
Increased MARCKS Activity in BRAF Inhibitor-Resistant Melanoma Cells Is Essential for Their Enhanced Metastatic Behavior Independent of Elevated WNT5A and IL-6 Signaling
 , ,
, ,  ,
,
Abstract
Simple Summary
Abstract
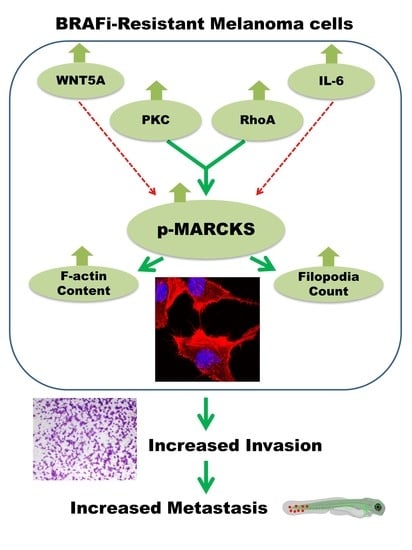
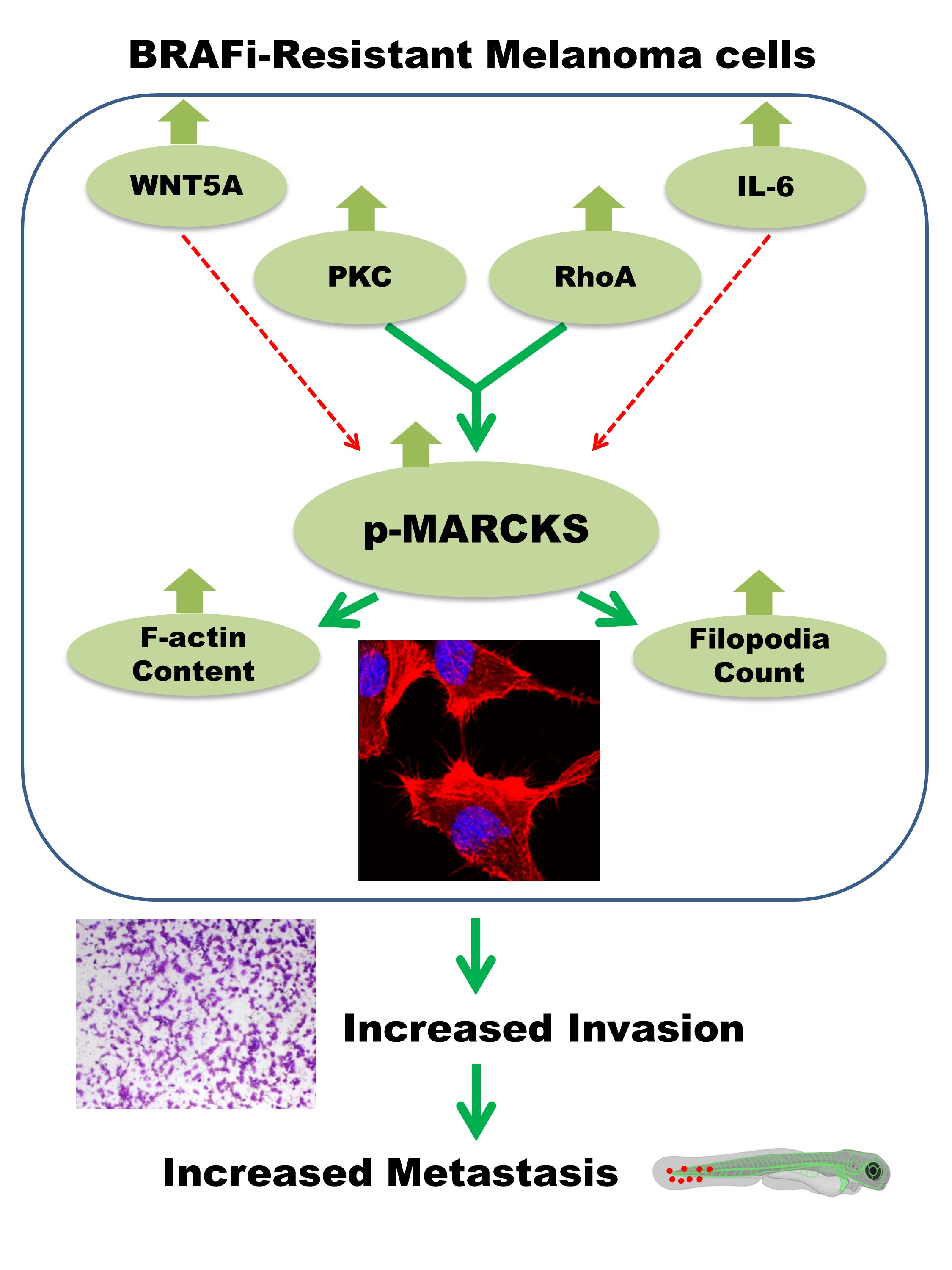{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Melanoma Cell Line Culture
2.2. Establishment of BRAFi-R Melanoma Cell Lines
2.3. MTT and WST1 Cell Proliferation Assays
2.4. Western Blotting
2.5. Migration and Invasion Assays
2.6. siRNA and shRNA Transfection
2.7. IL-6 ELISA and RhoA Activity Assay
2.8. Immunofluorescence Imaging
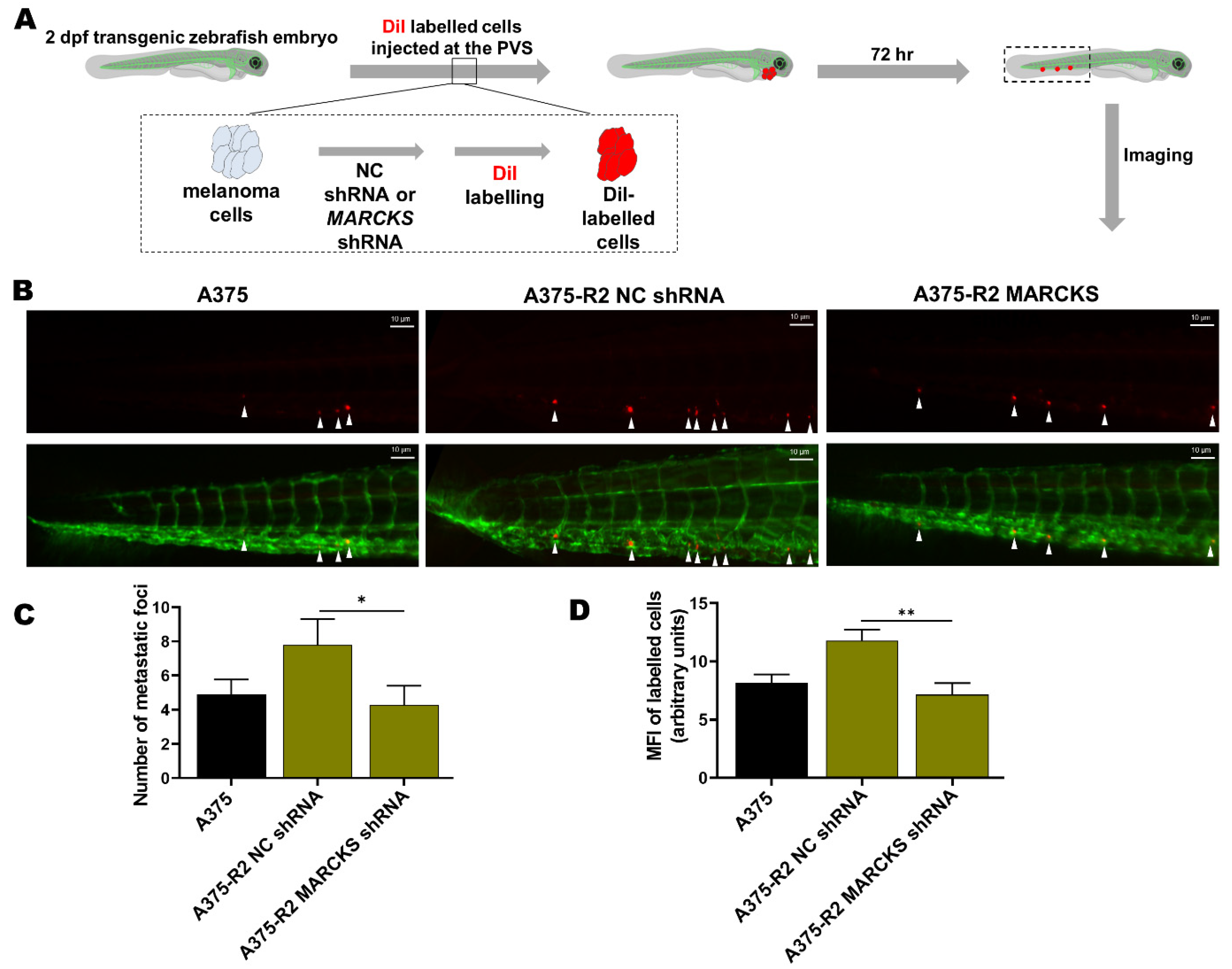
2.9. Evaluation of Tumor Cell Metastasis in a Zebrafish Model
2.10. Real-Time PCR
2.11. Statistical Analysis
3. Results
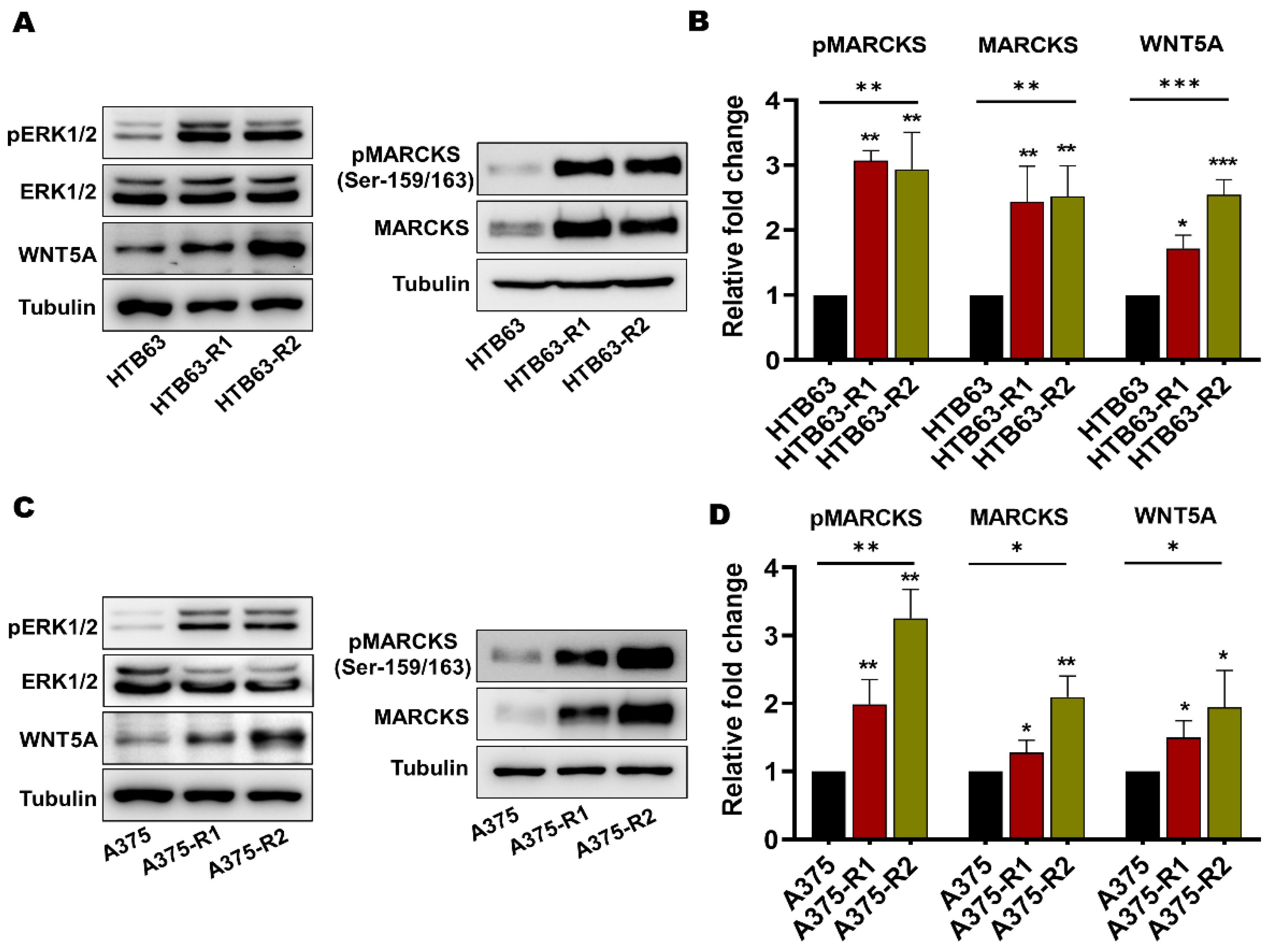
3.1. BRAFi-R Melanoma Cells Exhibit Enhanced MARCKS Expression and Phosphorylation of MARCKS at Serine-159 and Serine-163
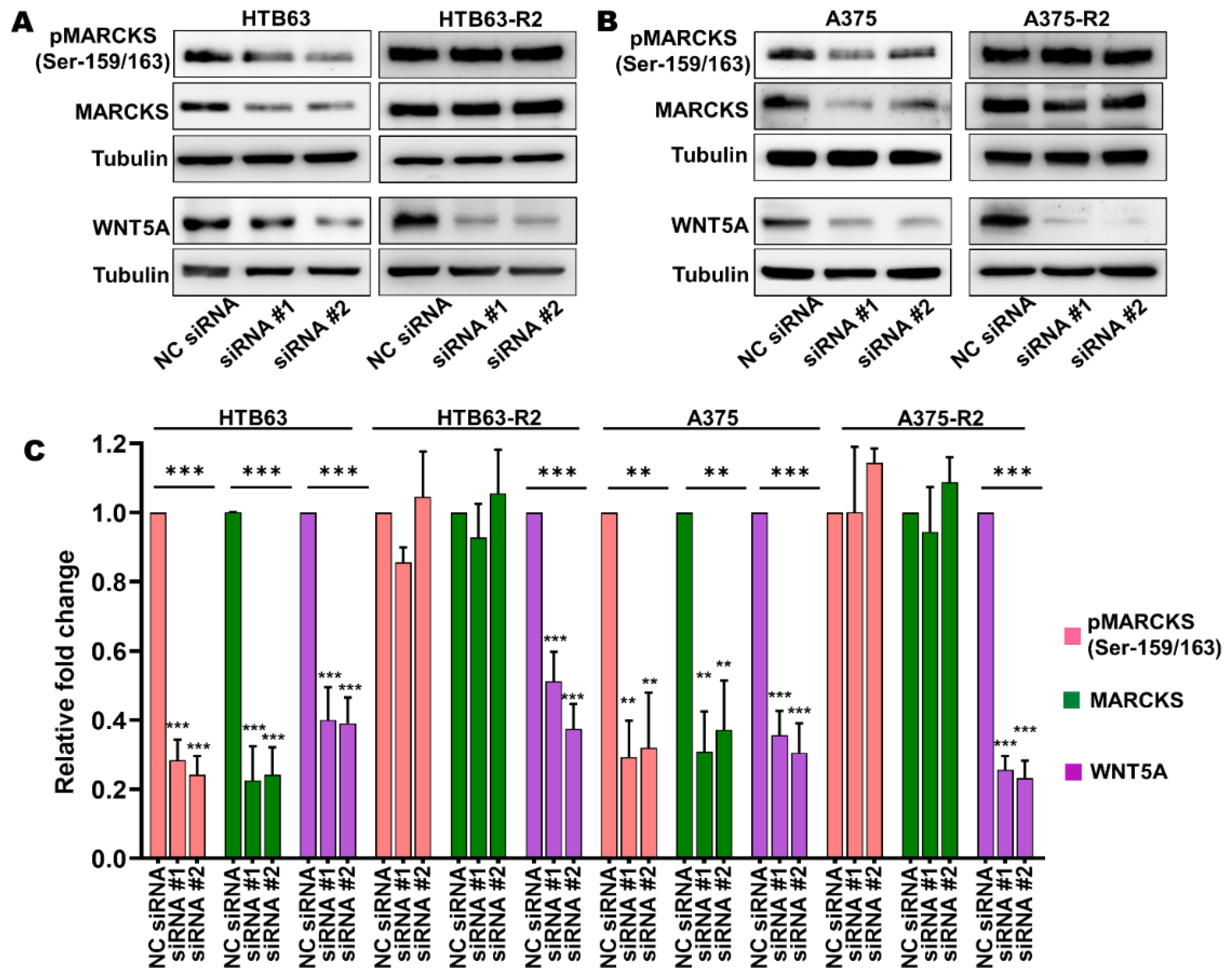
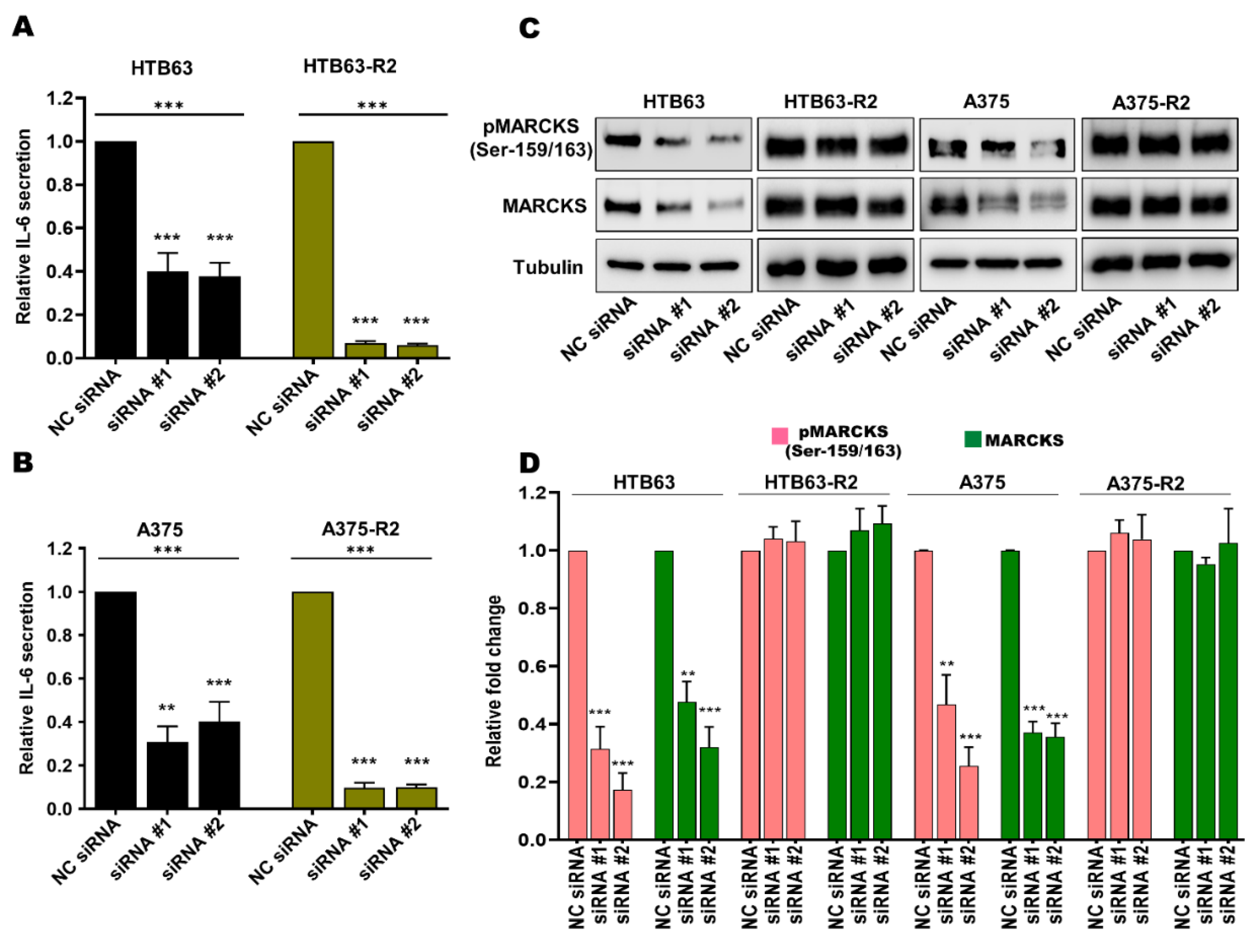
3.2. Enhanced MARCKS Expression and Activity Are Independent of WNT5A and IL-6 in BRAFi-R Melanoma Cells
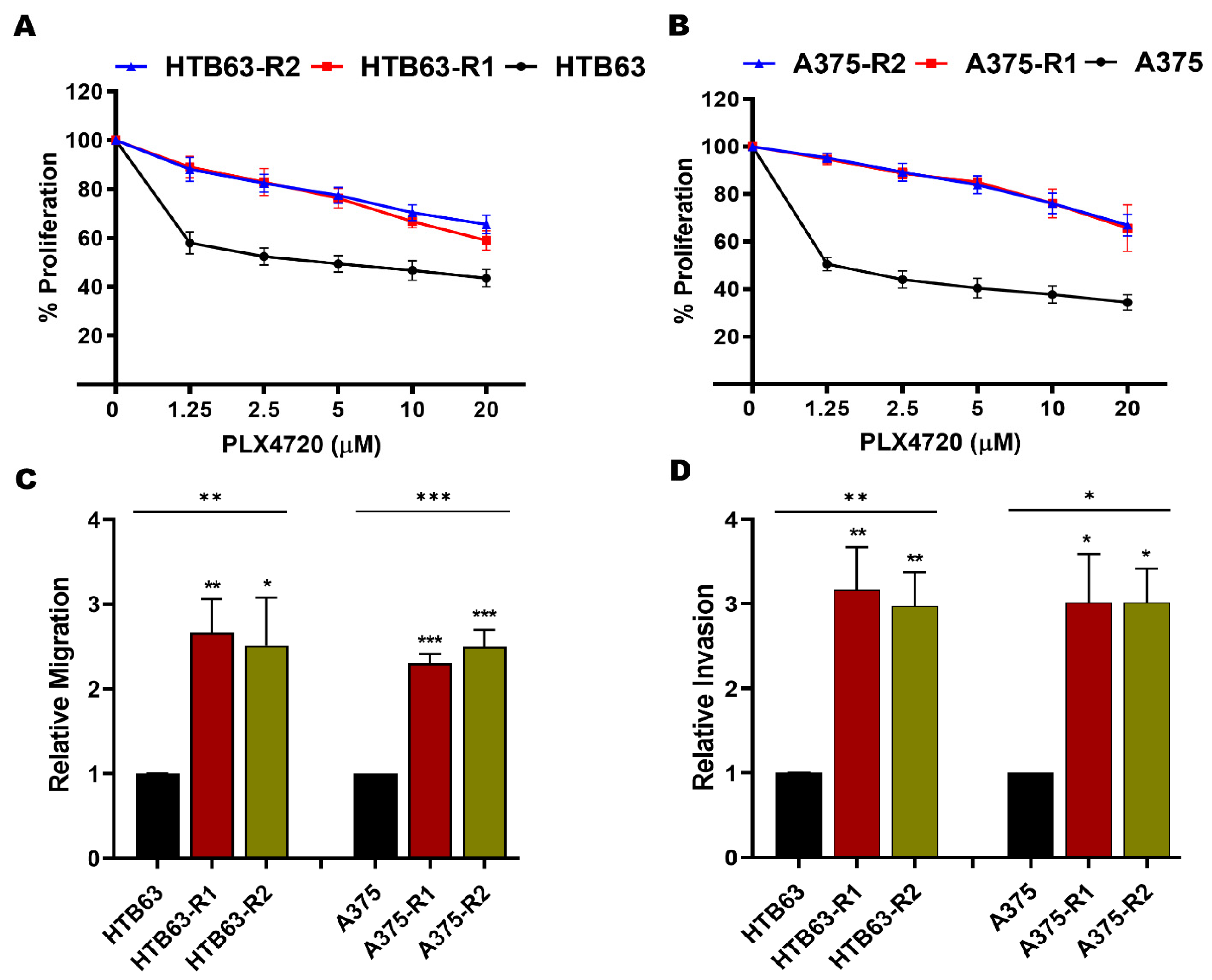
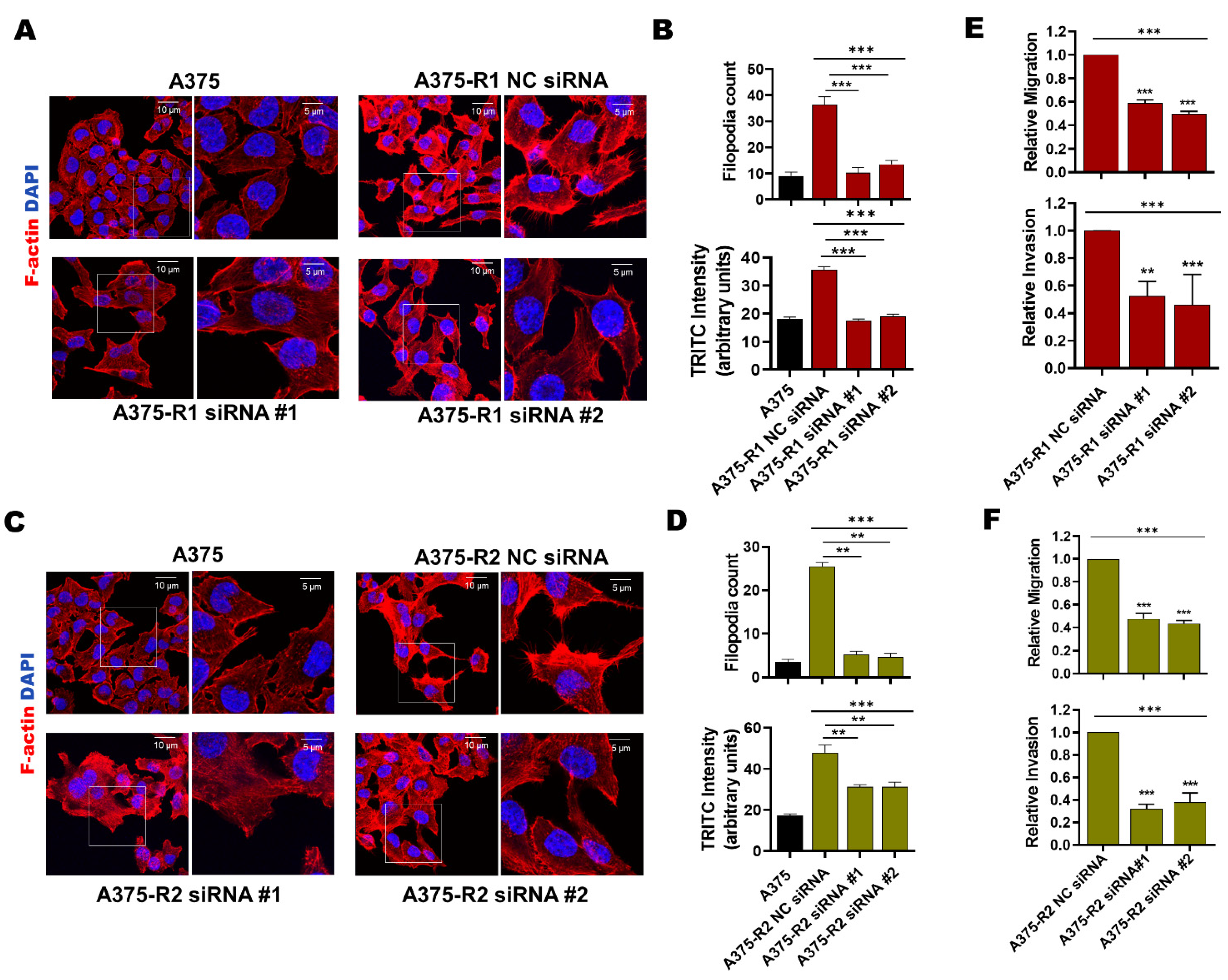
3.3. MARCKS Is an Attractive Antimetastatic Target in BRAFi-R Melanoma Cells
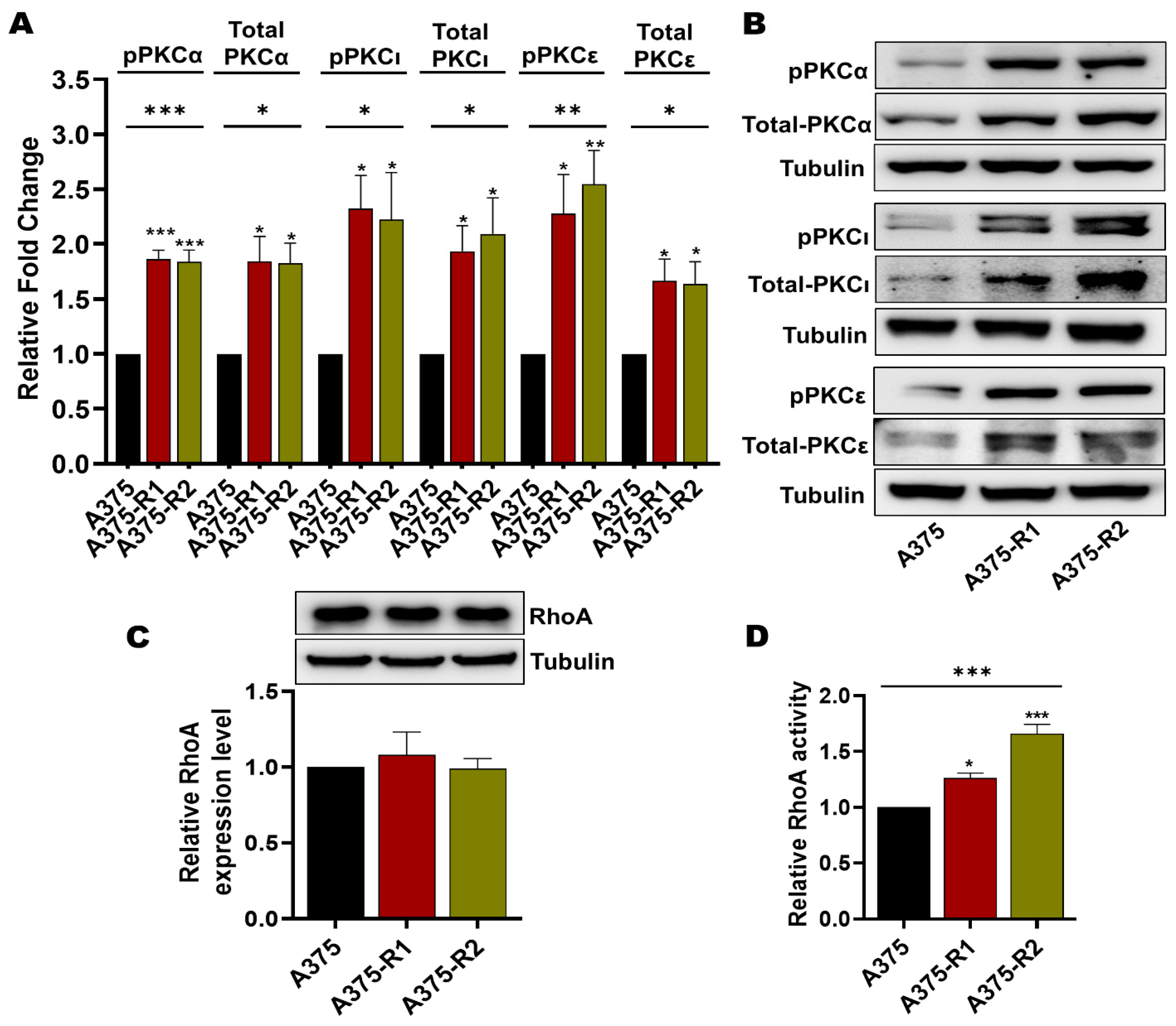
3.4. BRAFi-R Melanoma Cells Exhibit Enhanced PKC Isozyme and RhoA Activities
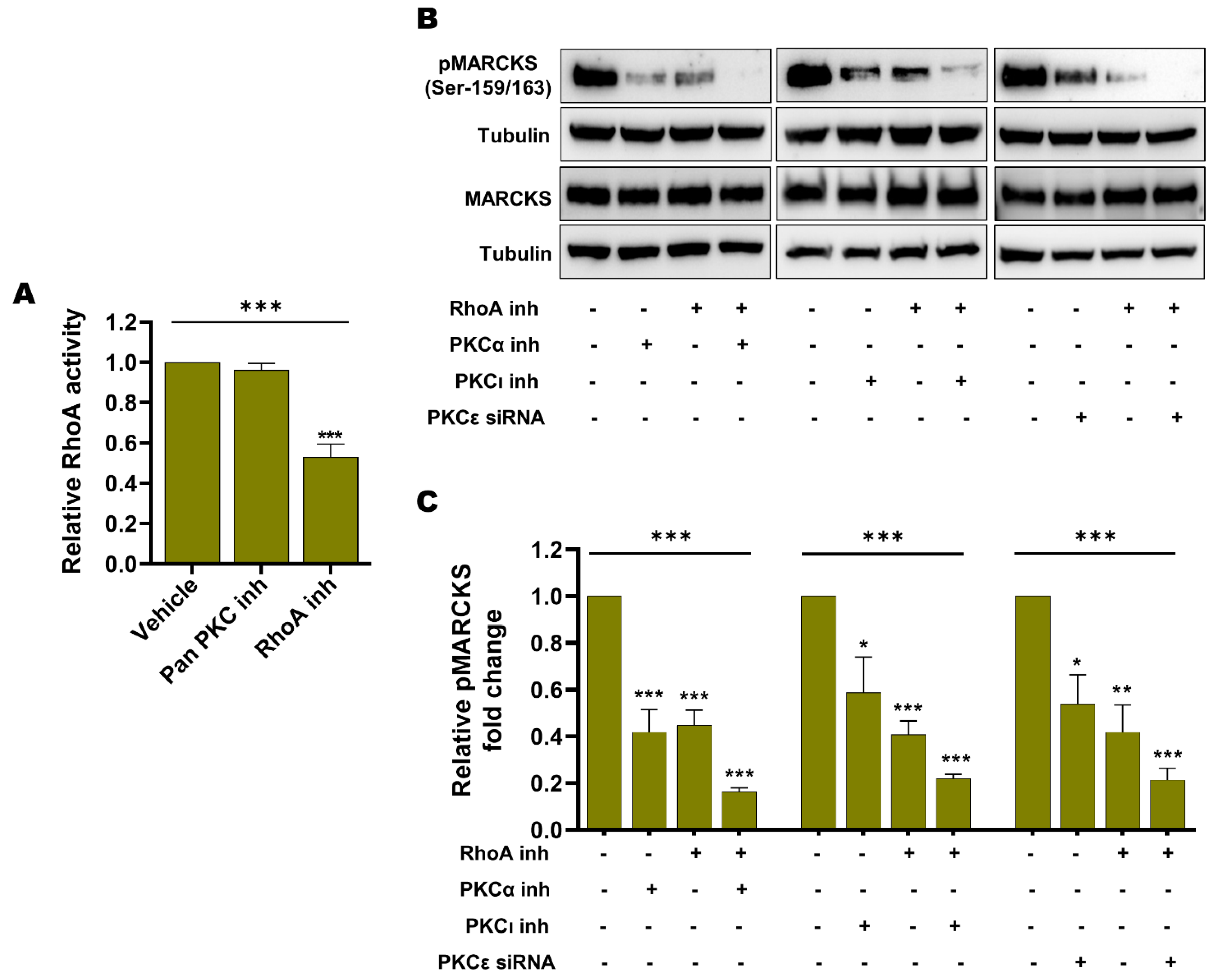
3.5. Endogenous Basal PKC Activity and RhoA Activity Independently Contribute to MARCKS Activation in BRAFi-R Melanoma Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Naik, P.P. Role of Biomarkers in the Integrated Management of Melanoma. Dis. Markers 2021, 2021, 6238317. [Google Scholar] [CrossRef] [PubMed]
- Luebker, S.A.; Koepsell, S.A. Diverse Mechanisms of BRAF Inhibitor Resistance in Melanoma Identified in Clinical and Preclinical Studies. Front. Oncol. 2019, 9, 268. [Google Scholar] [CrossRef] [PubMed]
- Zaman, A.; Wu, W.; Bivona, T.G. Targeting Oncogenic BRAF: Past, Present, and Future. Cancers 2019, 11, 1197. [Google Scholar] [CrossRef] [PubMed]
- Eddy, K.; Shah, R.; Chen, S. Decoding Melanoma Development and Progression: Identification of Therapeutic Vulnerabilities. Front. Oncol. 2020, 10, 626129. [Google Scholar] [CrossRef]
- Kim, A.; Cohen, M.S. The discovery of vemurafenib for the treatment of BRAF-mutated metastatic melanoma. Expert Opin. Drug Discov. 2016, 11, 907–916. [Google Scholar] [CrossRef]
- Jandova, J.; Wondrak, G.T. Vemurafenib Drives Epithelial-to-Mesenchymal Transition Gene Expression in BRAF InhibitorResistant BRAF(V600E)/NRAS(Q61K) Melanoma Enhancing Tumor Growth and Metastasis in a Bioluminescent Murine Model. J. Investig. Dermatol. 2022, 142, 1456–1465. [Google Scholar] [CrossRef]
- Li, F.Z.; Dhillon, A.S.; Anderson, R.L.; McArthur, G.; Ferrao, P.T. Phenotype switching in melanoma: Implications for progression and therapy. Front. Oncol. 2015, 5, 31. [Google Scholar] [CrossRef]
- Kakadia, S.; Yarlagadda, N.; Awad, R.; Kundranda, M.; Niu, J.; Naraev, B.; Mina, L.; Dragovich, T.; Gimbel, M.; Mahmoud, F. Mechanisms of resistance to BRAF and MEK inhibitors and clinical update of US Food and Drug Administration-approved targeted therapy in advanced melanoma. OncoTargets Ther. 2018, 11, 7095–7107. [Google Scholar] [CrossRef]
- Long, G.V.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandala, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A.; et al. Adjuvant Dabrafenib plus Trametinib in Stage III BRAF-Mutated Melanoma. N. Engl. J. Med. 2017, 377, 1813–1823. [Google Scholar] [CrossRef]
- Paris, A.; Tardif, N.; Baietti, F.M.; Berra, C.; Leclair, H.M.; Leucci, E.; Galibert, M.D.; Corre, S. The AhR-SRC axis as a therapeutic vulnerability in BRAFi-resistant melanoma. EMBO Mol. Med. 2022, 14, e15677. [Google Scholar] [CrossRef]
- Alqathama, A. BRAF in malignant melanoma progression and metastasis: Potentials and challenges. Am. J. Cancer Res. 2020, 10, 1103–1114. [Google Scholar] [PubMed]
- O’Connell, M.P.; Marchbank, K.; Webster, M.R.; Valiga, A.A.; Kaur, A.; Vultur, A.; Li, L.; Herlyn, M.; Villanueva, J.; Liu, Q.; et al. Hypoxia induces phenotypic plasticity and therapy resistance in melanoma via the tyrosine kinase receptors ROR1 and ROR2. Cancer Discov. 2013, 3, 1378–1393. [Google Scholar] [CrossRef]
- Anastas, J.N.; Kulikauskas, R.M.; Tamir, T.; Rizos, H.; Long, G.V.; von Euw, E.M.; Yang, P.T.; Chen, H.W.; Haydu, L.; Toroni, R.A.; et al. WNT5A enhances resistance of melanoma cells to targeted BRAF inhibitors. J. Clin. Investig. 2014, 124, 2877–2890. [Google Scholar] [CrossRef] [PubMed]
- Prasad, C.P.; Mohapatra, P.; Andersson, T. Therapy for BRAFi-Resistant Melanomas: Is WNT5A the Answer? Cancers 2015, 7, 1900–1924. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, P.; Prasad, C.P.; Andersson, T. Combination therapy targeting the elevated interleukin-6 level reduces invasive migration of BRAF inhibitor-resistant melanoma cells. Mol. Oncol. 2019, 13, 480–494. [Google Scholar] [CrossRef] [PubMed]
- Weeraratna, A.T.; Jiang, Y.; Hostetter, G.; Rosenblatt, K.; Duray, P.; Bittner, M.; Trent, J.M. Wnt5a signaling directly affects cell motility and invasion of metastatic melanoma. Cancer Cell 2002, 1, 279–288. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, M.P.; Fiori, J.L.; Xu, M.; Carter, A.D.; Frank, B.P.; Camilli, T.C.; French, A.D.; Dissanayake, S.K.; Indig, F.E.; Bernier, M.; et al. The orphan tyrosine kinase receptor, ROR2, mediates Wnt5A signaling in metastatic melanoma. Oncogene 2010, 29, 34–44. [Google Scholar] [CrossRef]
- Dissanayake, S.K.; Wade, M.; Johnson, C.E.; O’Connell, M.P.; Leotlela, P.D.; French, A.D.; Shah, K.V.; Hewitt, K.J.; Rosenthal, D.T.; Indig, F.E.; et al. The Wnt5A/protein kinase C pathway mediates motility in melanoma cells via the inhibition of metastasis suppressors and initiation of an epithelial to mesenchymal transition. J. Biol. Chem. 2007, 282, 17259–17271. [Google Scholar] [CrossRef]
- Da Forno, P.D.; Pringle, J.H.; Hutchinson, P.; Osborn, J.; Huang, Q.; Potter, L.; Hancox, R.A.; Fletcher, A.; Saldanha, G.S. WNT5A expression increases during melanoma progression and correlates with outcome. Clin. Cancer Res. 2008, 14, 5825–5832. [Google Scholar] [CrossRef]
- Webster, M.R.; Xu, M.; Kinzler, K.A.; Kaur, A.; Appleton, J.; O’Connell, M.P.; Marchbank, K.; Valiga, A.; Dang, V.M.; Perego, M.; et al. Wnt5A promotes an adaptive, senescent-like stress response, while continuing to drive invasion in melanoma cells. Pigment Cell Melanoma Res. 2015, 28, 184–195. [Google Scholar] [CrossRef]
- Mohapatra, P.; Yadav, V.; Toftdahl, M.; Andersson, T. WNT5A-Induced Activation of the Protein Kinase C Substrate MARCKS Is Required for Melanoma Cell Invasion. Cancers 2020, 12, 346. [Google Scholar] [CrossRef] [PubMed]
- Fong, L.W.R.; Yang, D.C.; Chen, C.H. Myristoylated alanine-rich C kinase substrate (MARCKS): A multirole signaling protein in cancers. Cancer Metastasis Rev. 2017, 36, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Doghri, R.; Manai, M.; Finetti, P.; Driss, M.; Agavnian, E.; Lopez, M.; Elghardallou, M.; Charafe-Jauffret, E.; Manai, M.; Chaffanet, M.; et al. Stromal Expression of MARCKS Protein in Ovarian Carcinomas Has Unfavorable Prognostic Value. Int. J. Mol. Sci. 2017, 19, 41. [Google Scholar] [CrossRef] [PubMed]
- Manai, M.; Thomassin-Piana, J.; Gamoudi, A.; Finetti, P.; Lopez, M.; Eghozzi, R.; Ayadi, S.; Lamine, O.B.; Manai, M.; Rahal, K.; et al. MARCKS protein overexpression in inflammatory breast cancer. Oncotarget 2017, 8, 6246–6257. [Google Scholar] [CrossRef]
- Li, C.; Xia, R.; Xue, H.; Hu, Y.; Sun, M.; Fang, D.; Yang, W.; Xiao, F.; Hou, J. Overexpression of MARCKS indicates a poor prognosis of oral squamous cell carcinoma. Oncol Lett 2018, 16, 5498–5504. [Google Scholar] [CrossRef]
- Iyer, D.N.; Faruq, O.; Zhang, L.; Rastgoo, N.; Liu, A.; Chang, H. Pathophysiological roles of myristoylated alanine-rich C-kinase substrate (MARCKS) in hematological malignancies. Biomark. Res. 2021, 9, 34. [Google Scholar] [CrossRef]
- Brudvig, J.J.; Weimer, J.M. X MARCKS the spot: Myristoylated alanine-rich C kinase substrate in neuronal function and disease. Front. Cell. Neurosci. 2015, 9, 407. [Google Scholar] [CrossRef]
- Su, D.M.; Zhang, Q.; Wang, X.; He, P.; Zhu, Y.J.; Zhao, J.; Rennert, O.M.; Su, Y.A. Two types of human malignant melanoma cell lines revealed by expression patterns of mitochondrial and survival-apoptosis genes: Implications for malignant melanoma therapy. Mol. Cancer Ther. 2009, 8, 1292–1304. [Google Scholar] [CrossRef]
- Szasz, I.; Koroknai, V.; Kiss, T.; Vizkeleti, L.; Adany, R.; Balazs, M. Molecular alterations associated with acquired resistance to BRAFV600E targeted therapy in melanoma cells. Melanoma Res. 2019, 29, 390–400. [Google Scholar] [CrossRef]
- Yadav, V.; Varshney, P.; Sultana, S.; Yadav, J.; Saini, N. Moxifloxacin and ciprofloxacin induces S-phase arrest and augments apoptotic effects of cisplatin in human pancreatic cancer cells via ERK activation. BMC Cancer 2015, 15, 581. [Google Scholar] [CrossRef]
- Linnskog, R.; Mohapatra, P.; Moradi, F.; Prasad, C.P.; Andersson, T. Demonstration of a WNT5A-IL-6 positive feedback loop in melanoma cells: Dual interference of this loop more effectively impairs melanoma cell invasion. Oncotarget 2016, 7, 37790–37802. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, G.; Ahmad, N. BRAF Inhibitors in Melanoma Management: When Friends Become Foes. J. Investig. Dermatol. 2021, 142, 1256–1259. [Google Scholar] [CrossRef] [PubMed]
- Linnskog, R.; Jonsson, G.; Axelsson, L.; Prasad, C.P.; Andersson, T. Interleukin-6 drives melanoma cell motility through p38alpha-MAPK-dependent up-regulation of WNT5A expression. Mol. Oncol. 2014, 8, 1365–1378. [Google Scholar] [CrossRef]
- Hoejberg, L.; Bastholt, L.; Schmidt, H. Interleukin-6 and melanoma. Melanoma Res. 2012, 22, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Na, Y.R.; Lee, J.S.; Lee, S.J.; Seok, S.H. Interleukin-6-induced Twist and N-cadherin enhance melanoma cell metastasis. Melanoma Res. 2013, 23, 434–443. [Google Scholar] [CrossRef]
- Kim, M.H.; Kim, J.; Hong, H.; Lee, S.H.; Lee, J.K.; Jung, E.; Kim, J. Actin remodeling confers BRAF inhibitor resistance to melanoma cells through YAP/TAZ activation. EMBO J. 2016, 35, 462–478. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Shiraishi, M.; Fukami, K.; Tanabe, A.; Ikeda-Matsuo, Y.; Naito, Y.; Sasaki, Y. MARCKS regulates lamellipodia formation induced by IGF-I via association with PIP2 and beta-actin at membrane microdomains. J. Cell. Physiol. 2009, 220, 748–755. [Google Scholar] [CrossRef]
- Satapathy, S.R.; Sjolander, A. Cysteinyl leukotriene receptor 1 promotes 5-fluorouracil resistance and resistance-derived stemness in colon cancer cells. Cancer Lett. 2020, 488, 50–62. [Google Scholar] [CrossRef]
- Verykiou, S.; Alexander, M.; Edwards, N.; Plummer, R.; Chaudhry, B.; Lovat, P.E.; Hill, D.S. Harnessing autophagy to overcome mitogen-activated protein kinase kinase inhibitor-induced resistance in metastatic melanoma. Br. J. Dermatol. 2019, 180, 346–356. [Google Scholar] [CrossRef]
- Spender, L.C.; Ferguson, G.J.; Liu, S.; Cui, C.; Girotti, M.R.; Sibbet, G.; Higgs, E.B.; Shuttleworth, M.K.; Hamilton, T.; Lorigan, P.; et al. Mutational activation of BRAF confers sensitivity to transforming growth factor beta inhibitors in human cancer cells. Oncotarget 2016, 7, 81995–82012. [Google Scholar] [CrossRef]
- Nagumo, H.; Ikenoya, M.; Sakurada, K.; Furuya, K.; Ikuhara, T.; Hiraoka, H.; Sasaki, Y. Rho-associated kinase phosphorylates MARCKS in human neuronal cells. Biochem. Biophys. Res. Commun. 2001, 280, 605–609. [Google Scholar] [CrossRef] [PubMed]
- El Amri, M.; Fitzgerald, U.; Schlosser, G. MARCKS and MARCKS-like proteins in development and regeneration. J. Biomed. Sci. 2018, 25, 43. [Google Scholar] [CrossRef]
- Jobe, N.P.; Asberg, L.; Andersson, T. Reduced WNT5A signaling in melanoma cells favors an amoeboid mode of invasion. Mol. Oncol. 2021, 15, 1835–1848. [Google Scholar] [CrossRef] [PubMed]
- Denning, M.F. Specifying protein kinase C functions in melanoma. Pigment. Cell. Melanoma Res. 2012, 25, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Oka, M.; Kikkawa, U. Protein kinase C in melanoma. Cancer Metastasis Rev. 2005, 24, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Putnam, A.J.; Schulz, V.V.; Freiter, E.M.; Bill, H.M.; Miranti, C.K. Src, PKCalpha, and PKCdelta are required for alphavbeta3 integrin-mediated metastatic melanoma invasion. Cell Commun. Signal. 2009, 7, 10. [Google Scholar] [CrossRef]
- Zhang, P.; Goodrich, C.; Fu, C.; Dong, C. Melanoma upregulates ICAM-1 expression on endothelial cells through engagement of tumor CD44 with endothelial E-selectin and activation of a PKCalpha-p38-SP-1 pathway. FASEB J. 2014, 28, 4591–4609. [Google Scholar] [CrossRef]
- Schonherr, M.; Bhattacharya, A.; Kottek, T.; Szymczak, S.; Koberle, M.; Wickenhauser, C.; Siebolts, U.; Saalbach, A.; Koczan, D.; Magin, T.M.; et al. Genomewide RNAi screen identifies protein kinase Cb and new members of mitogen-activated protein kinase pathway as regulators of melanoma cell growth and metastasis. Pigment Cell Melanoma Res. 2014, 27, 418–430. [Google Scholar] [CrossRef]
- Ratnayake, W.S.; Apostolatos, C.A.; Apostolatos, A.H.; Schutte, R.J.; Huynh, M.A.; Ostrov, D.A.; Acevedo-Duncan, M. Oncogenic PKC-iota activates Vimentin during epithelial-mesenchymal transition in melanoma; a study based on PKC-iota and PKC-zeta specific inhibitors. Cell Adh. Migr. 2018, 12, 447–463. [Google Scholar] [CrossRef]
- Lau, E.; Sedy, J.; Sander, C.; Shaw, M.A.; Feng, Y.; Scortegagna, M.; Claps, G.; Robinson, S.; Cheng, P.; Srivas, R.; et al. Transcriptional repression of IFNbeta1 by ATF2 confers melanoma resistance to therapy. Oncogene 2015, 34, 5739–5748. [Google Scholar] [CrossRef]
- Mehta, D.; Rahman, A.; Malik, A.B. Protein kinase C-alpha signals rho-guanine nucleotide dissociation inhibitor phosphorylation and rho activation and regulates the endothelial cell barrier function. J. Biol. Chem. 2001, 276, 22614–22620. [Google Scholar] [CrossRef] [PubMed]
- Dovas, A.; Yoneda, A.; Couchman, J.R. PKCbeta-dependent activation of RhoA by syndecan-4 during focal adhesion formation. J. Cell Sci. 2006, 119, 2837–2846. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; He, F.; Zhang, C.; Deng, X.; Yin, F. Protein kinase C-alpha signals P115RhoGEF phosphorylation and RhoA activation in TNF-alpha-induced mouse brain microvascular endothelial cell barrier dysfunction. J. Neuroinflammation 2011, 8, 28. [Google Scholar] [CrossRef] [PubMed]
- Cree, I.A.; Charlton, P. Molecular chess? Hallmarks of anti-cancer drug resistance. BMC Cancer 2017, 17, 10. [Google Scholar] [CrossRef]
- Beck, L.; Harel, M.; Yu, S.; Markovits, E.; Boursi, B.; Markel, G.; Geiger, T. Clinical Proteomics of Metastatic Melanoma Reveals Profiles of Organ Specificity and Treatment Resistance. Clin. Cancer Res. 2021, 27, 2074–2086. [Google Scholar] [CrossRef]
- Ottaviano, M.; Giunta, E.F.; Tortora, M.; Curvietto, M.; Attademo, L.; Bosso, D.; Cardalesi, C.; Rosanova, M.; De Placido, P.; Pietroluongo, E.; et al. BRAF Gene and Melanoma: Back to the Future. Int. J. Mol. Sci. 2021, 22, 3474. [Google Scholar] [CrossRef]
- Kim, Y.E.; Kim, E.K.; Song, M.J.; Kim, T.Y.; Jang, H.H.; Kang, D. SILAC-Based Quantitative Proteomic Analysis of Oxaliplatin-Resistant Pancreatic Cancer Cells. Cancers 2021, 13, 724. [Google Scholar] [CrossRef]
- Franke, N.E.; Kaspers, G.L.; Assaraf, Y.G.; van Meerloo, J.; Niewerth, D.; Kessler, F.L.; Poddighe, P.J.; Kole, J.; Smeets, S.J.; Ylstra, B.; et al. Exocytosis of polyubiquitinated proteins in bortezomib-resistant leukemia cells: A role for MARCKS in acquired resistance to proteasome inhibitors. Oncotarget 2016, 7, 74779–74796. [Google Scholar] [CrossRef]
- Muguruma, Y.; Yahata, T.; Warita, T.; Hozumi, K.; Nakamura, Y.; Suzuki, R.; Ito, M.; Ando, K. Jagged1-induced Notch activation contributes to the acquisition of bortezomib resistance in myeloma cells. Blood Cancer J. 2017, 7, 650. [Google Scholar] [CrossRef]
- Zhang, L.; Rastgoo, N.; Wu, J.; Zhang, M.; Pourabdollah, M.; Zacksenhaus, E.; Chen, Y.; Chang, H. MARCKS inhibition cooperates with autophagy antagonists to potentiate the effect of standard therapy against drug-resistant multiple myeloma. Cancer Lett. 2020, 480, 29–38. [Google Scholar] [CrossRef]
- Misek, S.A.; Appleton, K.M.; Dexheimer, T.S.; Lisabeth, E.M.; Lo, R.S.; Larsen, S.D.; Gallo, K.A.; Neubig, R.R. Rho-mediated signaling promotes BRAF inhibitor resistance in de-differentiated melanoma cells. Oncogene 2020, 39, 1466–1483. [Google Scholar] [CrossRef] [PubMed]
- Takashima, A.; English, B.; Chen, Z.; Cao, J.; Cui, R.; Williams, R.M.; Faller, D.V. Protein kinase Cdelta is a therapeutic target in malignant melanoma with NRAS mutation. ACS Chem. Biol. 2014, 9, 1003–1014. [Google Scholar] [CrossRef] [PubMed]
- Eckert, R.E.; Neuder, L.E.; Park, J.; Adler, K.B.; Jones, S.L. Myristoylated alanine-rich C-kinase substrate (MARCKS) protein regulation of human neutrophil migration. Am. J. Respir. Cell Mol. Biol. 2010, 42, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Rohrbach, T.D.; Jones, R.B.; Hicks, P.H.; Weaver, A.N.; Cooper, T.S.; Eustace, N.J.; Yang, E.S.; Jarboe, J.S.; Anderson, J.C.; Willey, C.D. MARCKS phosphorylation is modulated by a peptide mimetic of MARCKS effector domain leading to increased radiation sensitivity in lung cancer cell lines. Oncol. Lett. 2017, 13, 1216–1222. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Thai, P.; Yoneda, K.; Adler, K.B.; Yang, P.C.; Wu, R. A peptide that inhibits function of Myristoylated Alanine-Rich C Kinase Substrate (MARCKS) reduces lung cancer metastasis. Oncogene 2014, 33, 3696–3706. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Fong, L.W.R.; Yu, E.; Wu, R.; Trott, J.F.; Weiss, R.H. Upregulation of MARCKS in kidney cancer and its potential as a therapeutic target. Oncogene 2017, 36, 3588–3598. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yadav, V.; Jobe, N.; Satapathy, S.R.; Mohapatra, P.; Andersson, T. Increased MARCKS Activity in BRAF Inhibitor-Resistant Melanoma Cells Is Essential for Their Enhanced Metastatic Behavior Independent of Elevated WNT5A and IL-6 Signaling. Cancers 2022, 14, 6077. https://doi.org/10.3390/cancers14246077
Yadav V, Jobe N, Satapathy SR, Mohapatra P, Andersson T. Increased MARCKS Activity in BRAF Inhibitor-Resistant Melanoma Cells Is Essential for Their Enhanced Metastatic Behavior Independent of Elevated WNT5A and IL-6 Signaling. Cancers. 2022; 14(24):6077. https://doi.org/10.3390/cancers14246077
Chicago/Turabian StyleYadav, Vikas, Njainday Jobe, Shakti Ranjan Satapathy, Purusottam Mohapatra, and Tommy Andersson. 2022. "Increased MARCKS Activity in BRAF Inhibitor-Resistant Melanoma Cells Is Essential for Their Enhanced Metastatic Behavior Independent of Elevated WNT5A and IL-6 Signaling" Cancers 14, no. 24: 6077. https://doi.org/10.3390/cancers14246077
APA StyleYadav, V., Jobe, N., Satapathy, S. R., Mohapatra, P., & Andersson, T. (2022). Increased MARCKS Activity in BRAF Inhibitor-Resistant Melanoma Cells Is Essential for Their Enhanced Metastatic Behavior Independent of Elevated WNT5A and IL-6 Signaling. Cancers, 14(24), 6077. https://doi.org/10.3390/cancers14246077