Current Status of Novel Agents for the Treatment of B Cell Malignancies: What’s Coming Next?



Abstract
Simple Summary
Abstract
1. Background and Introduction

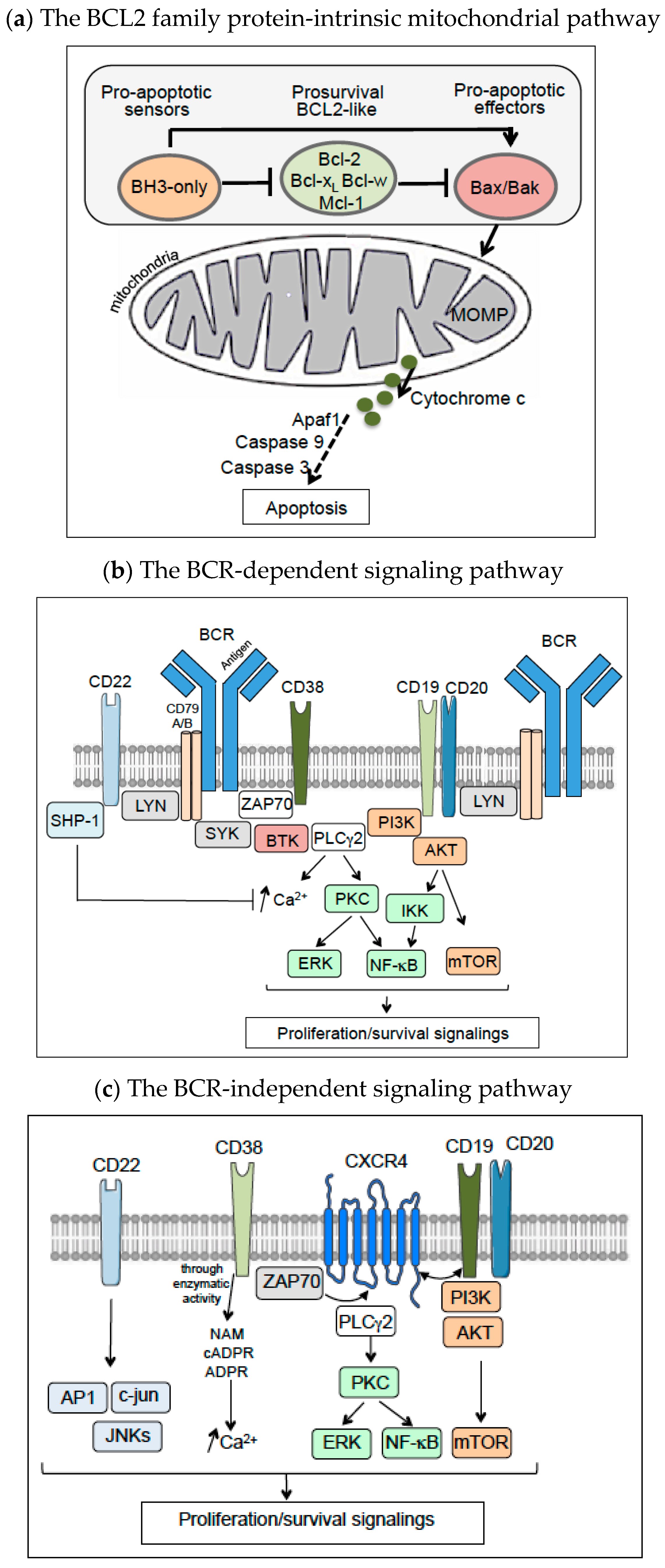{kind=link}
{kind=link}
{kind=link}
{kind=link}
| B Cell Malignancy | Pathophysiology | Expression of BCL2 Prosurvival Proteins | Active (Pre-)BCR Signaling | Surface Markers That Are Strongly Expressed | References |
|---|---|---|---|---|---|
| B-ALL | Transformation and expansion of lymphatic B progenitor cells | Bcl-2, Mcl-1, Bcl-xL | pre + | CD19 CD20 CD22 CXCR4 | [7,35,36,37,38,39,40,41] |
| HCL | Accumulation of CD5+ B cells most in the blood, bone marrow and spleen | Bcl-2 > Mcl-1 | + | CD19 CD20 CD22 CD38 CXCR4 | [2,11,40,42,43] |
| CLL | Accumulation of CD5+ B cells primarily in the blood and bone marrow | High Bcl-2 > Mcl-1 >> Bcl-xL | + | CD19 CD20 CD38 (poor prognosis) BAFF-R ROR1 CXCR4 | [3,7,36,38,40,41,44,45] |
| SLL (B-NHL) | Accumulation of CD5+ B cells most in the lymph nodes | Bcl-2, Mcl-1, Bcl-xL | + | CD19 CD20 CD38 (poor prognosis) ROR1 | [40,44,45,46,47] |
| MM | Accumulation of clonal, Ig secreting plasma cells in the bone marrow | High Bcl-2, Mcl-1, Bcl-xL, Bcl-w | + | CD19 CD22 CD38 BCMA CD13 ADAM17 ROR1 CXCR4 | [3,7,40,41,48,49,50,51] |
| FL (B-NHL) | Extensive proliferation and accumulation of abnormal B cells in lymph nodes | High Bcl-2, Bcl-w and Bcl-xL > Mcl-1 | −/+ | CD19 CD20 CD22 CD38 CXCR4 | [3,7,40,41,45,48,52] |
| MCL (B-NHL) | Development of abnormal B cells in the mantle zone of lymph nodes, spleen, bone marrow, blood, and gastrointestinal tract | High Bcl-w and Bcl-2 > Mcl-1 | + | CD19 CD20 CD22 CD38 ROR1 CXCR4 | [3,7,40,41,47,52,53] |
| MZL (B-NHL) | Development of abnormal B cells in the marginal zones of lymph tissue | High Bcl-2 and Bcl-w > Bcl-xL | + | CD19 CD20 CD22 CXCR4 | [3,10,40,41,52,54,55] |
| DLBCL (B-NHL) | Development of abnormal B cells in germinal centers of secondary lymphoid organs | Bcl-2 and Bcl-w > Mcl-1, Bcl-xL | + | CD19 CD20 CD22 CD38 CXCR4 | [3,7,36,40,41,45,52,56,57] |
| WM (B-NHL) | Proliferation of clonal, IgM-secreting plasma cells in the lymph nodes and bone marrow | High Bcl-2 > Bcl-xL and Mcl-1 | + | CD19 CD20 CD38 CXCR4 (mutation) | [8,40,46,58,59] |
2. Current Treatments and Clinical Trials of B Cell Malignancies Using Drugs That Target BCL2, BTK, PI3K Proteins, and TAAs (CD19, CD20, CD22, CD38)
2.1. BCL2 Inhibitors
| B Cell Malignancy | Current Treatment Options | References |
|---|---|---|
| B-ALL | Conventional chemotherapies * Tyrosine kinase inhibitors imatinib or dasatinib for patients with Philadelphia chromosome-positive Blinatumomab Tisagenlecleucel Inotuzumab ozogamycin (adults) | [85,86,87,88,89] |
| CLL/SLL | Fludarabine, cyclophosphamide and rituximab (FCR) Bendamustine and rituximab (BR) Rituximab and chlorambucil (RCb) Rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) Obinutuzumab and chlorambucil Ibrutinib or acalabrutinib, alone Idelalisib alone and with rituximab Venetoclax + rituximab (or obinituzumab) | [60,90,91,92,93,94,95,96,97] |
| HCL | Purine analogs (cladribine, pentostatin) alone and with rituximab Moxetumomab pasudotox | [98,99] |
| MM | Conventional chemotherapies ** Daratumumab (or isatuximab) + lenalidomide (or pomalidomide) + dexamethasone Belantamab mafodotin | [34,49,100,101,102] |
| FL | BR Rituximab, cyclophosphamide, vincristine, and prednisone (R-CVP) R-CHOP Rituximab + hyaluronidase human Rituximab + lenalidomide Obinutuzumab + bendamustine Idelalisib or copanlisib alone Ibritumomab tiuxetan (Yttrium-90) Axicabtagene ciloleucel | [103,104,105,106,107] |
| MCL | R-CVP, R-CHOP Rituximab + lenalidomide Rituximab + bortezomib Ibrutinib or acalabrutinib or zanubrutinib, alone Ibrutinib + rituximab | [53,108,109] |
| MZL | R-CVP, R-CHOP, FCR, BR Fludarabine and rituximab (FR) Rituximab + lenalidomide Ibrutinib or zanubrutinib | [54,110,111,112] |
| DLBCL | R-CHOP R-CHOP + etoposide Rituximab Tafasitamab + lenalidomide Loncastuximab teserine Tisagenleucel Axicabtagene ciloleucel | [89,113,114,115,116,117] |
| WM | Alkylating drugs and proteasome inhibitors both in combination with rituximab Ibrutinib or acalabrutinib or zanubrutinib, alone and with rituximab | [59,118,119,120,121,122] |
| Drug | Class | Disease Setting | Phase | Study | References |
|---|---|---|---|---|---|
| BGB-11417 | Bcl-2 inhibitor | CLL/SLL, B-NHL | I | NCT04277637 Recruiting, 2020–2023 Alone or combined with zanubrutinib | [76] |
| Lisaftoclax/ APG-2575 | Bcl-2/Bcl-xL inhibitor | CLL/SLL, MZL, MCL CLL/SLL | I Ib/II | NCT03913949 Recruiting, 2019–2023 NCT04494503 Recruiting, 2020–2023 Alone or combined with rituximab or ibrutinib | [77] [123] |
| PRT1419 | Mcl-1 inhibitor | R/R MM R/R B-NHL | I | NCT04543305 Active, 2020–2022 | [124] |
| S64351/ MIK-665 | Mcl-1 inhibitor | AML | I/II | NCT04629443 Recruiting, 2020–2024 + azacytidine | |
| Orelabrutinib /ICP-022 | BTK inhibitor | MCL CLL/SLL FL, MZL, MCL, CLL/SLL R/R MZL, R/R WM, DLBCL | I/II I/II II | NCT03494179 NCT03493217 Active, 2018–2022 NCT04014205 Recruiting, 2019- NCT03797456 NCT04440059 NCT04438005 Recruiting, 2020- | [125] |
| Nemtabrutinib/ MK-1026/ARQ-531 | BTK inhibitor | CLL/SLL, MCL, R/R MZL R/R FL, R/R WM | II | NCT04728893 Recruiting, 2021–2027 | |
| Pirtobrutinib/ LOX-305 | BK inhibitor | Previously treated CLL/SLL, WM, MCL, MZL Untreated CLL/SLL Previously treated CLL/SLL | I/II III III | NCT03740529 Recruiting, 2018–2023 Combined with venetoclax + rituximab NCT05023980 Recruiting 2021- NCT04965493 Recruiting, 2021- Combined with venetoclax + rituximab | |
| Parsaclisib/INCB500465/ IBI-376 | PI3Kδ inhibitor | CLL, DLBCL | I/II | NCT04809467 Recruiting, 2021–2023 Alone or combined with tafasitamab | |
| Zandelisib/PWT143/ME-401 | PI3Kδ inhibitor | R/R FL, R/R MZL R/R CLL | III II | NCT04745832 Recruiting, 2021–2031 Combined with rituximab NCT05209308 Recruiting, 2022–2026 Combined with rituximab + venetoclax | [126] |
| BGB-10188 | PI3Kδ inhibitor | R/R CLL, FL, MCL, DLBCL, MZL | I/II | NCT04282018 Recruiting, 2020–2025 Alone or combined with zanubrutinib and tiselizumab | |
| Umbralisib/ TGR-1202 | Dual PI3Kδ/CK1ε inhibitor | R/R CLL, R/R MCL R/R CLL, R/R B-NHL | I/Ib I | NCT02268851 Active, 2014–2023 Combined with ibrutinib NCT03283137 Active, 2017–2024 Combined with pembrolizumab | [127] |
| Mosunetuzumab/ BTC4465A | BiTE anti-CD20/CD3 | CLL, B-NHL B-NHL | I/II I/II | NCT02500407 Recruiting, 2015- Alone or combined with atezolizumab (anti-PD-L1) NCT03671018 Recruiting, 2018- Alone or combined with polatuzumab (anti-CD79B-monomethyl auristatin E) | [128] |
| Odronextamab/ REGN1979 | BiTE anti-CD20/CD3 | R/R DLBCL, FL, MZL, WM | I/II | NCT02651662 Active, 2016–2026 Alone or combined with cemiplimab (anti-PD-1) NCT03888105 Recruiting, 2019–2028 | [129] |
| Epcoritamab/ GEN301 | BiTE duoBody-anti-CD20/CD3 | R/R or progressive DLBCL, MCL, FL, MZL, SLL R/R DLBCL R/R CLL | I/II III I/II | NCT03625037 Recruiting, 2018–2024 NCT04628494 Recruiting, 2020–2024 NCT04623541 Recruiting, 2020–2024 | [130,131] |
| Glofitamab (RO7082859) | BiTE anti-CD20/CD3 | R/R B-NHL | I/II Ib/II | NCT03075696 Recruiting, 2017–2023 Alone or combined with obinutzumab NCT03533283 Recruiting, 2018–2024 Alone or combined with atezolizumab, obinutuzumab, tocilizumab polatuzumab vedotin | [132] |
| Tafasitamab/ MOR208/ | Anti-CD19 | R/R FL, R/R MZL DLBCL | III III | NCT04680052 Recruiting, 2020- Combined with rituximab + lenalidomide compared with rituximab + lenalidomide NCT04824092 Recruiting, 2021–2026 Combined with lenalidomide + R-CHOP | [133] |
| CAR-20.19-T | Anti-CD19/CD20 CAR-T/CD28/ 4-1BB/CD3ζ | CLL/SLL, R/R B-NHL | I | NCT03019055 Active, 2017–2022 | [134] |
| CD19-CAR-NK using blood cord NKs | Anti-CD19 CAR-NK/OX40/CD3ζ/IL-15 | B-ALL CLL/SLL, R/R B-NHL | I/II | NCT03056339 Active, 2017–2022 after treatment with fludarabine/cyclophosphamide lymphodepletion | [135,136] |
| NKX019 using peripheral blood NKs | Anti-CD19 CAR-NK/OX40/ CD3ζ/IL-15 | B-ALL CLL/SLL, R/R B-NHL | I | NCT05020678 Recruiting 2021–2023 | [137] |
| C-CAR066/CBM.20-CAR-T | Anti-CD20 CAR-T | NHLs resistant to rituximab or anti-CD19 CAR-T | I | NCT04036019 Recruiting, 2019–2021 NCT04316624 Recruiting, 2020- | [138] |
| Inotuzumab ozogamicin | ADC anti-CD22- calicheamicin | B-ALL, B-NHL | III | NCT03959085 Recruiting, 2019–2029 post chemotherapy | |
| JNJ-75348780 | BiTE anti-CD22/CD3 | CLL, B-NHL | I | NCT04540796 Recruiting, 2020–2024 | |
| CD22-CAR | Anti-CD22 CAR-T/CD8/4-1BB/CD3ζ | R/R B-ALL, B-NHL | I | NCT02315612 Recruiting, 2014–2040 | [139] |
| LV20.19 | Bi anti-CD19/CD22 CAR-T/4-1BB/CD3ζ | CLL/SLL, B-NHL | I | NCT03019055 Active, 2017–2022 | [134,140] |
| AUTO3 | Bi anti-CD19 (OX40 costim)/CD22 (4-1BB costim) CAR-T | R/R DLBCL | I/II | NCT03287817 Active 2017- | [141] |
| CD19-22.BB.z-CAR | Bi anti-CD19/ CD22 CAR-T | R/R B-ALL, DLBCL Pretreated B-ALL | I I | NCT03233854 Recruiting, 2017–2025 Combined with NKTR-255 (IL-15 receptor agonist) NCT03448393 Recruiting, 2018–2040 following cyclophosphamide/fludarabine treatment | [142] [143] |
| CD20-22 CAR | Bi anti-CD20/CD22 CAR-T | B cell malignancies | I | NCT04283006 Recruiting, 2020–2028 | |
| Felzartamab/ MOR202/TJ202 | Anti-CD38 | R/R MM | II III | NCT03860038 Active, 2019–2022 Combined with dexamethasone NCT03952091 Active, 2019–2022 Combined with lenalidomide + dexamethasone | |
| TAK-079 | Anti-CD38 | R/R MM MM | I/IIa I | NCT03499280 Completed, 2018–2022 Alone or combined with pomalidomide and dexamethasone NCT03984097 Active, 2019–2023 Combined with lenalidomide + dexamethasone | |
| TAK-573/ modakafusp | ADC anti-CD38 delivering attenuated IFN-α2b | R/R MM | I/II | NCT03215030 Recruiting, 2017- Alone or combined with dexamethasone | [144] |
| TAK-169/ MT-0169 | ADC anti-CD38 delivering Shiga-like toxin | R/R B-NHL | I | NCT04017130 Recruiting, 2019- | [145] |
| ISB 1342/ GBR 1342 | BiTE anti-CD38/CD3 | R/R MM | I | NCT03309111 Recruiting 2017–2024 |
2.2. BTK Inhibitors
2.3. PI3K Inhibitors
2.4. mAbs and CAR-T Cells
2.4.1. CD20 mAbs and CD20-Targeting CAR-T
2.4.2. CD19 mAbs and CD19-Targeting CAR-T
2.4.3. CD22 mAbs and CD22-Targeting CAR-T
2.4.4. CD38 mAbs and CD38-Targeting CAR-T
3. Clinical Trials of Novel Anti-TAA Inhibitors in the Treatment of B Cell Malignancies
3.1. B Cell Maturation Antigen (BCMA) and B-Cell Activation Factor Receptor (BAFF-R)
3.2. Receptor Tyrosine Kinase-like Orphan Receptor (ROR1)
3.3. Program Cell Death 1 (PD1) and PD-Ligand 1 (PD-L1)
3.4. C-X-C Chemokine Receptor Type 4 (CXCR4)
3.5. CS1/Signaling Lymphocyte Activation Molecule Family 7 (SLAMF7)
3.6. Other TAAS
3.6.1. CD13
3.6.2. CD16
3.6.3. CD30
3.6.4. CD37
3.6.5. CD46
3.6.6. CD47
3.6.7. CD56
3.6.8. CD74
3.6.9. CD79B
3.6.10. CD156/a Disintegrin and Metalloprotease 17 (ADAM17) (Also Referred to as TNF-α Converting Enzyme/TACE)
3.6.11. CD307/Fc Receptor-like 5 (FcRL5)
3.6.12. G-Protein Coupled Receptor Family C Group 5 Member D (GPRC5D)
4. Targeting Metabolic Reprogramming in B Cell Malignancies—Preclinical and Clinical Studies
4.1. Metabolic Reprogramming in B Cell Malignancies
4.1.1. The Metabolism in Cancer Cells, the Warburg Theory
4.1.2. Specific Metabolic Features of B Cell Malignancies
4.1.3. Involvement of the Tumor Microenvironment in the Adaptative Metabolism
4.2. Targeting Metabolism in B Cell Malignancies: Clinical Perspectives
4.2.1. Metabolic Features as Biomarkers of B Cell Malignancies
4.2.2. New Strategies for Targeting Metabolism in B Cell Malignancies and Optimizing Current Treatments
4.2.3. Metabolism as a Tool to Boost Immunotherapy
5. Concluding Remarks and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef] [PubMed]
- Tessoulin, B.; Papin, A.; Gomez-Bougie, P.; Bellanger, C.; Amiot, M.; Pellat-Deceunynck, C.; Chiron, D. BCL2-Family Dysregulation in B-Cell Malignancies: From Gene Expression Regulation to a Targeted Therapy Biomarker. Front. Oncol. 2018, 8, 645. [Google Scholar] [CrossRef]
- Adams, C.M.; Clark-Garvey, S.; Porcu, P.; Eischen, C.M. Targeting the Bcl-2 Family in B Cell Lymphoma. Front. Oncol. 2019, 8, 636. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, I.; Bodo, J.; Hill, B.T.; Hsi, E.D.; Almasan, A. Targeting BCL-2 in B-cell malignancies and overcoming therapeutic resistance. Cell Death Dis. 2020, 11, 941. [Google Scholar] [CrossRef] [PubMed]
- Morales-Martínez, M.; Vega, M.I. Roles and Regulation of BCL-xL in Hematological Malignancies. Int. J. Mol. Sci. 2022, 23, 2193. [Google Scholar] [CrossRef] [PubMed]
- Sulkshane, P.; Teni, T. Myeloid cell leukemia-1: A formidable barrier to anticancer therapeutics and the quest of targeting it. Explor. Target Antitumor Ther. 2022, 3, 278–296. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Huang, D. Targeting BCL2 With BH3 Mimetics: Basic Science and Clinical Application of Venetoclax in Chronic Lymphocytic Leukemia and Related B Cell Malignancies. Clin. Pharmacol. Ther. 2017, 101, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Argyropoulos, K.V.; Vogel, R.; Ziegler, C.; Altan-Bonnet, G.; Velardi, E.; Calafiore, M.; Dogan, A.; Arcila, M.; Patel, M.; Knapp, K.; et al. Clonal B cells in Waldenström’s macroglobulinemia exhibit functional features of chronic active B-cell receptor signaling. Leukemia 2016, 30, 1116–1125. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Saba, N.S.; Liu, D.; Herman, S.E.; Underbayev, C.; Tian, X.; Behrend, D.; Weniger, M.A.; Skarzynski, M.; Gyamfi, J.; Fontan, L.; et al. Pathogenic role of B-cell receptor signaling and canonical NF-κB activation in mantle cell lymphoma. Blood 2016, 128, 82–92. [Google Scholar] [CrossRef]
- Efremov, D.; Turkalj, S.; Laurenti, L. Mechanisms of B Cell Receptor Activation and Responses to B Cell Receptor Inhibitors in B Cell Malignancies. Cancers 2020, 12, 1396. [Google Scholar] [CrossRef]
- Paillassa, J.; Safa, F.; Troussard, X. Updates in hairy cell leukemia (HCL) and variant-type HCL (HCL-V): Rationale for targeted treatments with a focus on ibrutinib. Ther. Adv. Hematol. 2022, 13, 20406207221090886. [Google Scholar] [CrossRef] [PubMed]
- Profitós-Pelejà, N.; Santos, J.C.; Marín-Niebla, A.; Roué, G.; Ribeiro, M.L. Regulation of B-Cell Receptor Signaling and Its Therapeutic Relevance in Aggressive B-Cell Lymphomas. Cancers 2022, 14, 860. [Google Scholar] [CrossRef]
- Burger, J.A.; Wiestner, A. Targeting B cell receptor signalling in cancer: Preclinical and clinical advances. Nat. Rev. Cancer 2018, 18, 148–167. [Google Scholar] [CrossRef] [PubMed]
- Gambella, M.; Carlomagno, S.; Raiola, A.M.; Giannoni, L.; Ghiggi, C.; Setti, C.; Giordano, C.; Luchetti, S.; Serio, A.; Bo, A.; et al. CD19-Targeted Immunotherapies for Diffuse Large B-Cell Lymphoma. Front. Immunol. 2022, 13, 837457. [Google Scholar] [CrossRef]
- Pavlasova, G.; Mraz, M. The regulation and function of CD20: An “enigma” of B-cell biology and targeted therapy. Haematologica 2020, 105, 1494–1506. [Google Scholar] [CrossRef]
- Shah, N.N.; Sokol, L. Targeting CD22 for the Treatment of B-Cell Malignancies. Immunotargets Ther. 2021, 10, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Horenstein, A.L.; Faini, A.C.; Morandi, F.; Bracci, C.; Lanza, F.; Giuliani, N.; Paulus, A.; Malavasi, F. The Circular Life of Human CD38: From Basic Science to Clinics and Back. Molecules 2020, 25, 4844. [Google Scholar] [CrossRef] [PubMed]
- Buhl, A.M.; Pleiman, C.M.; Rickert, R.C.; Cambier, J.C. Qualitative regulation of B cell antigen receptor signaling by CD19: Selective requirement for PI3-kinase activation, inositol-1,4,5-trisphosphate production and Ca2+ mobilization. J. Exp. Med. 1997, 186, 1897–1910. [Google Scholar] [CrossRef]
- Deaglio, S.; Capobianco, A.; Bergui, L.; Dürig, J.; Morabito, F.; Dührsen, U.; Malavasi, F. CD38 is a signaling molecule in B-cell chronic lymphocytic leukemia cells. Blood 2003, 102, 2146–2155. [Google Scholar] [CrossRef]
- Polyak, M.J.; Li, H.; Shariat, N.; Deans, J.P. CD20 homo-oligomers physically associate with the B cell antigen receptor. Dissociation upon receptor engagement and recruitment of phosphoproteins and calmodulin-binding proteins. J. Biol. Chem. 2008, 283, 18545–18552. [Google Scholar] [CrossRef]
- Clark, E.A.; Giltiay, N.V. CD22: A Regulator of Innate and Adaptive B Cell Responses and Autoimmunity. Front. Immunol. 2018, 9, 2235. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wei, G.; Liu, D. CD19: A biomarker for B cell development, lymphoma diagnosis and therapy. Exp. Hematol. Oncol. 2012, 1, 36. [Google Scholar] [CrossRef]
- Sato, S.; Tuscano, J.M.; Inaoki, M.; Tedder, T.F. CD22 negatively and positively regulates signal transduction through the B lymphocyte antigen receptor. Semin. Immunol. 1998, 10, 287–297. [Google Scholar] [CrossRef]
- Tuscano, J.M.; Riva, A.; Toscano, S.N.; Tedder, T.F.; Kehrl, J.H. CD22 cross-linking generates B-cell antigen receptor-independent signals that activate the JNK/SAPK signaling cascade. Blood 1999, 94, 1382–1392. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.A.; Smith, K.G. CD22: An inhibitory enigma. Immunology 2008, 123, 314–325. [Google Scholar] [CrossRef]
- Vaisitti, T.; Audrito, V.; Serra, S.; Buonincontri, R.; Sociali, G.; Mannino, E.; Pagnani, A.; Zucchetto, A.; Tissino, E.; Vitale, C.; et al. The enzymatic activities of CD38 enhance CLL growth and trafficking: Implications for therapeutic targeting. Leukemia 2015, 29, 356–368. [Google Scholar] [CrossRef]
- Sochacka-Ćwikła, A.; Mączyński, M.; Regiec, A. FDA-Approved Drugs for Hematological Malignancies—The Last Decade Review. Cancers 2021, 14, 87. [Google Scholar] [CrossRef] [PubMed]
- Puła, B.; Gołos, A.; Górniak, P.; Jamroziak, K. Overcoming Ibrutinib Resistance in Chronic Lymphocytic Leukemia. Cancers 2019, 11, 1834. [Google Scholar] [CrossRef] [PubMed]
- Marine, J.C.; Dawson, S.J.; Dawson, M.A. Non-genetic mechanisms of therapeutic resistance in cancer. Nat. Rev. Cancer 2020, 20, 743–756. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Wei, A.H.; Huang, D.C.S. BCL2 and MCL1 inhibitors for hematologic malignancies. Blood 2021, 138, 1120–1136. [Google Scholar] [CrossRef] [PubMed]
- Mohty, R.; Gauthier, J. Current combinatorial CAR T cell strategies with Bruton tyrosine kinase inhibitors and immune checkpoint inhibitors. Bone Marrow Transplant. 2021, 56, 2630–2636. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, A.; Huang, Y.; Kittai, A.; Maakaron, J.E.; Saygin, C.; Brammer, J.; Penza, S.; Saad, A.; Jaglowski, S.M.; William, B.M. Cytopenias After CD19 Chimeric Antigen Receptor T-Cells (CAR-T) Therapy for Diffuse Large B-Cell Lymphomas or Transformed Follicular Lymphoma: A Single Institution Experience. Cancer Manag. Res. 2021, 13, 8901–8906. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Wu, G.; Huang, X.; Ma, Y.; Zhang, Y.; Song, Q.; Xie, M.; Sun, Y.; Huang, Y.; Huang, Z.; et al. Efficacy and safety of new anti-CD20 monoclonal antibodies versus rituximab for induction therapy of CD20(+) B-cell non-Hodgkin lymphomas: A systematic review and meta-analysis. Sci. Rep. 2021, 11, 3255. [Google Scholar] [CrossRef]
- Petrucci, M.T.; Vozella, F. The Anti-CD38 Antibody Therapy in Multiple Myeloma. Cells 2019, 8, 1629. [Google Scholar] [CrossRef] [PubMed]
- Stam, R.W.; Den Boer, M.L.; Schneider, P.; de Boer, J.; Hagelstein, J.; Valsecchi, M.G.; de Lorenzo, P.; Sallan, S.E.; Brady, H.J.; Armstrong, S.A.; et al. Association of high-level MCL-1 expression with in vitro and in vivo prednisone resistance in MLL-rearranged infant acute lymphoblastic leukemia. Blood 2010, 115, 1018–1025. [Google Scholar] [CrossRef] [PubMed]
- Rickert, R.C. New insights into pre-BCR and BCR signalling with relevance to B cell malignancies. Nat. Rev. Immunol. 2013, 13, 578–591. [Google Scholar] [CrossRef]
- Eswaran, J.; Sinclair, P.; Heidenreich, O.; Irving, J.; Russell, L.J.; Hall, A.; Calado, D.P.; Harrison, C.J.; Vormoor, J. The pre-B-cell receptor checkpoint in acute lymphoblastic leukaemia. Leukemia 2015, 29, 1623–1631. [Google Scholar] [CrossRef] [PubMed]
- Köhrer, S.; Havranek, O.; Seyfried, F.; Hurtz, C.; Coffey, G.P.; Kim, E.; Ten Hacken, E.; Jäger, U.; Vanura, K.; O’Brien, S.; et al. Pre-BCR signaling in precursor B-cell acute lymphoblastic leukemia regulates PI3K/AKT, FOXO1 and MYC, and can be targeted by SYK inhibition. Leukemia 2016, 30, 1246–1254. [Google Scholar] [CrossRef]
- Seyfried, F.; Stirnweiß, F.U.; Niedermayer, A.; Enzenmüller, S.; Hörl, R.L.; Münch, V.; Köhrer, S.; Debatin, K.M.; Meyer, L.H. Synergistic activity of combined inhibition of anti-apoptotic molecules in B-cell precursor ALL. Leukemia 2022, 36, 901–912. [Google Scholar] [CrossRef]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef]
- Vadillo, E.; Dorantes-Acosta, E.; Pelayo, R.; Schnoor, M. T cell acute lymphoblastic leukemia (T-ALL): New insights into the cellular origins and infiltration mechanisms common and unique among hematologic malignancies. Blood Rev. 2018, 32, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Maitre, E.; Cornet, E.; Salaün, V.; Kerneves, P.; Chèze, S.; Repesse, Y.; Damaj, G.; Troussard, X. Immunophenotypic Analysis of Hairy Cell Leukemia (HCL) and Hairy Cell Leukemia-like (HCL-like) Disorders. Cancers 2022, 14, 1050. [Google Scholar] [CrossRef] [PubMed]
- Zaja, F.; Di Loreto, C.; Amoroso, V.; Salmaso, F.; Russo, D.; Silvestri, F.; Fanin, R.; Damiani, D.; Infanti, L.; Mariuzzi, L.; et al. BCL-2 immunohistochemical evaluation in B-cell chronic lymphocytic leukemia and hairy cell leukemia before treatment with fludarabine and 2-chloro-deoxy-adenosine. Leuk. Lymphoma 1998, 28, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Batata, A.; Shen, B. Relationship between chronic lymphocytic leukemia and small lymphocytic lymphoma. A comparative study of membrane phenotypes in 270 cases. Cancer 1992, 70, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Myklebust, J.H.; Brody, J.; Kohrt, H.E.; Kolstad, A.; Czerwinski, D.K.; Wälchli, S.; Green, M.R.; Trøen, G.; Liestøl, K.; Beiske, K.; et al. Distinct patterns of B-cell receptor signaling in non-Hodgkin lymphomas identified by single-cell profiling. Blood 2017, 129, 759–770. [Google Scholar] [CrossRef]
- Pangalis, G.A.; Angelopoulou, M.K.; Vassilakopoulos, T.P.; Siakantaris, M.P.; Kittas, C. B-chronic lymphocytic leukemia, small lymphocytic lymphoma, and lymphoplasmacytic lymphoma, including Waldenström’s macroglobulinemia: A clinical, morphologic, and biologic spectrum of similar disorders. Semin. Hematol. 1999, 36, 104–114. [Google Scholar]
- Zhang, S.; Chen, L.; Wang-Rodriguez, J.; Zhang, L.; Cui, B.; Frankel, W.; Wu, R.; Kipps, T.J. The onco-embryonic antigen ROR1 is expressed by a variety of human cancers. Am. J. Pathol. 2012, 181, 1903–1910. [Google Scholar] [CrossRef]
- Cho-Vega, J.H.; Rassidakis, G.Z.; Admirand, J.H.; Oyarzo, M.; Ramalingam, P.; Paraguya, A.; McDonnell, T.J.; Amin, H.M.; Medeiros, L.J. MCL-1 expression in B-cell non-Hodgkin’s lymphomas. Hum. Pathol. 2004, 35, 1095–1100. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Kumar, S. Multiple myeloma current treatment algorithms. Blood Cancer J. 2020, 10, 94. [Google Scholar] [CrossRef]
- Ilić, V.; Milosević-Jovcić, N.; Petrović, S.; Marković, D.; Bila, J.; Bosković, D.; Stefanović, G.; Marković, O.; Glibetić, M. Signaling status of IgG B cell receptor (IgG BCR) is indicative for an activated state of circulating B cells in multiple myeloma. Ann. Hematol. 2007, 86, 905–912. [Google Scholar] [CrossRef]
- Leite, L.A.; Kerbauy, D.M.; Kimura, E.; Yamamoto, M. Multiples aberrant phenotypes in multiple myeloma patient expressing CD56(-), CD28(+),CD19(+). Rev. Bras. Hematol. Hemoter. 2012, 34, 66–67. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Horna, P.; Nowakowski, G.; Endell, J.; Boxhammer, R. Comparative Assessment of Surface CD19 and CD20 Expression on B-Cell Lymphomas from Clinical Biopsies: Implications for Targeted Therapies. Blood 2019, 34, 627. [Google Scholar] [CrossRef]
- Radhakrishnan, V.S.; Lokireddy, P.; Parihar, M.; Prakash, P.S.; Menon, H. Mantle cell lymphoma: A clinical review of the changing treatment paradigms with the advent of novel therapies, and an insight into Indian data. Cancer Rep. 2021, 5, e1590. [Google Scholar] [CrossRef] [PubMed]
- Cheah, C.Y.; Zucca, E.; Rossi, D.; Habermann, T.M. Marginal zone lymphoma: Present status and future perspectives. Haematologica 2022, 107, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.; Arber, D.A.; Chang, K.L.; Wilson, C.S.; Weiss, L.M. Frequency of bcl-2 expression in non-Hodgkin’s lymphoma: A study of 778 cases with comparison of marginal zone lymphoma and monocytoid B-cell hyperplasia. Mod. Pathol. 1998, 11, 864–869. [Google Scholar] [PubMed]
- Wada, F.; Shimomura, Y.; Kamijo, K.; Yamashita, D.; Ohno, A.; Himeno, M.; Maruoka, H.; Hara, S.; Ishikawa, T. Prognostic impact of CD38 expression in relapsed or refractory diffuse large B-cell lymphoma and follicular lymphoma transformation. Leuk. Lymphoma 2022, 63, 1484–1487. [Google Scholar] [CrossRef]
- Diepstraten, S.T.; Chang, C.; Tai, L.; Gong, J.N.; Lan, P.; Dowell, A.C.; Taylor, G.S.; Strasser, A.; Kelly, G.L. BCL-W is dispensable for the sustained survival of select Burkitt lymphoma and diffuse large B-cell lymphoma cell lines. Blood Adv. 2020, 4, 356–366. [Google Scholar] [CrossRef]
- Nichols, G.L.; Stein, C.A. Modulation of the activity of Bcl-2 in Waldenstrom’s macroglobulinemia using antisense oligonucleotides. Semin. Oncol. 2003, 30, 297–299. [Google Scholar] [CrossRef]
- Pratt, G.; El-Sharkawi, D.; Kothari, J.; D’Sa, S.; Auer, R.; McCarthy, H.; Krishna, R.; Miles, O.; Kyriakou, C.; Owen, R. Diagnosis and management of Waldenström macroglobulinaemia-A British Society for Haematology guideline. Br. J. Haematol. 2022, 197, 171–187. [Google Scholar] [CrossRef]
- Lew, T.E.; Seymour, J.F. Clinical experiences with venetoclax and other pro-apoptotic agents in lymphoid malignancies: Lessons from monotherapy and chemotherapy combination. J. Hematol. Oncol. 2022, 15, 75. [Google Scholar] [CrossRef]
- Zhu, H.; Almasan, A. Development of venetoclax for therapy of lymphoid malignancies. Drug Des. Dev. Ther. 2017, 11, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Hafezi, S.; Rahmani, M. Targeting BCL-2 in Cancer: Advances, Challenges, and Perspectives. Cancers 2021, 13, 1292. [Google Scholar] [CrossRef]
- Ferrarini, I.; Rigo, A.; Visco, C. The mitochondrial anti-apoptotic dependencies of hematologic malignancies: From disease biology to advances in precision medicine. Haematologica 2022, 107, 790–802. [Google Scholar] [CrossRef]
- Wierda, W.G.; Allan, J.N.; Siddiqi, T.; Kipps, T.J.; Opat, S.; Tedeschi, A.; Badoux, X.C.; Kuss, B.J.; Jackson, S.; Moreno, C.; et al. Ibrutinib Plus Venetoclax for First-Line Treatment of Chronic Lymphocytic Leukemia: Primary Analysis Results From the Minimal Residual Disease Cohort of the Randomized Phase II CAPTIVATE Study. J. Clin. Oncol. 2021, 39, 3853–3865. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Keating, M.; Thompson, P.; Ferrajoli, A.; Burger, J.; Borthakur, G.; Takahashi, K.; Estrov, Z.; Fowler, N.; Kadia, T.; et al. Ibrutinib and Venetoclax for First-Line Treatment of CLL. N. Engl. J. Med. 2019, 380, 2095–2103. [Google Scholar] [CrossRef]
- Davids, M.S.; Roberts, A.W.; Seymour, J.F.; Pagel, J.M.; Kahl, B.S.; Wierda, W.G.; Puvvada, S.; Kipps, T.J.; Anderson, M.A.; Salem, A.H.; et al. Phase I First-in-Human Study of Venetoclax in Patients With Relapsed or Refractory Non-Hodgkin Lymphoma. J. Clin. Oncol. 2017, 35, 826–833. [Google Scholar] [CrossRef]
- Wang, M.; Ramchandren, R.; Chen, R.; Karlin, L.; Chong, G.; Jurczak, W.; Wu, K.L.; Bishton, M.; Collins, G.P.; Eliadis, P.; et al. Concurrent ibrutinib plus venetoclax in relapsed/refractory mantle cell lymphoma: The safety run-in of the phase 3 SYMPATICO study. J. Hematol. Oncol. 2021, 14, 179. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Harrison, S.J.; Cavo, M.; de la Rubia, J.; Popat, R.; Gasparetto, C.; Hungria, V.; Salwender, H.; Suzuki, K.; Kim, I.; et al. Venetoclax or placebo in combination with bortezomib and dexamethasone in patients with relapsed or refractory multiple myeloma (BELLINI): A randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2020, 21, 1630–1642. [Google Scholar] [CrossRef] [PubMed]
- Morschhauser, F.; Feugier, P.; Flinn, I.W.; Gasiorowski, R.; Greil, R.; Illés, Á.; Johnson, N.A.; Larouche, J.F.; Lugtenburg, P.J.; Patti, C.; et al. A phase 2 study of venetoclax plus R-CHOP as first-line treatment for patients with diffuse large B-cell lymphoma. Blood 2021, 137, 600–609. [Google Scholar] [CrossRef]
- Del Gaizo Moore, V.; Schlis, K.D.; Sallan, S.E.; Armstrong, S.A.; Letai, A. BCL-2 dependence and ABT-737 sensitivity in acute lymphoblastic leukemia. Blood 2008, 111, 2300–2309. [Google Scholar] [CrossRef]
- Alford, S.E.; Kothari, A.; Loeff, F.C.; Eichhorn, J.M.; Sakurikar, N.; Goselink, H.M.; Saylors, R.L.; Jedema, I.; Falkenburg, J.H.; Chambers, T.C. BH3 Inhibitor Sensitivity and Bcl-2 Dependence in Primary Acute Lymphoblastic Leukemia Cells. Cancer Res. 2015, 75, 1366–1375. [Google Scholar] [CrossRef] [PubMed]
- Gibson, A.; Trabal, A.; McCall, D.; Khazal, S.; Toepfer, L.; Bell, D.H.; Roth, M.; Mahadeo, K.M.; Nunez, C.; Short, N.J.; et al. Venetoclax for Children and Adolescents with Acute Lymphoblastic Leukemia and Lymphoblastic Lymphoma. Cancers 2021, 14, 150. [Google Scholar] [CrossRef] [PubMed]
- Pullarkat, V.A.; Lacayo, N.J.; Jabbour, E.; Rubnitz, J.E.; Bajel, A.; Laetsch, T.W.; Leonard, J.; Colace, S.I.; Khaw, S.L.; Fleming, S.A.; et al. Venetoclax and Navitoclax in Combination with Chemotherapy in Patients with Relapsed or Refractory Acute Lymphoblastic Leukemia and Lymphoblastic Lymphoma. Cancer Discov. 2021, 11, 1440–1453. [Google Scholar] [CrossRef] [PubMed]
- Hunger, S.P.; Raetz, E.A. How I treat relapsed acute lymphoblastic leukemia in the pediatric population. Blood 2020, 136, 1803–1812. [Google Scholar] [CrossRef]
- Jain, N.; Stevenson, K.E.; Winer, E.S.; Garcia, J.S.; Stone, R.M.; Jabbour, E.; Ravandi, F.; Stewart, J.M.; Legg, D.R.; Kantarjian, H.M.; et al. A Multicenter Phase I Study Combining Venetoclax with Mini-Hyper-CVD in Older Adults with Untreated and Relapsed/Refractory Acute Lymphoblastic Leukemia. Blood 2019, 134, 3867. [Google Scholar] [CrossRef]
- Tam, C.S.; Verner, E.; Lasica, M.; Arbelaez, A.; Browett, P.J.; Soumerai, J.D.; Hilger, J.; Fang, Y.; Huang, J.; Simpson, D.; et al. Preliminary Safety and Efficacy Data from Patients (Pts) with Relapsed/Refractory (R/R) B-Cell Malignancies Treated with the Novel B-Cell Lymphoma 2 (BCL2) Inhibitor BGB-11417 in Monotherapy or in Combination with Zanubrutinib. Blood 2021, 138, 1419. [Google Scholar] [CrossRef]
- Sun, M.; Qi, J.; Chen, Z.; Zhang, H.; Song, Y.; Shen, A.; Liu, H.; Huang, J.; Zhou, F.; Jin, J.; et al. A Phase 1 Study to Evaluate the Safety, Pharmacokinetics (PK) and Pharmacodynamics (PD) of Lisaftoclax (APG-2575), a Novel BCL-2 Inhibitor (BCL-2i), in Patients (pts) with Certain Relapsed or Refractory (R/R) Hematologic Malignancies (HMs). Blood 2021, 138, 3730. [Google Scholar] [CrossRef]
- Yap, J.L.; Chen, L.; Lanning, M.E.; Fletcher, S. Expanding the Cancer Arsenal with Targeted Therapies: Disarmament of the Antiapoptotic Bcl-2 Proteins by Small Molecules. J. Med. Chem. 2017, 60, 821–838. [Google Scholar] [CrossRef]
- Wan, Y.; Dai, N.; Tang, Z.; Fang, H. Small-molecule Mcl-1 inhibitors: Emerging anti-tumor agents. Eur. J. Med. Chem. 2018, 146, 471–482. [Google Scholar] [CrossRef]
- Hird, A.W.; Tron, A.E. Recent advances in the development of Mcl-1 inhibitors for cancer therapy. Pharmacol. Ther. 2019, 198, 59–67. [Google Scholar] [CrossRef]
- Caenepeel, S.; Brown, S.P.; Belmontes, B.; Moody, G.; Keegan, K.S.; Chui, D.; Whittington, D.A.; Huang, X.; Poppe, L.; Cheng, A.C.; et al. AMG 176, a Selective MCL1 Inhibitor, Is Effective in Hematologic Cancer Models Alone and in Combination with Established Therapies. Cancer Discov. 2018, 8, 1582–1597. [Google Scholar] [CrossRef]
- Caenepeel, S.; Karen, R.; Belmontes, B.; Verlinsky, A.; Tan, H.; Yang, Y.; Chen, X.; Li, K.; Allen, J.; Wahlstrom, J.; et al. Discovery and preclinical evaluation of AMG 397, a potent, selective and orally bioavailable MCL1 inhibitor. Cancer Res. 2020, 180, 6218. [Google Scholar] [CrossRef]
- Szlavik, Z.; Csekei, M.; Paczal, A.; Szabo, Z.B.; Sipos, S.; Radics, G.; Proszenyak, A.; Balint, B.; Murray, J.; Davidson, J.; et al. Discovery of S64315, a Potent and Selective Mcl-1 Inhibitor. J. Med. Chem. 2020, 63, 13762–13795. [Google Scholar] [CrossRef] [PubMed]
- Tron, A.E.; Belmonte, M.A.; Adam, A.; Aquila, B.M.; Boise, L.H.; Chiarparin, E.; Cidado, J.; Embrey, K.J.; Gangl, E.; Gibbons, F.D.; et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat. Commun. 2018, 9, 5341. [Google Scholar] [CrossRef] [PubMed]
- Deak, D.; Pop, C.; Zimta, A.A.; Jurj, A.; Ghiaur, A.; Pasca, S.; Teodorescu, P.; Dascalescu, A.; Antohe, I.; Ionescu, B.; et al. Let’s Talk About BiTEs and Other Drugs in the Real-Life Setting for B-Cell Acute Lymphoblastic Leukemia. Front. Immunol. 2019, 10, 2856. [Google Scholar] [CrossRef] [PubMed]
- Hodby, K.A.; Marks, D.I. Recent Advances in the Management of Acute Lymphoblastic Leukaemia. Curr. Treat. Options Oncol. 2020, 21, 23. [Google Scholar] [CrossRef] [PubMed]
- Ratti, S.; Lonetti, A.; Follo, M.Y.; Paganelli, F.; Martelli, A.M.; Chiarini, F.; Evangelisti, C. B-ALL Complexity: Is Targeted Therapy Still A Valuable Approach for Pediatric Patients? Cancers 2020, 12, 3498. [Google Scholar] [CrossRef] [PubMed]
- Si Lim, S.J.; Grupp, S.A.; DiNofia, A.M. Tisagenlecleucel for treatment of children and young adults with relapsed/refractory B-cell acute lymphoblastic leukemia. Pediatr. Blood Cancer 2021, 68, e29123. [Google Scholar] [CrossRef] [PubMed]
- Zinzani, P.L.; Minotti, G. Anti-CD19 monoclonal antibodies for the treatment of relapsed or refractory B-cell malignancies: A narrative review with focus on diffuse large B-cell lymphoma. J. Cancer Res. Clin. Oncol. 2022, 148, 177–190. [Google Scholar] [CrossRef]
- Sanford, D.S.; Wierda, W.G.; Burger, J.A.; Keating, M.J.; O’Brien, S.M. Three newly approved drugs for chronic lymphocytic leukemia: Incorporating ibrutinib, idelalisib, and obinutuzumab into clinical practice. Clin. Lymphoma Myeloma Leuk. 2015, 15, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Bewarder, M.; Stilgenbauer, S.; Thurner, L.; Kaddu-Mulindwa, D. Current Treatment Options in CLL. Cancers 2021, 13, 2468. [Google Scholar] [CrossRef] [PubMed]
- Eichhorst, B.; Robak, T.; Montserrat, E.; Ghia, P.; Niemann, C.U.; Kater, A.P.; Gregor, M.; Cymbalista, F.; Buske, C.; Hillmen, P.; et al. Chronic lymphocytic leukaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2021, 32, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Hallek, M.; Al-Sawaf, O. Chronic lymphocytic leukemia: 2022 update on diagnostic and therapeutic procedures. Am. J. Hematol. 2021, 96, 1679–1705. [Google Scholar] [CrossRef]
- Patel, K.; Pagel, J.M. Current and future treatment strategies in chronic lymphocytic leukemia. J. Hematol. Oncol. 2021, 14, 69. [Google Scholar] [CrossRef]
- Marchetti, M.; Rivela, P.; Bertassello, C.; Canicattì, M. Comparative Clinical Value of Pharmacologic Therapies for B-Cell Chronic Lymphocytic Leukemia: An Umbrella Analysis. J. Clin. Med. 2022, 11, 1868. [Google Scholar] [CrossRef] [PubMed]
- Kay, N.E.; Hampel, P.J.; Van Dyke, D.L.; Parikh, S.A. CLL update 2022: A continuing evolution in care. Blood Rev 2022, 54, 100930. [Google Scholar] [CrossRef] [PubMed]
- Small, S.; Ma, S. Frontline Treatment for Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma (CLL/SLL): Targeted Therapy vs. Chemoimmunotherapy. Curr. Hematol. Malig. Rep. 2021, 16, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Sullivan-Chang, L.; O’Donnell, R.T.; Tuscano, J.M. Targeting CD22 in B-cell malignancies: Current status and clinical outlook. BioDrugs 2013, 27, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Kreitman, R.J.; Dearden, C.; Zinzani, P.L.; Delgado, J.; Robak, T.; le Coutre, P.D.; Gjertsen, B.T.; Troussard, X.; Roboz, G.J.; Karlin, L.; et al. Moxetumomab pasudotox in heavily pre-treated patients with relapsed/refractory hairy cell leukemia (HCL): Long-term follow-up from the pivotal trial. J. Hematol. Oncol. 2021, 14, 35. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, S.; Rendón-Huerta, E.P.; Ortiz-Navarrete, V.; Montaño, L.F. CD38 and Regulation of the Immune Response Cells in Cancer. J. Oncol. 2021, 2021, 6630295. [Google Scholar] [CrossRef]
- Jiao, Y.; Yi, M.; Xu, L.; Chu, Q.; Yan, Y.; Luo, S.; Wu, K. CD38: Targeted therapy in multiple myeloma and therapeutic potential for solid cancers. Expert Opin. Investig. Drugs 2020, 29, 1295–1308. [Google Scholar] [CrossRef] [PubMed]
- Abramson, H.N. B-Cell Maturation Antigen (BCMA) as a Target for New Drug Development in Relapsed and/or Refractory Multiple Myeloma. Int. J. Mol. Sci. 2020, 21, 5192. [Google Scholar] [CrossRef] [PubMed]
- Hanel, W.; Epperla, N. Evolving therapeutic landscape in follicular lymphoma: A look at emerging and investigational therapies. J. Hematol. Oncol. 2021, 14, 104. [Google Scholar] [CrossRef]
- Pongas, G.; Cheson, B. Recent Advances in the Management of Patients with Relapsed/Refractory Follicular Lymphoma. Blood Lymphat. Cancer 2021, 11, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Cahill, K.E.; Smith, S.M. Follicular Lymphoma: A Focus on Current and Emerging Therapies. Oncology 2022, 36, 97–106. [Google Scholar] [PubMed]
- Qualls, D.; Salles, G. Prospects in the management of patients with follicular lymphoma beyond first-line therapy. Haematologica 2022, 107, 19–34. [Google Scholar] [CrossRef]
- Narkhede, M.; Cheson, B.D. Copanlisib in the treatment of non-Hodgkin lymphoma. Future Oncol. 2020, 16, 1947–1955. [Google Scholar] [CrossRef]
- Salles, G.; Gopal, A.K.; Minnema, M.C.; Wakamiya, K.; Feng, H.; Schecter, J.M.; Wang, M. Phase 2 Study of Daratumumab in Relapsed/Refractory Mantle-Cell Lymphoma, Diffuse Large B-Cell Lymphoma, and Follicular Lymphoma. Clin. Lymphoma Myeloma Leuk. 2019, 19, 275–284. [Google Scholar] [CrossRef]
- Pu, J.J.; Savani, M.; Huang, N.; Epner, E.M. Mantle cell lymphoma management trends and novel agents: Where are we going? Ther. Adv. Hematol. 2022, 13, 20406207221080743. [Google Scholar] [CrossRef] [PubMed]
- Alderuccio, J.P.; Kahl, B.S. Current Treatments in Marginal Zone Lymphoma. Oncology 2022, 36, 206–215. [Google Scholar]
- Chilakamarri, N.O.K.; Brem, E.A. Current and Future Therapies for Marginal Zone Lymphoma. Oncol. Haematol. 2022, 2022, 18. [Google Scholar] [CrossRef]
- Opat, S.; Tedeschi, A.; Linton, K.; McKay, P.; Hu, B.; Chan, H.; Jin, J.; Sobieraj-Teague, M.; Zinzani, P.L.; Coleman, M.; et al. The MAGNOLIA Trial: Zanubrutinib, a Next-Generation Bruton Tyrosine Kinase Inhibitor, Demonstrates Safety and Efficacy in Relapsed/Refractory Marginal Zone Lymphoma. Clin. Cancer Res. 2021, 27, 6323–6332. [Google Scholar] [CrossRef] [PubMed]
- Micallef, I.N.; Maurer, M.J.; Wiseman, G.A.; Nikcevich, D.A.; Kurtin, P.J.; Cannon, M.W.; Perez, D.G.; Soori, G.S.; Link, B.K.; Habermann, T.M.; et al. Epratuzumab with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone chemotherapy in patients with previously untreated diffuse large B-cell lymphoma. Blood 2011, 118, 4053–4061. [Google Scholar] [CrossRef]
- Cheson, B.D.; Nowakowski, G.; Salles, G. Diffuse large B-cell lymphoma: New targets and novel therapies. Blood Cancer J. 2021, 11, 68. [Google Scholar] [CrossRef] [PubMed]
- Spinner, M.A.; Advani, R.H. Current Frontline Treatment of Diffuse Large B-Cell Lymphoma. Oncology 2022, 36, 51–58. [Google Scholar]
- Davis, J.A.; Shockley, A.; Glode, A.E. Newly approved anti-CD19 monoclonal antibodies for the treatment of relapsed or refractory diffuse large B-cell lymphoma. J. Oncol. Pharm. Pract. 2022, 28, 686–690. [Google Scholar] [CrossRef]
- Kahl, B.S.; Hamadani, M.; Radford, J.; Carlo-Stella, C.; Caimi, P.; Reid, E.; Feingold, J.M.; Ardeshna, K.M.; Solh, M.; Heffner, L.T.; et al. A Phase I Study of ADCT-402 (Loncastuximab Tesirine), a Novel Pyrrolobenzodiazepine-Based Antibody-Drug Conjugate, in Relapsed/Refractory B-Cell Non-Hodgkin Lymphoma. Clin. Cancer Res. 2019, 25, 6986–6994. [Google Scholar] [CrossRef]
- Castillo, J.J.; Advani, R.H.; Branagan, A.R.; Buske, C.; Dimopoulos, M.A.; D’Sa, S.; Kersten, M.J.; Leblond, V.; Minnema, M.C.; Owen, R.G.; et al. Consensus treatment recommendations from the tenth International Workshop for Waldenström Macroglobulinaemia. Lancet Haematol. 2020, 7, e827–e837. [Google Scholar] [CrossRef]
- Grimont, C.N.; Castillo Almeida, N.E.; Gertz, M.A. Current and Emerging Treatments for Waldenström Macroglobulinemia. Acta Haematol. 2021, 144, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.K. SOHO State of the Art Updates and Next Questions: Waldenström Macroglobulinemia-2021 Update on Management and Future Directions. Clin. Lymphoma Myeloma Leuk. 2022, 22, 347–355. [Google Scholar] [CrossRef]
- Gertz, M.A. Waldenström macroglobulinemia: 2021 update on diagnosis, risk stratification, and management. Am. J. Hematol. 2021, 96, 258–269. [Google Scholar] [CrossRef]
- Castillo, J.J.; Treon, S.P. Management of Waldenström macroglobulinemia in 2020. Hematol. Am. Soc. Hematol. Educ. Program 2020, 1, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhou, K.; Nian, W.; Cui, G.; Wang, J.; Zhang, X.; Cen, H.; Li, F.; Yi, S.; Feng, R.; et al. A phase Ib/II study of lisaftoclax (APG-2575), a novel BCL-2 inhibitor (BCL-2i), in patients (pts) with relapsed/refractory chronic lymphocytic leukemia or small lymphocytic lymphoma (R/R CLL/SLL). J. Clin. Oncol. 2022, 40, 7543. [Google Scholar] [CrossRef]
- Bhagwat, N.; Grego, A.; Gowen-MacDonald, W.; Wang, M.; Cowart, M.; Wu, X.; Zhuo, J.; Combs, A.; Ruggeri, B.; Scherle, P.; et al. Preclinical characterization of PRT1419, a potent, selective and orally available inhibitor of MCL1. Cancer Res. 2021, 81, 983. [Google Scholar] [CrossRef]
- Dhillon, S. Orelabrutinib: First Approval. Drugs 2021, 81, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Jurczak, W.; Zinzani, P.L.L.; Cunningham, D.; Azoulay, M.; Huang, W.; Xu, W.; Ribrag, V. Coastal: A phase 3 study of the PI3Kδ inhibitor zandelisib with rituximab (R) versus immunochemotherapy in patients with relapsed indolent non-Hodgkin’s lymphoma (iNHL). J. Clin. Oncol. 2021, 39, 7573. [Google Scholar] [CrossRef]
- Davids, M.S.; Kim, H.T.; Nicotra, A.; Savell, A.; Francoeur, K.; Hellman, J.M.; Bazemore, J.; Miskin, H.P.; Sportelli, P.; Stampleman, L.; et al. Umbralisib in combination with ibrutinib in patients with relapsed or refractory chronic lymphocytic leukaemia or mantle cell lymphoma: A multicentre phase 1-1b study. Lancet Haematol. 2019, 6, e38–e47. [Google Scholar] [CrossRef]
- Budde, L.E.; Assouline, S.; Sehn, L.H.; Schuster, S.J.; Yoon, S.S.; Yoon, D.H.; Matasar, M.J.; Bosch, F.; Kim, W.S.; Nastoupil, L.J.; et al. Single-Agent Mosunetuzumab Shows Durable Complete Responses in Patients With Relapsed or Refractory B-Cell Lymphomas: Phase I Dose-Escalation Study. J. Clin. Oncol. 2022, 40, 481–491. [Google Scholar] [CrossRef]
- Bannerji, R.; Allan, J.N.; Arnason, J.E.; Brown, J.R.; Advani, R.H.; Barnes, J.A.; Ansell, S.M.; O’Brien, S.M.; Chavez, J.; Duell, J.; et al. Clinical Activity of REGN1979, a Bispecific Human, Anti-CD20 x Anti-CD3 Antibody, in Patients with Relapsed/Refractory (R/R) B-Cell Non-Hodgkin Lymphoma (B-NHL). Blood 2019, 134, 762. [Google Scholar] [CrossRef]
- Van der Horst, H.J.; de Jonge, A.V.; Hiemstra, I.H.; Gelderloos, A.T.; Berry, D.; Hijmering, N.J.; van Essen, H.F.; de Jong, D.; Chamuleau, M.E.D.; Zweegman, S.; et al. Epcoritamab induces potent anti-tumor activity against malignant B-cells from patients with DLBCL, FL and MCL, irrespective of prior CD20 monoclonal antibody treatment. Blood Cancer J. 2021, 11, 38. [Google Scholar] [CrossRef] [PubMed]
- Engelberts, P.J.; Hiemstra, I.H.; de Jong, B.; Schuurhuis, D.H.; Meesters, J.; Beltran Hernandez, I.; Oostindie, S.C.; Neijssen, J.; van den Brink, E.N.; Horbach, G.J.; et al. DuoBody-CD3xCD20 induces potent T-cell-mediated killing of malignant B cells in preclinical models and provides opportunities for subcutaneous dosing. eBioMedicine 2020, 52, 102625. [Google Scholar] [CrossRef] [PubMed]
- Hutchings, M.; Morschhauser, F.; Iacoboni, G.; Carlo-Stella, C.; Offner, F.C.; Sureda, A.; Salles, G.; Martínez-Lopez, J.; Crump, M.; Thomas, D.N.; et al. Glofitamab, a Novel, Bivalent CD20-Targeting T-Cell-Engaging Bispecific Antibody, Induces Durable Complete Remissions in Relapsed or Refractory B-Cell Lymphoma: A Phase I Trial. J. Clin. Oncol. 2021, 39, 1959–1970. [Google Scholar] [CrossRef] [PubMed]
- Salles, G.; Duell, J.; González Barca, E.; Tournilhac, O.; Jurczak, W.; Liberati, A.M.; Nagy, Z.; Obr, A.; Gaidano, G.; André, M.; et al. Tafasitamab plus lenalidomide in relapsed or refractory diffuse large B-cell lymphoma (L-MIND): A multicentre, prospective, single-arm, phase 2 study. Lancet Oncol. 2020, 21, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.N.; Johnson, B.D.; Schneider, D.; Zhu, F.; Szabo, A.; Keever-Taylor, C.A.; Krueger, W.; Worden, A.A.; Kadan, M.J.; Yim, S.; et al. Bispecific anti-CD20, anti-CD19 CAR T cells for relapsed B cell malignancies: A phase 1 dose escalation and expansion trial. Nat. Med. 2020, 26, 1569–1575. [Google Scholar] [CrossRef]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, M.; Hamad, N.; Bryant, C.E.; Borthakur, G.; Hosing, C.; Shook, D.; Tan, J.; Rajangam, K.; Liu, H.; Kennedy, G.A.; et al. A Phase 1 Study of NKX019, a CD19 Chimeric Antigen Receptor Natural Killer (CAR NK) Cell Therapy, in Subjects with B-Cell Malignancies. Blood 2021, 138, 3868. [Google Scholar] [CrossRef]
- Cheng, Q.; Tan, J.; Liu, R.; Kang, L.; Zhang, Y.; Wang, E.; Li, Y.; Zhang, J.; Xiao, H.; Xu, N.; et al. CD20-specific chimeric antigen receptor-expressing T cells as salvage therapy in rituximab-refractory/relapsed B-cell non-Hodgkin lymphoma. Cytotherapy 2022, 24, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018, 24, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.N.; Zhu, F.; Taylor, C.; Schneider, D.; Krueger, W.; Worden, A.; Yim, S.; Fenske, T.S.; Hamadani, M.; Johnson, B.; et al. A Phase 1 Study with Point-of-Care Manufacturing of Dual Targeted, Tandem Anti-CD19, Anti-CD20 Chimeric Antigen Receptor Modified T (CAR-T) Cells for Relapsed, Refractory, Non-Hodgkin Lymphoma. Blood 2018, 132, 4193. [Google Scholar] [CrossRef]
- Osborne, W.; Marzolini, M.; Tholouli, E.; Ramakrishnan, A.; Bachier, C.R.; McSweeney, P.A.; Irvine, D.; Zhang, M.; Al-Hajj, M.A.; Pule, M.; et al. Phase I Alexander study of AUTO3, the first CD19/22 dual targeting CAR T cell therapy, with pembrolizumab in patients with relapsed/refractory (r/r) DLBCL. J. Clin. Oncol. 2020, 38, 8001. [Google Scholar] [CrossRef]
- Spiegel, J.Y.; Patel, S.; Muffly, L.; Hossain, N.M.; Oak, J.; Baird, J.H.; Frank, M.J.; Shiraz, P.; Sahaf, B.; Craig, J.; et al. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: A phase 1 trial. Nat. Med. 2021, 27, 1419–1431. [Google Scholar] [CrossRef]
- Shalabi, H.; Nellan, A.; Shah, N.N.; Gust, J. Immunotherapy Associated Neurotoxicity in Pediatric Oncology. Front. Oncol. 2022, 12, 836452. [Google Scholar] [CrossRef] [PubMed]
- Vogl, D.T.; Kaufman, J.L.; Holstein, S.A.; Atrash, S.; Nadeem, O.; Janakiram, M.; Suryanarayan, K.; Liu, Y.; Collins, S.; Parot, X.; et al. Modakafusp Alfa (TAK-573), an Immunocytokine, Shows Clinical Activity in Patients with Relapsed/Refractory Multiple Myeloma; Updated Results from a First-in-Human Phase 1 Study. Blood 2021, 138, 898. [Google Scholar] [CrossRef]
- Kumar, S.A.M.; Dabovic, K.; Wang, J.; Anand, B.; Yuet, A.; Dholaria, B.; Roy, V. 447 Interim results of a phase 1 study of the novel engineered toxin body TAK-169 in patients with relapsed or refractory multiple myeloma. J. Immunother. Cancer 2021, 9, A475. [Google Scholar] [CrossRef]
- Byrd, J.C.; Hillmen, P.; Ghia, P.; Kater, A.P.; Chanan-Khan, A.; Furman, R.R.; O’Brien, S.; Yenerel, M.N.; Illés, A.; Kay, N.; et al. Acalabrutinib Versus Ibrutinib in Previously Treated Chronic Lymphocytic Leukemia: Results of the First Randomized Phase III Trial. J. Clin. Oncol. 2021, 39, 3441–3452. [Google Scholar] [CrossRef]
- Danilov, A.V.; Persky, D.O. Incorporating acalabrutinib, a selective next-generation Bruton tyrosine kinase inhibitor, into clinical practice for the treatment of haematological malignancies. Br. J. Haematol. 2021, 193, 15–25. [Google Scholar] [CrossRef]
- Zhu, S.; Jung, J.; Victor, E.; Arceo, J.; Gokhale, S.; Xie, P. Clinical Trials of the BTK Inhibitors Ibrutinib and Acalabrutinib in Human Diseases Beyond B Cell Malignancies. Front. Oncol. 2021, 11, 737943. [Google Scholar] [CrossRef]
- Ran, F.; Liu, Y.; Wang, C.; Xu, Z.; Zhang, Y.; Liu, Y.; Zhao, G.; Ling, Y. Review of the development of BTK inhibitors in overcoming the clinical limitations of ibrutinib. Eur. J. Med. Chem. 2022, 229, 114009. [Google Scholar] [CrossRef]
- Muñoz, J.; Wang, Y.; Jain, P.; Wang, M. Zanubrutinib in lymphoproliferative disorders: A comprehensive review. Ther. Adv. Hematol. 2022, 13, 20406207221093980. [Google Scholar] [CrossRef]
- Campbell, R.; Chong, G.; Hawkes, E.A. Novel Indications for Bruton’s Tyrosine Kinase Inhibitors, beyond Hematological Malignancies. J. Clin. Med. 2018, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Von Suskil, M.; Sultana, K.N.; Elbezanti, W.O.; Al-Odat, O.S.; Chitren, R.; Tiwari, A.K.; Challagundla, K.B.; Srivastava, S.K.; Jonnalagadda, S.C.; Budak-Alpdogan, T.; et al. Bruton’s Tyrosine Kinase Targeting in Multiple Myeloma. Int. J. Mol. Sci. 2021, 22, 5707. [Google Scholar] [CrossRef]
- Wilson, W.H.; Wright, G.W.; Huang, D.W.; Hodkinson, B.; Balasubramanian, S.; Fan, Y.; Vermeulen, J.; Shreeve, M.; Staudt, L.M. Effect of ibrutinib with R-CHOP chemotherapy in genetic subtypes of DLBCL. Cancer Cell 2021, 39, 1643–1653.e3. [Google Scholar] [CrossRef] [PubMed]
- Gopal, A.K.; Schuster, S.J.; Fowler, N.H.; Trotman, J.; Hess, G.; Hou, J.Z.; Yacoub, A.; Lill, M.; Martin, P.; Vitolo, U.; et al. Ibrutinib as Treatment for Patients With Relapsed/Refractory Follicular Lymphoma: Results From the Open-Label, Multicenter, Phase II DAWN Study. J. Clin. Oncol. 2018, 36, 2405–2412. [Google Scholar] [CrossRef] [PubMed]
- Mozas, P.; Delgado, J. Zanubrutinib joins the CLL treatment buffet. Lancet Oncol. 2022, 23, 965–967. [Google Scholar] [CrossRef]
- Zinzani, P.L.; Mayer, J.; Auer, R.; Bijou, F.; de Oliveira, A.C.; Flowers, C.; Merli, M.; Bouabdallah, K.; Ganly, P.S.; Johnson, R.; et al. Zanubrutinib plus obinutuzumab (ZO) versus obinutuzumab (O) monotherapy in patients (pts) with relapsed or refractory (R/R) follicular lymphoma (FL): Primary analysis of the phase 2 randomized ROSEWOOD trial. J. Clin. Oncol. 2022, 40, 7510. [Google Scholar] [CrossRef]
- Yang, H.; Xiang, B.; Song, Y.; Zhang, H.; Zhao, W.; Zou, D.; Lv, F.; Guo, W.; Liu, A.; Li, C.; et al. Zanubrutinib monotherapy for relapsed or refractory non-germinal center diffuse large B-cell lymphoma. Blood Adv. 2022, 6, 1629–1636. [Google Scholar] [CrossRef]
- Ferrer, G.; Montserrat, E. Critical molecular pathways in CLL therapy. Mol. Med. 2018, 24, 9. [Google Scholar] [CrossRef]
- Byrd, J.C.; Smith, S.; Wagner-Johnston, N.; Sharman, J.; Chen, A.I.; Advani, R.; Augustson, B.; Marlton, P.; Renee Commerford, S.; Okrah, K.; et al. First-in-human phase 1 study of the BTK inhibitor GDC-0853 in relapsed or refractory B-cell NHL and CLL. Oncotarget 2018, 9, 13023–13035. [Google Scholar] [CrossRef]
- Phillips, T.J.; Michot, J.M.; Ribrag, V. Can Next-Generation PI3K Inhibitors Unlock the Full Potential of the Class in Patients With B-Cell Lymphoma? Clin. Lymphoma Myeloma Leuk. 2021, 21, 8–20.e3. [Google Scholar] [CrossRef] [PubMed]
- Hus, I.; Puła, B.; Robak, T. PI3K Inhibitors for the Treatment of Chronic Lymphocytic Leukemia: Current Status and Future Perspectives. Cancers 2022, 14, 1571. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Barrientos, J.C. Oral PI3K-δ,γ Inhibitor for the Management of People with Chronic Lymphocytic Leukemia and Small Lymphocytic Lymphoma: A Narrative Review on Duvelisib. Onco Targets Ther. 2021, 14, 2109–2119. [Google Scholar] [CrossRef] [PubMed]
- Shouse, G.; Danilova, O.V.; Danilov, A.V. Current status of phosphoinotiside-3 kinase inhibitors in blood cancers. Curr. Opin. Oncol. 2022, 34, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Le, T.; Jerel, D.; Bryan, L.J. Update on the role of copanlisib in hematologic malignancies. Ther. Adv. Hematol. 2021, 12, 20406207211006027. [Google Scholar] [CrossRef] [PubMed]
- Gopal, A.K.; Kahl, B.S.; de Vos, S.; Wagner-Johnston, N.D.; Schuster, S.J.; Jurczak, W.J.; Flinn, I.W.; Flowers, C.R.; Martin, P.; Viardot, A.; et al. PI3Kδ inhibition by idelalisib in patients with relapsed indolent lymphoma. N. Engl. J. Med. 2014, 370, 1008–1018. [Google Scholar] [CrossRef]
- Chauhan, A.F.; Cheson, B.D. Copanlisib in the Treatment of Relapsed Follicular Lymphoma: Utility and Experience from the Clinic. Cancer Manag. Res. 2021, 13, 677–692. [Google Scholar] [CrossRef]
- Casadei, B.; Argnani, L.; Broccoli, A.; Patti, C.; Stefani, P.M.; Cuneo, A.; Margiotta Casaluci, G.; Visco, C.; Gini, G.; Pane, F.; et al. Treatment with Idelalisib in Patients with Relapsed or Refractory Follicular Lymphoma: The Observational Italian Multicenter FolIdela Study. Cancers 2022, 14, 654. [Google Scholar] [CrossRef]
- Rodrigues, D.A.; Sagrillo, F.S.; Fraga, C.A.M. Duvelisib: A 2018 Novel FDA-Approved Small Molecule Inhibiting Phosphoinositide 3-Kinases. Pharmaceuticals 2019, 12, 69. [Google Scholar] [CrossRef]
- Tucker, N. Duvelisib for R/R Follicular Lymphoma Voluntarily Pulled from the US Market. Target. Oncol. 2021. Available online: https://www.targetedonc.com/view/duvelisib-for-r-r-follicular-lymphoma-voluntarily-pulled-from-the-us-market (accessed on 6 December 2021).
- Matasar, M.J.; Capra, M.; Özcan, M.; Lv, F.; Li, W.; Yañez, E.; Sapunarova, K.; Lin, T.; Jin, J.; Jurczak, W.; et al. Copanlisib plus rituximab versus placebo plus rituximab in patients with relapsed indolent non-Hodgkin lymphoma (CHRONOS-3): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2021, 22, 678–689. [Google Scholar] [CrossRef]
- Forero-Torres, A.; Ramchandren, R.; Yacoub, A.; Wertheim, M.S.; Edenfield, W.J.; Caimi, P.; Gutierrez, M.; Akard, L.; Escobar, C.; Call, J.; et al. Parsaclisib, a potent and highly selective PI3Kδ inhibitor, in patients with relapsed or refractory B-cell malignancies. Blood 2019, 133, 1742–1752. [Google Scholar] [CrossRef] [PubMed]
- Coleman, M.; Belada, D.; Casasnovas, R.O.; Gressin, R.; Lee, H.P.; Mehta, A.; Munoz, J.; Verhoef, G.; Corrado, C.; DeMarini, D.J.; et al. Phase 2 study of parsaclisib (INCB050465), a highly selective, next-generation PI3Kδ inhibitor, in relapsed or refractory diffuse large B-cell lymphoma (CITADEL-202). Leuk Lymphoma 2021, 62, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Joszt, L. Incyte Withdraws FDA Submission for Parsaclisib for FL, MZL, MCL. AJMC 2022. Available online: https://www.ajmc.com/view/incyte-withdraws-fda-submission-for-parsaclisib-for-fl-mzl-mcl (accessed on 28 January 2022).
- Pagel, J.M.; Soumerai, J.D.; Reddy, N.; Jagadeesh, D.; Stathis, A.; Asch, A.; Salman, H.; Kenkre, V.P.; Iasonos, A.; Llorin-Sangalang, J.; et al. Zandelisib with continuous or intermittent dosing as monotherapy or in combination with rituximab in patients with relapsed or refractory B-cell malignancy: A multicentre, first-in-patient, dose-escalation and dose-expansion, phase 1b trial. Lancet Oncol. 2022, 23, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Maharaj, K.; Powers, J.J.; Achille, A.; Mediavilla-Varela, M.; Gamal, W.; Burger, K.L.; Fonseca, R.; Jiang, K.; Miskin, H.P.; Maryanski, D.; et al. The dual PI3Kδ/CK1ε inhibitor umbralisib exhibits unique immunomodulatory effects on CLL T cells. Blood Adv. 2020, 4, 3072–3084. [Google Scholar] [CrossRef]
- Burris, H.A., 3rd; Flinn, I.W.; Patel, M.R.; Fenske, T.S.; Deng, C.; Brander, D.M.; Gutierrez, M.; Essell, J.H.; Kuhn, J.G.; Miskin, H.P.; et al. Umbralisib, a novel PI3Kδ and casein kinase-1ε inhibitor, in relapsed or refractory chronic lymphocytic leukaemia and lymphoma: An open-label, phase 1, dose-escalation, first-in-human study. Lancet Oncol. 2018, 19, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Mato, A.R.; Ghosh, N.; Schuster, S.J.; Lamanna, N.; Pagel, J.M.; Flinn, I.W.; Barrientos, J.C.; Rai, K.R.; Reeves, J.A.; Cheson, B.D.; et al. Phase 2 study of the safety and efficacy of umbralisib in patients with CLL who are intolerant to BTK or PI3Kδ inhibitor therapy. Blood 2021, 137, 2817–2826. [Google Scholar] [CrossRef]
- Lunning, M.; Vose, J.; Nastoupil, L.; Fowler, N.; Burger, J.A.; Wierda, W.G.; Schreeder, M.T.; Siddiqi, T.; Flowers, C.R.; Cohen, J.B.; et al. Ublituximab and umbralisib in relapsed/refractory B-cell non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood 2019, 134, 1811–1820. [Google Scholar] [CrossRef]
- Virgil, H. FDA Places Partial Clinical Hold on Studies Featuring Umbralisib and Ublituximab in CLL and Non-Hodgkin Lymphoma. Cancer Netwok 2022. Available online: https://www.cancerneetwork.com/view/fda-places-partial-clinical-hold-on-studies-featuring-umbralisib-and-ublituximab-in-cll-and-non-hodgkin-lymphoma (accessed on 1 February 2022).
- Lu, R.M.; Hwang, Y.C.; Liu, I.J.; Lee, C.C.; Tsai, H.Z.; Li, H.J.; Wu, H.C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef]
- Fishelson, Z.; Kirschfink, M. Complement C5b-9 and Cancer: Mechanisms of Cell Damage, Cancer Counteractions, and Approaches for Intervention. Front. Immunol. 2019, 10, 752. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Kankala, R.K.; Yang, Z.; Li, W.; Xie, S.; Li, H.; Chen, A.Z.; Zou, L. Antibody-based drug delivery systems for cancer therapy: Mechanisms, challenges, and prospects. Theranostics 2022, 12, 3719–3746. [Google Scholar] [CrossRef]
- Hedrich, W.D.; Fandy, T.E.; Ashour, H.M.; Wang, H.; Hassan, H.E. Antibody-Drug Conjugates: Pharmacokinetic/Pharmacodynamic Modeling, Preclinical Characterization, Clinical Studies, and Lessons Learned. Clin. Pharmacokinet. 2018, 57, 687–703. [Google Scholar] [CrossRef] [PubMed]
- Lejeune, M.; Köse, M.C.; Duray, E.; Einsele, H.; Beguin, Y.; Caers, J. Bispecific, T-Cell-Recruiting Antibodies in B-Cell Malignancies. Front. Immunol. 2020, 11, 762. [Google Scholar] [CrossRef] [PubMed]
- Morcos, P.N.; Li, J.; Hosseini, I.; Li, C.C. Quantitative Clinical Pharmacology of T-Cell Engaging Bispecifics: Current Perspectives and Opportunities. Clin. Transl. Sci. 2021, 14, 75–85. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef]
- Spiess, C.; Zhai, Q.; Carter, P.J. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol. Immunol. 2015, 67, 95–106. [Google Scholar] [CrossRef]
- Keane, J.T.; Posey, A.D., Jr. Chimeric Antigen Receptors Expand the Repertoire of Antigenic Macromolecules for Cellular Immunity. Cells 2021, 10, 3356. [Google Scholar] [CrossRef] [PubMed]
- Abrantes, R.; Duarte, H.O.; Gomes, C.; Wälchli, S.; Reis, C.A. CAR-Ts: New perspectives in cancer therapy. FEBS Lett. 2022, 596, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.W.; Bhattarai, N. CAR-T Cell Therapy: Mechanism, Management, and Mitigation of Inflammatory Toxicities. Front. Immunol. 2021, 12, 693016. [Google Scholar] [CrossRef] [PubMed]
- Moreno, C.; Haynie, C.; Johnson, A.; Weber, K.S. Alternative CAR Therapies: Recent Approaches in Engineering Chimeric Antigen Receptor Immune Cells to Combat Cancer. Biomedicines 2022, 10, 1493. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.H.; Beers, S.A.; French, R.R.; Johnson, P.W.; Glennie, M.J.; Cragg, M.S. Anti-CD20 monoclonal antibodies: Historical and future perspectives. Haematologica 2010, 95, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Tuscano, J.M.; Maverakis, E.; Groshen, S.; Tsao-Wei, D.; Luxardi, G.; Merleev, A.A.; Beaven, A.; DiPersio, J.F.; Popplewell, L.; Chen, R.; et al. A Phase I Study of the Combination of Rituximab and Ipilimumab in Patients with Relapsed/Refractory B-Cell Lymphoma. Clin. Cancer Res. 2019, 25, 7004–7013. [Google Scholar] [CrossRef] [PubMed]
- Marshall, M.J.E.; Stopforth, R.J.; Cragg, M.S. Therapeutic Antibodies: What Have We Learnt from Targeting CD20 and Where Are We Going? Front. Immunol. 2017, 8, 1245. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Kumar, N.K.; Dwiwedi, P.; Charan, J.; Kaur, R.; Sidhu, P.; Chugh, V.K. Monoclonal Antibodies: A Review. Curr. Clin. Pharmacol. 2018, 13, 85–99. [Google Scholar] [CrossRef]
- Bannerji, R.; Arnason, J.E.; Advani, R.H.; Brown, J.R.; Allan, J.N.; Ansell, S.M.; Barnes, J.A.; O’Brien, S.M.; Chávez, J.C.; Duell, J.; et al. Odronextamab, a human CD20×CD3 bispecific antibody in patients with CD20-positive B-cell malignancies (ELM-1): Results from the relapsed or refractory non-Hodgkin lymphoma cohort in a single-arm, multicentre, phase 1 trial. Lancet Haematol. 2022, 9, e327–e339. [Google Scholar] [CrossRef]
- Zhao, Z.; Chen, Y.; Francisco, N.M.; Zhang, Y.; Wu, M. The application of CAR-T cell therapy in hematological malignancies: Advantages and challenges. Acta Pharm. Sin. B 2018, 8, 539–551. [Google Scholar] [CrossRef]
- Aleksandrova, K.; Leise, J.; Priesner, C.; Melk, A.; Kubaink, F.; Abken, H.; Hombach, A.; Aktas, M.; Essl, M.; Bürger, I.; et al. Functionality and Cell Senescence of CD4/CD8-Selected CD20 CAR T Cells Manufactured Using the Automated CliniMACS Prodigy® Platform. Transfus. Med. Hemother. 2019, 46, 47–54. [Google Scholar] [CrossRef]
- Naddafi, F.; Davami, F. Anti-CD19 Monoclonal Antibodies: A New Approach to Lymphoma Therapy. Int. J. Mol. Cell. Med. 2015, 4, 143–151. [Google Scholar]
- Watkins, M.P.; Bartlett, N.L. CD19-targeted immunotherapies for treatment of patients with non-Hodgkin B-cell lymphomas. Expert Opin. Investig. Drugs 2018, 27, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Ribera, J.M.; Genescà, E.; Ribera, J. Bispecific T-cell engaging antibodies in B-cell precursor acute lymphoblastic leukemias: Focus on blinatumomab. Ther. Adv. Hematol. 2020, 11, 2040620720919632. [Google Scholar] [CrossRef]
- Xu, B. Loncastuximab tesirine: An effective therapy for relapsed or refractory diffuse large B-cell lymphoma. Eur. J. Clin. Pharmacol. 2022, 78, 707–719. [Google Scholar] [CrossRef]
- Sesques, P.; Ferrant, E.; Safar, V.; Wallet, F.; Tordo, J.; Dhomps, A.; Karlin, L.; Brisou, G.; Vercasson, M.; Hospital-Gustem, C.; et al. Commercial anti-CD19 CAR T cell therapy for patients with relapsed/refractory aggressive B cell lymphoma in a European center. Am. J. Hematol. 2020, 95, 1324–1333. [Google Scholar] [CrossRef]
- Sharma, P.; King, G.T.; Shinde, S.S.; Purev, E.; Jimeno, A. Axicabtagene ciloleucel for the treatment of relapsed/refractory B-cell non-Hodgkin’s lymphomas. Drugs Today 2018, 54, 187–198. [Google Scholar] [CrossRef]
- Abramson, J.S. Anti-CD19 CAR T-Cell Therapy for B-Cell Non-Hodgkin Lymphoma. Transfus. Med. Rev. 2020, 34, 29–33. [Google Scholar] [CrossRef]
- Dhakal, B.; Hari, P.N.; Usmani, S.Z.; Hamadani, M. Chimeric antigen receptor T cell therapy in multiple myeloma: Promise and challenges. Bone Marrow Transplant. 2021, 56, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Rambaldi, A.; Ribera, J.M.; Kantarjian, H.M.; Dombret, H.; Ottmann, O.G.; Stein, A.S.; Tuglus, C.A.; Zhao, X.; Kim, C.; Martinelli, G. Blinatumomab compared with standard of care for the treatment of adult patients with relapsed/refractory Philadelphia chromosome-positive B-precursor acute lymphoblastic leukemia. Cancer 2020, 126, 304–310. [Google Scholar] [CrossRef]
- Horton, H.M.; Bernett, M.J.; Pong, E.; Peipp, M.; Karki, S.; Chu, S.Y.; Richards, J.O.; Vostiar, I.; Joyce, P.F.; Repp, R.; et al. Potent in vitro and in vivo activity of an Fc-engineered anti-CD19 monoclonal antibody against lymphoma and leukemia. Cancer Res. 2008, 68, 8049–8057. [Google Scholar] [CrossRef]
- Awan, F.T.; Lapalombella, R.; Trotta, R.; Butchar, J.P.; Yu, B.; Benson, D.M., Jr.; Roda, J.M.; Cheney, C.; Mo, X.; Lehman, A.; et al. CD19 targeting of chronic lymphocytic leukemia with a novel Fc-domain-engineered monoclonal antibody. Blood 2010, 115, 1204–1213. [Google Scholar] [CrossRef] [PubMed]
- Caimi, P.F.; Ai, W.; Alderuccio, J.P.; Ardeshna, K.M.; Hamadani, M.; Hess, B.; Kahl, B.S.; Radford, J.; Solh, M.; Stathis, A.; et al. Loncastuximab tesirine in relapsed or refractory diffuse large B-cell lymphoma (LOTIS-2): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2021, 22, 790–800. [Google Scholar] [CrossRef]
- Halford, Z.; Anderson, M.K.; Bennett, L.L. Axicabtagene Ciloleucel: Clinical Data for the Use of CAR T-cell Therapy in Relapsed and Refractory Large B-cell Lymphoma. Ann. Pharmacother. 2021, 55, 390–405. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.D.; Bachmeier, C.A.; Phuoc, V.H.; Chavez, J.C. Axicabtagene ciloleucel (KTE-C19), an anti-CD19 CAR T therapy for the treatment of relapsed/refractory aggressive B-cell non-Hodgkin’s lymphoma. Ther. Clin. Risk Manag. 2018, 14, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Vairy, S.; Garcia, J.L.; Teira, P.; Bittencourt, H. CTL019 (tisagenlecleucel): CAR-T therapy for relapsed and refractory B-cell acute lymphoblastic leukemia. Drug Des. Dev. Ther. 2018, 12, 3885–3898. [Google Scholar] [CrossRef] [PubMed]
- Kamdar, M.; Solomon, S.R.; Arnason, J.; Johnston, P.B.; Glass, B.; Bachanova, V.; Ibrahimi, S.; Mielke, S.; Mutsaers, P.; Hernandez-Ilizaliturri, F.; et al. Lisocabtagene maraleucel versus standard of care with salvage chemotherapy followed by autologous stem cell transplantation as second-line treatment in patients with relapsed or refractory large B-cell lymphoma (TRANSFORM): Results from an interim analysis of an open-label, randomised, phase 3 trial. Lancet 2022, 399, 2294–2308. [Google Scholar]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Laetsch, T.W.; Maude, S.L.; Balduzzi, A.; Rives, S.; Bittencourt, H.; Boyer, M.W.; Buechner, J.; De Moerloose, B.; Qayed, M.; Phillips, C.L.; et al. Tisagenlecleucel in pediatric and young adult patients with Down syndrome-associated relapsed/refractory acute lymphoblastic leukemia. Leukemia 2022, 36, 1508–1515. [Google Scholar] [CrossRef]
- Moskop, A.; Pommert, L.; Baggott, C.; Prabhu, S.; Pacenta, H.L.; Phillips, C.L.; Rossoff, J.; Stefanski, H.E.; Talano, J.A.; Margossian, S.P.; et al. Real-world use of tisagenlecleucel in infant acute lymphoblastic leukemia. Blood Adv. 2022, 6, 4251–4255. [Google Scholar] [CrossRef]
- Kwon, M.; Iacoboni, G.; Reguera, J.L.; Corral, L.L.; Morales, R.H.; Ortiz-Maldonado, V.; Guerreiro, M.; Caballero, A.C.; Domínguez, M.L.G.; Pina, J.M.S.; et al. Axicabtagene ciloleucel compared to tisagenlecleucel for the treatment of aggressive B-cell lymphoma. Haematologica 2022. Online ahead of print. [Google Scholar] [CrossRef]
- Ghione, P.; Palomba, M.L.; Patel, A.; Bobillo, S.; Deighton, K.; Jacobson, C.A.; Nahas, M.; Hatswell, A.J.; Jung, A.S.; Kanters, S.; et al. Comparative effectiveness of ZUMA-5 (axi-cel) vs. SCHOLAR-5 external control in relapsed/refractory follicular lymphoma. Blood 2022, 140, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Locke, F.L.; Miklos, D.B.; Jacobson, C.A.; Perales, M.A.; Kersten, M.J.; Oluwole, O.O.; Ghobadi, A.; Rapoport, A.P.; McGuirk, J.; Pagel, J.M.; et al. Axicabtagene Ciloleucel as Second-Line Therapy for Large B-Cell Lymphoma. N. Engl. J. Med. 2022, 386, 640–654. [Google Scholar] [CrossRef]
- Siddiqi, T.; Soumerai, J.D.; Dorritie, K.A.; Stephens, D.M.; Riedell, P.A.; Arnason, J.; Kipps, T.J.; Gillenwater, H.H.; Gong, L.; Yang, L.; et al. Phase 1 TRANSCEND CLL 004 study of lisocabtagene maraleucel in patients with relapsed/refractory CLL or SLL. Blood 2022, 139, 1794–1806. [Google Scholar] [CrossRef]
- Chavez, J.C.; Locke, F.L. CAR T cell therapy for B-cell lymphomas. Best Pract. Res. Clin. Haematol. 2018, 31, 135–146. [Google Scholar] [CrossRef]
- Shah, N.N.; Maatman, T.; Hari, P.; Johnson, B. Multi Targeted CAR-T Cell Therapies for B-Cell Malignancies. Front. Oncol. 2019, 9, 146. [Google Scholar] [CrossRef] [PubMed]
- Messéant, O.; Houot, R. CAR-T cells in lymphomas: Current and evolving role. Bull. Cancer 2021, 108, S28–S39. [Google Scholar] [CrossRef]
- Martyniszyn, A.; Krahl, A.C.; André, M.C.; Hombach, A.A.; Abken, H. CD20-CD19 Bispecific CAR T Cells for the Treatment of B-Cell Malignancies. Hum. Gene Ther. 2017, 28, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Tong, C.; Zhang, Y.; Liu, Y.; Ji, X.; Zhang, W.; Guo, Y.; Han, X.; Ti, D.; Dai, H.; Wang, C.; et al. Optimized tandem CD19/CD20 CAR-engineered T cells in refractory/relapsed B-cell lymphoma. Blood 2020, 136, 1632–1644. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Hu, Y.; Shi, C. Targeting Natural Killer Cells for Tumor Immunotherapy. Front. Immunol. 2020, 11, 60. [Google Scholar] [CrossRef] [PubMed]
- Heipertz, E.L.; Zynda, E.R.; Stav-Noraas, T.E.; Hungler, A.D.; Boucher, S.E.; Kaur, N.; Vemuri, M.C. Current Perspectives on “Off-The-Shelf” Allogeneic NK and CAR-NK Cell Therapies. Front. Immunol. 2021, 12, 732135. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Su, Y.; Jabbour, E.J.; Bhattacharyya, H.; Yan, E.; Cappelleri, J.C.; Marks, D.I. Patient-reported outcomes from a phase 3 randomized controlled trial of inotuzumab ozogamicin versus standard therapy for relapsed/refractory acute lymphoblastic leukemia. Cancer 2018, 124, 2151–2160. [Google Scholar] [CrossRef]
- Jabbour, E.; Stelljes, M.; Advani, A.S.; DeAngelo, D.J.; Wang, T.; Neuhof, A.; Vandendries, E.; Kantarjian, H.M. Time from randomization to first subsequent induction/salvage therapy (ST) in patients with relapsed/refractory (R/R) acute lymphoblastic leukemia (ALL) treated with inotuzumab ozogamicin (InO) in the phase 3 INO-VATE trial. J. Clin. Oncol. 2019, 373, 7025. [Google Scholar] [CrossRef]
- Shah, N.N.; Bhojwani, D.; August, K.; Baruchel, A.; Bertrand, Y.; Boklan, J.; Dalla-Pozza, L.; Dennis, R.; Hijiya, N.; Locatelli, F.; et al. Results from an international phase 2 study of the anti-CD22 immunotoxin moxetumomab pasudotox in relapsed or refractory childhood B-lineage acute lymphoblastic leukemia. Pediatr Blood Cancer 2020, 67, e28112. [Google Scholar] [CrossRef]
- Contreras, C.F.; Higham, C.S.; Behnert, A.; Kim, K.; Stieglitz, E.; Tasian, S.K. Clinical utilization of blinatumomab and inotuzumab immunotherapy in children with relapsed or refractory B-acute lymphoblastic leukemia. Pediatr. Blood Cancer 2021, 68, e28718. [Google Scholar] [CrossRef] [PubMed]
- Kreitman, R.J.; Pastan, I. Antibody fusion proteins: Anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clin. Cancer Res. 2011, 17, 6398–6405. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.; Sasaki, K.; Ravandi, F.; Huang, X.; Short, N.J.; Khouri, M.; Kebriaei, P.; Burger, J.; Khoury, J.; Jorgensen, J.; et al. Chemoimmunotherapy with inotuzumab ozogamicin combined with mini-hyper-CVD, with or without blinatumomab, is highly effective in patients with Philadelphia chromosome-negative acute lymphoblastic leukemia in first salvage. Cancer 2018, 124, 4044–4055. [Google Scholar] [CrossRef] [PubMed]
- Pennesi, E.; Michels, N.; Brivio, E.; van der Velden, V.H.J.; Jiang, Y.; Thano, A.; Ammerlaan, A.J.C.; Boer, J.M.; Beverloo, H.B.; Sleight, B.; et al. Inotuzumab ozogamicin as single agent in pediatric patients with relapsed and refractory acute lymphoblastic leukemia: Results from a phase II trial. Leukemia 2022, 36, 1516–1524. [Google Scholar] [CrossRef] [PubMed]
- Advani, A.; Coiffier, B.; Czuczman, M.S.; Dreyling, M.; Foran, J.; Gine, E.; Gisselbrecht, C.; Ketterer, N.; Nasta, S.; Rohatiner, A.; et al. Safety, pharmacokinetics, and preliminary clinical activity of inotuzumab ozogamicin, a novel immunoconjugate for the treatment of B-cell non-Hodgkin’s lymphoma: Results of a phase I study. J. Clin. Oncol. 2010, 28, 2085–2093. [Google Scholar] [CrossRef] [PubMed]
- Ogura, M.; Tobinai, K.; Hatake, K.; Uchida, T.; Kasai, M.; Oyama, T.; Suzuki, T.; Kobayashi, Y.; Watanabe, T.; Azuma, T.; et al. Phase I study of inotuzumab ozogamicin (CMC-544) in Japanese patients with follicular lymphoma pretreated with rituximab-based therapy. Cancer Sci. 2010, 101, 1840–1845. [Google Scholar] [CrossRef]
- Fayad, L.; Offner, F.; Smith, M.R.; Verhoef, G.; Johnson, P.; Kaufman, J.L.; Rohatiner, A.; Advani, A.; Foran, J.; Hess, G.; et al. Safety and clinical activity of a combination therapy comprising two antibody-based targeting agents for the treatment of non-Hodgkin lymphoma: Results of a phase I/II study evaluating the immunoconjugate inotuzumab ozogamicin with rituximab. J. Clin. Oncol. 2013, 31, 573–583. [Google Scholar] [CrossRef]
- Short, N.J.; Kantarjian, H.; Jabbour, E.; Cortes, J.E.; Thomas, D.A.; Rytting, M.E.; Daver, N.; Alvarado, Y.; Konopleva, M.; Kebriaei, P.; et al. A phase I study of moxetumomab pasudotox in adults with relapsed or refractory B-cell acute lymphoblastic leukaemia. Br. J. Haematol. 2018, 182, 442–444. [Google Scholar] [CrossRef]
- Haso, W.; Lee, D.W.; Shah, N.N.; Stetler-Stevenson, M.; Yuan, C.M.; Pastan, I.H.; Dimitrov, D.S.; Morgan, R.A.; FitzGerald, D.J.; Barrett, D.M.; et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood 2013, 121, 1165–1174. [Google Scholar] [CrossRef]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Shalabi, H.; Qin, H.; Su, A.; Yates, B.; Wolters, P.L.; Steinberg, S.M.; Ligon, J.A.; Silbert, S.; DéDé, K.; Benzaoui, M.; et al. CD19/22 CAR T-cells in Children and Young Adults with B-ALL: Phase I Results and Development of a Novel Bicistronic CAR. Blood 2022, 140, 451–463. [Google Scholar] [CrossRef]
- Usmani, S.Z.; Quach, H.; Mateos, M.V.; Landgren, O.; Leleu, X.; Siegel, D.; Weisel, K.; Gavriatopoulou, M.; Oriol, A.; Rabin, N.; et al. Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): Updated outcomes from a randomised, multicentre, open-label, phase 3 study. Lancet Oncol. 2022, 23, 65–76. [Google Scholar] [CrossRef]
- Dimopoulos, M.; Bringhen, S.; Anttila, P.; Capra, M.; Cavo, M.; Cole, C.; Gasparetto, C.; Hungria, V.; Jenner, M.; Vorobyev, V.; et al. Isatuximab as monotherapy and combined with dexamethasone in patients with relapsed/refractory multiple myeloma. Blood 2021, 137, 1154–1165. [Google Scholar] [CrossRef]
- Moreau, P.; Dimopoulos, M.A.; Mikhael, J.; Yong, K.; Capra, M.; Facon, T.; Hajek, R.; Špička, I.; Baker, R.; Kim, K.; et al. Isatuximab, carfilzomib, and dexamethasone in relapsed multiple myeloma (IKEMA): A multicentre, open-label, randomised phase 3 trial. Lancet 2021, 397, 2361–2371. [Google Scholar] [CrossRef]
- Richardson, P.G.; Harrison, S.J.; Bringhen, S.; Schjesvold, F.; Yong, K.; Campana, F.; Le-Guennec, S.; Macé, S.; Dimopoulos, M.A. Isatuximab for relapsed/refractory multiple myeloma: Review of key subgroup analyses from the Phase III ICARIA-MM study. Future Oncol. 2021, 17, 4797–4812. [Google Scholar] [CrossRef] [PubMed]
- Calabretta, E.; Carlo-Stella, C. The Many Facets of CD38 in Lymphoma: From Tumor-Microenvironment Cell Interactions to Acquired Resistance to Immunotherapy. Cells 2020, 9, 802. [Google Scholar] [CrossRef]
- Gozzetti, A.; Ciofini, S.; Simoncelli, M.; Santoni, A.; Pacelli, P.; Raspadori, D.; Bocchia, M. Anti CD38 monoclonal antibodies for multiple myeloma treatment. Hum. Vaccines Immunother. 2022, 18, 2052658. [Google Scholar] [CrossRef]
- Jiang, H.; Acharya, C.; An, G.; Zhong, M.; Feng, X.; Wang, L.; Dasilva, N.; Song, Z.; Yang, G.; Adrian, F.; et al. SAR650984 directly induces multiple myeloma cell death via lysosomal-associated and apoptotic pathways, which is further enhanced by pomalidomide. Leukemia 2016, 30, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Franssen, L.E.; Stege, C.A.M.; Zweegman, S.; van de Donk, N.; Nijhof, I.S. Resistance Mechanisms Towards CD38-Directed Antibody Therapy in Multiple Myeloma. J. Clin. Med. 2020, 9, 1195. [Google Scholar] [CrossRef]
- Yang, Y.; Li, Y.; Gu, H.; Dong, M.; Cai, Z. Emerging agents and regimens for multiple myeloma. J. Hematol. Oncol. 2020, 13, 150. [Google Scholar] [CrossRef]
- Usmani, S.Z.; Karanes, C.; Bensinger, W.I.; D’Souza, A.; Raje, N.; Tuchman, S.A.; Sborov, D.; Laubach, J.P.; Bianchi, G.; Kanagavel, D.; et al. Final results of a phase 1b study of isatuximab short-duration fixed-volume infusion combination therapy for relapsed/refractory multiple myeloma. Leukemia 2021, 35, 3526–3533. [Google Scholar] [CrossRef] [PubMed]
- Voorhees, P.M.; Kaufman, J.L.; Laubach, J.; Sborov, D.W.; Reeves, B.; Rodriguez, C.; Chari, A.; Silbermann, R.; Costa, L.J.; Anderson, L.D., Jr.; et al. Daratumumab, lenalidomide, bortezomib, and dexamethasone for transplant-eligible newly diagnosed multiple myeloma: The GRIFFIN trial. Blood 2020, 136, 936–945. [Google Scholar] [CrossRef] [PubMed]
- Matas-Céspedes, A.; Vidal-Crespo, A.; Rodriguez, V.; Villamor, N.; Delgado, J.; Giné, E.; Roca-Ho, H.; Menéndez, P.; Campo, E.; López-Guillermo, A.; et al. The Human CD38 Monoclonal Antibody Daratumumab Shows Antitumor Activity and Hampers Leukemia-Microenvironment Interactions in Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2017, 23, 1493–1505. [Google Scholar] [CrossRef]
- Manna, A.; Aulakh, S.; Jani, P.; Ahmed, S.; Akhtar, S.; Coignet, M.; Heckman, M.; Meghji, Z.; Bhatia, K.; Sharma, A.; et al. Targeting CD38 Enhances the Antileukemic Activity of Ibrutinib in Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2019, 25, 3974–3985. [Google Scholar] [CrossRef]
- Wang, A.; Song, Z.; Zheng, G.; Nicolazzi, C.; Fromm, J.R.; Shehu, E.; Srinivasan, S.; Chen, X.; Zhu, C.; Blondel, M.C.; et al. Evaluation of Preclinical Activity of Isatuximab in Patients with Acute Lymphoblastic Leukemia. Mol. Cancer Ther. 2021, 20, 1916–1925. [Google Scholar] [CrossRef] [PubMed]
- Baruchel, A.; Abrahamsson, J.; Bertrand, Y.; Gonzalez, O.; Nysom, K.; Quinones, W.; Rizzari, C.; Buechner, J.; Cesaro, S.; Duarte, J.; et al. Isatuximab in Combination with Chemotherapy in Pediatric Patients with Relapsed/Refractory Acute Lymphoblastic Leukemia or Acute Myeloid Leukemia (ISAKIDS): Interim Analysis. Blood 2021, 138, 516. [Google Scholar] [CrossRef]
- Hashmi, H.; Husnain, M.; Khan, A.; Usmani, S.Z. CD38-Directed Therapies for Management of Multiple Myeloma. Immunotargets Ther. 2021, 10, 201–211. [Google Scholar] [CrossRef]
- Abramson, H.N. Emerging Monoclonal Antibodies for the Treatment of Multiple Myeloma; IntechOpen: London, UK, 2020; pp. 1–20. [Google Scholar]
- Raab, M.S.; Engelhardt, M.; Blank, A.; Goldschmidt, H.; Agis, H.; Blau, I.W.; Einsele, H.; Ferstl, B.; Schub, N.; Röllig, C.; et al. MOR202, a novel anti-CD38 monoclonal antibody, in patients with relapsed or refractory multiple myeloma: A first-in-human, multicentre, phase 1-2a trial. Lancet Haematol. 2020, 7, e381–e394. [Google Scholar] [CrossRef]
- Krishnan, A.Y.; Patel, K.K.; Hari, P.; Jagannath, S.; Niesvizky, R.; Silbermann, R.W.; Berg, D.T.; Li, Q.; Allikmets, K.; Stockerl-Goldstein, K. A phase Ib study of TAK-079, an investigational anti-CD38 monoclonal antibody (mAb) in patients with relapsed/refractory multiple myeloma (RRMM): Preliminary results. J. Clin. Oncol. 2020, 38, 8539. [Google Scholar] [CrossRef]
- Zuch de Zafra, C.L.; Fajardo, F.; Zhong, W.; Bernett, M.J.; Muchhal, U.S.; Moore, G.L.; Stevens, J.; Case, R.; Pearson, J.T.; Liu, S.; et al. Targeting Multiple Myeloma with AMG 424, a Novel Anti-CD38/CD3 Bispecific T-cell-recruiting Antibody Optimized for Cytotoxicity and Cytokine Release. Clin. Cancer Res. 2019, 25, 3921–3933. [Google Scholar] [CrossRef] [PubMed]
- Doucey, M.A.; Estoppey, C.; Stutz, C.; Croset, A.; Laurendon, A.; Monney, T.; Pluess, M.; Ries-Fecourt, C.; Macoin, J.; Turrini, R.; et al. ISB 1342: A first-in-class CD38 T cell engager for the treatment of relapsed refractory multiple myeloma. J. Clin. Oncol. 2021, 39, 8044. [Google Scholar] [CrossRef]
- Guo, Y.; Feng, K.; Tong, C.; Jia, H.; Liu, Y.; Wang, Y.; Ti, D.; Yang, Q.; Wu, Z.; Han, W. Efficiency and side effects of anti-CD38 CAR T cells in an adult patient with relapsed B-ALL after failure of bi-specific CD19/CD22 CAR T cell treatment. Cell. Mol. Immunol. 2020, 17, 430–432. [Google Scholar] [CrossRef]
- Duggal, R.; Santara, S.S.; Gordon, M.; Kilgallon, A.; Hermanson, D.; Childs, R.W.; O’Dwyer, M.E. Promising Preliminary Activity of Optimized Affinity, CD38 CAR NK Cells Generated Using a Non-Viral Engineering Approach in Gene Edited Cord Blood Derived NK Cells for the Treatment of Multiple Myeloma. Blood 2021, 138, 4793. [Google Scholar] [CrossRef]
- Stikvoort, A.; van der Schans, J.; Sarkar, S.; Poels, R.; Ruiter, R.; Naik, J.; Yuan, H.; de Bruijn, J.D.; van de Donk, N.W.; Zweegman, S.; et al. CD38-specific Chimeric Antigen Receptor Expressing Natural Killer KHYG-1 Cells: A Proof of Concept for an “Off the Shelf” Therapy for Multiple Myeloma. HemaSphere 2021, 5, e596. [Google Scholar] [CrossRef]
- Cho, S.F.; Anderson, K.C.; Tai, Y.T. Targeting B Cell Maturation Antigen (BCMA) in Multiple Myeloma: Potential Uses of BCMA-Based Immunotherapy. Front. Immunol. 2018, 9, 1821. [Google Scholar] [CrossRef]
- Zhou, X.; Mulazzani, M.; von Mücke-Heim, I.A.; Langer, S.; Zhang, W.; Ishikawa-Ankerhold, H.; Dreyling, M.; Straube, A.; von Baumgarten, L. The Role of BAFF-R Signaling in the Growth of Primary Central Nervous System Lymphoma. Front. Oncol. 2020, 10, 682. [Google Scholar] [CrossRef]
- Sanchez, L.; Dardac, A.; Madduri, D.; Richard, S.; Richter, J. B-cell maturation antigen (BCMA) in multiple myeloma: The new frontier of targeted therapies. Ther. Adv. Hematol. 2021, 12, 2040620721989585. [Google Scholar] [CrossRef]
- Lancman, G.; Richter, J.; Chari, A. Bispecifics, trispecifics, and other novel immune treatments in myeloma. Hematol. Am. Soc. Hematol. Educ. Program 2020, 2020, 264–271. [Google Scholar] [CrossRef]
- Strassl, I.; Schreder, M.; Steiner, N.; Rudzki, J.; Agis, H.; Künz, T.; Müser, N.; Willenbacher, W.; Petzer, A.; Neumeister, P.; et al. The Agony of Choice-Where to Place the Wave of BCMA-Targeted Therapies in the Multiple Myeloma Treatment Puzzle in 2022 and Beyond. Cancers 2021, 13, 4701. [Google Scholar] [CrossRef]
- Sheikh, S.; Lebel, E.; Trudel, S. Belantamab mafodotin in the treatment of relapsed or refractory multiple myeloma. Future Oncol. 2020, 16, 2783–2798. [Google Scholar] [CrossRef] [PubMed]
- Trudel, S.; Nooka, A.; Fecteau, D.; Talekar, M.; Jewell, R.C.; Williams, D.; Evans, J.; Opalinska, J. DREAMM 4: A phase I/II single-arm open-label study to explore safety and clinical activity of belantamab mafodotin (GSK2857916) administered in combination with pembrolizumab in patients with relapsed/refractory multiple myeloma (RRMM). Ann. Oncol. 2019, 30, v447. [Google Scholar] [CrossRef]
- Xing, L.; Lin, L.; Yu, T.; Li, Y.; Cho, S.F.; Liu, J.; Wen, K.; Hsieh, P.A.; Kinneer, K.; Munshi, N.; et al. A novel BCMA PBD-ADC with ATM/ATR/WEE1 inhibitors or bortezomib induce synergistic lethality in multiple myeloma. Leukemia 2020, 34, 2150–2162. [Google Scholar] [CrossRef]
- Kumar, S.; Migkou, M.; Bhutani, M.; Spencer, A.; Ailawadhi, S.; Kalff, A.; Walcott, F.; Pore, N.; Gibson, D.; Wang, F.; et al. Phase 1, First-in-Human Study of MEDI2228, a BCMA-Targeted ADC in Patients with Relapsed/Refractory Multiple Myeloma. Blood 2020, 136, 26–27. [Google Scholar] [CrossRef]
- Trudel, S.; Lendvai, N.; Popat, R.; Voorhees, P.M.; Reeves, B.; Libby, E.N.; Richardson, P.G.; Hoos, A.; Gupta, I.; Bragulat, V.; et al. Antibody-drug conjugate, GSK2857916, in relapsed/refractory multiple myeloma: An update on safety and efficacy from dose expansion phase I study. Blood Cancer J. 2019, 9, 37. [Google Scholar] [CrossRef]
- Lee, H.C.; Raje, N.S.; Landgren, O.; Upreti, V.V.; Wang, J.; Avilion, A.A.; Hu, X.; Rasmussen, E.; Ngarmchamnanrith, G.; Fujii, H.; et al. Phase 1 study of the anti-BCMA antibody-drug conjugate AMG 224 in patients with relapsed/refractory multiple myeloma. Leukemia 2021, 35, 255–258. [Google Scholar] [CrossRef]
- Usmani, S.Z.; Garfall, A.L.; van de Donk, N.; Nahi, H.; San-Miguel, J.F.; Oriol, A.; Rosinol, L.; Chari, A.; Bhutani, M.; Karlin, L.; et al. Teclistamab, a B-cell maturation antigen × CD3 bispecific antibody, in patients with relapsed or refractory multiple myeloma (MajesTEC-1): A multicentre, open-label, single-arm, phase 1 study. Lancet 2021, 398, 665–674. [Google Scholar] [CrossRef]
- Munshi, N.C.; Anderson, L.D., Jr.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef]
- Costa, L.J.; Lin, Y.; Cornell, R.F.; Martin, T.; Chhabra, S.; Usmani, S.Z.; Jagannath, S.; Callander, N.S.; Berdeja, J.G.; Kang, Y.; et al. Comparison of Cilta-cel, an Anti-BCMA CAR-T Cell Therapy, Versus Conventional Treatment in Patients With Relapsed/Refractory Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2022, 22, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Yin, H.; Zhao, X.; Jin, D.; Liang, Y.; Xiong, T.; Li, L.; Tang, W.; Zhang, J.; Liu, M.; et al. High efficacy and safety of CD38 and BCMA bispecific CAR-T in relapsed or refractory multiple myeloma. J. Exp. Clin. Cancer Res. 2022, 41, 2. [Google Scholar] [CrossRef] [PubMed]
- Ng, Y.Y.; Du, Z.; Zhang, X.; Chng, W.J.; Wang, S. CXCR4 and anti-BCMA CAR co-modified natural killer cells suppress multiple myeloma progression in a xenograft mouse model. Cancer Gene Ther. 2022, 29, 475–483. [Google Scholar] [CrossRef]
- Ding, W.; LaPlant, B.R.; Call, T.G.; Parikh, S.A.; Leis, J.F.; He, R.; Shanafelt, T.D.; Sinha, S.; Le-Rademacher, J.; Feldman, A.L.; et al. Pembrolizumab in patients with CLL and Richter transformation or with relapsed CLL. Blood 2017, 129, 3419–3427. [Google Scholar] [CrossRef]
- Sandhu, K.S.; Huynh-Tran, Q.; Cooper, E.E.; Palmer, J.; Tsai, N.C.; Thomas, S.; Robbins, M.; Aribi, A.; Salhotra, A.; Mei, M.; et al. ALL-440: Promising Safety and Efficacy Results from an Ongoing Phase 1/2 Study of Pembrolizumab in Combination with Blinatumomab in Patients (pts) with Relapsed or Refractory (R/R) Acute Lymphoblastic Leukemia (ALL). Clin. Lymphoma Leukemia 2021, 21, 276. [Google Scholar] [CrossRef]
- Carlo-Stella, C.; Zinzani, P.L.L.; Sureda, A.; Araújo, L.F.; Casasnovas, O.; Carpio, C.; Yeh, S.P.; Bouabdallah, K.; Cartron, G.; Kim, W.S.; et al. A Phase 1/2, Open-Label, Multicenter Study of Isatuximab in Combination with Cemiplimab in Patients with Lymphoma. Blood 2021, 138, 4362. [Google Scholar] [CrossRef]
- Jacobson, C.S.J. End of Phase 1 Results from Zuma-6: Axicabtagene Ciloleucel (Axi-Cel) in Combination with Atezolizumab for the Treatment of Patients with Refractory Diffuse Large B Cell Lymphoma. Blood 2018, 132, 4192. [Google Scholar] [CrossRef]
- Treon, S.P.; Meid, K.; Hunter, Z.R.; Flynn, C.A.; Sarosiek, S.R.; Leventoff, C.R.; White, T.P.; Cao, Y.; Roccaro, A.M.; Sacco, A.; et al. Phase 1 study of ibrutinib and the CXCR4 antagonist ulocuplumab in CXCR4-mutated Waldenström macroglobulinemia. Blood 2021, 138, 1535–1539. [Google Scholar] [CrossRef]
- Schjesvold, F.; Robak, P.; Pour, L.; Aschan, J.; Sonneveld, P. OCEAN: A randomized Phase III study of melflufen + dexamethasone to treat relapsed refractory multiple myeloma. Future Oncol. 2020, 16, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Kolstad, A.; Illidge, T.; Bolstad, N.; Spetalen, S.; Madsbu, U.; Stokke, C.; Blakkisrud, J.; Løndalen, A.; O’Rourke, N.; Beasley, M.; et al. Phase 1/2a study of 177Lu-lilotomab satetraxetan in relapsed/refractory indolent non-Hodgkin lymphoma. Blood Adv. 2020, 4, 4091–4101. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.M.; Wu, Z.Q.; Wang, Y.; Guo, Y.L.; Dai, H.R.; Wang, X.H.; Li, X.; Zhang, Y.J.; Zhang, W.Y.; Chen, M.X.; et al. Autologous T Cells Expressing CD30 Chimeric Antigen Receptors for Relapsed or Refractory Hodgkin Lymphoma: An Open-Label Phase I Trial. Clin. Cancer Res. 2017, 23, 1156–1166. [Google Scholar] [CrossRef]
- Ramos, C.A.; Ballard, B.; Zhang, H.; Dakhova, O.; Gee, A.P.; Mei, Z.; Bilgi, M.; Wu, M.F.; Liu, H.; Grilley, B.; et al. Clinical and immunological responses after CD30-specific chimeric antigen receptor-redirected lymphocytes. J. Clin. Investig. 2017, 127, 3462–3471. [Google Scholar] [CrossRef]
- Advani, R.H.; Flinn, I.; Popplewell, L.; Forero-Torres, A.; Bartlett, N.L.; Ghosh, N.; Kline, J.P.; Tran, T.; Lynn, J.; Chen, J.Y.; et al. Activity and tolerabilty of the first-in-class anti-CD47 antibody Hu5F9-G4 with rituximab tolerated in relapsed/refractory non-Hodgkin lymphoma: Initial phase 1b/2 results. J. Clin. Oncol. 2018, 36, 7504. [Google Scholar] [CrossRef]
- Chauchet, X.; Cons, L.; Chatel, L.; Daubeuf, B.; Didelot, G.; Moine, V.; Chollet, D.; Malinge, P.; Pontini, G.; Masternak, K.; et al. CD47xCD19 bispecific antibody triggers recruitment and activation of innate immune effector cells in a B-cell lymphoma xenograft model. Exp. Hematol. Oncol. 2022, 11, 26. [Google Scholar] [CrossRef]
- Krishnan, A.Y.; Shah, N.; Spira, A.I.; Kaufman, J.L.; Niesvizky, R.; Shah, N.N.; Burke, J.M.; Popplewell, L.; Martin, T.G.; Cheung, J.; et al. A phase 1 open-label, safety, pharmacokinetic, and preliminary efficacy study of STRO-001, an anti-CD74 antibody drug conjugate, in patients with advanced B-cell malignancies. J. Clin. Oncol. 2018, 36, 7586. [Google Scholar] [CrossRef]
- Cohen, A.D.; Harrison, S.J.; Krishnan, A.; Fonseca, R.; Forsberg, P.A.; Spencer, A.; Berdeja, J.G.; Laubach, J.P.; Li, M.; Choeurng, V.; et al. Initial clinical activity and safety of BFCR4350A, a FcRH5/CD3 T-cell-engaging bispecific antibody, in relapsed/refractory multiple myeloma. Blood 2020, 136, 42–43. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Krishnan, A.Y.; Oriol, A.; van de Donk, N.W.; Rodríguez-Otero, P.; Askari, E.; Mateos, M.V.; Minnema, M.C.; Costa, L.J.; Verona, R.; et al. Updated results of a phase 1, first-in-human study of talquetamab, a G protein-coupled receptor family C group 5 member D (GPRC5D) × CD3 bispecific antibody, in relapsed/refractory multiple myeloma (MM). J. Clin. Oncol. 2021, 39, 8008. [Google Scholar] [CrossRef]
- Cui, B.; Ghia, E.M.; Chen, L.; Rassenti, L.Z.; DeBoever, C.; Widhopf, G.F., 2nd; Yu, J.; Neuberg, D.S.; Wierda, W.G.; Rai, K.R.; et al. High-level ROR1 associates with accelerated disease progression in chronic lymphocytic leukemia. Blood 2016, 128, 2931–2940. [Google Scholar] [CrossRef]
- Fukuda, T.; Chen, L.; Endo, T.; Tang, L.; Lu, D.; Castro, J.E.; Widhopf, G.F., 2nd; Rassenti, L.Z.; Cantwell, M.J.; Prussak, C.E.; et al. Antisera induced by infusions of autologous Ad-CD154-leukemia B cells identify ROR1 as an oncofetal antigen and receptor for Wnt5a. Proc. Natl. Acad. Sci. USA 2008, 105, 3047–3052. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Chen, L.; Cui, B.; Widhopf, G.F., 2nd; Shen, Z.; Wu, R.; Zhang, L.; Zhang, S.; Briggs, S.P.; Kipps, T.J. Wnt5a induces ROR1/ROR2 heterooligomerization to enhance leukemia chemotaxis and proliferation. J. Clin. Investig. 2016, 126, 585–598. [Google Scholar] [CrossRef]
- Ghia, E.M.; Rassenti, L.Z.; Choi, M.Y.; Quijada-Álamo, M.; Chu, E.; Widhopf, G.F., 2nd; Kipps, T.J. High expression level of ROR1 and ROR1-signaling associates with venetoclax resistance in chronic lymphocytic leukemia. Leukemia 2022, 36, 1609–1618. [Google Scholar] [CrossRef]
- Choi, M.Y.; Widhopf, G.F., 2nd; Ghia, E.M.; Kidwell, R.L.; Hasan, M.K.; Yu, J.; Rassenti, L.Z.; Chen, L.; Chen, Y.; Pittman, E.; et al. Phase I Trial: Cirmtuzumab Inhibits ROR1 Signaling and Stemness Signatures in Patients with Chronic Lymphocytic Leukemia. Cell Stem Cell 2018, 22, 951–959.e3. [Google Scholar] [CrossRef]
- Beenen, A.C.; Sauerer, T.; Schaft, N.; Dörrie, J. Beyond Cancer: Regulation and Function of PD-L1 in Health and Immune-Related Diseases. Int. J. Mol. Sci. 2022, 23, 8599. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Bao, Y.; Meng, B.; Xu, Z.; Li, S.; Wang, X.; Hou, R.; Ma, W.; Liu, D.; Zheng, J.; et al. From rough to precise: PD-L1 evaluation for predicting the efficacy of PD-1/PD-L1 blockades. Front. Immunol. 2022, 13, 920021. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, H.; Liu, C.; Wang, Z.; Wu, W.; Zhang, N.; Zhang, L.; Hu, J.; Luo, P.; Zhang, J.; et al. Immune checkpoint modulators in cancer immunotherapy: Recent advances and emerging concepts. J. Hematol. Oncol. 2022, 15, 111. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- Boyiadzis, M.M.; Kirkwood, J.M.; Marshall, J.L.; Pritchard, C.C.; Azad, N.S.; Gulley, J.L. Significance and implications of FDA approval of pembrolizumab for biomarker-defined disease. J. Immunother. Cancer 2018, 6, 35. [Google Scholar] [CrossRef] [PubMed]
- Lesokhin, A.M.; Ansell, S.M.; Armand, P.; Scott, E.C.; Halwani, A.; Gutierrez, M.; Millenson, M.M.; Cohen, A.D.; Schuster, S.J.; Lebovic, D.; et al. Nivolumab in Patients With Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J. Clin. Oncol. 2016, 34, 2698–2704. [Google Scholar] [CrossRef] [PubMed]
- Armand, P.; Lesokhin, A.; Borrello, I.; Timmerman, J.; Gutierrez, M.; Zhu, L.; Popa McKiver, M.; Ansell, S.M. A phase 1b study of dual PD-1 and CTLA-4 or KIR blockade in patients with relapsed/refractory lymphoid malignancies. Leukemia 2021, 35, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Bertamini, L.; Gay, F. Checkpoint inhibitors and myeloma: Promises, deadlocks and new directions. Ann. Transl. Med. 2020, 8, 777. [Google Scholar] [CrossRef]
- Casulo, C.; Santoro, A.; Ando, K.; Le Gouill, S.; Ruan, J.; Radford, J.; Arcaini, L.; Pinto, A.; Bouabdallah, R.; Izutsu, K.; et al. Durvalumab (Anti PD-L1) As Monotherapy or in Combination Therapy for Relapsed/Refractory (r/r) Diffuse Large B-Cell Lymphoma (DLBCL) and Follicular Lymphoma (FL): A Subgroup Analysis from the Phase 1/2 Fusion NHL-001 Global Multicenter Trial. Blood 2019, 134, 5320. [Google Scholar] [CrossRef]
- Ribrag, V.; Lee, S.T.; Rizzieri, D.; Dyer, M.J.S.; Fayad, L.; Kurzrock, R.; Andritsos, L.; Bouabdallah, R.; Hayat, A.; Bacon, L.; et al. A Phase 1b Study to Evaluate the Safety and Efficacy of Durvalumab in Combination with Tremelimumab or Danvatirsen in Patients with Relapsed or Refractory Diffuse Large B-Cell Lymphoma. Clin. Lymphoma Myeloma Leuk. 2021, 21, 309–317.e3. [Google Scholar] [CrossRef]
- Hirayama, A.V.; Gauthier, J.; Hay, K.A.; Sheih, A.; Cherian, S.; Chen, X.; Pender, B.S.; Hawkins, R.M.; Vakil, A.; Steinmetz, R.N.; et al. Efficacy and Toxicity of JCAR014 in Combination with Durvalumab for the Treatment of Patients with Relapsed/Refractory Aggressive B-Cell Non-Hodgkin Lymphoma. Blood 2018, 132, 1680. [Google Scholar] [CrossRef]
- Kaiser, L.M.; Hunter, Z.R.; Treon, S.P.; Buske, C. CXCR4 in Waldenström’s Macroglobulinema: Chances and challenges. Leukemia 2021, 35, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, M.K.; Kumar, D.; Jones, H.; Amaya-Chanaga, C.I.; Choi, M.Y.; Melo-Cardenas, J.; Ale-Ali, A.; Kuhne, M.R.; Sabbatini, P.; Cohen, L.J.; et al. Ulocuplumab (BMS-936564/MDX1338): A fully human anti-CXCR4 antibody induces cell death in chronic lymphocytic leukemia mediated through a reactive oxygen species-dependent pathway. Oncotarget 2016, 7, 2809–2822. [Google Scholar] [CrossRef]
- Roccaro, A.M.; Sacco, A.; Jimenez, C.; Maiso, P.; Moschetta, M.; Mishima, Y.; Aljawai, Y.; Sahin, I.; Kuhne, M.; Cardarelli, P.; et al. C1013G/CXCR4 acts as a driver mutation of tumor progression and modulator of drug resistance in lymphoplasmacytic lymphoma. Blood 2014, 123, 4120–4131. [Google Scholar] [CrossRef]
- Ghobrial, I.M.; Liu, C.J.; Redd, R.A.; Perez, R.P.; Baz, R.; Zavidij, O.; Sklavenitis-Pistofidis, R.; Richardson, P.G.; Anderson, K.C.; Laubach, J.; et al. A Phase Ib/II Trial of the First-in-Class Anti-CXCR4 Antibody Ulocuplumab in Combination with Lenalidomide or Bortezomib Plus Dexamethasone in Relapsed Multiple Myeloma. Clin. Cancer Res. 2020, 26, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Rasche, L.; Wäsch, R.; Munder, M.; Goldschmidt, H.; Raab, M.S. Novel immunotherapies in multiple myeloma—Chances and challenges. Haematologica 2021, 106, 2555–2565. [Google Scholar] [CrossRef] [PubMed]
- Gormley, N.J.; Ko, C.W.; Deisseroth, A.; Nie, L.; Kaminskas, E.; Kormanik, N.; Goldberg, K.B.; Farrell, A.T.; Pazdur, R. FDA Drug Approval: Elotuzumab in Combination with Lenalidomide and Dexamethasone for the Treatment of Relapsed or Refractory Multiple Myeloma. Clin. Cancer Res. 2017, 23, 6759–6763. [Google Scholar] [CrossRef]
- Bauvois, B. Transmembrane proteases in focus: Diversity and redundancy? J. Leukoc. Biol. 2001, 70, 11–17. [Google Scholar] [CrossRef]
- Verhulst, E.; Garnier, D.; De Meester, I.; Bauvois, B. Validating Cell Surface Proteases as Drug Targets for Cancer Therapy: What Do We Know, and Where Do We Go? Cancers 2022, 14, 624. [Google Scholar] [CrossRef] [PubMed]
- Schliemann, C.; Gerwing, M.; Heinzow, H.; Harrach, S.; Schwöppe, C.; Wildgruber, M.; Hansmeier, A.A.; Angenendt, L.; Berdel, A.F.; Stalmann, U.; et al. First-In-Class CD13-Targeted Tissue Factor tTF-NGR in Patients with Recurrent or Refractory Malignant Tumors: Results of a Phase I Dose-Escalation Study. Cancers 2020, 12, 1488. [Google Scholar] [CrossRef] [PubMed]
- Berglund, Å.; Ullén, A.; Lisyanskaya, A.; Orlov, S.; Hagberg, H.; Tholander, B.; Lewensohn, R.; Nygren, P.; Spira, J.; Harmenberg, J.; et al. First-in-human, phase I/IIa clinical study of the peptidase potentiated alkylator melflufen administered every three weeks to patients with advanced solid tumor malignancies. Investig. New Drugs 2015, 33, 1232–1241. [Google Scholar] [CrossRef]
- Richardson, P.G.; Oriol, A.; Larocca, A.; Bladé, J.; Cavo, M.; Rodriguez-Otero, P.; Leleu, X.; Nadeem, O.; Hiemenz, J.W.; Hassoun, H.; et al. Melflufen and Dexamethasone in Heavily Pretreated Relapsed and Refractory Multiple Myeloma. J. Clin. Oncol. 2021, 39, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Kudo, K.; Imai, C.; Lorenzini, P.; Kamiya, T.; Kono, K.; Davidoff, A.M.; Chng, W.J.; Campana, D. T lymphocytes expressing a CD16 signaling receptor exert antibody-dependent cancer cell killing. Cancer Res. 2014, 74, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Munoz, J.; Jaglowski, S.; McKinney, M.S.; Isufi, I.; Stiff, P.J.; Sachs, J.; Ranger, A.; Harris, P.; Payumo, F.; Akard, L.P. A Phase 1 Study of ACTR087 in Combination with Rituximab, in Subjects with Relapsed or Refractory CD20-Positive B-Cell Lymphoma. Blood 2019, 134, 244. [Google Scholar] [CrossRef]
- Merli, M.; Ferrario, A.; Maffioli, M.; Olivares, C.; Stasia, A.; Arcaini, L.; Passamonti, F. New uses for brentuximab vedotin and novel antibody drug conjugates in lymphoma. Expert Rev. Hematol. 2016, 9, 767–780. [Google Scholar] [CrossRef]
- Grover, N.S.; Savoldo, B. Challenges of driving CD30-directed CAR-T cells to the clinic. BMC Cancer 2019, 19, 203. [Google Scholar] [CrossRef]
- Rodrigues-Fernandes, C.I.; Abreu, L.G.; Radhakrishnan, R.; Perez, D.; Amaral-Silva, G.K.; Gondak, R.O.; Rahimi, S.; Brennan, P.A.; Fonseca, F.P.; Vargas, P.A. Prognostic significance of CD30 expression in diffuse large B-cell lymphoma: A systematic review with meta-analysis. J. Oral Pathol. Med. 2021, 50, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Atrash, S.; Moyo, T.K. A Review of Chimeric Antigen Receptor T-Cell Therapy for Myeloma and Lymphoma. OncoTargets Ther. 2021, 14, 2185–2201. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.; Ruella, M.; Levine, B.L.; Svoboda, J. Adoptive T-cell therapy for Hodgkin lymphoma. Blood Adv. 2021, 5, 4291–4302. [Google Scholar] [CrossRef]
- Witkowska, M.; Smolewski, P.; Robak, T. Investigational therapies targeting CD37 for the treatment of B-cell lymphoid malignancies. Expert Opin. Investig. Drugs 2018, 27, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Payandeh, Z.; Noori, E.; Khalesi, B.; Mard-Soltani, M.; Abdolalizadeh, J.; Khalili, S. Anti-CD37 targeted immunotherapy of B-Cell malignancies. Biotechnol. Lett. 2018, 40, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Bobrowicz, M.; Kubacz, M.; Slusarczyk, A.; Winiarska, M. CD37 in B Cell Derived Tumors-More than Just a Docking Point for Monoclonal Antibodies. Int. J. Mol. Sci. 2020, 21, 9531. [Google Scholar] [CrossRef] [PubMed]
- Danilov, A.V.; Spurgeon, S.E.; Siddiqi, T.; Quinson, A.M.; Maier, D.; Smith, D.; Brown, J.R. A phase Ib, open label, dose escalation trial of the anti-CD37 monoclonal antibody, BI 836826, in combination with ibrutinib in patients with relapsed/refractory chronic lymphocytic leukemia. Investig. New Drugs 2021, 39, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Zhang, Y.; Wang, X. Advances in chimeric antigen receptor T-cell therapy for B-cell non-Hodgkin lymphoma. Biomark Res. 2021, 9, 58. [Google Scholar] [CrossRef]
- Kroschinsky, F.; Middeke, J.M.; Janz, M.; Lenz, G.; Witzens-Harig, M.; Bouabdallah, R.; La Rosée, P.; Viardot, A.; Salles, G.; Kim, S.J.; et al. Phase I dose escalation study of BI 836826 (CD37 antibody) in patients with relapsed or refractory B-cell non-Hodgkin lymphoma. Investig. New Drugs 2020, 38, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- Stathis, A.; Flinn, I.W.; Madan, S.; Maddocks, K.; Freedman, A.; Weitman, S.; Zucca, E.; Munteanu, M.C.; Lia Palomba, M. Safety, tolerability, and preliminary activity of IMGN529, a CD37-targeted antibody-drug conjugate, in patients with relapsed or refractory B-cell non-Hodgkin lymphoma: A dose-escalation, phase I study. Investig. New Drugs 2018, 36, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.Y.; Jagadeesh, D.; Grudeva-Popova, Z.; Trněný, M.; Jurczak, W.; Pylypenko, H.; André, M.; Nasta, S.D.; Rechavi-Robinson, D.; Toffanin, S.; et al. Safety and Efficacy of CD37-Targeting Naratuximab Emtansine PLUS Rituximab in Diffuse Large B-Cell Lymphoma and Other NON-Hodgkin’S B-Cell Lymphomas-a Phase 2 Study. Blood 2021, 138, 526. [Google Scholar] [CrossRef]
- Sawas, A.; Savage, K.J.; Perez, R.P.; Advani, R.H.; Melhem-Bertrandt, A.; Lackey, J.; Trave, F.; Anand, B.; Huang, Y.; Vincent, M.; et al. A first in human experience of the anti-CD37 antibody-drug conjugate AGS67E in lymphoid malignancies. J. Clin. Oncol. 2016, 34, 7549. [Google Scholar] [CrossRef]
- Elvington, M.; Liszewski, M.K.; Atkinson, J.P. CD46 and Oncologic Interactions: Friendly Fire against Cancer. Antibodies 2020, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.; Imus, P.; Mark, T.; Kaufman, J.; Imus, A.; Zonder, J.A.; Walker, Z.; Sherbenou, D.; Schroeder, M.; Abbey, J.; et al. P-225: A first-in-human study of FOR46 in patients with triple refractory Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2021, 21, 164. [Google Scholar] [CrossRef]
- Sick, E.; Jeanne, A.; Schneider, C.; Dedieu, S.; Takeda, K.; Martiny, L. CD47 update: A multifaceted actor in the tumour microenvironment of potential therapeutic interest. Br. J. Pharmacol. 2012, 167, 1415–1430. [Google Scholar] [CrossRef] [PubMed]
- Maute, R.; Xu, J.; Weissman, I.L. CD47-SIRPα-targeted therapeutics: Status and prospects. Immuno. Oncol. Technol. 2022, 13, 100070. [Google Scholar] [CrossRef]
- Jiang, Z.; Sun, H.; Yu, J.; Tian, W.; Song, Y. Targeting CD47 for cancer immunotherapy. J. Hematol. Oncol. 2021, 14, 180. [Google Scholar] [CrossRef]
- Saygin, C.; Carraway, H.E. Current and emerging strategies for management of myelodysplastic syndromes. Blood Rev. 2021, 48, 100791. [Google Scholar] [CrossRef]
- Gilead. Safety Concerns Prompt Pause of Magrolimab Trials. Cancer Discov. 2022, 12, 877–878. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M.; O’Leary, J.; Whiteman, K.R.; Goldmacher, V.S. Targeting CD56 (NCAM)-Expressing Neoplasms with Lorvotuzumab Mertansine. In Antibody-Drug Conjugates and Immunotoxins; Springer: New York, NY, USA, 2012; pp. 273–293. [Google Scholar]
- Ailawadhi, S.; Kelly, K.R.; Vescio, R.A.; Jagannath, S.; Wolf, J.; Gharibo, M.; Sher, T.; Bojanini, L.; Kirby, M.; Chanan-Khan, A. A Phase I Study to Assess the Safety and Pharmacokinetics of Single-agent Lorvotuzumab Mertansine (IMGN901) in Patients with Relapsed and/or Refractory CD-56-positive Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2019, 19, 29–34. [Google Scholar] [CrossRef]
- Gil-Yarom, N.; Radomir, L.; Sever, L.; Kramer, M.P.; Lewinsky, H.; Bornstein, C.; Blecher-Gonen, R.; Barnett-Itzhaki, Z.; Mirkin, V.; Friedlander, G.; et al. CD74 is a novel transcription regulator. Proc. Natl. Acad. Sci. USA 2017, 114, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Stein, R.; Mattes, M.J.; Cardillo, T.M.; Hansen, H.J.; Chang, C.H.; Burton, J.; Govindan, S.; Goldenberg, D.M. CD74: A new candidate target for the immunotherapy of B-cell neoplasms. Clin. Cancer Res. 2007, 13, 5556s–5563s. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, J.L.; Niesvizky, R.; Stadtmauer, E.A.; Chanan-Khan, A.; Siegel, D.; Horne, H.; Wegener, W.A.; Goldenberg, D.M. Phase I, multicentre, dose-escalation trial of monotherapy with milatuzumab (humanized anti-CD74 monoclonal antibody) in relapsed or refractory multiple myeloma. Br. J. Haematol. 2013, 163, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, C.L.; Li, X.; Embry, M.; Yu, A.; Krimm, S.; Krueger, S.; Greenland, N.Y.; Wen, K.W.; Jones, C.; DeAlmeida, V.; et al. Targeting CD74 in multiple myeloma with the novel, site-specific antibody-drug conjugate STRO-001. Oncotarget 2018, 9, 37700–37714. [Google Scholar] [CrossRef] [PubMed]
- Sawalha, Y.; Maddocks, K. Profile of Polatuzumab Vedotin in the Treatment of Patients with Relapsed/Refractory Non-Hodgkin Lymphoma: A Brief Report on the Emerging Clinical Data. Onco Targets Ther. 2020, 13, 5123–5133. [Google Scholar] [CrossRef] [PubMed]
- Arribas, J.; Esselens, C. ADAM17 as a therapeutic target in multiple diseases. Curr. Pharm. Des. 2009, 15, 2319–2335. [Google Scholar] [CrossRef]
- Witters, L.; Scherle, P.; Friedman, S.; Fridman, J.; Caulder, E.; Newton, R.; Lipton, A. Synergistic inhibition with a dual epidermal growth factor receptor/HER-2/neu tyrosine kinase inhibitor and a disintegrin and metalloprotease inhibitor. Cancer Res. 2008, 68, 7083–7089. [Google Scholar] [CrossRef] [PubMed]
- Bachanova, V.; Kolla, B.; Cao, Q.; Weisdorf, D.; Rashidi, A.; Warlick, E.; El Jurdi, N.; Wangen, R.; Arora, M.; Brunstein, C.; et al. ADAM17 Inhibitor INCB7839 With Rituximab As Consolidation After Autologous Hct For Diffuse Large B Cell Lymphoma: A Novel Relapse Prevention Strategy. In Proceedings of the Virtual 46th Annual Meeting of the EBMT, Virtual, 29 August–1 September 2020; p. 133. Available online: https://www.ebmt.org/46th-annual-meeting-ebmt (accessed on 6 November 2022).
- Polson, A.G.; Zheng, B.; Elkins, K.; Chang, W.; Du, C.; Dowd, P.; Yen, L.; Tan, C.; Hongo, J.A.; Koeppen, H.; et al. Expression pattern of the human FcRH/IRTA receptors in normal tissue and in B-chronic lymphocytic leukemia. Int. Immunol. 2006, 18, 1363–1373. [Google Scholar] [CrossRef]
- Ise, T.; Nagata, S.; Kreitman, R.J.; Wilson, W.H.; Wayne, A.S.; Stetler-Stevenson, M.; Bishop, M.R.; Scheinberg, D.A.; Rassenti, L.; Kipps, T.J.; et al. Elevation of soluble CD307 (IRTA2/FcRH5) protein in the blood and expression on malignant cells of patients with multiple myeloma, chronic lymphocytic leukemia, and mantle cell lymphoma. Leukemia 2007, 21, 169–174. [Google Scholar] [CrossRef]
- Smith, E.L.; Harrington, K.; Staehr, M.; Masakayan, R.; Jones, J.; Long, T.J.; Ng, K.Y.; Ghoddusi, M.; Purdon, T.J.; Wang, X.; et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci. Transl. Med. 2019, 11, eaau7746. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Kirsch, B.J.; Chang, S.J.; Betenbaugh, M.J.; Le, A. Non-Hodgkin Lymphoma Metabolism. Adv. Exp. Med. Biol. 2021, 1311, 103–116. [Google Scholar]
- Bhalla, K.; Jaber, S.; Nahid, M.N.; Underwood, K.; Beheshti, A.; Landon, A.; Bhandary, B.; Bastian, P.; Evens, A.M.; Haley, J.; et al. Role of hypoxia in Diffuse Large B-cell Lymphoma: Metabolic repression and selective translation of HK2 facilitates development of DLBCL. Sci. Rep. 2018, 8, 744. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, K.; Kawashima, I.; Koshiisi, M.; Kumagai, T.; Suzuki, M.; Suzuki, J.; Mitsumori, T.; Kirito, K. Glycolytic enzyme hexokinase II is a putative therapeutic target in B-cell malignant lymphoma. Exp. Hematol. 2019, 78, 46–55.e3. [Google Scholar] [CrossRef]
- Lu, J.; Böttcher, M.; Walther, T.; Mougiakakos, D.; Zenz, T.; Huber, W. Energy metabolism is co-determined by genetic variants in chronic lymphocytic leukemia and influences drug sensitivity. Haematologica 2019, 104, 1830–1840. [Google Scholar] [CrossRef] [PubMed]
- Adekola, K.U.; Dalva Aydemir, S.; Ma, S.; Zhou, Z.; Rosen, S.T.; Shanmugam, M. Investigating and targeting chronic lymphocytic leukemia metabolism with the human immunodeficiency virus protease inhibitor ritonavir and metformin. Leuk. Lymphoma 2015, 56, 450–459. [Google Scholar] [CrossRef]
- Dalva-Aydemir, S.; Bajpai, R.; Martinez, M.; Adekola, K.U.; Kandela, I.; Wei, C.; Singhal, S.; Koblinski, J.E.; Raje, N.S.; Rosen, S.T.; et al. Targeting the metabolic plasticity of multiple myeloma with FDA-approved ritonavir and metformin. Clin. Cancer Res. 2015, 21, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Swanson, K.D.; Zheng, B. Therapeutic Repurposing of Biguanides in Cancer. Trends Cancer 2021, 7, 714–730. [Google Scholar] [CrossRef]
- Hartmann, S.; Agostinelli, C.; Diener, J.; Döring, C.; Fanti, S.; Zinzani, P.L.; Gallamini, A.; Bergmann, L.; Pileri, S.; Hansmann, M.L. GLUT1 expression patterns in different Hodgkin lymphoma subtypes and progressively transformed germinal centers. BMC Cancer 2012, 12, 586. [Google Scholar] [CrossRef]
- Afonso, J.; Pinto, T.; Simões-Sousa, S.; Schmitt, F.; Longatto-Filho, A.; Pinheiro, C.; Marques, H.; Baltazar, F. Clinical significance of metabolism-related biomarkers in non-Hodgkin lymphoma-MCT1 as potential target in diffuse large B cell lymphoma. Cell Oncol. (Dordr.) 2019, 42, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Noble, R.A.; Bell, N.; Blair, H.; Sikka, A.; Thomas, H.; Phillips, N.; Nakjang, S.; Miwa, S.; Crossland, R.; Rand, V.; et al. Inhibition of monocarboxyate transporter 1 by AZD3965 as a novel therapeutic approach for diffuse large B-cell lymphoma and Burkitt lymphoma. Haematologica 2017, 102, 1247–1257. [Google Scholar] [CrossRef]
- Halford, S.E.; Walter, H.; McKay, P.; Townsend, W.; Linton, K.; Heinzmann, K.; Dragoni, I.; Brotherton, L.; Veal, G.; Siskos, A.; et al. Phase I expansion study of the first-in-class monocarboxylate transporter 1 (MCT1) inhibitor AZD3965 in patients with diffuse large B-cell lymphoma (DLBCL) and Burkitt lymphoma (BL). J. Clin. Oncol. 2021, 39, 3115. [Google Scholar] [CrossRef]
- Xiao, G.; Chan, L.N.; Klemm, L.; Braas, D.; Chen, Z.; Geng, H.; Zhang, Q.C.; Aghajanirefah, A.; Cosgun, K.N.; Sadras, T.; et al. B-Cell-Specific Diversion of Glucose Carbon Utilization Reveals a Unique Vulnerability in B Cell Malignancies. Cell 2018, 173, 470–484.e18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yao, Y.; Zhang, S.; Liu, Y.; Guo, H.; Ahmed, M.; Bell, T.; Zhang, H.; Han, G.; Lorence, E.; et al. Metabolic reprogramming toward oxidative phosphorylation identifies a therapeutic target for mantle cell lymphoma. Sci. Transl. Med. 2019, 11, eaau1167. [Google Scholar] [CrossRef]
- Zhan, X.; Yu, W.; Franqui-Machin, R.; Bates, M.L.; Nadiminti, K.; Cao, H.; Amendt, B.A.; Jethava, Y.; Frech, I.; Zhan, F.; et al. Alteration of mitochondrial biogenesis promotes disease progression in multiple myeloma. Oncotarget 2017, 8, 111213–111224. [Google Scholar] [CrossRef] [PubMed]
- Galicia-Vázquez, G.; Aloyz, R. Metabolic rewiring beyond Warburg in chronic lymphocytic leukemia: How much do we actually know? Crit. Rev Oncol. Hematol. 2019, 134, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Yun, X.; Zhang, Y.; Wang, X. Targeting metabolic reprogramming in chronic lymphocytic leukemia. Exp. Hematol. Oncol. 2022, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Jitschin, R.; Hofmann, A.D.; Bruns, H.; Gießl, A.; Bricks, J.; Berger, J.; Saul, D.; Eckart, M.J.; Mackensen, A.; Mougiakakos, D. Mitochondrial metabolism contributes to oxidative stress and reveals therapeutic targets in chronic lymphocytic leukemia. Blood 2014, 123, 2663–2672. [Google Scholar] [CrossRef]
- Ecker, V.; Stumpf, M.; Brandmeier, L.; Neumayer, T.; Pfeuffer, L.; Engleitner, T.; Ringshausen, I.; Nelson, N.; Jücker, M.; Wanninger, S.; et al. Targeted PI3K/AKT-hyperactivation induces cell death in chronic lymphocytic leukemia. Nat. Commun. 2021, 12, 3526. [Google Scholar] [CrossRef]
- Noble, R.A.; Thomas, H.; Zhao, Y.; Herendi, L.; Howarth, R.; Dragoni, I.; Keun, H.C.; Vellano, C.P.; Marszalek, J.R.; Wedge, S.R. Simultaneous targeting of glycolysis and oxidative phosphorylation as a therapeutic strategy to treat diffuse large B-cell lymphoma. Br. J. Cancer 2022, 127, 937–947. [Google Scholar] [CrossRef]
- Zachar, Z.; Marecek, J.; Maturo, C.; Gupta, S.; Stuart, S.D.; Howell, K.; Schauble, A.; Lem, J.; Piramzadian, A.; Karnik, S.; et al. Non-redox-active lipoate derivates disrupt cancer cell mitochondrial metabolism and are potent anticancer agents in vivo. J. Mol. Med. 2011, 89, 1137–1148. [Google Scholar] [CrossRef]
- Stuart, S.D.; Schauble, A.; Gupta, S.; Kennedy, A.D.; Keppler, B.R.; Bingham, P.M.; Zachar, Z. A strategically designed small molecule attacks alpha-ketoglutarate dehydrogenase in tumor cells through a redox process. Cancer Metab. 2014, 2, 4. [Google Scholar] [CrossRef]
- Ambrosio, M.R.; Piccaluga, P.P.; Ponzoni, M.; Rocca, B.J.; Malagnino, V.; Onorati, M.; De Falco, G.; Calbi, V.; Ogwang, M.; Naresh, K.N.; et al. The alteration of lipid metabolism in Burkitt lymphoma identifies a novel marker: Adipophilin. PLoS ONE 2012, 7, e44315. [Google Scholar] [CrossRef] [PubMed]
- Heintel, D.; Kienle, D.; Shehata, M.; Kröber, A.; Kroemer, E.; Schwarzinger, I.; Mitteregger, D.; Le, T.; Gleiss, A.; Mannhalter, C.; et al. High expression of lipoprotein lipase in poor risk B-cell chronic lymphocytic leukemia. Leukemia 2005, 19, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Rozovski, U.; Grgurevic, S.; Bueso-Ramos, C.; Harris, D.M.; Li, P.; Liu, Z.; Wu, J.Y.; Jain, P.; Wierda, W.; Burger, J.; et al. Aberrant LPL expression, driven by STAT3, Mediates free fatty acid metabolism in cll cells. Mol. Cancer Res. 2015, 13, 944–953. [Google Scholar] [CrossRef] [PubMed]
- Pallasch, C.P.; Schwamb, J.; Königs, S.; Schulz, A.; Debey, S.; Kofler, D.; Schultze, J.L.; Hallek, M.; Ultsch, A.; Wendtner, C.M. Targeting lipid metabolism by the lipoprotein lipase inhibitor orlistat results in apoptosis of B-cell chronic lymphocytic leukemia cells. Leukemia 2008, 22, 585–592. [Google Scholar] [CrossRef]
- Gelebart, P.; Zak, Z.; Anand, M.; Belch, A.; Lai, R. Blockade of fatty acid synthase triggers significant apoptosis in mantle cell lymphoma. PLoS ONE 2012, 7, e33738. [Google Scholar] [CrossRef]
- Uddin, S.; Hussain, A.R.; Ahmed, M.; Abubaker, J.; Al-Sanea, N.; Abduljabbar, A.; Ashari, L.H.; Alhomoud, S.; Al-Dayel, F.; Bavi, P.; et al. High prevalence of fatty acid synthase expression in colorectal cancers in Middle Eastern patients and its potential role as a therapeutic target. Am. J. Gastroenterol. 2009, 104, 1790–1801. [Google Scholar] [CrossRef]
- Rozovski, U.; Harris, D.M.; Li, P.; Liu, Z.; Jain, P.; Ferrajoli, A.; Burger, J.; Thompson, P.; Jain, N.; Wierda, W.; et al. STAT3-activated CD36 facilitates fatty acid uptake in chronic lymphocytic leukemia cells. Oncotarget 2018, 9, 21268–21280. [Google Scholar] [CrossRef]
- Liu, P.P.; Liu, J.; Jiang, W.Q.; Carew, J.S.; Ogasawara, M.A.; Pelicano, H.; Croce, C.M.; Estrov, Z.; Xu, R.H.; Keating, M.J.; et al. Elimination of chronic lymphocytic leukemia cells in stromal microenvironment by targeting CPT with an antiangina drug perhexiline. Oncogene 2016, 35, 5663–5673. [Google Scholar] [CrossRef]
- Bolzoni, M.; Chiu, M.; Accardi, F.; Vescovini, R.; Airoldi, I.; Storti, P.; Todoerti, K.; Agnelli, L.; Missale, G.; Andreoli, R.; et al. Dependence on glutamine uptake and glutamine addiction characterize myeloma cells: A new attractive target. Blood 2016, 128, 667–679. [Google Scholar] [CrossRef]
- Galicia-Vázquez, G.; Smith, S.; Aloyz, R. Del11q-positive CLL lymphocytes exhibit altered glutamine metabolism and differential response to GLS1 and glucose metabolism inhibition. Blood Cancer J. 2018, 8, 13. [Google Scholar] [CrossRef]
- Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H.; et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012, 15, 110–121. [Google Scholar] [CrossRef]
- Anastasiou, D. Tumour microenvironment factors shaping the cancer metabolism landscape. Br. J. Cancer 2017, 116, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhou, X.; Wang, X. Targeting the tumor microenvironment in B-cell lymphoma: Challenges and opportunities. J. Hematol. Oncol. 2021, 14, 125. [Google Scholar] [CrossRef] [PubMed]
- Vangapandu, H.V.; Ayres, M.L.; Bristow, C.A.; Wierda, W.G.; Keating, M.J.; Balakrishnan, K.; Stellrecht, C.M.; Gandhi, V. The Stromal Microenvironment Modulates Mitochondrial Oxidative Phosphorylation in Chronic Lymphocytic Leukemia Cells. Neoplasia 2017, 19, 762–771. [Google Scholar] [CrossRef] [PubMed]
- Yosifov, D.Y.; Idler, I.; Bhattacharya, N.; Reichenzeller, M.; Close, V.; Ezerina, D.; Scheffold, A.; Jebaraj, B.M.C.; Kugler, S.; Bloehdorn, J.; et al. Oxidative stress as candidate therapeutic target to overcome microenvironmental protection of CLL. Leukemia 2020, 34, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Trachootham, D.; Liu, J.; Chen, G.; Pelicano, H.; Garcia-Prieto, C.; Lu, W.; Burger, J.A.; Croce, C.M.; Plunkett, W.; et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat. Cell Biol. 2012, 14, 276–286. [Google Scholar] [CrossRef]
- Chen, Z.; Simon-Molas, H.; Cretenet, G.; Valle-Argos, B.; Smith, L.D.; Forconi, F.; Schomakers, B.V.; van Weeghel, M.; Bryant, D.J.; van Bruggen, J.A.C.; et al. Characterization of metabolic alterations of chronic lymphocytic leukemia in the lymph node microenvironment. Blood 2022, 140, 630–643. [Google Scholar] [CrossRef]
- Burt, R.; Dey, A.; Aref, S.; Aguiar, M.; Akarca, A.; Bailey, K.; Day, W.; Hooper, S.; Kirkwood, A.; Kirschner, K.; et al. Activated stromal cells transfer mitochondria to rescue acute lymphoblastic leukemia cells from oxidative stress. Blood 2019, 134, 1415–1429. [Google Scholar] [CrossRef]
- Marlein, C.R.; Piddock, R.E.; Mistry, J.J.; Zaitseva, L.; Hellmich, C.; Horton, R.H.; Zhou, Z.; Auger, M.J.; Bowles, K.M.; Rushworth, S.A. CD38-Driven Mitochondrial Trafficking Promotes Bioenergetic Plasticity in Multiple Myeloma. Cancer Res. 2019, 79, 2285–2297. [Google Scholar] [CrossRef]
- Panaroni, C.; Fulzele, K.; Mori, T.; Siu, K.T.; Onyewadume, C.; Maebius, A.; Raje, N. Multiple myeloma cells induce lipolysis in adipocytes and uptake fatty acids through fatty acid transporter proteins. Blood 2022, 139, 876–888. [Google Scholar] [CrossRef]
- Pavlides, S.; Tsirigos, A.; Vera, I.; Flomenberg, N.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; Pestell, R.G.; et al. Loss of stromal caveolin-1 leads to oxidative stress, mimics hypoxia and drives inflammation in the tumor microenvironment, conferring the “reverse Warburg effect”: A transcriptional informatics analysis with validation. Cell Cycle 2010, 9, 2201–2219. [Google Scholar] [CrossRef] [PubMed]
- Sotgia, F.; Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Flomenberg, N.; Birbe, R.C.; Witkiewicz, A.K.; Howell, A.; Philp, N.J.; Pestell, R.G.; Lisanti, M.P. Mitochondrial metabolism in cancer metastasis: Visualizing tumor cell mitochondria and the “reverse Warburg effect” in positive lymph node tissue. Cell Cycle 2012, 11, 1445–1454. [Google Scholar] [CrossRef] [PubMed]
- Gooptu, M.; Whitaker-Menezes, D.; Sprandio, J.; Domingo-Vidal, M.; Lin, Z.; Uppal, G.; Gong, J.; Fratamico, R.; Leiby, B.; Dulau-Florea, A.; et al. Mitochondrial and glycolytic metabolic compartmentalization in diffuse large B-cell lymphoma. Semin. Oncol. 2017, 44, 204–217. [Google Scholar] [PubMed]
- Sakamoto, A.; Kunou, S.; Shimada, K.; Tsunoda, M.; Aoki, T.; Iriyama, C.; Tomita, A.; Nakamura, S.; Hayakawa, F.; Kiyoi, H. Pyruvate secreted from patient-derived cancer-associated fibroblasts supports survival of primary lymphoma cells. Cancer Sci. 2019, 110, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Mikkilineni, L.; Whitaker-Menezes, D.; Domingo-Vidal, M.; Sprandio, J.; Avena, P.; Cotzia, P.; Dulau-Florea, A.; Gong, J.; Uppal, G.; Zhan, T.; et al. Hodgkin lymphoma: A complex metabolic ecosystem with glycolytic reprogramming of the tumor microenvironment. Semin. Oncol. 2017, 44, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Alfaifi, A.; Bahashwan, S.; Alsaadi, M.; Malhan, H.; Aqeel, A.; Al-Kahiry, W.; Almehdar, H.; Qadri, I. Metabolic Biomarkers in B-Cell Lymphomas for Early Diagnosis and Prediction, as Well as Their Influence on Prognosis and Treatment. Diagnostics 2022, 12, 394. [Google Scholar] [CrossRef]
- Liu, X.; Wang, L.; Jiang, W.; Lu, W.; Yang, J.; Yang, W. B cell lymphoma with different metabolic characteristics show distinct sensitivities to metabolic inhibitors. J. Cancer 2018, 9, 1582–1591. [Google Scholar] [CrossRef]
- Barberini, L.; Noto, A.; Fattuoni, C.; Satta, G.; Zucca, M.; Cabras, M.G.; Mura, E.; Cocco, P. The Metabolomic Profile of Lymphoma Subtypes: A Pilot Study. Molecules 2019, 24, 2367. [Google Scholar] [CrossRef]
- Puchades-Carrasco, L.; Lecumberri, R.; Martínez-López, J.; Lahuerta, J.J.; Mateos, M.V.; Prósper, F.; San-Miguel, J.F.; Pineda-Lucena, A. Multiple myeloma patients have a specific serum metabolomic profile that changes after achieving complete remission. Clin. Cancer Res. 2013, 19, 4770–4779. [Google Scholar] [CrossRef]
- Bai, Y.; Zhang, H.; Sun, X.; Sun, C.; Ren, L. Biomarker identification and pathway analysis by serum metabolomics of childhood acute lymphoblastic leukemia. Clin. Chim. Acta 2014, 436, 207–216. [Google Scholar] [CrossRef]
- MacIntyre, D.A.; Jiménez, B.; Lewintre, E.J.; Martín, C.R.; Schäfer, H.; Ballesteros, C.G.; Mayans, J.R.; Spraul, M.; García-Conde, J.; Pineda-Lucena, A. Serum metabolome analysis by 1H-NMR reveals differences between chronic lymphocytic leukaemia molecular subgroups. Leukemia 2010, 24, 788–797. [Google Scholar] [CrossRef] [PubMed]
- Monti, S.; Savage, K.J.; Kutok, J.L.; Feuerhake, F.; Kurtin, P.; Mihm, M.; Wu, B.; Pasqualucci, L.; Neuberg, D.; Aguiar, R.C.; et al. Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood 2005, 105, 1851–1861. [Google Scholar] [CrossRef] [PubMed]
- Caro, P.; Kishan, A.U.; Norberg, E.; Stanley, I.A.; Chapuy, B.; Ficarro, S.B.; Polak, K.; Tondera, D.; Gounarides, J.; Yin, H.; et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell 2012, 22, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Monti, S.; Juszczynski, P.; Daley, J.; Chen, W.; Witzig, T.E.; Habermann, T.M.; Kutok, J.L.; Shipp, M.A. SYK-dependent tonic B-cell receptor signaling is a rational treatment target in diffuse large B-cell lymphoma. Blood 2008, 111, 2230–2237. [Google Scholar] [CrossRef]
- Chiche, J.; Reverso-Meinietti, J.; Mouchotte, A.; Rubio-Patiño, C.; Mhaidly, R.; Villa, E.; Bossowski, J.P.; Proics, E.; Grima-Reyes, M.; Paquet, A.; et al. GAPDH Expression Predicts the Response to R-CHOP, the Tumor Metabolic Status, and the Response of DLBCL Patients to Metabolic Inhibitors. Cell Metab. 2019, 29, 1243–1257.e10. [Google Scholar] [CrossRef]
- Lemberg, K.M.; Gori, S.S.; Tsukamoto, T.; Rais, R.; Slusher, B.S. Clinical development of metabolic inhibitors for oncology. J. Clin. Investig. 2022, 132, e148550. [Google Scholar] [CrossRef]
- Stine, Z.E.; Schug, Z.T.; Salvino, J.M.; Dang, C.V. Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 2022, 21, 141–162. [Google Scholar] [CrossRef]
- Pi, M.; Kuang, H.; Yue, C.; Yang, Q.; Wu, A.; Li, Y.; Assaraf, Y.G.; Yang, D.H.; Wu, S. Targeting metabolism to overcome cancer drug resistance: A promising therapeutic strategy for diffuse large B cell lymphoma. Drug Resist. Updates 2022, 61, 100822. [Google Scholar] [CrossRef]
- Gdynia, G.; Robak, T.; Kopitz, J.; Heller, A.; Grekova, S.; Duglova, K.; Laukemper, G.; Heinzel-Gutenbrunner, M.; Gutenbrunner, C.; Roth, W.; et al. Distinct Activities of Glycolytic Enzymes Identify Chronic Lymphocytic Leukemia Patients with a more Aggressive Course and Resistance to Chemo-Immunotherapy. eBioMedicine 2018, 32, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Guièze, R.; Liu, V.M.; Rosebrock, D.; Jourdain, A.A.; Hernández-Sánchez, M.; Martinez Zurita, A.; Sun, J.; Ten Hacken, E.; Baranowski, K.; Thompson, P.A.; et al. Mitochondrial Reprogramming Underlies Resistance to BCL-2 Inhibition in Lymphoid Malignancies. Cancer Cell 2019, 36, 369–384.e13. [Google Scholar] [CrossRef]
- Bajpai, R.; Sharma, A.; Achreja, A.; Edgar, C.L.; Wei, C.; Siddiqa, A.A.; Gupta, V.A.; Matulis, S.M.; McBrayer, S.K.; Mittal, A.; et al. Electron transport chain activity is a predictor and target for venetoclax sensitivity in multiple myeloma. Nat. Commun. 2020, 11, 1228. [Google Scholar] [CrossRef]
- Chukkapalli, V.; Gordon, L.I.; Venugopal, P.; Borgia, J.A.; Karmali, R. Metabolic changes associated with metformin potentiates Bcl-2 inhibitor, Venetoclax, and CDK9 inhibitor, BAY1143572 and reduces viability of lymphoma cells. Oncotarget 2018, 9, 21166–21181. [Google Scholar] [CrossRef] [PubMed]
- Ravera, S.; Ghiotto, F.; Tenca, C.; Gugiatti, E.; Santamaria, S.; Ledda, B.; Ibatici, A.; Cutrona, G.; Mazzarello, A.N.; Bagnara, D.; et al. Berberine affects mitochondrial activity and cell growth of leukemic cells from chronic lymphocytic leukemia patients. Sci. Rep. 2020, 10, 16519. [Google Scholar] [CrossRef]
- Vangapandu, H.V.; Havranek, O.; Ayres, M.L.; Kaipparettu, B.A.; Balakrishnan, K.; Wierda, G.D.W.; Keating, M.J.; Eric Davis, R.; Stellrecht, C.M.; Gandhi, V.; et al. B-cell receptor signaling regulates metabolism in chronic lymphocytic leukemia. Mol. Cancer Res. 2017, 15, 1692–1703. [Google Scholar] [CrossRef]
- Chowdhury, R.S.; Bouchard, E.D.J.; Saleh, R.; Nugent, Z.; Peltier, C.; Mejia, E.; Hou, S.; McFall, C.; Squires, M.; Hewitt, D.; et al. Mitochondrial Respiration Correlates with Prognostic Markers in Chronic Lymphocytic Leukemia and Is Normalized by Ibrutinib Treatment. Cancers 2020, 12, 650. [Google Scholar] [CrossRef]
- Lee, S.C.; Shestov, A.A.; Guo, L.; Zhang, Q.; Roman, J.C.; Liu, X.; Wang, H.Y.; Pickup, S.; Nath, K.; Lu, P.; et al. Metabolic Detection of Bruton’s Tyrosine Kinase Inhibition in Mantle Cell Lymphoma Cells. Mol. Cancer Res. 2019, 17, 1365–1377. [Google Scholar] [CrossRef] [PubMed]
- Yang, M. B cell lymphoma R-CHOPped by metabolic inhibitors. Sci. Transl. Med. 2019, 11, 117. [Google Scholar] [CrossRef]
- Gu, J.J.; Singh, A.; Xue, K.; Mavis, C.; Barth, M.; Yanamadala, V.; Lenz, P.; Grau, M.; Lenz, G.; Czuczman, M.S.; et al. Up-regulation of hexokinase II contributes to rituximab-chemotherapy resistance and is a clinically relevant target for therapeutic development. Oncotarget 2018, 9, 4020–4033. [Google Scholar] [CrossRef] [PubMed]
- Curtis, N.J.; Mooney, L.; Hopcroft, L.; Michopoulos, F.; Whalley, N.; Zhong, H.; Murray, C.; Logie, A.; Revill, M.; Byth, K.F.; et al. Pre-clinical pharmacology of AZD3965, a selective inhibitor of MCT1: DLBCL, NHL and Burkitt’s lymphoma anti-tumor activity. Oncotarget 2017, 8, 69219–69236. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.R.; Gu, J.J.; Zhang, Q.; Torka, P.; Sundaram, S.; Mavis, C.; Hernandez-Ilizaliturri, F.J. Metformin sensitizes therapeutic agents and improves outcome in pre-clinical and clinical diffuse large B-cell lymphoma. Cancer Metab. 2020, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Mo, Q.; Sun, X.; Wang, X.; Dong, M.; Zhang, G.; Chen, F.; Zhao, Q. Pyruvate dehydrogenase kinase 4-mediated metabolic reprogramming is involved in rituximab resistance in diffuse large B-cell lymphoma by affecting the expression of MS4A1/CD20. Cancer Sci. 2021, 112, 3585–3597. [Google Scholar] [CrossRef] [PubMed]
- Ricci, J.E.; Chiche, J. Metabolic Reprogramming of Non-Hodgkin’s B-Cell Lymphomas and Potential Therapeutic Strategies. Front. Oncol. 2018, 8, 556. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.M.; Dytfeld, D.; Reyes, L.; Robinson, R.M.; Smith, B.; Manevich, Y.; Jakubowiak, A.; Komarnicki, M.; Przybylowicz-Chalecka, A.; Szczepaniak, T.; et al. Glutaminase inhibitor CB-839 synergizes with carfilzomib in resistant multiple myeloma cells. Oncotarget 2017, 8, 35863. [Google Scholar] [CrossRef]
- Sanchez, W.Y.; McGee, S.L.; Connor, T.; Mottram, B.; Wilkinson, A.; Whitehead, J.P.; Vuckovic, S.; Catley, L. Dichloroacetate inhibits aerobic glycolysis in multiple myeloma cells and increases sensitivity to bortezomib. Br. J. Cancer 2013, 108, 1624–1633. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, M.; Baur, R.; Stoll, A.; Mackensen, A.; Mougiakakos, D. Linking Immunoevasion and Metabolic Reprogramming in B-Cell-Derived Lymphomas. Front. Oncol. 2020, 10, 594782. [Google Scholar] [CrossRef]
- Beielstein, A.C.; Pallasch, C.P. Tumor Metabolism as a Regulator of Tumor-Host Interactions in the B-Cell Lymphoma Microenvironment-Fueling Progression and Novel Brakes for Therapy. Int. J. Mol. Sci. 2019, 20, 4158. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wenes, M.; Romero, P.; Huang, S.C.; Fendt, S.M.; Ho, P.C. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 425–441. [Google Scholar] [CrossRef]
- Xu, X.; Gnanaprakasam, J.N.R.; Sherman, J.; Wang, R. A Metabolism Toolbox for CAR T Therapy. Front. Oncol. 2019, 9, 322. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Zhou, W.; Guo, C.; Fu, Z.; Zhu, L.; Li, P.; Xu, Y.; Zheng, L.; Zhang, H.; Shan, C.; et al. Glutaminolysis Mediated by MALT1 Protease Activity Facilitates PD-L1 Expression on ABC-DLBCL Cells and Contributes to Their Immune Evasion. Front. Oncol. 2018, 8, 632. [Google Scholar] [CrossRef]
- Blombery, P. Mechanisms of intrinsic and acquired resistance to venetoclax in B-cell lymphoproliferative disease. Leuk. Lymphoma 2020, 61, 257–262. [Google Scholar] [CrossRef]
- Diepstraten, S.T.; Anderson, M.A.; Czabotar, P.E.; Lessene, G.; Strasser, A.; Kelly, G.L. The manipulation of apoptosis for cancer therapy using BH3-mimetic drugs. Nat. Rev. Cancer 2022, 22, 45–64. [Google Scholar] [CrossRef]
- Lipsky, A.; Lamanna, N. Managing toxicities of Bruton tyrosine kinase inhibitors. Hematol. Am. Soc. Hematol. Educ. Program 2020, 2020, 336–345. [Google Scholar] [CrossRef] [PubMed]
- George, B.; Chowdhury, S.M.; Hart, A.; Sircar, A.; Singh, S.K.; Nath, U.K.; Mamgain, M.; Singhal, N.K.; Sehgal, L.; Jain, N. Ibrutinib Resistance Mechanisms and Treatment Strategies for B-Cell lymphomas. Cancers 2020, 12, 1328. [Google Scholar] [CrossRef] [PubMed]
- Wright, S.C.E.; Vasilevski, N.; Serra, V.; Rodon, J.; Eichhorn, P.J.A. Mechanisms of Resistance to PI3K Inhibitors in Cancer: Adaptive Responses, Drug Tolerance and Cellular Plasticity. Cancers 2021, 13, 1538. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Liu, Z.; Liu, J.; Xia, Y.; Hu, K.; Yu, J. Application of Chimeric Antigen Receptor T Cells in the Treatment of Hematological Malignancies. Biomed. Res. Int. 2020, 2020, 4241864. [Google Scholar] [CrossRef] [PubMed]
- Edeline, J.; Houot, R.; Marabelle, A.; Alcantara, M. CAR-T cells and BiTEs in solid tumors: Challenges and perspectives. J. Hematol. Oncol. 2021, 14, 65. [Google Scholar] [CrossRef]
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson, R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B.; et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 2019, 37, 1049–1058. [Google Scholar] [CrossRef]
- Yin, Y.; Rodriguez, J.L.; Li, N.; Thokala, R.; Nasrallah, M.P.; Hu, L.; Zhang, L.; Zhang, J.V.; Logun, M.T.; Kainth, D.; et al. Locally secreted BiTEs complement CAR T cells by enhancing killing of antigen heterogeneous solid tumors. Mol. Ther. 2022, 30, 2537–2553. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shmuel, A.; Biber, G.; Barda-Saad, M. Unleashing Natural Killer Cells in the Tumor Microenvironment-The Next Generation of Immunotherapy? Front. Immunol. 2020, 11, 275. [Google Scholar] [CrossRef]
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J. CAR-NK cells: A promising cellular immunotherapy for cancer. eBioMedicine 2020, 59, 102975. [Google Scholar] [CrossRef]
- Islam, R.; Pupovac, A.; Evtimov, V.; Boyd, N.; Shu, R.; Boyd, R.; Trounson, A. Enhancing a Natural Killer: Modification of NK Cells for Cancer Immunotherapy. Cells 2021, 10, 1058. [Google Scholar] [CrossRef] [PubMed]
- Berenson, J.R.; Martinez, D.; Safaie, T.; Silagan, N.; To, J.; Spektor, T.M.; Forouzan, E.; Swift, R.; Eades, B.; Eshaghian, S.; et al. Efficacy and Safety of Ruxolitinib and Steroids for Treating Patients with Relapsed or Refractory Multiple Myeloma. Blood 2021, 138, 2727. [Google Scholar] [CrossRef]
- Italiano, A.; Soria, J.C.; Toulmonde, M.; Michot, J.M.; Lucchesi, C.; Varga, A.; Coindre, J.M.; Blakemore, S.J.; Clawson, A.; Suttle, B.; et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: A first-in-human, open-label, phase 1 study. Lancet Oncol. 2018, 19, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Sarkozy, C.; Morschhauser, F.; Dubois, S.; Molina, T.; Michot, J.M.; Cullières-Dartigues, P.; Suttle, B.; Karlin, L.; Le Gouill, S.; Picquenot, J.M.; et al. A LYSA Phase Ib Study of Tazemetostat (EPZ-6438) plus R-CHOP in Patients with Newly Diagnosed Diffuse Large B-Cell Lymphoma (DLBCL) with Poor Prognosis Features. Clin. Cancer Res. 2020, 26, 3145–3153. [Google Scholar] [CrossRef] [PubMed]
- Morschhauser, F.; Tilly, H.; Chaidos, A.; McKay, P.; Phillips, T.; Assouline, S.; Batlevi, C.L.; Campbell, P.; Ribrag, V.; Damaj, G.L.; et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: An open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol. 2020, 21, 1433–1442. [Google Scholar] [CrossRef]
- Major, A.; Kline, J.; Karrison, T.G.; Fishkin, P.A.S.; Kimball, A.S.; Petrich, A.M.; Nattam, S.; Rao, K.; Sleckman, B.G.; Cohen, K.; et al. Phase I/II clinical trial of temsirolimus and lenalidomide in patients with relapsed and refractory lymphomas. Haematologica 2022, 107, 1608–1618. [Google Scholar] [CrossRef]
- Tan, F.H.; Putoczki, T.L.; Stylli, S.S.; Luwor, R.B. Ponatinib: A novel multi-tyrosine kinase inhibitor against human malignancies. Onco Targets Ther. 2019, 12, 635–645. [Google Scholar] [CrossRef]
- Cortes, J.E.; Kim, D.W.; Pinilla-Ibarz, J.; le Coutre, P.D.; Paquette, R.; Chuah, C.; Nicolini, F.E.; Apperley, J.F.; Khoury, H.J.; Talpaz, M.; et al. Ponatinib efficacy and safety in Philadelphia chromosome-positive leukemia: Final 5-year results of the phase 2 PACE trial. Blood 2018, 132, 393–404. [Google Scholar] [CrossRef]
- Martinelli, G.; Papayannidis, C.; Piciocchi, A.; Robustelli, V.; Soverini, S.; Terragna, C.; Marconi, G.; Lemoli, R.M.; Guolo, F.; Fornaro, A.; et al. INCB84344-201: Ponatinib and steroids in frontline therapy for unfit patients with Ph+ acute lymphoblastic leukemia. Blood Adv. 2022, 6, 1742–1753. [Google Scholar] [CrossRef]
- Ribera, J.M.; García-Calduch, O.; Ribera, J.; Montesinos, P.; Cano-Ferri, I.; Martínez, P.; Esteve, J.; Esteban, D.; García-Fortes, M.; Alonso, N.; et al. Ponatinib, chemotherapy, and transplant in adults with Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood Adv. 2022, 6, 5395–5402. [Google Scholar] [CrossRef]
- Short, N.J.; Kantarjian, H.M.; Konopleva, M.; Jain, N.; Huang, X.; Ravandi, F.; Wierda, W.G.; Borthakur, G.; Sasaki, K.; Issa, G.C.; et al. Combination of ponatinib and blinatumomab in Philadelphia chromosome-positive acute lymphoblastic leukemia: Early results from a phase II study. J. Clin. Oncol. 2021, 39, 7001. [Google Scholar] [CrossRef]
- Peterson, T.J.; Orozco, J.; Buege, M. Selinexor: A First-in-Class Nuclear Export Inhibitor for Management of Multiply Relapsed Multiple Myeloma. Ann. Pharmacother. 2020, 54, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Sarnik, J.; Popławski, T.; Tokarz, P. BET Proteins as Attractive Targets for Cancer Therapeutics. Int. J. Mol. Sci. 2021, 22, 11102. [Google Scholar] [CrossRef]
- Burgoyne, A.M.; Vann, K.R.; Joshi, S.; Morales, G.A.; Vega, F.M.; Singh, A.; Pal, D.; Merati, A.B.; Kutateladze, T.G.; Durden, D.L. A triple action CDK4/6-PI3K-BET inhibitor with augmented cancer cell cytotoxicity. Cell Discov. 2020, 6, 49. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.L.; Eiken, A.P.; Skupa, S.A.; Moore, D.Y.; Umeta, L.T.; Smith, L.M.; Lyden, E.R.; D’Angelo, C.R.; Kallam, A.; Vose, J.M.; et al. A Novel Triple-Action Inhibitor Targeting B-Cell Receptor Signaling and BRD4 Demonstrates Preclinical Activity in Chronic Lymphocytic Leukemia. Int. J. Mol. Sci. 2022, 23, 6712. [Google Scholar] [CrossRef] [PubMed]
- Vann, K.R.; Pal, D.; Smith, A.L.; Sahar, N.E.; Krishnaiah, M.; El-Gamal, D.; Kutateladze, T.G. Combinatorial inhibition of BTK, PI3K-AKT and BRD4-MYC as a strategy for treatment of mantle cell lymphoma. Mol. Biomed. 2022, 3, 2. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Peiris-Pagés, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14, 113. [Google Scholar] [CrossRef]




| Drug | Class | Disease Setting | Phase | Study | References |
|---|---|---|---|---|---|
| Belantamab mafodotin/ GSK2857916 | ADC anti-BCMA-monomethyl auristatin F | R/R MM | I/II I/II | NCT03848845 Active, 2018- Alone or combined with pembrolizumab NCT04126200 NCT03544281 NCT03715478 Recruiting, 2018- Alone or combined with current therapies | [273] |
| Teclistamab/ JNJ-64007957 | BiTE anti-BCMA/CD3 | R/R MM | I/II I III | NCT04557098 Active, 2017–2024 NCT04586426 Recruiting, 2019–2024 Combined with talquetamab or daratumuab or pomalidomide NCT05083169 Recruiting, 2021–2026 daratumumab compared with current therapies | [278] |
| Elranatamab/ PF-06863135 | BiTE anti-BCMA/CD3 | Refractory MM | II | NCT04649359 Active, 2020–2024 | |
| Idecabtagene vicleucel/ bb2121 | Anti-BCMA CAR-T | R/R MM | II | NCT03361748 Active, 2017–2024 | [279] |
| BM38 | Dual anti-BCMA/CD38 CAR-T | R/R MM | I | ChiCTR1900026286 Active, 2019- | [281] |
| BCMA-CAR-NK92 | Anti-BCMA CAR-NK | R/R MM | I/II | NCT03940833 Recruiting, 2019–2022 | [282] |
| Lanalumab/VAY-736 | Anti-BAFF-R Ab | CLL | I | NCT03400176, Recruiting, 2018- Alone or combined with ibrutinib | |
| BAFFR-CAR T | Anti-BAFF-R CAR-T | B-ALL, R/R MCL | I | NCT04690595, NCT05370430 Recruiting, 2020–2024 | |
| Cirmtuzumab/ UC961 newly renamed Zilovertamab | Anti-ROR1 Ab | CLL CLL, SLL, MCL R/R MCL | II I/II III | NCT04501939 Recruiting, 2020- Combined with venetoclax NCT03088878 Active, 2018–2025 Combined with ibrutinib NCT05431179 Recruiting, 2022- Combined with ibrutinib | |
| Pembrolizumab/MK-3475 | Anti-PD1 Ab recruiting immune cells to attack tumor B cells | R/R CLL, B-NHL | II | NCT02332980 Active, 2015–2023 Alone or combined with ibrutinib or idelalisib | [283] |
| High-Risk or R/R CLL/SLL | II |
NCT03204188 Active, 2017–2030 Combined with ibrutinib, fludarabine | |||
| R/R WM | II |
NCT03630042 Recruiting, 2018- Combined with rituximab | |||
| R/R B-ALL | I/II |
NCT03512405 Recruiting, 2018- Combined with blinatumomab | [284] | ||
| R/R DLBCL | I |
NCT03340766 Active, 2017- Combined with blinatumomab | |||
| Cemiplimab/ REGN2810 | Anti-PD1 Ab recruiting immune cells to attack tumor B cells | B-NHL R/R MM DLBCL | I I/II I/II | NCT02651662 Active, 2016- Combined with REGN1979 (anti-CD20-CD3) NCT03194867 Active, 2016- Combined with isatuximab NCT03769181 Active, 2018- Combined with isatuximab | [285] |
| Atezolizumab | Anti-PD-L1 Ab | DLBCL | I/II | NCT02926833 Active, 2016–2022 Alone or combined with axicabtagene ciloleucel | [286] |
| Ulocuplumab/ BMS-936564 | Anti-CXCR4 Ab | WM | I/II | NCT03225716 Active, 2017–2025 Alone or combined with ibrutinib | [287] |
| Mavorixafor/ AMD070 | CXCR4 antagonist (blocks CXCL12 binding) | WM | I | NCT04274738 Active, 2020–2025 Alone or combined with ibrutinib | |
| CS1-CAR-T | CS1-CAR-T/IL7- CCL19 | R/R MM | I | NCT03778346 Recruiting, 2018–2022 | |
| CS1-CAR-T | CS1-CAR-T/41BB- tEGFR | R/R MM | I | NCT03710421 Recruiting, 2018–2023 following chemotherapy | |
| J1/Melflufen | Melphalan-p-fluoro-L-phenylalalnine (aminopeptidase substrat) | MM refractory to lenalidomide | III | NCT03151811 Active, 2017- combined with dexamethasone compared to pomalidomide + dexamethasone | [288] |
| Brentuximab vedotin | ADC anti-CD30-mono methyl auristatin E | R/R B-NHL | II | NCT03646123 Active, 2018–2026 | |
| Lilotomab/ tetulomab/ betalutin | ARC anti-CD37-177lutetium | Relapsed B-NHL | I/II | NCT01796171 Recruiting, 2013–2026 | [289] |
| GEN3009 | DuoHexaBody-CD37 | R/R B-NHL | I | NCT04358458 Recruiting, 2020–2025 | |
| CART30 | Anti-CD30 CAR-T | B-NHL | I | NCT02259556 Recruiting, 2016–2029 Combined with cyclophosphamide and fludarabine | [290] |
| CD30.CAR-Ts | Anti-CD30 CAR-T | B-NHL | I/II | NCT02690545 Recruiting, 2016–2038 Combined with fludarabine and bendamustine | [291] |
| CD30.CAR T | Anti-CD30 CAR-T | B-NHL | I | NCT02917083 Recruiting, 2018–2036 | |
| ICAR30 | Anti-CD30 CAR-T | B-NHL | I | NCT03383965 Recruiting, 2017–2025 | |
| ATLCAR.CD30 | Anti-CD30 CAR-T | B-NHL | I | NCT026663297 Active, 2016–2037 | |
| Magrolimab/ Hu5F9-G4 | Anti-CD47 | R/R B-NHL | I/II | NCT02953509 Active, 2016–2026 Combined with rituximab and (in some cases) with chemotherapy | [292] |
| R/R MM | II |
NCT04892446 Recruiting, 2021–2024 Combined with daratumumab, or pomalidomide and dexamethasone, or bortezomib and dexamethasone | |||
| R/R B-NHL | II |
NCT04788043 Recruiting, 2021–2027 Combined with pembrolizumab | |||
| IBI322 | Bi anti-CD47/PD-L1 | ALL, CLL/SLL B-NHL | I | NCT04795128 Recruiting, 2021–2024 | |
| TG-1801/ NI-1701 | Bi anti-CD47/CD19 | B-NHL B-NHL, CLL | I Ib | NCT03804996 Active, 2019–2022 Combined with ublituximab NCT04806035 Recruiting, 2021–2023 Alone or combined with ublituximab | [293] |
| CPO107/ JMT601 | Fusion protein Anti-CD20-SIRPα | Advanced B-NHL | I | NCT04853329 Recruiting, 2021–2024 | |
| STRO-001 | ADC anti-CD74- maytansoid | DLBCL, FL, MM | I | NCT03424603 Recruiting, 2018–2023 | [294] |
| Polatuzumab vedotin | ADC anti-CD79b-monomethyl auristatin E | DLBCL | I | NCT04231877 Recruiting, 2020–2031 Combined with rituximab, prednisone, etoposide, doxorubicin, cyclophosphamide | |
| R/R DLBCL | I |
NCT04739813 Recruiting, 2021–2027 Combined with venetoclax, ibrutinib, prednisone, obinutuzumab, and lenalidomide | |||
| R/R B-NHL | Ib/II |
NCT03533283 Recruiting, 2018–2024 Combined with glofitamab | |||
| Cevostomab/ BFCR4350A | BiTE-anti-FcRL5/CD3 | R/R MM | I | NCT03275103 Recruiting, 2017–2023 | [295] |
| Talquetamab/ JNJ-64407564 | BiTE duoBody-anti-GPRC5D/CD3 | R/R MM | I/II | NCT03399799 Recruiting, 2018–2025 | [296] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tannoury, M.; Garnier, D.; Susin, S.A.; Bauvois, B. Current Status of Novel Agents for the Treatment of B Cell Malignancies: What’s Coming Next? Cancers 2022, 14, 6026. https://doi.org/10.3390/cancers14246026
Tannoury M, Garnier D, Susin SA, Bauvois B. Current Status of Novel Agents for the Treatment of B Cell Malignancies: What’s Coming Next? Cancers. 2022; 14(24):6026. https://doi.org/10.3390/cancers14246026
Chicago/Turabian StyleTannoury, Mariana, Delphine Garnier, Santos A. Susin, and Brigitte Bauvois. 2022. "Current Status of Novel Agents for the Treatment of B Cell Malignancies: What’s Coming Next?" Cancers 14, no. 24: 6026. https://doi.org/10.3390/cancers14246026
APA StyleTannoury, M., Garnier, D., Susin, S. A., & Bauvois, B. (2022). Current Status of Novel Agents for the Treatment of B Cell Malignancies: What’s Coming Next? Cancers, 14(24), 6026. https://doi.org/10.3390/cancers14246026