A.W. NOD2 Ligation Subverts IFN-α Production by Liver Plasmacytoid Dendritic Cells and Inhibits Their T Cell Allostimulatory Activity via B7-H1 Up-Regulation. J. Immunol. 2009, 183, 6922–6932. [Google Scholar] [CrossRef]
- Herber, D.L.; Cao, W.; Nefedova, Y.; Novitskiy, S.V.; Nagaraj, S.; Tyurin, V.A.; Corzo, A.; Cho, H.I.; Celis, E.; Lennox, B.; et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat. Med. 2010, 16, 880–886. [Google Scholar] [CrossRef]
- Ibrahim, J.; Nguyen, A.H.; Rehman, A.; Ochi, A.; Jamal, M.; Graffeo, C.S.; Henning, J.R.; Zambirinis, C.P.; Fallon, N.C.; Barilla, R.; et al. Dendritic cell populations with different concentrations of lipid regulate tolerance and immunity in mouse and human liver. Gastroenterology 2012, 143, 1061–1072. [Google Scholar] [CrossRef]
- Deczkowska, A.; David, E.; Ramadori, P.; Pfister, D.; Safran, M.; Li, B.; Giladi, A.; Jaitin, D.A.; Barboy, O.; Cohen, M.; et al. XCR1(+) type 1 conventional dendritic cells drive liver pathology in non-alcoholic steatohepatitis. Nat. Med. 2021, 27, 1043–1054. [Google Scholar] [CrossRef]
- Ruiz de Galarreta, M.; Bresnahan, E.; Molina-Sánchez, P.; Lindblad, K.E.; Maier, B.; Sia, D.; Puigvehi, M.; Miguela, V.; Casanova-Acebes, M.; Dhainaut, M.; et al. β-Catenin Activation Promotes Immune Escape and Resistance to Anti-PD-1 Therapy in Hepatocellular Carcinoma. Cancer Discov. 2019, 9, 1124–1141. [Google Scholar] [CrossRef]
- Zhang, Q.; He, Y.; Luo, N.; Patel, S.J.; Han, Y.; Gao, R.; Modak, M.; Carotta, S.; Haslinger, C.; Kind, D.; et al. Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma. Cell 2019, 179, 829–845.e20. [Google Scholar] [CrossRef]
- Maier, B.; Leader, A.M.; Chen, S.T.; Tung, N.; Chang, C.; LeBerichel, J.; Chudnovskiy, A.; Maskey, S.; Walker, L.; Finnigan, J.P.; et al. A conserved dendritic-cell regulatory program limits antitumour immunity. Nature 2020, 580, 257–262. [Google Scholar] [CrossRef]
- Di Pilato, M.; Kfuri-Rubens, R.; Pruessmann, J.N.; Ozga, A.J.; Messemaker, M.; Cadilha, B.L.; Sivakumar, R.; Cianciaruso, C.; Warner, R.D.; Marangoni, F.; et al. CXCR6 positions cytotoxic T cells to receive critical survival signals in the tumor microenvironment. Cell 2021, 184, 4512–4530.e22. [Google Scholar] [CrossRef]
- Gerhard, G.M.; Bill, R.; Messemaker, M.; Klein, A.M.; Pittet, M.J. Tumor-infiltrating dendritic cell states are conserved across solid human cancers. J. Exp. Med. 2020, 218, e20200264. [Google Scholar] [CrossRef]
- Sutti, S.; Bruzzì, S.; Heymann, F.; Liepelt, A.; Krenkel, O.; Toscani, A.; Ramavath, N.N.; Cotella, D.; Albano, E.; Tacke, F. CX(3)CR1 Mediates the Development of Monocyte-Derived Dendritic Cells during Hepatic Inflammation. Cells 2019, 8, 1099. [Google Scholar] [CrossRef]
- Sutti, S.; Locatelli, I.; Bruzzì, S.; Jindal, A.; Vacchiano, M.; Bozzola, C.; Albano, E. CX3CR1-expressing inflammatory dendritic cells contribute to the progression of steatohepatitis. Clin. Sci. 2015, 129, 797–808. [Google Scholar] [CrossRef]
- Pradere, J.-P.; Kluwe, J.; De Minicis, S.; Jiao, J.-J.; Gwak, G.-Y.; Dapito, D.H.; Jang, M.-K.; Guenther, N.D.; Mederacke, I.; Friedman, R.; et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 2013, 58, 1461–1473. [Google Scholar] [CrossRef]
- Blois, S.M.; Piccioni, F.; Freitag, N.; Tirado-González, I.; Moschansky, P.; Lloyd, R.; Hensel-Wiegel, K.; Rose, M.; Garcia, M.G.; Alaniz, L.D.; et al. Dendritic cells regulate angiogenesis associated with liver fibrogenesis. Angiogenesis 2014, 17, 119–128. [Google Scholar] [CrossRef]
- Ramakrishnan, R.; Tyurin, V.A.; Veglia, F.; Condamine, T.; Amoscato, A.; Mohammadyani, D.; Johnson, J.J.; Zhang, L.M.; Klein-Seetharaman, J.; Celis, E.; et al. Oxidized lipids block antigen cross-presentation by dendritic cells in cancer. J. Immunol. 2014, 192, 2920–2931. [Google Scholar] [CrossRef]
- Veglia, F.; Tyurin, V.A.; Mohammadyani, D.; Blasi, M.; Duperret, E.K.; Donthireddy, L.; Hashimoto, A.; Kapralov, A.; Amoscato, A.; Angelini, R.; et al. Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat. Commun. 2017, 8, 2122. [Google Scholar] [CrossRef]
- Wiedemann, G.M.; Knott, M.M.; Vetter, V.K.; Rapp, M.; Haubner, S.; Fesseler, J.; Kühnemuth, B.; Layritz, P.; Thaler, R.; Kruger, S.; et al. Cancer cell-derived IL-1α induces CCL22 and the recruitment of regulatory T cells. Oncoimmunology 2016, 5, e1175794. [Google Scholar] [CrossRef]
- Ouyang, F.-Z.; Wu, R.-Q.; Wei, Y.; Liu, R.-X.; Yang, D.; Xiao, X.; Zheng, L.; Li, B.; Lao, X.-M.; Kuang, D.-M. Dendritic cell-elicited B-cell activation fosters immune privilege via IL-10 signals in hepatocellular carcinoma. Nat. Commun. 2016, 7, 13453. [Google Scholar] [CrossRef]
- Bonnardel, J.; T’Jonck, W.; Gaublomme, D.; Browaeys, R.; Scott, C.L.; Martens, L.; Vanneste, B.; De Prijck, S.; Nedospasov, S.A.; Kremer, A.; et al. Stellate Cells, Hepatocytes, and Endothelial Cells Imprint the Kupffer Cell Identity on Monocytes Colonizing the Liver Macrophage Niche. Immunity 2019, 51, 638–654.e9. [Google Scholar] [CrossRef]
- Crispe, I.N. Liver antigen-presenting cells. J. Hepatol. 2011, 54, 357–365. [Google Scholar] [CrossRef]
- Liu, Z.; Gu, Y.; Chakarov, S.; Bleriot, C.; Kwok, I.; Chen, X.; Shin, A.; Huang, W.; Dress, R.J.; Dutertre, C.A.; et al. Fate Mapping via Ms4a3-Expression History Traces Monocyte-Derived Cells. Cell 2019, 178, 1509–1525.e19. [Google Scholar] [CrossRef]
- Sierro, F.; Evrard, M.; Rizzetto, S.; Melino, M.; Mitchell, A.J.; Florido, M.; Beattie, L.; Walters, S.B.; Tay, S.S.; Lu, B.; et al. A Liver Capsular Network of Monocyte-Derived Macrophages Restricts Hepatic Dissemination of Intraperitoneal Bacteria by Neutrophil Recruitment. Immunity 2017, 47, 374–388.e376. [Google Scholar] [CrossRef]
- Seidman, J.S.; Troutman, T.D.; Sakai, M.; Gola, A.; Spann, N.J.; Bennett, H.; Bruni, C.M.; Ouyang, Z.; Li, R.Z.; Sun, X.; et al. Niche-Specific Reprogramming of Epigenetic Landscapes Drives Myeloid Cell Diversity in Nonalcoholic Steatohepatitis. Immunity 2020, 52, 1057–1074.e1057. [Google Scholar] [CrossRef]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef]
- Ginhoux, F.; Guilliams, M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef]
- Geissmann, F.; Jung, S.; Littman, D.R. Blood Monocytes Consist of Two Principal Subsets with Distinct Migratory Properties. Immunity 2003, 19, 71–82. [Google Scholar] [CrossRef]
- Sunderkötter, C.; Nikolic, T.; Dillon, M.J.; van Rooijen, N.; Stehling, M.; Drevets, D.A.; Leenen, P.J.M. Subpopulations of Mouse Blood Monocytes Differ in Maturation Stage and Inflammatory Response. J. Immunol. 2004, 172, 4410–4417. [Google Scholar] [CrossRef]
- Tran, S.; Baba, I.; Poupel, L.; Dussaud, S.; Moreau, M.; Gélineau, A.; Marcelin, G.; Magréau-Davy, E.; Ouhachi, M.; Lesnik, P.; et al. Impaired Kupffer Cell Self-Renewal Alters the Liver Response to Lipid Overload during Non-alcoholic Steatohepatitis. Immunity 2020, 53, 627–640.e625. [Google Scholar] [CrossRef]
- Zigmond, E.; Samia-Grinberg, S.; Pasmanik-Chor, M.; Brazowski, E.; Shibolet, O.; Halpern, Z.; Varol, C. Infiltrating monocyte-derived macrophages and resident kupffer cells display different ontogeny and functions in acute liver injury. J. Immunol. 2014, 193, 344–353. [Google Scholar] [CrossRef]
- van de Laar, L.; Saelens, W.; De Prijck, S.; Martens, L.; Scott, C.L.; Van Isterdael, G.; Hoffmann, E.; Beyaert, R.; Saeys, Y.; Lambrecht, B.N.; et al. Yolk Sac Macrophages, Fetal Liver, and Adult Monocytes Can Colonize an Empty Niche and Develop into Functional Tissue-Resident Macrophages. Immunity 2016, 44, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Karlmark, K.R.; Weiskirchen, R.; Zimmermann, H.W.; Gassler, N.; Ginhoux, F.; Weber, C.; Merad, M.; Luedde, T.; Trautwein, C.; Tacke, F. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology 2009, 50, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, H.W.; Seidler, S.; Nattermann, J.; Gassler, N.; Hellerbrand, C.; Zernecke, A.; Tischendorf, J.J.; Luedde, T.; Weiskirchen, R.; Trautwein, C.; et al. Functional contribution of elevated circulating and hepatic non-classical CD14CD16 monocytes to inflammation and human liver fibrosis. PLoS ONE 2010, 5, e11049. [Google Scholar] [CrossRef] [PubMed]
- Daemen, S.; Gainullina, A.; Kalugotla, G.; He, L.; Chan, M.M.; Beals, J.W.; Liss, K.H.; Klein, S.; Feldstein, A.E.; Finck, B.N.; et al. Dynamic Shifts in the Composition of Resident and Recruited Macrophages Influence Tissue Remodeling in NASH. Cell Rep. 2021, 34, 108626. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.; Dobie, R.; Wilson-Kanamori, J.R.; Dora, E.F.; Henderson, B.E.P.; Luu, N.T.; Portman, J.R.; Matchett, K.P.; Brice, M.; Marwick, J.A.; et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 2019, 575, 512–518. [Google Scholar] [CrossRef]
- MacParland, S.A.; Liu, J.C.; Ma, X.Z.; Innes, B.T.; Bartczak, A.M.; Gage, B.K.; Manuel, J.; Khuu, N.; Echeverri, J.; Linares, I.; et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 2018, 9, 4383. [Google Scholar] [CrossRef]
- Huang, W.; Metlakunta, A.; Dedousis, N.; Zhang, P.; Sipula, I.; Dube, J.J.; Scott, D.K.; O’Doherty, R.M. Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes 2010, 59, 347–357. [Google Scholar] [CrossRef]
- Morinaga, H.; Mayoral, R.; Heinrichsdorff, J.; Osborn, O.; Franck, N.; Hah, N.; Walenta, E.; Bandyopadhyay, G.; Pessentheiner, A.R.; Chi, T.J.; et al. Characterization of distinct subpopulations of hepatic macrophages in HFD/obese mice. Diabetes 2015, 64, 1120–1130. [Google Scholar] [CrossRef]
- Remmerie, A.; Martens, L.; Thoné, T.; Castoldi, A.; Seurinck, R.; Pavie, B.; Roels, J.; Vanneste, B.; De Prijck, S.; Vanhockerhout, M.; et al. Osteopontin Expression Identifies a Subset of Recruited Macrophages Distinct from Kupffer Cells in the Fatty Liver. Immunity 2020, 53, 641–657.e614. [Google Scholar] [CrossRef]
- Morgantini, C.; Jager, J.; Li, X.; Levi, L.; Azzimato, V.; Sulen, A.; Barreby, E.; Xu, C.; Tencerova, M.; Näslund, E.; et al. Liver macrophages regulate systemic metabolism through non-inflammatory factors. Nat. Metab. 2019, 1, 445–459. [Google Scholar] [CrossRef]
- Tencerova, M.; Aouadi, M.; Vangala, P.; Nicoloro, S.M.; Yawe, J.C.; Cohen, J.L.; Shen, Y.; Garcia-Menendez, L.; Pedersen, D.J.; Gallagher-Dorval, K.; et al. Activated Kupffer cells inhibit insulin sensitivity in obese mice. Faseb J. 2015, 29, 2959–2969. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Benkdane, M.; Teixeira-Clerc, F.; Bonnafous, S.; Louvet, A.; Lafdil, F.; Pecker, F.; Tran, A.; Gual, P.; Mallat, A.; et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: A protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 2014, 59, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.; Pellicoro, A.; Vernon, M.A.; Boulter, L.; Aucott, R.L.; Ali, A.; Hartland, S.N.; Snowdon, V.K.; Cappon, A.; Gordon-Walker, T.T.; et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, E3186–E3195. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Cassatella, M.A.; Costantini, C.; Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011, 11, 519–531. [Google Scholar] [CrossRef]
- Das, S.; Maras, J.S.; Hussain, M.S.; Sharma, S.; David, P.; Sukriti, S.; Shasthry, S.M.; Maiwall, R.; Trehanpati, N.; Singh, T.P.; et al. Hyperoxidized albumin modulates neutrophils to induce oxidative stress and inflammation in severe alcoholic hepatitis. Hepatology 2017, 65, 631–646. [Google Scholar] [CrossRef]
- Forrest, E.H.; Storey, N.; Sinha, R.; Atkinson, S.R.; Vergis, N.; Richardson, P.; Masson, S.; Ryder, S.; Thursz, M.R.; Allison, M.; et al. Baseline neutrophil-to-lymphocyte ratio predicts response to corticosteroids and is associated with infection and renal dysfunction in alcoholic hepatitis. Aliment. Pharmacol. Ther. 2019, 50, 442–453. [Google Scholar] [CrossRef]
- Tranah, T.H.; Vijay, G.K.M.; Ryan, J.M.; Abeles, R.D.; Middleton, P.K.; Shawcross, D.L. Dysfunctional neutrophil effector organelle mobilization and microbicidal protein release in alcohol-related cirrhosis. Am. J. Physiol. -Gastrointest. Liver Physiol. 2017, 313, G203–G211. [Google Scholar] [CrossRef]
- Bukong, T.N.; Cho, Y.; Iracheta-Vellve, A.; Saha, B.; Lowe, P.; Adejumo, A.; Furi, I.; Ambade, A.; Gyongyosi, B.; Catalano, D.; et al. Abnormal neutrophil traps and impaired efferocytosis contribute to liver injury and sepsis severity after binge alcohol use. J. Hepatol. 2018, 69, 1145–1154. [Google Scholar] [CrossRef]
- Alkhouri, N.; Morris-Stiff, G.; Campbell, C.; Lopez, R.; Tamimi, T.A.-R.; Yerian, L.; Zein, N.N.; Feldstein, A.E. Neutrophil to lymphocyte ratio: A new marker for predicting steatohepatitis and fibrosis in patients with nonalcoholic fatty liver disease. Liver Int. 2012, 32, 297–302. [Google Scholar] [CrossRef]
- Tritto, G.; Bechlis, Z.; Stadlbauer, V.; Davies, N.; Francés, R.; Shah, N.; Mookerjee, R.P.; Such, J.; Jalan, R. Evidence of neutrophil functional defect despite inflammation in stable cirrhosis. J. Hepatol. 2011, 55, 574–581. [Google Scholar] [CrossRef]
- Zang, S.; Wang, L.; Ma, X.; Zhu, G.; Zhuang, Z.; Xun, Y.; Zhao, F.; Yang, W.; Liu, J.; Luo, Y.; et al. Neutrophils Play a Crucial Role in the Early Stage of Nonalcoholic Steatohepatitis via Neutrophil Elastase in Mice. Cell Biochem. Biophys. 2015, 73, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Rensen, S.S.; Slaats, Y.; Nijhuis, J.; Jans, A.; Bieghs, V.; Driessen, A.; Malle, E.; Greve, J.W.; Buurman, W.A. Increased Hepatic Myeloperoxidase Activity in Obese Subjects with Nonalcoholic Steatohepatitis. Am. J. Pathol. 2009, 175, 1473–1482. [Google Scholar] [CrossRef] [PubMed]
- Fabre, T.; Molina, M.F.; Soucy, G.; Goulet, J.-P.; Willems, B.; Villeneuve, J.-P.; Bilodeau, M.; Shoukry, N.H. Type 3 cytokines IL-17A and IL-22 drive TGF–β–dependent liver fibrosis. Sci. Immunol. 2018, 3, eaar7754. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Xu, M.-J.; Cai, Y.; Wang, W.; Jiang, J.X.; Varga, Z.V.; Feng, D.; Pacher, P.; Kunos, G.; Torok, N.J.; et al. Neutrophil–Hepatic Stellate Cell Interactions Promote Fibrosis in Experimental Steatohepatitis. Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 399–413. [Google Scholar] [CrossRef]
- Khanam, A.; Trehanpati, N.; Riese, P.; Rastogi, A.; Guzman, C.A.; Sarin, S.K. Blockade of Neutrophil’s Chemokine Receptors CXCR1/2 Abrogate Liver Damage in Acute-on-Chronic Liver Failure. Front. Immunol. 2017, 8, 464. [Google Scholar] [CrossRef]
- Wilson, C.L.; Jurk, D.; Fullard, N.; Banks, P.; Page, A.; Luli, S.; Elsharkawy, A.M.; Gieling, R.G.; Chakraborty, J.B.; Fox, C.; et al. NFκB1 is a suppressor of neutrophil-driven hepatocellular carcinoma. Nat. Commun. 2015, 6, 6818. [Google Scholar] [CrossRef]
- He, M.; Peng, A.; Huang, X.-Z.; Shi, D.-C.; Wang, J.-C.; Zhao, Q.; Lin, H.; Kuang, D.-M.; Ke, P.-F.; Lao, X.-M. Peritumoral stromal neutrophils are essential for c-Met-elicited metastasis in human hepatocellular carcinoma. OncoImmunology 2016, 5, e1219828. [Google Scholar] [CrossRef]
- Motomura, T.; Shirabe, K.; Mano, Y.; Muto, J.; Toshima, T.; Umemoto, Y.; Fukuhara, T.; Uchiyama, H.; Ikegami, T.; Yoshizumi, T.; et al. Neutrophil–lymphocyte ratio reflects hepatocellular carcinoma recurrence after liver transplantation via inflammatory microenvironment. J. Hepatol. 2013, 58, 58–64. [Google Scholar] [CrossRef]
- Masucci, M.T.; Minopoli, M.; Carriero, M.V. Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Front. Oncol. 2019, 9, 1146. [Google Scholar] [CrossRef]
- Zhou, S.-L.; Zhou, Z.-J.; Hu, Z.-Q.; Huang, X.-W.; Wang, Z.; Chen, E.-B.; Fan, J.; Cao, Y.; Dai, Z.; Zhou, J. Tumor-Associated Neutrophils Recruit Macrophages and T-Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology 2016, 150, 1646–1658.e1617. [Google Scholar] [CrossRef]
- Leslie, J.; Mackey, J.B.G.; Jamieson, T.; Ramon-Gil, E.; Drake, T.M.; Fercoq, F.; Clark, W.; Gilroy, K.; Hedley, A.; Nixon, C.; et al. CXCR2 inhibition enables NASH-HCC immunotherapy. Gut 2022, 71, 1937–1938. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Tao, Y.; Wu, Y.; Zhao, X.; Ye, W.; Zhao, D.; Fu, L.; Tian, C.; Yang, J.; He, F.; et al. Neutrophils promote the development of reparative macrophages mediated by ROS to orchestrate liver repair. Nat. Commun. 2019, 10, 1076. [Google Scholar] [CrossRef] [PubMed]
- Saijou, E.; Enomoto, Y.; Matsuda, M.; Yuet-Yin Kok, C.; Akira, S.; Tanaka, M.; Miyajima, A. Neutrophils alleviate fibrosis in the CCl4-induced mouse chronic liver injury model. Hepatol. Commun. 2018, 2, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Takano, T.; Chiang, N.; Gronert, K.; Clish, C.B. Formation of Endogenous “Antiinflammatory” Lipid Mediators by Transcellular Biosynthesis. Am. J. Respir. Crit. Care Med. 2000, 161, S95–S101. [Google Scholar] [CrossRef]
- Kaech, S.M.; Cui, W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol. 2012, 12, 749–761. [Google Scholar] [CrossRef]
- Warren, A.; Le Couteur, D.G.; Fraser, R.; Bowen, D.G.; McCaughan, G.W.; Bertolino, P. T lymphocytes interact with hepatocytes through fenestrations in murine liver sinusoidal endothelial cells. Hepatology 2006, 44, 1182–1190. [Google Scholar] [CrossRef]
- Guidotti, L.G.; Inverso, D.; Sironi, L.; Di Lucia, P.; Fioravanti, J.; Ganzer, L.; Fiocchi, A.; Vacca, M.; Aiolfi, R.; Sammicheli, S.; et al. Immunosurveillance of the liver by intravascular effector CD8(+) T cells. Cell 2015, 161, 486–500. [Google Scholar] [CrossRef]
- Andreas Limmer, J.O.; Christian, K.; Hans-Gustaf, L.; Yuval Reiss, M.G.; Frank, M.; Bernd, A.; Percy, A.K. Efficient presentation of exogenous antigen by liver endothelial cells to CD8+ T cells results in antigen-specific T-cell tolerance. Nat. Med. 2000, 6, 1348–1354. [Google Scholar] [CrossRef]
- Klein, I.; Crispe, I.N. Complete differentiation of CD8+ T cells activated locally within the transplanted liver. J. Exp. Med. 2006, 203, 437–447. [Google Scholar] [CrossRef]
- Bertolino, P.; Bowen, D.G.; McCaughan, G.W.; Fazekas de St Groth, B. Antigen-specific primary activation of CD8+ T cells within the liver. J. Immunol. 2001, 166, 5430–5438. [Google Scholar] [CrossRef]
- Holz, L.E.; Benseler, V.; Bowen, D.G.; Bouillet, P.; Strasser, A.; O’Reilly, L.; d’Avigdor, W.M.; Bishop, A.G.; McCaughan, G.W.; Bertolino, P. Intrahepatic murine CD8 T-cell activation associates with a distinct phenotype leading to Bim-dependent death. Gastroenterology 2008, 135, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Benseler, V.; Warren, A.; Vo, M.; Holz, L.E.; Tay, S.S.; Le Couteur, D.G.; Breen, E.; Allison, A.C.; van Rooijen, N.; McGuffog, C.; et al. Hepatocyte entry leads to degradation of autoreactive CD8 T cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16735–16740. [Google Scholar] [CrossRef] [PubMed]
- Benechet, A.P.; De Simone, G.; Di Lucia, P.; Cilenti, F.; Barbiera, G.; Le Bert, N.; Fumagalli, V.; Lusito, E.; Moalli, F.; Bianchessi, V.; et al. Dynamics and genomic landscape of CD8(+) T cells undergoing hepatic priming. Nature 2019, 574, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Steinert, E.M.; Schenkel, J.M.; Fraser, K.A.; Beura, L.K.; Manlove, L.S.; Igyarto, B.Z.; Southern, P.J.; Masopust, D. Quantifying Memory CD8 T Cells Reveals Regionalization of Immunosurveillance. Cell 2015, 161, 737–749. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Burrows, P.D.; Wang, J.-Y. B Cell Development and Maturation. In B Cells in Immunity and Tolerance; Wang, J.-Y., Ed.; Springer: Singapore, 2020; pp. 1–22. [Google Scholar]
- Barrow, F.; Khan, S.; Wang, H.; Revelo, X.S. The Emerging Role of B Cells in the Pathogenesis of NAFLD. Hepatology 2021, 74, 2277–2286. [Google Scholar] [CrossRef]
- Wu, Z.; Xu, J.; Tan, J.; Song, Y.; Liu, L.; Zhang, F.; Zhang, Y.; Li, X.; Chi, Y.; Liu, Y. Mesenteric adipose tissue B lymphocytes promote local and hepatic inflammation in non-alcoholic fatty liver disease mice. J. Cell Mol. Med. 2019, 23, 3375–3385. [Google Scholar] [CrossRef]
- Bruzzi, S.; Sutti, S.; Giudici, G.; Burlone, M.E.; Ramavath, N.N.; Toscani, A.; Bozzola, C.; Schneider, P.; Morello, E.; Parola, M.; et al. B2-Lymphocyte responses to oxidative stress-derived antigens contribute to the evolution of nonalcoholic fatty liver disease (NAFLD). Free Radic. Biol. Med. 2018, 124, 249–259. [Google Scholar] [CrossRef]
- Barrow, F.; Khan, S.; Fredrickson, G.; Wang, H.; Dietsche, K.; Parthiban, P.; Robert, S.; Kaiser, T.; Winer, S.; Herman, A.; et al. Microbiota-Driven Activation of Intrahepatic B Cells Aggravates NASH Through Innate and Adaptive Signaling. Hepatology 2021, 74, 704–722. [Google Scholar] [CrossRef]
- Nakamura, Y.; Abe, M.; Kawasaki, K.; Miyake, T.; Watanabe, T.; Yoshida, O.; Hirooka, M.; Matsuura, B.; Hiasa, Y. Depletion of B cell-activating factor attenuates hepatic fat accumulation in a murine model of nonalcoholic fatty liver disease. Sci. Rep. 2019, 9, 977. [Google Scholar] [CrossRef]
- Spruss, A.; Kanuri, G.; Wagnerberger, S.; Haub, S.; Bischoff, S.C.; Bergheim, I. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology 2009, 50, 1094–1104. [Google Scholar] [CrossRef]
- Tarrats, N.; Moles, A.; Morales, A.; Garcia-Ruiz, C.; Fernandez-Checa, J.C.; Mari, M. Critical role of tumor necrosis factor receptor 1, but not 2, in hepatic stellate cell proliferation, extracellular matrix remodeling, and liver fibrogenesis. Hepatology 2011, 54, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Faggioli, F.; Palagano, E.; Di Tommaso, L.; Donadon, M.; Marrella, V.; Recordati, C.; Mantero, S.; Villa, A.; Vezzoni, P.; Cassani, B. B lymphocytes limit senescence-driven fibrosis resolution and favor hepatocarcinogenesis in mouse liver injury. Hepatology 2018, 67, 1970–1985. [Google Scholar] [CrossRef] [PubMed]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Highton, A.J.; Schuster, I.S.; Degli-Esposti, M.A.; Altfeld, M. The role of natural killer cells in liver inflammation. Semin. Immunopathol. 2021, 43, 519–533. [Google Scholar] [CrossRef] [PubMed]
- Mikulak, J.; Bruni, E.; Oriolo, F.; Di Vito, C.; Mavilio, D. Hepatic Natural Killer Cells: Organ-Specific Sentinels of Liver Immune Homeostasis and Physiopathology. Front. Immunol. 2019, 10, 946. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef]
- Klose, C.S.N.; Flach, M.; Mohle, L.; Rogell, L.; Hoyler, T.; Ebert, K.; Fabiunke, C.; Pfeifer, D.; Sexl, V.; Fonseca-Pereira, D.; et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell 2014, 157, 340–356. [Google Scholar] [CrossRef]
- Fuchs, A.; Vermi, W.; Lee, J.S.; Lonardi, S.; Gilfillan, S.; Newberry, R.D.; Cella, M.; Colonna, M. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-gamma-producing cells. Immunity 2013, 38, 769–781. [Google Scholar] [CrossRef]
- Bernink, J.H.; Peters, C.P.; Munneke, M.; te Velde, A.A.; Meijer, S.L.; Weijer, K.; Hreggvidsdottir, H.S.; Heinsbroek, S.E.; Legrand, N.; Buskens, C.J.; et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol. 2013, 14, 221–229. [Google Scholar] [CrossRef]
- Tu, Z.; Bozorgzadeh, A.; Pierce, R.H.; Kurtis, J.; Crispe, I.N.; Orloff, M.S. TLR-dependent cross talk between human Kupffer cells and NK cells. J. Exp. Med. 2008, 205, 233–244. [Google Scholar] [CrossRef]
- Chong, W.P.; Zhou, J.; Law, H.K.; Tu, W.; Lau, Y.L. Natural killer cells become tolerogenic after interaction with apoptotic cells. Eur. J. Immunol. 2010, 40, 1718–1727. [Google Scholar] [CrossRef] [PubMed]
- Molgora, M.; Bonavita, E.; Ponzetta, A.; Riva, F.; Barbagallo, M.; Jaillon, S.; Popović, B.; Bernardini, G.; Magrini, E.; Gianni, F.; et al. IL-1R8 is a checkpoint in NK cells regulating anti-tumour and anti-viral activity. Nature 2017, 551, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Peng, H.; Li, K.; Qu, K.; Wang, B.; Wu, Y.; Ye, L.; Dong, Z.; Wei, H.; Sun, R.; et al. Liver-Resident NK Cells Control Antiviral Activity of Hepatic T Cells via the PD-1-PD-L1 Axis. Immunity 2019, 50, 403–417.e4. [Google Scholar] [CrossRef]
- Zhao, J.; Li, Y.; Jin, L.; Zhang, S.; Fan, R.; Sun, Y.; Zhou, C.; Shang, Q.; Li, W.; Zhang, Z.; et al. Natural killer cells are characterized by the concomitantly increased interferon-gamma and cytotoxicity in acute resolved hepatitis B patients. PLoS ONE 2012, 7, e49135. [Google Scholar] [CrossRef]
- Hengst, J.; Klein, A.L.; Lunemann, S.; Deterding, K.; Hardtke, S.; Falk, C.S.; Manns, M.P.; Cornberg, M.; Schlaphoff, V.; Wedemeyer, H. Role of soluble inflammatory mediators and different immune cell populations in early control of symptomatic acute hepatitis C virus infection. J. Viral. Hepat. 2019, 26, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Lunemann, S.; Malone, D.F.; Hengst, J.; Port, K.; Grabowski, J.; Deterding, K.; Markova, A.; Bremer, B.; Schlaphoff, V.; Cornberg, M.; et al. Compromised function of natural killer cells in acute and chronic viral hepatitis. J. Infect. Dis. 2014, 209, 1362–1373. [Google Scholar] [CrossRef]
- Oliviero, B.; Varchetta, S.; Paudice, E.; Michelone, G.; Zaramella, M.; Mavilio, D.; De Filippi, F.; Bruno, S.; Mondelli, M.U. Natural killer cell functional dichotomy in chronic hepatitis B and chronic hepatitis C virus infections. Gastroenterology 2009, 137, 1151–1160.e7. [Google Scholar] [CrossRef]
- Diedrich, T.; Kummer, S.; Galante, A.; Drolz, A.; Schlicker, V.; Lohse, A.W.; Kluwe, J.; Eberhard, J.M.; Schulze Zur Wiesch, J. Characterization of the immune cell landscape of patients with NAFLD. PLoS ONE 2020, 15, e0230307. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, Z.; Luan, Y.; Zou, Z.; Sun, Y.; Li, Y.; Jin, L.; Zhou, C.; Fu, J.; Gao, B.; et al. Pathological functions of interleukin-22 in chronic liver inflammation and fibrosis with hepatitis B virus infection by promoting T helper 17 cell recruitment. Hepatology 2014, 59, 1331–1342. [Google Scholar] [CrossRef]
- Wu, Y.; Kuang, D.M.; Pan, W.D.; Wan, Y.L.; Lao, X.M.; Wang, D.; Li, X.F.; Zheng, L. Monocyte/macrophage-elicited natural killer cell dysfunction in hepatocellular carcinoma is mediated by CD48/2B4 interactions. Hepatology 2013, 57, 1107–1116. [Google Scholar] [CrossRef]
- Zecca, A.; Barili, V.; Rizzo, D.; Olivani, A.; Biasini, E.; Laccabue, D.; Dalla Valle, R.; Ferrari, C.; Cariani, E.; Missale, G. Intratumor Regulatory Noncytotoxic NK Cells in Patients with Hepatocellular Carcinoma. Cells 2021, 10, 614. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Xu, J.; Huang, Q.; Huang, M.; Wen, H.; Zhang, C.; Wang, J.; Song, J.; Zheng, M.; Sun, H.; et al. High NKG2A expression contributes to NK cell exhaustion and predicts a poor prognosis of patients with liver cancer. Oncoimmunology 2017, 6, e1264562. [Google Scholar] [CrossRef] [PubMed]
- Chew, V.; Lai, L.; Pan, L.; Lim, C.J.; Li, J.; Ong, R.; Chua, C.; Leong, J.Y.; Lim, K.H.; Toh, H.C.; et al. Delineation of an immunosuppressive gradient in hepatocellular carcinoma using high-dimensional proteomic and transcriptomic analyses. Proc. Natl. Acad. Sci. USA 2017, 114, E5900–E5909. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yang, Y.; Hua, X.; Wang, G.; Liu, W.; Jia, C.; Tai, Y.; Zhang, Q.; Chen, G. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett. 2012, 318, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Langhans, B.; Nischalke, H.D.; Kramer, B.; Dold, L.; Lutz, P.; Mohr, R.; Vogt, A.; Toma, M.; Eis-Hubinger, A.M.; Nattermann, J.; et al. Role of regulatory T cells and checkpoint inhibition in hepatocellular carcinoma. Cancer Immunol. Immunother. 2019, 68, 2055–2066. [Google Scholar] [CrossRef]
- Langhans, B.; Alwan, A.W.; Kramer, B.; Glassner, A.; Lutz, P.; Strassburg, C.P.; Nattermann, J.; Spengler, U. Regulatory CD4+ T cells modulate the interaction between NK cells and hepatic stellate cells by acting on either cell type. J. Hepatol. 2015, 62, 398–404. [Google Scholar] [CrossRef]
- Sun, H.; Huang, Q.; Huang, M.; Wen, H.; Lin, R.; Zheng, M.; Qu, K.; Li, K.; Wei, H.; Xiao, W.; et al. Human CD96 Correlates to Natural Killer Cell Exhaustion and Predicts the Prognosis of Human Hepatocellular Carcinoma. Hepatology 2019, 70, 168–183. [Google Scholar] [CrossRef]
- Terabe, M.; Berzofsky, J.A. Tissue-Specific Roles of NKT Cells in Tumor Immunity. Front. Immunol. 2018, 9, 1838. [Google Scholar] [CrossRef]
- Kitamura, H.; Iwakabe, K.; Yahata, T.; Nishimura, S.-i.; Ohta, A.; Ohmi, Y.; Sato, M.; Takeda, K.; Okumura, K.; Van Kaer, L.; et al. The Natural Killer T (NKT) Cell Ligand α-Galactosylceramide Demonstrates Its Immunopotentiating Effect by Inducing Interleukin (IL)-12 Production by Dendritic Cells and IL-12 Receptor Expression on NKT Cells. J. Exp. Med. 1999, 189, 1121–1128. [Google Scholar] [CrossRef]
- Smyth, M.J.; Wallace, M.E.; Nutt, S.L.; Yagita, H.; Godfrey, D.I.; Hayakawa, Y. Sequential activation of NKT cells and NK cells provides effective innate immunotherapy of cancer. J. Exp. Med. 2005, 201, 1973–1985. [Google Scholar] [CrossRef]
- Kawano, T.; Nakayama, T.; Kamada, N.; Kaneko, Y.; Harada, M.; Ogura, N.; Akutsu, Y.; Motohashi, S.; Iizasa, T.; Endo, H.; et al. Antitumor cytotoxicity mediated by ligand-activated human V alpha24 NKT cells. Cancer Res. 1999, 59, 5102–5105. [Google Scholar] [PubMed]
- Wingender, G.; Krebs, P.; Beutler, B.; Kronenberg, M. Antigen-Specific Cytotoxicity by Invariant NKT Cells In Vivo Is CD95/CD178-Dependent and Is Correlated with Antigenic Potency. J. Immunol. 2010, 185, 2721–2729. [Google Scholar] [CrossRef] [PubMed]
- Crowe, N.Y.; Smyth, M.J.; Godfrey, D.I. A Critical Role for Natural Killer T Cells in Immunosurveillance of Methylcholanthrene-induced Sarcomas. J. Exp. Med. 2002, 196, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Giaccone, G.; Punt, C.J.; Ando, Y.; Ruijter, R.; Nishi, N.; Peters, M.; von Blomberg, B.M.; Scheper, R.J.; van der Vliet, H.J.; van den Eertwegh, A.J.; et al. A phase I study of the natural killer T-cell ligand alpha-galactosylceramide (KRN7000) in patients with solid tumors. Clin. Cancer Res. 2002, 8, 3702–3709. [Google Scholar] [PubMed]
- Dhodapkar, M.V.; Kumar, V. Type II NKT Cells and Their Emerging Role in Health and Disease. J. Immunol. 2017, 198, 1015–1021. [Google Scholar] [CrossRef]
- Terabe, M.; Swann, J.; Ambrosino, E.; Sinha, P.; Takaku, S.; Hayakawa, Y.; Godfrey, D.I.; Ostrand-Rosenberg, S.; Smyth, M.J.; Berzofsky, J.A. A nonclassical non-Vα14Jα18 CD1d-restricted (type II) NKT cell is sufficient for down-regulation of tumor immunosurveillance. J. Exp. Med. 2005, 202, 1627–1633. [Google Scholar] [CrossRef]
- Renukaradhya, G.J.; Khan, M.A.; Vieira, M.; Du, W.; Gervay-Hague, J.; Brutkiewicz, R.R. Type I NKT cells protect (and type II NKT cells suppress) the host’s innate antitumor immune response to a B-cell lymphoma. Blood 2008, 111, 5637–5645. [Google Scholar] [CrossRef]
- Marrero, I.; Maricic, I.; Feldstein, A.E.; Loomba, R.; Schnabl, B.; Rivera-Nieves, J.; Eckmann, L.; Kumar, V. Complex Network of NKT Cell Subsets Controls Immune Homeostasis in Liver and Gut. Front. Immunol. 2018, 9, 2082. [Google Scholar] [CrossRef]
- Maricic, I.; Girardi, E.; Zajonc, D.M.; Kumar, V. Recognition of Lysophosphatidylcholine by Type II NKT Cells and Protection from an Inflammatory Liver Disease. J. Immunol. 2014, 193, 4580–4589. [Google Scholar] [CrossRef]
- Maricic, I.; Sheng, H.; Marrero, I.; Seki, E.; Kisseleva, T.; Chaturvedi, S.; Molle, N.; Mathews, S.A.; Gao, B.; Kumar, V. Inhibition of type I natural killer T cells by retinoids or following sulfatide-mediated activation of type II natural killer T cells attenuates alcoholic liver disease in mice. Hepatology 2015, 61, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Halder, R.C.; Aguilera, C.; Maricic, I.; Kumar, V. Type II NKT cell–mediated anergy induction in type I NKT cells prevents inflammatory liver disease. J. Clin. Investig. 2007, 117, 2302–2312. [Google Scholar] [CrossRef] [PubMed]
- Arrenberg, P.; Maricic, I.; Kumar, V. Sulfatide-Mediated Activation of Type II Natural Killer T Cells Prevents Hepatic Ischemic Reperfusion Injury In Mice. Gastroenterology 2011, 140, 646–655. [Google Scholar] [CrossRef]
- Kumar, V. NKT-cell subsets: Promoters and protectors in inflammatory liver disease. J. Hepatol. 2013, 59, 618–620. [Google Scholar] [CrossRef]
- Mathews, S.; Feng, D.; Maricic, I.; Ju, C.; Kumar, V.; Gao, B. Invariant natural killer T cells contribute to chronic-plus-binge ethanol-mediated liver injury by promoting hepatic neutrophil infiltration. Cell. Mol. Immunol. 2016, 13, 206–216. [Google Scholar] [CrossRef]
- Maricic, I.; Marrero, I.; Eguchi, A.; Nakamura, R.; Johnson, C.D.; Dasgupta, S.; Hernandez, C.D.; Nguyen, P.S.; Swafford, A.D.; Knight, R.; et al. Differential Activation of Hepatic Invariant NKT Cell Subsets Plays a Key Role in Progression of Nonalcoholic Steatohepatitis. J. Immunol. 2018, 201, 3017–3035. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Jeong, W.-I.; Tian, Z. Liver: An organ with predominant innate immunity. Hepatology 2008, 47, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Lombes, A.; Durand, A.; Charvet, C.; Rivière, M.; Bonilla, N.; Auffray, C.; Lucas, B.; Martin, B. Adaptive Immune-like γ/δ T Lymphocytes Share Many Common Features with Their α/β T Cell Counterparts. J. Immunol. 2015, 195, 1449–1458. [Google Scholar] [CrossRef]
- Ribeiro, S.; Ribot, J.; Silva-Santos, B. Five layers of receptor signalling in γδ T cell differentiation and activation. Front. Immunol. 2015, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.; Graffeo, C.S.; Gulati, R.; Jamal, M.; Narayan, S.; Zambirinis, C.P.; Barilla, R.; Deutsch, M.; Greco, S.H.; Ochi, A.; et al. Interleukin 17–Producing γδT Cells Promote Hepatic Regeneration in Mice. Gastroenterology 2014, 147, 473–484.e472. [Google Scholar] [CrossRef]
- Hammerich, L.; Bangen, J.M.; Govaere, O.; Zimmermann, H.W.; Gassler, N.; Huss, S.; Liedtke, C.; Prinz, I.; Lira, S.A.; Luedde, T.; et al. Chemokine receptor CCR6-dependent accumulation of γδ T cells in injured liver restricts hepatic inflammation and fibrosis. Hepatology 2014, 59, 630–642. [Google Scholar] [CrossRef]
- Zhao, N.; Hao, J.; Ni, Y.; Luo, W.; Liang, R.; Cao, G.; Zhao, Y.; Wang, P.; Zhao, L.; Tian, Z.; et al. Vγ4 γδ T Cell-Derived IL-17A Negatively Regulates NKT Cell Function in Con A-Induced Fulminant Hepatitis. J. Immunol. 2011, 187, 5007–5014. [Google Scholar] [CrossRef] [PubMed]
- Seo, W.; Eun, H.S.; Kim, S.Y.; Yi, H.-S.; Lee, Y.-S.; Park, S.-H.; Jang, M.-J.; Jo, E.; Kim, S.C.; Han, Y.-M.; et al. Exosome-mediated activation of toll-like receptor 3 in stellate cells stimulates interleukin-17 production by γδ T cells in liver fibrosis. Hepatology 2016, 64, 616–631. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Wang, K.; Aoyama, T.; Grivennikov, S.I.; Paik, Y.; Scholten, D.; Cong, M.; Iwaisako, K.; Liu, X.; Zhang, M.; et al. Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology 2012, 143, 765–776.e763. [Google Scholar] [CrossRef] [PubMed]
- Harley, I.T.; Stankiewicz, T.E.; Giles, D.A.; Softic, S.; Flick, L.M.; Cappelletti, M.; Sheridan, R.; Xanthakos, S.A.; Steinbrecher, K.A.; Sartor, R.B.; et al. IL-17 signaling accelerates the progression of nonalcoholic fatty liver disease in mice. Hepatology 2014, 59, 1830–1839. [Google Scholar] [CrossRef]
- Torres-Hernandez, A.; Wang, W.; Nikiforov, Y.; Tejada, K.; Torres, L.; Kalabin, A.; Adam, S.; Wu, J.; Lu, L.; Chen, R.; et al. gammadelta T Cells Promote Steatohepatitis by Orchestrating Innate and Adaptive Immune Programming. Hepatology 2020, 71, 477–494. [Google Scholar] [CrossRef]
- Yi, Y.; He, H.-W.; Wang, J.-X.; Cai, X.-Y.; Li, Y.-W.; Zhou, J.; Cheng, Y.-F.; Jin, J.-J.; Fan, J.; Qiu, S.-J. The functional impairment of HCC-infiltrating γδ T cells, partially mediated by regulatory T cells in a TGFβ- and IL-10-dependent manner. J. Hepatol. 2013, 58, 977–983. [Google Scholar] [CrossRef]
- Zakeri, N.; Hall, A.; Swadling, L.; Pallett, L.J.; Schmidt, N.M.; Diniz, M.O.; Kucykowicz, S.; Amin, O.E.; Gander, A.; Pinzani, M.; et al. Characterisation and induction of tissue-resident gamma delta T-cells to target hepatocellular carcinoma. Nat. Commun. 2022, 13, 1372. [Google Scholar] [CrossRef]
- Alberti, K.G.M.M.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.-C.; James, W.P.T.; Loria, C.M.; Smith, S.C. Harmonizing the Metabolic Syndrome. Circulation 2009, 120, 1640–1645. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Angulo, P. Nonalcoholic Fatty Liver Disease. N. Engl. J. Med. 2002, 346, 1221–1231. [Google Scholar] [CrossRef]
- Michelotti, G.A.; Machado, M.V.; Diehl, A.M. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef]
- Yki-Järvinen, H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. 2014, 2, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Cusi, K. Role of Insulin Resistance and Lipotoxicity in Non-Alcoholic Steatohepatitis. Clin. Liver Dis. 2009, 13, 545–563. [Google Scholar] [CrossRef] [PubMed]
- Kirpich, I.A.; Marsano, L.S.; McClain, C.J. Gut–liver axis, nutrition, and non-alcoholic fatty liver disease. Clin. Biochem. 2015, 48, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Pessayre, D.; Fromenty, B. NASH: A mitochondrial disease. J. Hepatol. 2005, 42, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef]
- Bouman, L.; Schlierf, A.; Lutz, A.K.; Shan, J.; Deinlein, A.; Kast, J.; Galehdar, Z.; Palmisano, V.; Patenge, N.; Berg, D.; et al. Parkin is transcriptionally regulated by ATF4: Evidence for an interconnection between mitochondrial stress and ER stress. Cell Death Differ. 2011, 18, 769–782. [Google Scholar] [CrossRef]
- Wei, C.; Yang, X.; Liu, N.; Geng, J.; Tai, Y.; Sun, Z.; Mei, G.; Zhou, P.; Peng, Y.; Wang, C.; et al. Tumor Microenvironment Regulation by the Endoplasmic Reticulum Stress Transmission Mediator Golgi Protein 73 in Mice. Hepatology 2019, 70, 851–870. [Google Scholar] [CrossRef]
- Li, X.; Ramadori, P.; Pfister, D.; Seehawer, M.; Zender, L.; Heikenwalder, M. The immunological and metabolic landscape in primary and metastatic liver cancer. Nat. Rev. Cancer 2021, 21, 541–557. [Google Scholar] [CrossRef]
- Wang, M.; Han, J.; Xing, H.; Zhang, H.; Li, Z.; Liang, L.; Li, C.; Dai, S.; Wu, M.; Shen, F.; et al. Dysregulated fatty acid metabolism in hepatocellular carcinoma. Hepat. Oncol. 2016, 3, 241–251. [Google Scholar] [CrossRef]
- Xia, S.; Pan, Y.; Liang, Y.; Xu, J.; Cai, X. The microenvironmental and metabolic aspects of sorafenib resistance in hepatocellular carcinoma. EBioMedicine 2020, 51, 102610. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Li, J.; Zhang, W.; Xiao, C.; Zhang, S.; Nian, C.; Li, J.; Su, D.; Chen, L.; Zhao, Q.; et al. Glycogen accumulation and phase separation drives liver tumor initiation. Cell 2021, 184, 5559–5576.e5519. [Google Scholar] [CrossRef] [PubMed]
- Li, T.-E.; Wang, S.; Shen, X.-T.; Zhang, Z.; Chen, M.; Wang, H.; Zhu, Y.; Xu, D.; Hu, B.-Y.; Wei, R.; et al. PKM2 Drives Hepatocellular Carcinoma Progression by Inducing Immunosuppressive Microenvironment. Front. Immunol. 2020, 11, 589997. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.P.; Ning, W.R.; Jiang, Z.Z.; Peng, Z.P.; Zhu, L.Y.; Zhuang, S.M.; Kuang, D.M.; Zheng, L.; Wu, Y. Glycolytic activation of peritumoral monocytes fosters immune privilege via the PFKFB3-PD-L1 axis in human hepatocellular carcinoma. J. Hepatol. 2019, 71, 333–343. [Google Scholar] [CrossRef]
- Pacella, I.; Procaccini, C.; Focaccetti, C.; Miacci, S.; Timperi, E.; Faicchia, D.; Severa, M.; Rizzo, F.; Coccia, E.M.; Bonacina, F.; et al. Fatty acid metabolism complements glycolysis in the selective regulatory T cell expansion during tumor growth. Proc. Natl. Acad. Sci. USA 2018, 115, E6546–E6555. [Google Scholar] [CrossRef]
- Masetti, M.; Carriero, R.; Portale, F.; Marelli, G.; Morina, N.; Pandini, M.; Iovino, M.; Partini, B.; Erreni, M.; Ponzetta, A.; et al. Lipid-loaded tumor-associated macrophages sustain tumor growth and invasiveness in prostate cancer. J. Exp. Med. 2022, 219, e20210564. [Google Scholar] [CrossRef]
- Zeyda, M.; Säemann, M.D.; Stuhlmeier, K.M.; Mascher, D.G.; Nowotny, P.N.; Zlabinger, G.J.; Waldhäusl, W.; Stulnig, T.M. Polyunsaturated Fatty Acids Block Dendritic Cell Activation and Function Independently of NF-κB Activation*. J. Biol. Chem. 2005, 280, 14293–14301. [Google Scholar] [CrossRef]
- Shamshiev, A.T.; Ampenberger, F.; Ernst, B.; Rohrer, L.; Marsland, B.J.; Kopf, M. Dyslipidemia inhibits Toll-like receptor–induced activation of CD8α-negative dendritic cells and protective Th1 type immunity. J. Exp. Med. 2007, 204, 441–452. [Google Scholar] [CrossRef]
- Angeli, V.; Llodrá, J.; Rong, J.X.; Satoh, K.; Ishii, S.; Shimizu, T.; Fisher, E.A.; Randolph, G.J. Dyslipidemia Associated with Atherosclerotic Disease Systemically Alters Dendritic Cell Mobilization. Immunity 2004, 21, 561–574. [Google Scholar] [CrossRef]
- Aliberti, J.; Hieny, S.; Reis e Sousa, C.; Serhan, C.N.; Sher, A. Lipoxin-mediated inhibition of IL-12 production by DCs: A mechanism for regulation of microbial immunity. Nat. Immunol. 2002, 3, 76–82. [Google Scholar] [CrossRef]
- Gallage, S.; Garcia-Beccaria, M.; Szydlowska, M.; Rahbari, M.; Mohr, R.; Tacke, F.; Heikenwalder, M. The therapeutic landscape of hepatocellular carcinoma. Med 2021, 2, 505–552. [Google Scholar] [CrossRef] [PubMed]
- Tabrizian, P.; Jibara, G.; Shrager, B.; Schwartz, M.; Roayaie, S. Recurrence of hepatocellular cancer after resection: Patterns, treatments, and prognosis. Ann. Surg. 2015, 261, 947–955. [Google Scholar] [CrossRef]
- Lencioni, R.; Crocetti, L. Local-regional treatment of hepatocellular carcinoma. Radiology 2012, 262, 43–58. [Google Scholar] [CrossRef]
- Nault, J.C.; Sutter, O.; Nahon, P.; Ganne-Carrié, N.; Séror, O. Percutaneous treatment of hepatocellular carcinoma: State of the art and innovations. J. Hepatol. 2018, 68, 783–797. [Google Scholar] [CrossRef]
- Ding, J.; Jing, X.; Liu, J.; Wang, Y.; Wang, F.; Wang, Y.; Du, Z. Comparison of two different thermal techniques for the treatment of hepatocellular carcinoma. Eur. J. Radiol. 2013, 82, 1379–1384. [Google Scholar] [CrossRef]
- Yeung, R.H.; Chapman, T.R.; Bowen, S.R.; Apisarnthanarax, S. Proton beam therapy for hepatocellular carcinoma. Expert. Rev. Anticancer Ther. 2017, 17, 911–924. [Google Scholar] [CrossRef]
- Raoul, J.L.; Adhoute, X.; Penaranda, G.; Perrier, H.; Castellani, P.; Oules, V.; Bourlière, M. Sorafenib: Experience and Better Manage-ment of Side Effects Improve Overall Survival in Hepatocellular Carcinoma Patients: A Real-Life Retrospective Analysis. Liver Cancer 2019, 8, 457–467. [Google Scholar] [CrossRef]
- Strumberg, D.; Richly, H.; Hilger, R.A.; Schleucher, N.; Korfee, S.; Tewes, M.; Faghih, M.; Brendel, E.; Voliotis, D.; Haase, C.G.; et al. Phase I Clinical and Pharmacokinetic Study of the Novel Raf Kinase and Vascular Endothelial Growth Factor Receptor Inhibitor BAY 43-9006 in Patients With Advanced Refractory Solid Tumors. J. Clin. Oncol. 2005, 23, 965–972. [Google Scholar] [CrossRef]
- Lavi, O. Redundancy: A critical obstacle to improving cancer therapy. Cancer Res. 2015, 75, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Tsai, C.J.; Jang, H. Are Parallel Proliferation Pathways Redundant? Trends Biochem. Sci. 2020, 45, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Roderburg, C.; Özdirik, B.; Wree, A.; Demir, M.; Tacke, F. Systemic treatment of hepatocellular carcinoma: From sorafenib to combination therapies. Hepatic Oncol. 2020, 7, HEP20. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H., 3rd; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.; Kang, Y.-K.; Kim, T.-Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; Matilla, A.; et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients With Advanced Hepatocellular Carcinoma Previously Treated With Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.K.; Gane, E.; Assenat, E.; Siebler, J.; Galle, P.R.; Merle, P.; Hourmand, I.O.; Cleverly, A.; Zhao, Y.; Gueorguieva, I.; et al. A Phase 2 Study of Galunisertib (TGF-β1 Receptor Type I Inhibitor) and Sorafenib in Patients With Advanced Hepatocellular Carcinoma. Clin. Transl. Gastroenterol. 2019, 10, e00056. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Kang, Y.K.; Yen, C.J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Yau, T.; Park, J.W.; Finn, R.S.; Cheng, A.L.; Mathurin, P.; Edeline, J.; Kudo, M.; Harding, J.J.; Merle, P.; Rosmorduc, O.; et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): A randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2022, 23, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Li, Y.; Feng, J.; Li, J.; Ji, J.; Wu, L.; Yu, Q.; Dai, W.; Wu, J.; Zhou, Y.; et al. Cellular based immunotherapy for primary liver cancer. J. Exp. Clin. Cancer Res. 2021, 40, 250. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Qin, L.X. Strategies for improving the efficacy of immunotherapy in hepatocellular carcinoma. Hepatobiliary Pancreat Dis. Int. 2022, 21, 420–429. [Google Scholar] [CrossRef]


{kind=link}
| Category | Clinical Trial Summary | Conditions | Interventions | URL/Identifier Accessed on 10 October 2022 |
|---|---|---|---|---|
| Monoclonal Antibodies | Durvalumab and Tremelimumab in Resectable HCC | Hepatocellular Carcinoma | Drug: Tremelimumab | https://ClinicalTrials.gov/show/NCT05440864 |
| A Study to Evaluate Tislelizumab Combined With Sitravatinib as Adjuvant Therapy in Participants With HCC at High Risk of Recurrence After Curative Resection | Hepatocellular Carcinoma | Drug: Tislelizumab + Sitravatinib | https://ClinicalTrials.gov/show/NCT05407519 | |
| Tislelizumab in the Systematic Treatment of Advanced Hepatocellular Carcinoma | Hepatocellular Carcinoma | Drug: Tislelizumab | https://ClinicalTrials.gov/show/NCT04996459 | |
| Camrelizumab in Patients With Unresectable Hepatocellular Carcinoma | Unresectable Hepatocellular Carcinoma | Drug: Camrelizumab | https://ClinicalTrials.gov/show/NCT04947956 | |
| A Trial of Hepatic Arterial Infusion Combined With Apatinib and Camrelizumab Versus Apatinib and Camrelizumab for C-Staged Hepatocellular Carcinoma in BCLC Classification | C-staged Hepatocellular Carcinoma in BCLC Classification | Combination Product: Hepatic Arterial Infusion combined with Apatinib and Camrelizumab|Combination Product: Apatinib combined with Camrelizumab | https://ClinicalTrials.gov/show/NCT05313282 | |
| Trial of Atezolizumab and Bevacizumab With SRF388 or Placebo in Patients With Hepatocellular Carcinoma | Hepatocellular Carcinoma | Drug: SRF388|Drug: Atezolizumab|Drug: Bevacizumab|Drug: Placebo | https://ClinicalTrials.gov/show/NCT05359861 | |
| Adjuvant Immunotherapy With Toripalimab Following Curative-Intent Ablation for Recurrent Hepatocarcinoma | Hepatocellular Carcinoma | Drug: Toripalimab|Procedure: Thermal ablation | https://ClinicalTrials.gov/show/NCT05240404 | |
| Camrelizumab Combined With Apatinib Mesylate for Perioperative Treatment of Resectable Hepatocellular Carcinoma | Hepatocellular Carcinoma|Immunotherapy|Molecular Targeted Therapy | Drug: Camrelizumab|Drug: Apatinib Mesylate|Procedure: TACE treatment|Procedure: Radical surgery | https://ClinicalTrials.gov/show/NCT04521153 | |
| Anti-CTLA4-NF mAb (BMS986218), Nivolumab, and Stereotactic Body Radiation Therapy for the Treatment of Metastatic Solid Malignancies | Advanced Lung and Liver cancer | Biological: Anti-CTLA4 Monoclonal Antibody BMS-986218|Biological: Nivolumab|Radiation: Stereotactic Body Radiation Therapy | https://ClinicalTrials.gov/show/NCT04785287 | |
| A Study to Determine Whether Chemotherapy, Bevazicumab, and Atezolizumab is Better Than Chemotherapy Alone in Patients With Advanced Liver Cancer | Combined Hepatocellular Carcinoma and Cholangiocarcinoma|Stage III Liver Cancer|Stage IV Liver Cancer | Biological: Atezolizumab|Biological: Bevacizumab|Procedure: Biospecimen Collection|Drug: Cisplatin|Procedure: Computed Tomography|Procedure: Conventional Magnetic Resonance Imaging|Drug: Gemcitabine Hydrochloride | https://ClinicalTrials.gov/show/NCT05211323 | |
| Feasibility and Efficacy of Perioperative Nivolumab With or Without Relatlimab for Patients With Potentially Resectable Hepatocellular Carcinoma (HCC) | Hepatocellular Carcinoma | Drug: Nivolumab|Drug: Relatlimab | https://ClinicalTrials.gov/show/NCT04658147 | |
| Camrelizumab, Apatinib Plus HAIC Versus Camrelizumab and Apatinib for HCC With Portal Vein Invasion: a Randomized Trial | Hepatocellular Carcinoma | Procedure: FOLFOX-HAIC|Drug: Camrelizumab|Drug: Apatinib | https://ClinicalTrials.gov/show/NCT05198609 | |
| A Phase II Study of Nivolumab + Ipilimumab in Advanced HCC Patients Who Have Progressed on First Line Atezolizumab + Bevacizumab | Advanced Hepatocellular Carcinoma |Unresectable Hepatocellular Carcinoma | Biological: Ipilimumab|Biological: Nivolumab | https://ClinicalTrials.gov/show/NCT05199285 | |
| Futibatinib and Pembrolizumab for the Treatment of Advanced or Metastatic FGF19 Positive BCLC Stage A, B, or C Liver Cancer | Advanced Hepatocellular Carcinoma | Drug: Futibatinib|Biological: Pembrolizumab|Other: Quality-of-Life Assessment | https://ClinicalTrials.gov/show/NCT04828486 | |
| Neoadjuvant Regorafenib Plus Durvalumab (MEDI4736) in Patients With High-Risk Hepatocellular Carcinoma | Stage IB Hepatocellular Carcinoma AJCC v8|Stage II Hepatocellular Carcinoma AJCC v8|Stage IIIA Hepatocellular Carcinoma AJCC v8 | Biological: Durvalumab|Drug: Regorafenib | https://ClinicalTrials.gov/show/NCT05194293 | |
| Durvalumab (MEDI4736) and Tremelimumab in Combination With Either Y-90 SIRT or TACE for Intermediate Stage HCC With Pick-the-winner Design | Hepatocellular Carcinoma Non-resectable | Drug: Tremelimumab|Drug: Durvalumab|Procedure: Y-90 SIRT|Procedure: TACE | https://ClinicalTrials.gov/show/NCT04522544 | |
| Atezolizumab in Combination With a Multi-Kinase Inhibitor for the Treatment of Unresectable, Locally Advanced, or Metastatic Liver Cancer | Advanced Hepatocellular Carcinoma|Metastatic Hepatocellular Carcinoma| | Biological: Atezolizumab|Drug: Cabozantinib|Drug: Lenvatinib | https://ClinicalTrials.gov/show/NCT05168163 | |
| HAIC Combined With Camrelizumab and TKI for Unresectable Hepatocellular Carcinoma After TACE Failure | Unresectable Hepatocellular Carcinoma | Drug: Camrelizumab|Drug: HAIC|Drug: TKI | https://ClinicalTrials.gov/show/NCT05135364 | |
| Hyperbaric Oxygen Therapy Combined Camrelizumab in Patients With Advanced/Metastatic Hepatocellular Carcinoma | Combinational Immunotherapy|Hepatocellular Carcinoma Non-Resectable|Hyperbaric Oxygen Therapy | Combination Product: Hyperbaric Oxygen Therapy plus Camrelizumab | https://ClinicalTrials.gov/show/NCT05031949 | |
| Nivolumab and ADI-PEG 20 Before Surgery for the Treatment of Resectable Liver Cancer | Resectable Hepatocellular Carcinoma | Biological: Nivolumab|Biological: Pegargiminase|Procedure: Resection | https://ClinicalTrials.gov/show/NCT04965714 | |
| TSR-022 (Anti-TIM-3 Antibody) and TSR-042 (Anti-PD-1 Antibody) in Patients With Liver Cancer | Adult Primary Liver Cancer|Advanced Adult Primary Liver Cancer|Localized Unresectable Adult Primary Liver Cancer | Drug: TSR-022 and TSR-042 | https://ClinicalTrials.gov/show/NCT03680508 | |
| Pembrolizumab With or Without Elbasvir/Grazoprevir and Ribavirin in Treating Patients With Advanced Refractory Liver Cancer | Hepatocellular Carcinoma|Hepatitis C Infection|Refractory Liver Carcinoma | Drug: Elbasvir/Grazoprevir|Other: Laboratory Biomarker Analysis|Biological: Pembrolizumab|Drug: Ribavirin | https://ClinicalTrials.gov/show/NCT02940496 | |
| Nivolumab With or Without Ipilimumab in Treating Patients With Resectable Liver Cancer | Hepatocellular Carcinoma|Resectable Hepatocellular Carcinoma | Biological: Ipilimumab|Biological: Nivolumab | https://ClinicalTrials.gov/show/NCT03222076 | |
| Tremelimumab With Chemoembolization or Ablation for Liver Cancer | Heptocellular Cancer|Biliary Tract Neoplasms|Liver Cancer|Hepatocellular Carcinoma|Biliary Cancer | Drug: Tremelimumab|Procedure: RFA|Procedure: TACE|Procedure: Cryoablation | https://ClinicalTrials.gov/show/NCT01853618 | |
| Lenvatinib Combined Anti-PD1 Antibody for the Advanced Hepatocellular Carcinoma | Hepatocellular Carcinoma|Anti-PD1 Antibody|Liver Diseases | Drug: Lenvatinib|Drug: Opdivo|Drug: Camrelizumab|Drug: Keytruda|Drug: Toripalimab|Drug: Sintilimab|Drug: Tislelizumab | https://ClinicalTrials.gov/show/NCT04627012 | |
| Study of Safety and Tolerability of PDR001 in Combination With Sorafenib and to Identify the Maximum Tolerated Dose and/or Phase 2 Dose for This Combination in Advanced Hepatocellular Patients | Hepatocellular Carcinoma | Drug: PDR001|Drug: Sorafenib | https://ClinicalTrials.gov/show/NCT02988440 | |
| Drugs/molecules/particles | Yttrium Y 90 Glass Microspheres, Atezolizumab, and Cabozantinib for the Treatment of Unresectable or Locally Advanced Hepatocellular Carcinoma | Locally Advanced Hepatocellular Carcinoma|Recurrent Hepatocellular Carcinoma|Unresectable Hepatocellular Carcinoma | Biological: Atezolizumab|Procedure: Biopsy|Drug: Cabozantinib S-malate|Radiation: Yttrium Y 90 Glass Microspheres | https://ClinicalTrials.gov/show/NCT05327738 |
| TheraSphere With and Without Durvalumab and Tremelimumab for HCC | Hepatocellular Carcinoma | Device: TheraSphere Y-90 Glass Microsphere Therapy|Drug: Durvalumab (Imfinzi) Immunotherapy|Drug: Tremelimumab Immunotherapy | https://ClinicalTrials.gov/show/NCT05063565 | |
| GEN2 Directed Cancer Immunotherapy Trial | Hepatocellular Carcinoma|Metastatic Cancer | Drug: GEN2 (HSV-Thymidine Kinase-m2 and hGM-CSF Genes) | https://ClinicalTrials.gov/show/NCT04313868 | |
| IRX-2, Cyclophosphamide, and Nivolumab in Treating Patients With Recurrent or Metastatic and Refractory Liver Cancer | Recurrent Hepatocellular Carcinoma|Refractory Liver Carcinoma | Drug: Cyclophosphamide|Biological: Cytokine-based Biologic Agent IRX-2|Biological: Nivolumab | https://ClinicalTrials.gov/show/NCT03655002 | |
| BO-112 and Pembrolizumab for the Treatment of PD-1/PD-L1 Refractory Liver Cancer | Advanced Hepatocellular Carcinoma |Refractory Hepatocellular Carcinoma | Biological: Nanoplexed Poly I:C BO-112|Biological: Pembrolizumab | https://ClinicalTrials.gov/show/NCT04777708 | |
| NBTXR3, Radiation Therapy, Ipilimumab, and Nivolumab for the Treatment of Lung and/or Liver Metastases From Solid Malignancy | Metastatic Malignant Neoplasm in the Liver|Metastatic Malignant Neoplasm in the Lung | Other: Hafnium Oxide-Containing Nanoparticles NBTXR3|Biological: Ipilimumab|Biological: Nivolumab|Radiation: Radiation Therapy | https://ClinicalTrials.gov/show/NCT05039632 | |
| Study Of OX40 Agonist PF-04518600 Alone And In Combination With 4-1BB Agonist PF-05082566 | Neoplasms | Drug: PF-04518600|Drug: PF-04518600 plus PF-05082566 | Completed | |
| Doxorubicin and Interleukin-2 in Treating Patients With Liver Cancer That Cannot Be Removed by Surgery | Liver Cancer | Biological: Aldesleukin|Drug: Doxorubicin Hydrochloride | https://ClinicalTrials.gov/show/NCT00004248 | |
| Study Of OX40 Agonist PF-04518600 Alone And In Combination With 4-1BB Agonist PF-05082566 | Neoplasms | Drug: PF-04518600|Drug: PF-04518600 plus PF-05082566 | https://ClinicalTrials.gov/show/NCT02315066 | |
| TACE-HAIC Combined With TKIs and Immunotherapy Versus TACE Alone for Hepatocellular Carcinoma With PVTT | Hepatocellular Carcinoma | Procedure: TACE-HAIC|Procedure: TACE|Drug: Targeted Therapy|Drug: PD-1 Inhibitors | https://ClinicalTrials.gov/show/NCT05535998 | |
| Safety and Immune Response to a Multi-component Immune Based Therapy (MKC1106-PP) for Patients With Advanced Cancer | Solid cancers | Biological: PSMA/PRAME | https://ClinicalTrials.gov/show/NCT00423254 | |
| Vaccines | Neoantigen Dendritic Cell Vaccine and Nivolumab in HCC and Liver Metastases From CRC | Hepatocellular Carcinoma|Hepatocellular Cancer|Colorectal Cancer|Colorectal Carcinoma|Liver Metastases | Biological: Neoantigen Dendritic Cell Vaccine|Drug: Nivolumab | https://ClinicalTrials.gov/show/NCT04912765 |
| Vaccine Therapy in Treating Patients With Liver or Lung Metastases From Colorectal Cancer | Colorectal Cancer|Metastatic Cancer | Biological: Falimarev|Biological: Inalimarev|Biological: Sargramostim|Biological: Therapeutic Autologous Dendritic Cells | https://ClinicalTrials.gov/show/NCT00103142 | |
| GP96 Heat Shock Protein-Peptide Complex Vaccine in Treating Patients With Liver Cancer | Liver Cancer | Biological: gp96 | https://ClinicalTrials.gov/show/NCT04206254 | |
| DNAJB1-PRKACA Fusion Kinase Peptide Vaccine Combined With Nivolumab and Ipilimumab for Patients With Fibrolamellar Hepatocellular Carcinoma | Fibrolamellar Hepatocellular Carcinoma (FLC) | Drug: DNAJB1-PRKACA Peptide Vaccine|Drug: Nivolumab|Drug: Ipilimumab | https://ClinicalTrials.gov/show/NCT04248569 | |
| Safety and Efficacy Study of Mix Vaccine in Hepatocyte Carcinoma Patient | Liver Neoplasms | Biological: MV|Other: Standard Treatment | https://ClinicalTrials.gov/show/NCT02338778 | |
| Vaccine Therapy in Treating Patients With Advanced or Metastatic Cancer | Breast Cancer|Colorectal Cancer|Gallbladder Cancer|Gastric Cancer|Head and Neck Cancer|Liver Cancer|Ovarian Cancer|Pancreatic Cancer|Testicular Germ Cell Tumor | Biological: TRICOM-CEA(6D) | https://ClinicalTrials.gov/show/NCT00027534 | |
| Vaccine Therapy and Radiation to Liver Metastasis in Patients With CEA-Positive Solid Tumors | Liver Neoplasms | Drug: rV-CEA(6D)/TRICOM-rF-CEA(6D)/TRICOM|Drug: rF-CEA(6D)/TRICOM|Drug: Recombinant Fowlpox-GM-CSF|Drug: Celecoxib | https://ClinicalTrials.gov/show/NCT00081848 | |
| Vaccine Therapy in Treating Patients With Liver or Lung Metastases From Colorectal Cancer | Colorectal Cancer|Metastatic Cancer | Biological: Falimarev|Biological: Inalimarev|Biological: Sargramostim|Biological: Therapeutic Autologous Dendritic Cells | https://ClinicalTrials.gov/show/NCT00103142 | |
| Immunotherapy in Treating Patients With Resected Liver Metastases From Colon Cancer | Colorectal Cancer|Metastatic Cancer | Biological: Carcinoembryonic Antigen RNA-Pulsed DC Cancer Vaccine | https://ClinicalTrials.gov/show/NCT00003433 | |
| Vaccine Therapy With or Without Sirolimus in Treating Patients With NY-ESO-1 Expressing Solid Tumors | Solid neoplasms | Biological: DEC-205/NY-ESO-1 Fusion Protein CDX-1401|Other: Laboratory Biomarker Analysis|Other: Pharmacological Study|Drug: Sirolimus | https://ClinicalTrials.gov/show/NCT01522820 | |
| CAR T cells | Anti-CEA CAR-T Cells to Treat Colorectal Liver Metastases | Colorectal Cancer|Metastatic Liver Cancer | Drug: Anti-CEA CAR-T Cells | https://ClinicalTrials.gov/show/NCT05240950 |
| Interleukin-15 Armored Glypican 3-specific Chimeric Antigen Receptor Expressed in Autologous T Cells for Hepatocellular Carcinoma | Liver Cell Carcinoma | Genetic: CATCH T Cells|Drug: Cytoxan|Drug: Fludara | https://ClinicalTrials.gov/show/NCT05103631 | |
| Interleukin-15 and -21 Armored Glypican-3-specific Chimeric Antigen Receptor Expressed in T Cells for Pediatric Solid Tumors | Liver Cancer|Rhabdomyosarcoma|Malignant Rhabdoid Tumor|Liposarcoma|Wilms Tumor|Yolk Sac Tumor | Genetic: CARE T Cells|Drug: Cytoxan|Drug: Fludara | https://ClinicalTrials.gov/show/NCT04715191 | |
| ECT204 T-Cell Therapy in Adults With Advanced HCC | Hepatocellular Carcinoma|Liver Cancer, Adult|Liver Neoplasm|Metastatic Liver Cancer | Biological: ECT204 T Cells | https://ClinicalTrials.gov/show/NCT04864054 | |
| GPC3 Targeted CAR-T Cell Therapy in Advanced GPC3 Expressing Hepatocellular Carcinoma (HCC) | Hepatocellular Carcinoma|Hepatocellular Cancer|Metastatic Hepatocellular Carcinoma | Drug: Cyclophosphamide|Biological: CAR-T Cells|Drug: Fludarabine | https://ClinicalTrials.gov/show/NCT05003895 | |
| Novel GPC3 CAR-T Cell Therapy for Hepatocellular Carcinoma | Hepatocellular Carcinoma | Biological: GPC3-CAR-T Cells | https://ClinicalTrials.gov/show/NCT05344664 | |
| GPC3-Targeted CAR-T Cell for Treating GPC3 Positive Advanced HCC | Hepatocellular Carcinoma | Biological: CAR-T Cell Immunotherapy | https://ClinicalTrials.gov/show/NCT04121273 | |
| Study of ET140203 T Cells in Adults With Advanced Hepatocellular Carcinoma (ARYA-1) | Hepatocellular Carcinoma|Liver Cancer|Liver Neoplasm|Metastatic Liver Cancer | Biological: ET140203 Autologous T Cell Product | https://ClinicalTrials.gov/show/NCT04502082 | |
| Interleukin-15 Armored Glypican 3-specific Chimeric Antigen Receptor Expressed in T Cells for Pediatric Solid Tumors | Liver Cancer|Rhabdomyosarcoma|Malignant Rhabdoid Tumor|Liposarcoma|Wilms Tumor|Yolk Sac Tumor | Genetic: AGAR T Cells|Drug: Cytoxan|Drug: Fludara | https://ClinicalTrials.gov/show/NCT04377932 | |
| GPC3-CAR-T Cells for Immunotherapy of Cancer With GPC3 Expression | Hepatocellular Carcinoma|Immunotherapy|CAR|GPC3 Gene Inactivation|T Cell|Squamous Cell Lung Cancer | Biological: GPC3 and/or TGFβ Targeting CAR-T Cells | https://ClinicalTrials.gov/show/NCT03198546 | |
| Glypican 3-Specific Chimeric Antigen Receptor Expressed in T Cells for Patients With Pediatric Solid Tumors (GAP) | Liver Cancer | Genetic: GAP T Cells|Drug: Cytoxan|Drug: Fludara | https://ClinicalTrials.gov/show/NCT02932956 | |
| Glypican 3-Specific Chimeric Antigen Receptor Expressing T Cells for Hepatocellular Carcinoma (GLYCAR) | Hepatocellular Carcinoma | Genetic: GLYCAR T Cells|Drug: Cytoxan|Drug: Fludarabine | https://ClinicalTrials.gov/show/NCT02905188 | |
| AFPc332T in Advanced HCC | Hepatocellular Cancer|AFP Expressing Tumors | Genetic: Autologous Genetically Modified AFPc332T Cells | https://ClinicalTrials.gov/show/NCT03132792 | |
| T Cell Receptor Immunotherapy Targeting NY-ESO-1 for Patients With NY-ESO-1 Expressing Cancer | Melanoma|Meningioma|Breast Cancer|Non-Small-Cell Lung Cancer|Hepatocellular Cancer | Biological: Anti-NY ESO-1 mTCR PBL|Drug: Cyclophosphamide|Drug: Fludarabine|Drug: Aldesleukin | https://ClinicalTrials.gov/show/NCT01967823 | |
| CEA-Expressing Liver Metastases Safety Study of Intrahepatic Infusions of Anti-CEA Designer T Cells | Liver Metastases | Biological: Anti-CEA 2nd-Generation Designer T Cells | https://ClinicalTrials.gov/show/NCT01373047 | |
| CAR-T Hepatic Artery Infusions and Sir-Spheres for Liver Metastases | Liver Metastases | Biological: Anti-CEA CAR-T Cells|Device: Sir-Spheres | https://ClinicalTrials.gov/show/NCT02416466 | |
| CAR-T Hepatic Artery Infusions or Pancreatic Venous Infusions for CEA-Expressing Liver Metastases or Pancreas Cancer | Liver Metastases | Biological: Anti-CEA CAR-T Cells | https://ClinicalTrials.gov/show/NCT02850536 | |
| Other Cell-based | Combination of Cryosurgery and NK Immunotherapy for Tumors in Transplanted Liver | Liver Tumor|Evidence of Liver Transplantation | Device: Cryosurgery|Biological: NK Immunotherapy | https://ClinicalTrials.gov/show/NCT02849015 |
| Combination of Irreversible Electroporation and NK Immunotherapy for Recurrent Liver Cancer | Recurrent Liver Carcinoma | Device: Irreversible Electroporation|Biological: Natural Killer | https://ClinicalTrials.gov/show/NCT03008343 | |
| Safety Study of Liver Natural Killer Cell Therapy for Hepatoma Liver Transplantation | Liver Cirrhosis|Hepatocellular Carcinoma|Evidence of Liver Transplantation | Biological: Liver NK Cell Inoculation | https://ClinicalTrials.gov/show/NCT01147380 | |
| Safety and Efficiency of γδ T Cell Against Liver Cancer | Liver Cancer | Procedure: Cryosurgery or IRE Surgery|Biological: γδ T cell|Other: γδ T cells/ A Cryosurgery or IRE | https://ClinicalTrials.gov/show/NCT03183219 | |
| Safety and Efficiency of γδ T Cell Against Hepatocellular Liver Cancer | Liver Cancer | Biological: DC-CIK Cells|Biological: γδ T Cells|Biological: γδ T/DC-CIK Cells | https://ClinicalTrials.gov/show/NCT02425735 | |
| A Study of DC-CIK Immunotherapy in the Treatment of Solid Tumors | Liver Cancer|Kidney Cancer|Nasopharyngeal Cancer|Lung Cancer|Colorectal Cancer|Breast Cancer | Other: CELL | https://ClinicalTrials.gov/show/NCT04476641 | |
| Safety and Efficacy of “Immuncell-LC” in TACE Therapy | Carcinoma, Hepatocellular | Biological: Immuncell-LC | https://ClinicalTrials.gov/show/NCT02856815 | |
| RFA or Surgical Resection Combined With Neo-MASCT for Primary HCC: a Phase II Trial | Primary Liver Cancer|Radiofrequency Ablation|Immunotherapy|Hepatectomy | Biological: Neo-MASCT | https://ClinicalTrials.gov/show/NCT03067493 | |
| Biological Therapy in Treating Patients With Metastatic Cancer | Solid Neoplasms | Biological: CEA RNA-pulsed DC Cancer Vaccine | https://ClinicalTrials.gov/show/NCT00004604 | |
| Efficacy and Safety of Immuncell-LC Group and Non-Treatment Group in Hepatocellular Carcinoma Patients | Hepatocellular Carcinoma | Biological: Immuncell-LC | https://ClinicalTrials.gov/show/NCT00699816 | |
| CIK Treatment for HCC Patient Underwent Radical Resection | Carcinoma, Hepatocellular | Biological: Cytokine-Induced Killer Cells | https://ClinicalTrials.gov/show/NCT01749865 | |
| Autologous Immune Cell Therapy in Primary Hepatocellular Carcinoma Patients Following Resection and TACE Therapy | Primary Hepatocellular Carcinoma | Biological: DC-TC+GM-CSF | https://ClinicalTrials.gov/show/NCT01828762 | |
| Combination Therapy of Microwave Ablation and Cellular Immunotherapy for Hepatocellular Carcinoma | Hepatocellular Carcinoma | Biological: Adoptive Immunotherapy|Procedure: MWA | https://ClinicalTrials.gov/show/NCT02851784 | |
| Combine TACE and Autologous Tcm Immunotherapy Versus TACE Alone for HCC With MVI After Radical Resection | Hepatocellular Carcinoma|Malignant Neoplasm | Combination Product: TACE plus Autologous Tcm Immunotherapy|Procedure: TACE | https://ClinicalTrials.gov/show/NCT03575806 | |
| Oncolytic viruses | Study of Talimogene Laherparepvec With Atezolizumab for Triple Negative Breast Cancer and Colorectal Cancer With Liver Metastases | Metastatic Triple-Negative Breast Cancer|Metastatic Colorectal Cancer | Biological: Talimogene Laherparepvec|Biological: Atezolizumab | https://ClinicalTrials.gov/show/NCT03256344 |
| Hepatocellular Carcinoma Study Comparing Vaccinia Virus Based Immunotherapy Plus Sorafenib vs. Sorafenib Alone | Hepatocellular Carcinoma (HCC) | Biological: Pexastimogene Devacirepvec (Pexa Vec)|Drug: Sorafenib | https://ClinicalTrials.gov/show/NCT02562755 | |
| Study of Talimogene Laherparepvec With Atezolizumab for Triple Negative Breast Cancer and Colorectal Cancer With Liver Metastases | Metastatic Triple-Negative Breast Cancer|Metastatic Colorectal Cancer | Biological: Talimogene Laherparepvec|Biological: Atezolizumab | https://ClinicalTrials.gov/show/NCT03256344 | |
| Alternative approaches | Huaier Granule for Prevention of Recurrence and Metastasis of Hepatocarcinoma After Radical Hepatectomy | Hepatic Carcinoma | Drug: Huaier Granule | https://ClinicalTrials.gov/show/NCT01770431 |
| Prediction/biomarkers | Predicting Response to Systemic Therapies for Hepatocellular Carcinoma (HCC) | Hepatocellular Carcinoma Non-Resectable|Effect of Drug | Diagnostic Test: Radiological Evaluation | https://ClinicalTrials.gov/show/NCT05543304 |
| Evaluation of Treatment Predictors Reflecting Beta-catenin Activation in Hepatocellular Carcinoma | Hepatocellular Carcinoma Non-resectable | Combination Product: Fluorine-18 Fluorocholine | https://ClinicalTrials.gov/show/NCT04965454 | |
| Immune Cells as a New Biomarker of Response in Patients Treated by Immunotherapy for Advanced Hepatocellular Carcinoma | Hepatocellular Carcinoma | Other: Patients with Hepatocellular Carcinoma | https://ClinicalTrials.gov/show/NCT05044676 | |
| Liquid Biopsy in Hepatocellular Carcinoma | Hepatic Carcinoma Malignant Primary Non-Resectable | Diagnostic Test: Cell-Free DNA | https://ClinicalTrials.gov/show/NCT04111029 | |
| Identification of Image Phenotypes to Predict Recurrence After Resection of Hepatocellular Carcinoma | CT Scans Prior to Surgery With a Least 2 Years of Follow-up | Other: Non-Intervention | https://ClinicalTrials.gov/show/NCT05235490 | |
| Analysis of Expression of Specific Markers and Their Prognostic Significance in Hepatocellular Carcinoma | Hepatocellular Carcinoma | Other: Retrospective Analysis of Already Archived Samples | https://ClinicalTrials.gov/show/NCT00911196 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Volponi, C.; Gazzillo, A.; Bonavita, E. The Tumor Microenvironment of Hepatocellular Carcinoma: Untying an Intricate Immunological Network. Cancers 2022, 14, 6151. https://doi.org/10.3390/cancers14246151
Volponi C, Gazzillo A, Bonavita E. The Tumor Microenvironment of Hepatocellular Carcinoma: Untying an Intricate Immunological Network. Cancers. 2022; 14(24):6151. https://doi.org/10.3390/cancers14246151
Chicago/Turabian StyleVolponi, Camilla, Aurora Gazzillo, and Eduardo Bonavita. 2022. "The Tumor Microenvironment of Hepatocellular Carcinoma: Untying an Intricate Immunological Network" Cancers 14, no. 24: 6151. https://doi.org/10.3390/cancers14246151
APA StyleVolponi, C., Gazzillo, A., & Bonavita, E. (2022). The Tumor Microenvironment of Hepatocellular Carcinoma: Untying an Intricate Immunological Network. Cancers, 14(24), 6151. https://doi.org/10.3390/cancers14246151