

Proteome-Wide Identification of RNA-Dependent Proteins in Lung Cancer Cells
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Cell Culture
2.2. Cell Harvest
2.3. Lysate Preparation
2.4. RNase Treatment
2.5. Ultracentrifugation and Fractionation
2.6. SDS-PAGE and Western Blot Analysis
2.7. Quantitative Analysis of the Western Blot Images
2.8. Individual Nucleotide Resolution and UV Crosslinked Immunoprecipitation (iCLIP2)
2.9. Sample Preparation for Mass Spectrometry
3. Results
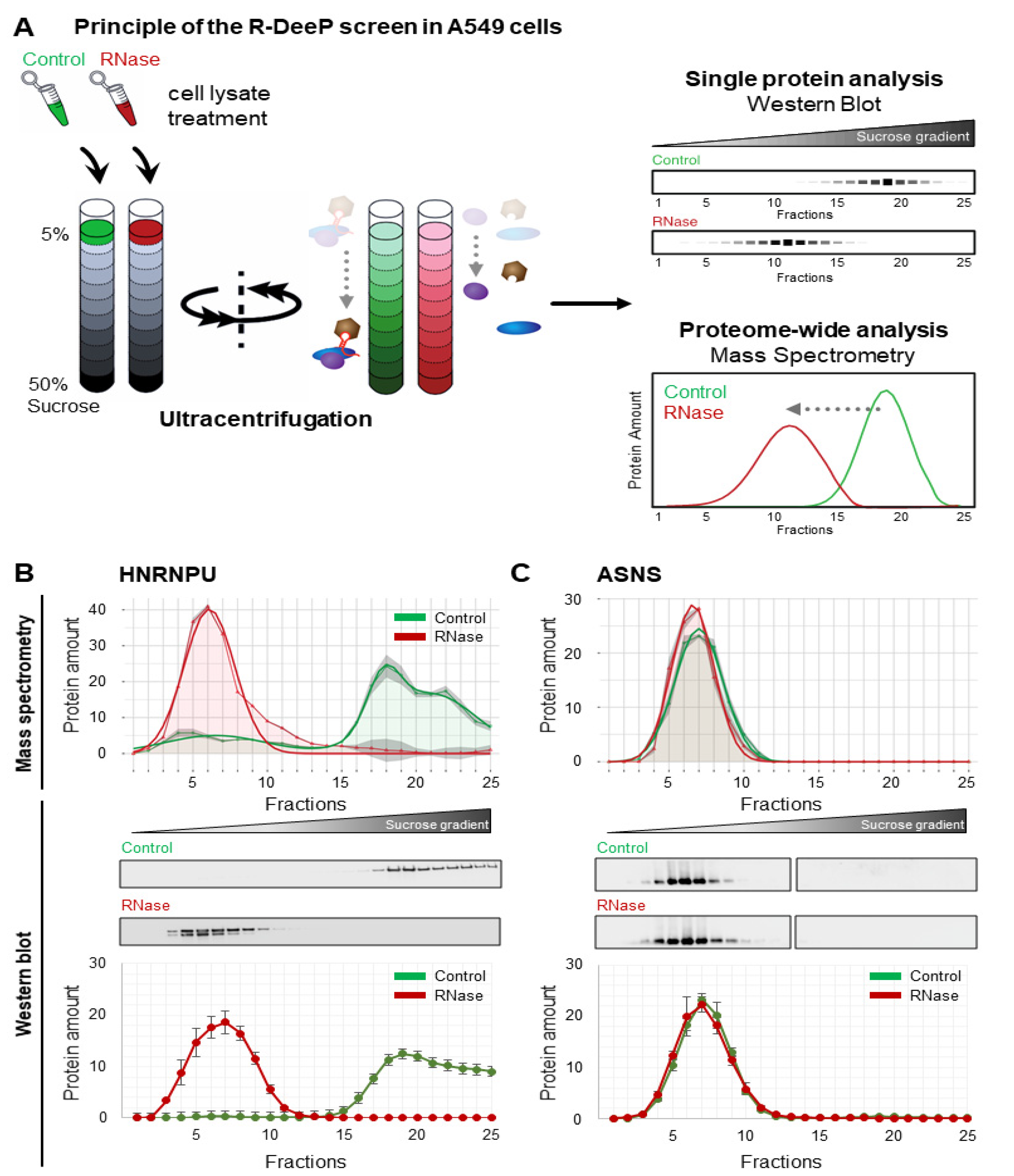
3.1. Proteome-Wide Identification of RNA-Dependent Proteins in Lung Cancer Cells Using the R-DeeP Approach
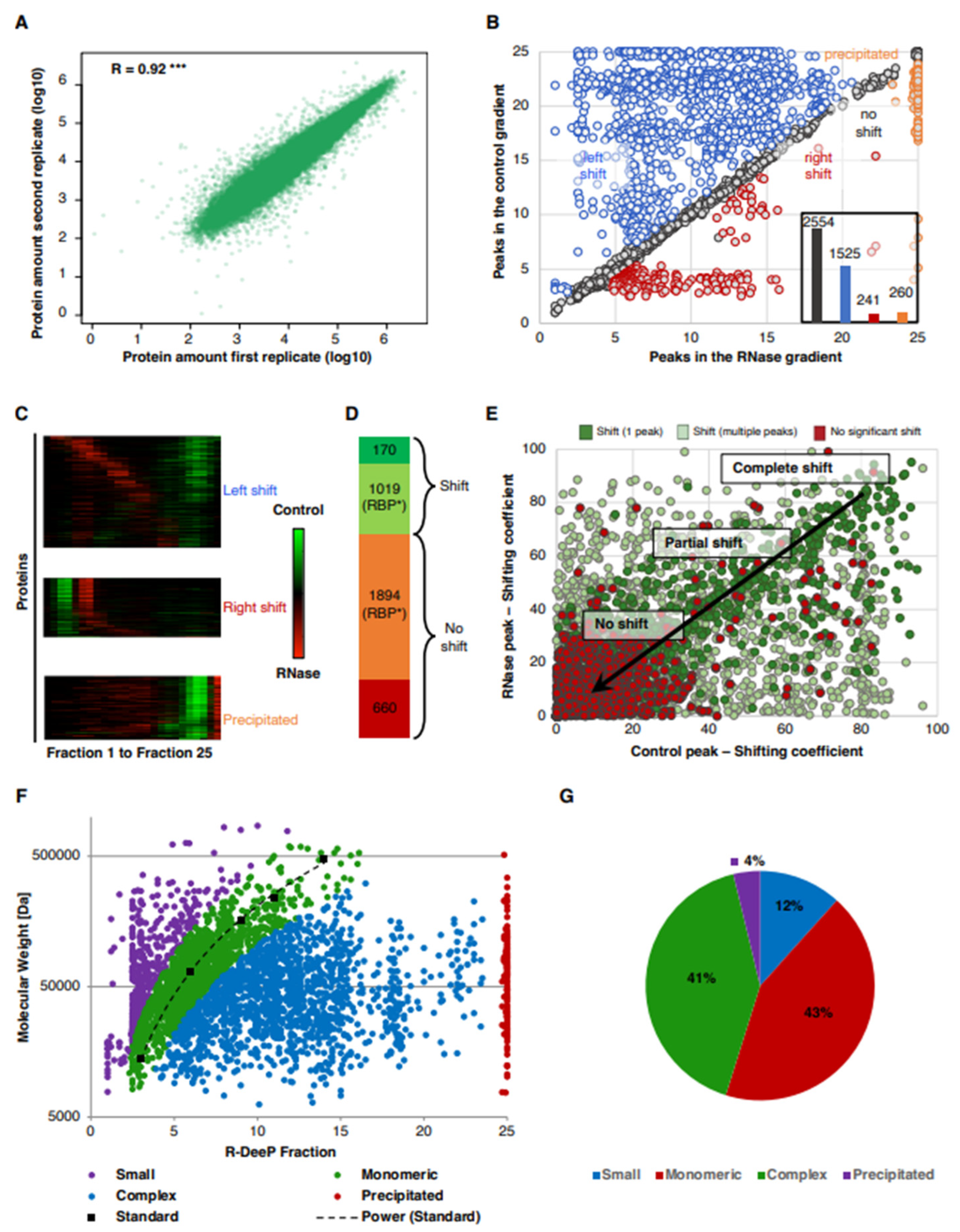
3.2. Analysis of the RNA-Dependent Shifts in the A549 R-DeeP Screen
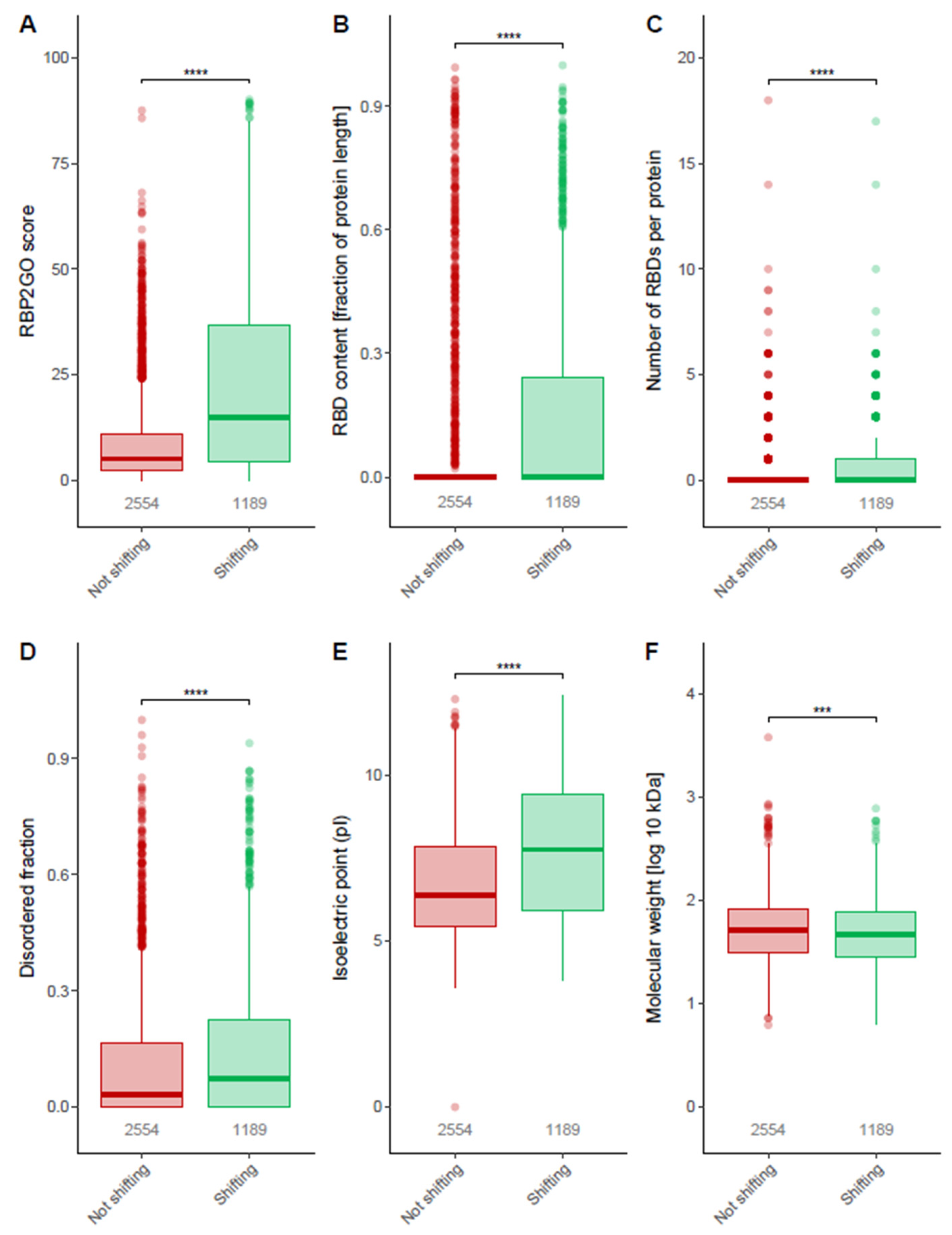
3.3. Properties of Shifting Proteins
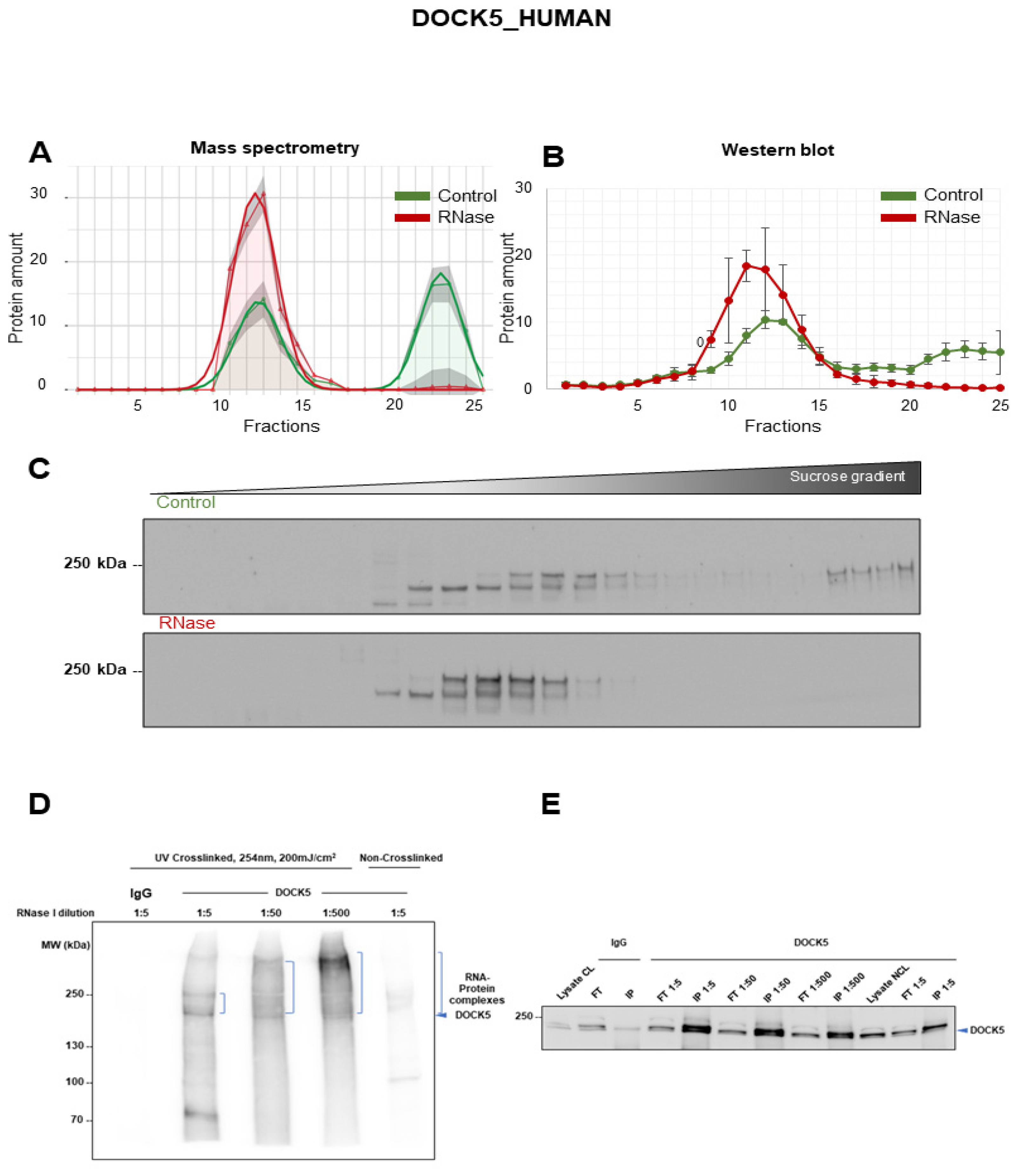
3.4. Validation of DOCK5 as RNA-Dependent Protein
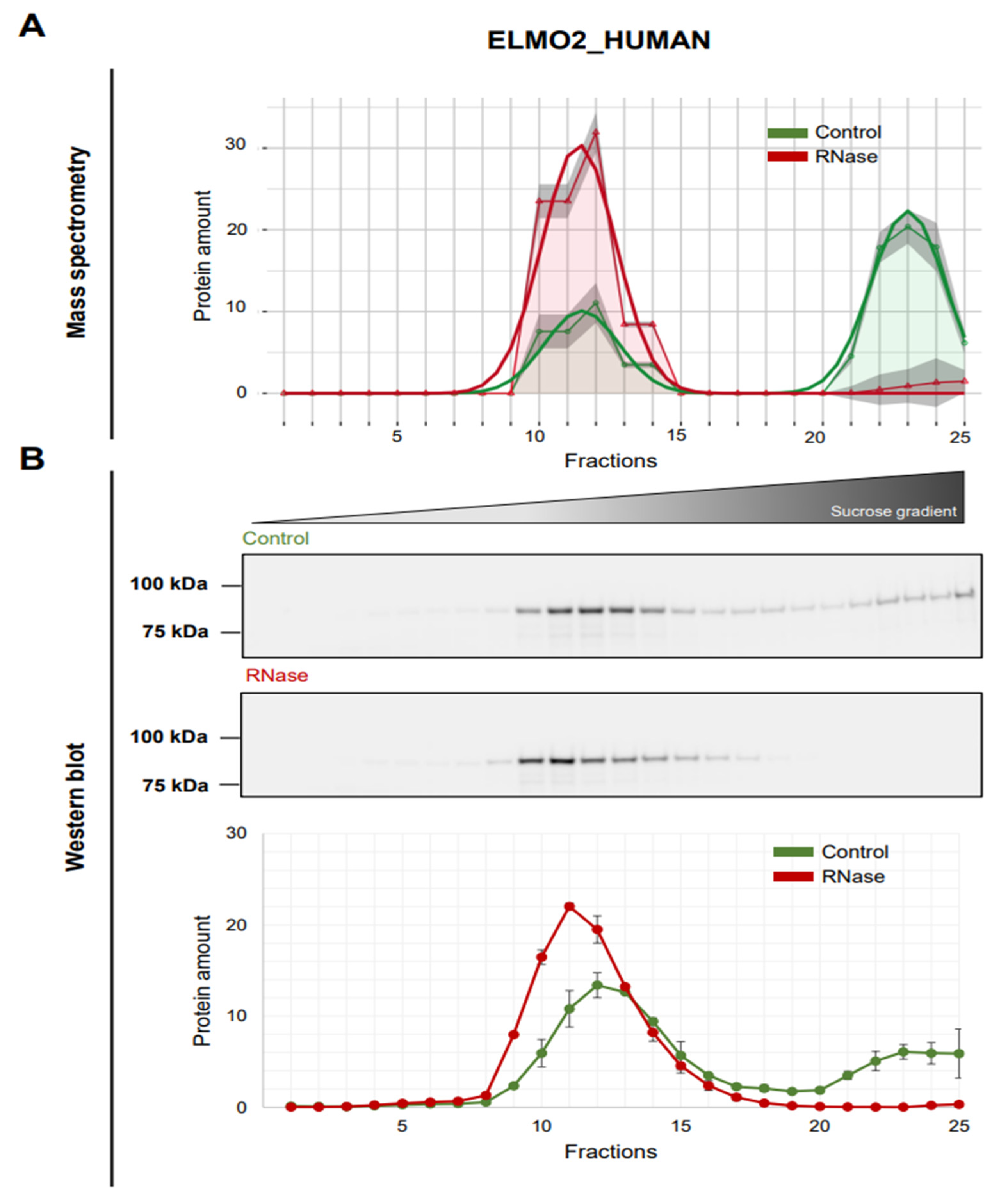
3.5. Validation of ELMO2 as RNA-Dependent Protein
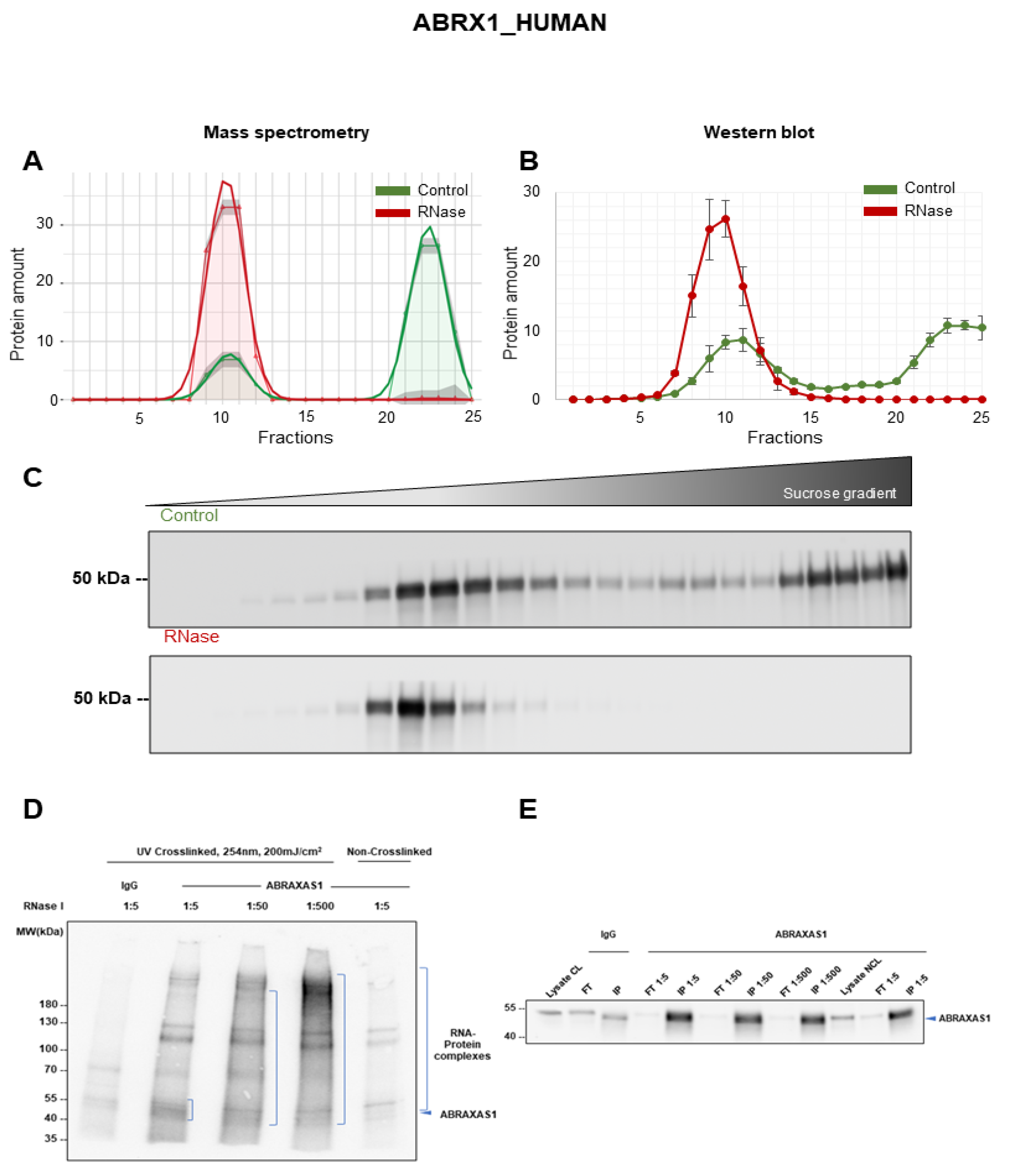
3.6. Validation of ABRAXAS1 as RNA-Dependent Protein
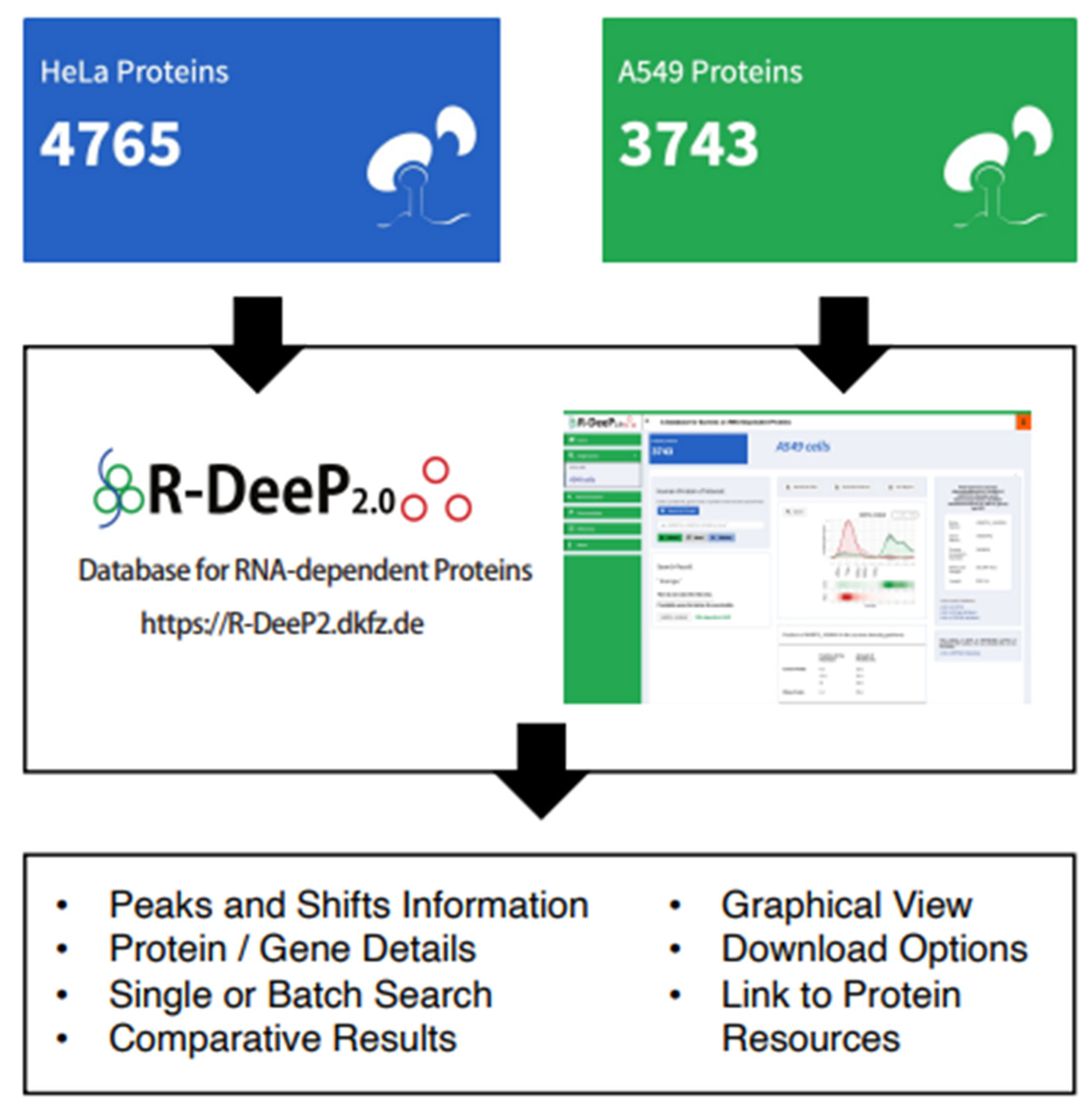
3.7. R-DeeP 2.0 Database as a Versatile Resource to Analyze RNA-Dependent Proteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Esteller, M. Non-Coding RNAs in Human Disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef]
- Gutschner, T.; Diederichs, S. The Hallmarks of Cancer. RNA Biol. 2012, 9, 703–719. [Google Scholar] [CrossRef]
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, S. Many Roads to Maturity: MicroRNA Biogenesis Pathways and Their Regulation. Nat. Cell Biol. 2009, 11, 228–234. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA Therapeutics: Towards a New Era for the Management of Cancer and Other Diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—MicroRNAs with a Role in Cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long Non-Coding RNAs: Insights into Functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef]
- Castello, A.; Fischer, B.; Eichelbaum, K.; Horos, R.; Beckmann, B.M.; Strein, C.; Davey, N.E.; Humphreys, D.T.; Preiss, T.; Steinmetz, L.M.; et al. Insights into RNA Biology from an Atlas of Mammalian MRNA-Binding Proteins. Cell 2012, 149, 1393–1406. [Google Scholar] [CrossRef]
- Lekka, E.; Hall, J. Noncoding RNAs in Disease. FEBS Lett. 2018, 592, 2884–2900. [Google Scholar] [CrossRef]
- Huarte, M. The Emerging Role of LncRNAs in Cancer. Nat. Med. 2015, 21, 1253–1261. [Google Scholar] [CrossRef]
- Cancer (IARC), T.I.A. for R. on Global Cancer Observatory. Available online: https://gco.iarc.fr/ (accessed on 19 January 2022).
- Liu, X.; Zou, H.; Zhao, Y.; Chen, H.; Liu, T.; Wu, Z.; Yang, C.; Li, Q.; Li, Y. Tanshinone Inhibits NSCLC by Downregulating AURKA Through Let-7a-5p. Front. Genet. 2020, 11, 838. [Google Scholar] [CrossRef]
- Jiang, L.; Li, Z.; Wang, R. Long Non-Coding RNAs in Lung Cancer: Regulation Patterns, Biologic Function and Diagnosis Implications (Review). Int. J. Oncol. 2019, 55, 585–596. [Google Scholar] [CrossRef]
- Ji, P.; Diederichs, S.; Wang, W.; Böing, S.; Metzger, R.; Schneider, P.M.; Tidow, N.; Brandt, B.; Buerger, H.; Bulk, E.; et al. MALAT-1, a Novel Noncoding RNA, and Thymosin Β4 Predict Metastasis and Survival in Early-Stage Non-Small Cell Lung Cancer. Oncogene 2003, 22, 8031–8041. [Google Scholar] [CrossRef]
- Ma, J.; Wu, K.; Liu, K.; Miao, R. Effects of MALAT1 on Proliferation and Apo- Ptosis of Human Non-Small Cell Lung Cancer A549 Cells in Vitro and Tumor Xenograft Growth in Vivo by Modulating Autophagy. Cancer Biomark. 2018, 22, 63–72. [Google Scholar] [CrossRef]
- Chen, Z.; Li, J.-L.; Lin, S.; Cao, C.; Gimbrone, N.T.; Yang, R.; Fu, D.A.; Carper, M.B.; Haura, E.B.; Schabath, M.B.; et al. CAMP/CREB-Regulated LINC00473 Marks LKB1-Inactivated Lung Cancer and Mediates Tumor Growth. J. Clin. Investig. 2016, 126, 2267–2279. [Google Scholar] [CrossRef]
- Qin, P.; Li, Y.; Liu, J.; Wang, N. Knockdown of LINC00473 Promotes Radiosensitivity of Non-Small Cell Lung Cancer Cells via Sponging MiR-513a-3p. Free Radic. Res. 2020, 54, 756–764. [Google Scholar] [CrossRef]
- Seiler, J.; Breinig, M.; Caudron-Herger, M.; Polycarpou-Schwarz, M.; Boutros, M.; Diederichs, S. The LncRNA VELUCT Strongly Regulates Viability of Lung Cancer Cells despite Its Extremely Low Abundance. Nucleic Acids Res. 2017, 45, 5458–5469. [Google Scholar] [CrossRef]
- Gandhi, M.; Groß, M.; Holler, J.M.; Coggins, S.A.; Patil, N.; Leupold, J.H.; Munschauer, M.; Schenone, M.; Hartigan, C.R.; Allgayer, H.; et al. The LncRNA LincNMR Regulates Nucleotide Metabolism via a YBX1—RRM2 Axis in Cancer. Nat. Commun. 2020, 11, 3214. [Google Scholar] [CrossRef]
- Roth, A.; Boulay, K.; Groß, M.; Polycarpou-Schwarz, M.; Mallette, F.A.; Regnier, M.; Bida, O.; Ginsberg, D.; Warth, A.; Schnabel, P.A.; et al. Targeting LINC00673 Expression Triggers Cellular Senescence in Lung Cancer. RNA Biol. 2018, 15, 1499–1511. [Google Scholar] [CrossRef]
- Qiu, C.; Li, S.; Sun, D.; Yang, S. LncRNA PVT1 Accelerates Progression of Non-small Cell Lung Cancer via Targeting MiRNA-526b/EZH2 Regulatory Loop. Oncol. Lett. 2020, 19, 1267–1272. [Google Scholar] [CrossRef]
- Pan, Y.; Liu, L.; Cheng, Y.; Yu, J.; Feng, Y. Amplified LncRNA PVT1 Promotes Lung Cancer Proliferation and Metastasis by Facilitating VEGFC Expression. Biochem. Cell Biol. Biochim. Biol. Cell. 2020, 98, 676–682. [Google Scholar] [CrossRef]
- Peng, Y.; Croce, C.M. The Role of MicroRNAs in Human Cancer. Signal Transduct. Target. Ther. 2016, 1, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Takamizawa, J.; Konishi, H.; Yanagisawa, K.; Tomida, S.; Osada, H.; Endoh, H.; Harano, T.; Yatabe, Y.; Nagino, M.; Nimura, Y.; et al. Reduced Expression of the Let-7 MicroRNAs in Human Lung Cancers in Association with Shortened Postoperative Survival. Cancer Res. 2004, 64, 3753–3756. [Google Scholar] [CrossRef] [PubMed]
- Karube, Y.; Tanaka, H.; Osada, H.; Tomida, S.; Tatematsu, Y.; Yanagisawa, K.; Yatabe, Y.; Takamizawa, J.; Miyoshi, S.; Mitsudomi, T.; et al. Reduced Expression of Dicer Associated with Poor Prognosis in Lung Cancer Patients. Cancer Sci. 2005, 96, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Hayashita, Y.; Osada, H.; Tatematsu, Y.; Yamada, H.; Yanagisawa, K.; Tomida, S.; Yatabe, Y.; Kawahara, K.; Sekido, Y.; Takahashi, T. A Polycistronic MicroRNA Cluster, MiR-17-92, Is Overexpressed in Human Lung Cancers and Enhances Cell Proliferation. Cancer Res. 2005, 65, 9628–9632. [Google Scholar] [CrossRef]
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. RAS Is Regulated by the Let-7 MicroRNA Family. Cell 2005, 120, 635–647. [Google Scholar] [CrossRef]
- Lemjabbar-Alaoui, H.; Hassan, O.; Yang, Y.-W.; Buchanan, P. Lung Cancer: Biology and Treatment Options. Biochim. Biophys. Acta 2015, 1856, 189–210. [Google Scholar] [CrossRef]
- Baltz, A.G.; Munschauer, M.; Schwanhäusser, B.; Vasile, A.; Murakawa, Y.; Schueler, M.; Youngs, N.; Penfold-Brown, D.; Drew, K.; Milek, M.; et al. The MRNA-Bound Proteome and Its Global Occupancy Profile on Protein-Coding Transcripts. Mol. Cell 2012, 46, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Caudron-Herger, M.; Jansen, R.E.; Wassmer, E.; Diederichs, S. RBP2GO: A Comprehensive Pan-Species Database on RNA-Binding Proteins, Their Interactions and Functions. Nucleic Acids Res. 2021, 49, D425–D436. [Google Scholar] [CrossRef]
- Caudron-Herger, M.; Rusin, S.F.; Adamo, M.E.; Seiler, J.; Schmid, V.K.; Barreau, E.; Kettenbach, A.N.; Diederichs, S. R-DeeP: Proteome-Wide and Quantitative Identification of RNA-Dependent Proteins by Density Gradient Ultracentrifugation. Mol. Cell 2019, 75, 184–199.e10. [Google Scholar] [CrossRef]
- Corley, M.; Burns, M.C.; Yeo, G.W. How RNA-Binding Proteins Interact with RNA: Molecules and Mechanisms. Mol. Cell 2020, 78, 9–29. [Google Scholar] [CrossRef]
- Gebauer, F.; Schwarzl, T.; Valcárcel, J.; Hentze, M.W. RNA-Binding Proteins in Human Genetic Disease. Nat. Rev. Genet. 2021, 22, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Ni, H.; Liu, Y.; Yuan, Y.; Xi, T.; Li, X.; Zheng, L. RNA-Binding Proteins in Tumor Progression. J. Hematol. Oncol. 2020, 13, 90. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.; Billaud, M.; Almeida, R. RNA-Binding Proteins in Cancer: Old Players and New Actors. Trends Cancer 2017, 3, 506–528. [Google Scholar] [CrossRef] [PubMed]
- Hong, S. RNA Binding Protein as an Emerging Therapeutic Target for Cancer Prevention and Treatment. J. Cancer Prev. 2017, 22, 203–210. [Google Scholar] [CrossRef]
- Treiber, T.; Treiber, N.; Plessmann, U.; Harlander, S.; Daiß, J.-L.; Eichner, N.; Lehmann, G.; Schall, K.; Urlaub, H.; Meister, G. A Compendium of RNA-Binding Proteins That Regulate MicroRNA Biogenesis. Mol. Cell 2017, 66, 270–284.e13. [Google Scholar] [CrossRef]
- Dreyfuss, G.; Kim, V.N.; Kataoka, N. Messenger-RNA-Binding Proteins and the Messages They Carry. Nat. Rev. Mol. Cell Biol. 2002, 3, 195–205. [Google Scholar] [CrossRef]
- Keene, J.D. RNA Regulons: Coordination of Post-Transcriptional Events. Nat. Rev. Genet. 2007, 8, 533–543. [Google Scholar] [CrossRef]
- Conrad, T.; Albrecht, A.-S.; de Melo Costa, V.R.; Sauer, S.; Meierhofer, D.; Ørom, U.A. Serial Interactome Capture of the Human Cell Nucleus. Nat. Commun. 2016, 7, 11212. [Google Scholar] [CrossRef]
- Beckmann, B.M.; Horos, R.; Fischer, B.; Castello, A.; Eichelbaum, K.; Alleaume, A.-M.; Schwarzl, T.; Curk, T.; Foehr, S.; Huber, W.; et al. The RNA-Binding Proteomes from Yeast to Man Harbour Conserved EnigmRBPs. Nat. Commun. 2015, 6, 10127. [Google Scholar] [CrossRef]
- RNA-Binding Proteins: Modular Design for Efficient Function | Nature Reviews Molecular Cell Biology. Available online: https://www.nature.com/articles/nrm2178 (accessed on 20 January 2022).
- Trendel, J.; Schwarzl, T.; Horos, R.; Prakash, A.; Bateman, A.; Hentze, M.W.; Krijgsveld, J. The Human RNA-Binding Proteome and Its Dynamics during Translational Arrest. Cell 2019, 176, 391–403.e19. [Google Scholar] [CrossRef]
- Bao, X.; Guo, X.; Yin, M.; Tariq, M.; Lai, Y.; Kanwal, S.; Zhou, J.; Li, N.; Lv, Y.; Pulido-Quetglas, C.; et al. Capturing the Interactome of Newly Transcribed RNA. Nat. Methods 2018, 15, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Butter, F.; Scheibe, M.; Mörl, M.; Mann, M. Unbiased RNA–Protein Interaction Screen by Quantitative Proteomics. Proc. Natl. Acad. Sci. USA 2009, 106, 10626–10631. [Google Scholar] [CrossRef] [PubMed]
- Mallam, A.L.; Sae-Lee, W.; Schaub, J.M.; Tu, F.; Battenhouse, A.; Jang, Y.J.; Kim, J.; Wallingford, J.B.; Finkelstein, I.J.; Marcotte, E.M.; et al. Systematic Discovery of Endogenous Human Ribonucleoprotein Complexes. Cell Rep. 2019, 29, 1351–1368.e5. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.-Y.; Yang, B.; Zhang, Y.-C.; Wang, X.-J.; Ye, Y.; Peng, J.-W.; Yang, Z.-Z.; He, J.-H.; Zhang, Y.; Hu, K.; et al. EuRBPDB: A Comprehensive Resource for Annotation, Functional and Oncological Investigation of Eukaryotic RNA Binding Proteins (RBPs). Nucleic Acids Res. 2020, 48, D307–D313. [Google Scholar] [CrossRef] [PubMed]
- Gerstberger, S.; Hafner, M.; Tuschl, T. A Census of Human RNA-Binding Proteins. Nat. Rev. Genet. 2014, 15, 829–845. [Google Scholar] [CrossRef]
- Cook, K.B.; Kazan, H.; Zuberi, K.; Morris, Q.; Hughes, T.R. RBPDB: A Database of RNA-Binding Specificities. Nucleic Acids Res. 2011, 39, D301–D308. [Google Scholar] [CrossRef]
- Caudron-Herger, M.; Wassmer, E.; Nasa, I.; Schultz, A.-S.; Seiler, J.; Kettenbach, A.N.; Diederichs, S. Identification, Quantification and Bioinformatic Analysis of RNA-Dependent Proteins by RNase Treatment and Density Gradient Ultracentrifugation Using R-DeeP. Nat. Protoc. 2020, 15, 1338–1370. [Google Scholar] [CrossRef]
- Buchbender, A.; Mutter, H.; Sutandy, F.X.R.; Körtel, N.; Hänel, H.; Busch, A.; Ebersberger, S.; König, J. Improved Library Preparation with the New ICLIP2 Protocol. Methods 2020, 178, 33–48. [Google Scholar] [CrossRef]
- Calabretta, S.; Richard, S. Emerging Roles of Disordered Sequences in RNA-Binding Proteins. Trends Biochem. Sci. 2015, 40, 662–672. [Google Scholar] [CrossRef]
- Hentze, M.W.; Castello, A.; Schwarzl, T.; Preiss, T. A Brave New World of RNA-Binding Proteins. Nat. Rev. Mol. Cell Biol. 2018, 19, 327–341. [Google Scholar] [CrossRef]
- Gadea, G.; Blangy, A. Dock-Family Exchange Factors in Cell Migration and Disease. Eur. J. Cell Biol. 2014, 93, 466–477. [Google Scholar] [CrossRef]
- Frank, S.R.; Köllmann, C.P.; van Lidth de Jeude, J.F.; Thiagarajah, J.R.; Engelholm, L.H.; Frödin, M.; Hansen, S.H. The Focal Adhesion-Associated Proteins DOCK5 and GIT2 Comprise a Rheostat in Control of Epithelial Invasion. Oncogene 2017, 36, 1816–1828. [Google Scholar] [CrossRef]
- Weng, Z.; Situ, C.; Lin, L.; Wu, Z.; Zhu, J.; Zhang, R. Structure of BAI1/ELMO2 Complex Reveals an Action Mechanism of Adhesion GPCRs via ELMO Family Scaffolds. Nat. Commun. 2019, 10, 51. [Google Scholar] [CrossRef]
- Xu, X.; Jin, T. ELMO Proteins Transduce G Protein-Coupled Receptor Signal to Control Reorganization of Actin Cytoskeleton in Chemotaxis of Eukaryotic Cells. Small GTPases 2017, 10, 271–279. [Google Scholar] [CrossRef]
- Wang, Y.; Li, H.; Li, F. ELMO2 Association with Gαi2 Regulates Pancreatic Cancer Cell Chemotaxis and Metastasis. PeerJ 2020, 8, e8910. [Google Scholar] [CrossRef]
- Mok, M.T.S.; Henderson, B.R. The In Vivo Dynamic Organization of BRCA1-A Complex Proteins at DNA Damage-Induced Nuclear Foci. Traffic 2012, 13, 800–814. [Google Scholar] [CrossRef]
- Castillo, A.; Paul, A.; Sun, B.; Huang, T.H.; Wang, Y.; Yazinski, S.A.; Tyler, J.; Li, L.; You, M.J.; Zou, L.; et al. The BRCA1-Interacting Protein Abraxas Is Required for Genomic Stability and Tumor Suppression. Cell Rep. 2014, 8, 807–817. [Google Scholar] [CrossRef][Green Version]
- Queiroz, R.M.L.; Smith, T.; Villanueva, E.; Marti-Solano, M.; Monti, M.; Pizzinga, M.; Mirea, D.-M.; Ramakrishna, M.; Harvey, R.F.; Dezi, V.; et al. Comprehensive Identification of RNA–Protein Interactions in Any Organism Using Orthogonal Organic Phase Separation (OOPS). Nat. Biotechnol. 2019, 37, 169–178. [Google Scholar] [CrossRef]
- Lunde, B.M.; Moore, C.; Varani, G. RNA-Binding Proteins: Modular Design for Efficient Function. Nat. Rev. Mol. Cell Biol. 2007, 8, 479–490. [Google Scholar] [CrossRef]
- Yamanaka, Y.; Faghihi, M.A.; Magistri, M.; Alvarez-Garcia, O.; Lotz, M.; Wahlestedt, C. Antisense RNA Controls LRP1 Sense Transcript Expression through Interaction with a Chromatin-Associated Protein, HMGB2. Cell Rep. 2015, 11, 967–976. [Google Scholar] [CrossRef]
- Dominguez, D.; Freese, P.; Alexis, M.S.; Su, A.; Hochman, M.; Palden, T.; Bazile, C.; Lambert, N.J.; Van Nostrand, E.L.; Pratt, G.A.; et al. Sequence, Structure, and Context Preferences of Human RNA Binding Proteins. Mol. Cell 2018, 70, 854–867.e9. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [CrossRef] [PubMed]
- Giurgiu, M.; Reinhard, J.; Brauner, B.; Dunger-Kaltenbach, I.; Fobo, G.; Frishman, G.; Montrone, C.; Ruepp, A. CORUM: The Comprehensive Resource of Mammalian Protein Complexes-2019. Nucleic Acids Res. 2019, 47, D559–D563. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING V10: Protein–Protein Interaction Networks, Integrated over the Tree of Life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajagopal, V.; Loubal, A.-S.; Engel, N.; Wassmer, E.; Seiler, J.; Schilling, O.; Caudron-Herger, M.; Diederichs, S. Proteome-Wide Identification of RNA-Dependent Proteins in Lung Cancer Cells. Cancers 2022, 14, 6109. https://doi.org/10.3390/cancers14246109
Rajagopal V, Loubal A-S, Engel N, Wassmer E, Seiler J, Schilling O, Caudron-Herger M, Diederichs S. Proteome-Wide Identification of RNA-Dependent Proteins in Lung Cancer Cells. Cancers. 2022; 14(24):6109. https://doi.org/10.3390/cancers14246109
Chicago/Turabian StyleRajagopal, Varshni, Astrid-Solveig Loubal, Niklas Engel, Elsa Wassmer, Jeanette Seiler, Oliver Schilling, Maiwen Caudron-Herger, and Sven Diederichs. 2022. "Proteome-Wide Identification of RNA-Dependent Proteins in Lung Cancer Cells" Cancers 14, no. 24: 6109. https://doi.org/10.3390/cancers14246109
APA StyleRajagopal, V., Loubal, A.-S., Engel, N., Wassmer, E., Seiler, J., Schilling, O., Caudron-Herger, M., & Diederichs, S. (2022). Proteome-Wide Identification of RNA-Dependent Proteins in Lung Cancer Cells. Cancers, 14(24), 6109. https://doi.org/10.3390/cancers14246109