
Secondary Malignancies after Ewing Sarcoma—Epidemiological and Clinical Analysis of an International Trial Registry


 , , and
, , and 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Cohorts and Eligibility Criteria
2.2. Follow-Up and Statistical Analysis
2.3. Literature Search
3. Results
3.1. Patient Characteristics and Clinical Features of Primary Ewing Sarcoma
3.2. Epidemiology of Secondary Malignant Neoplasms
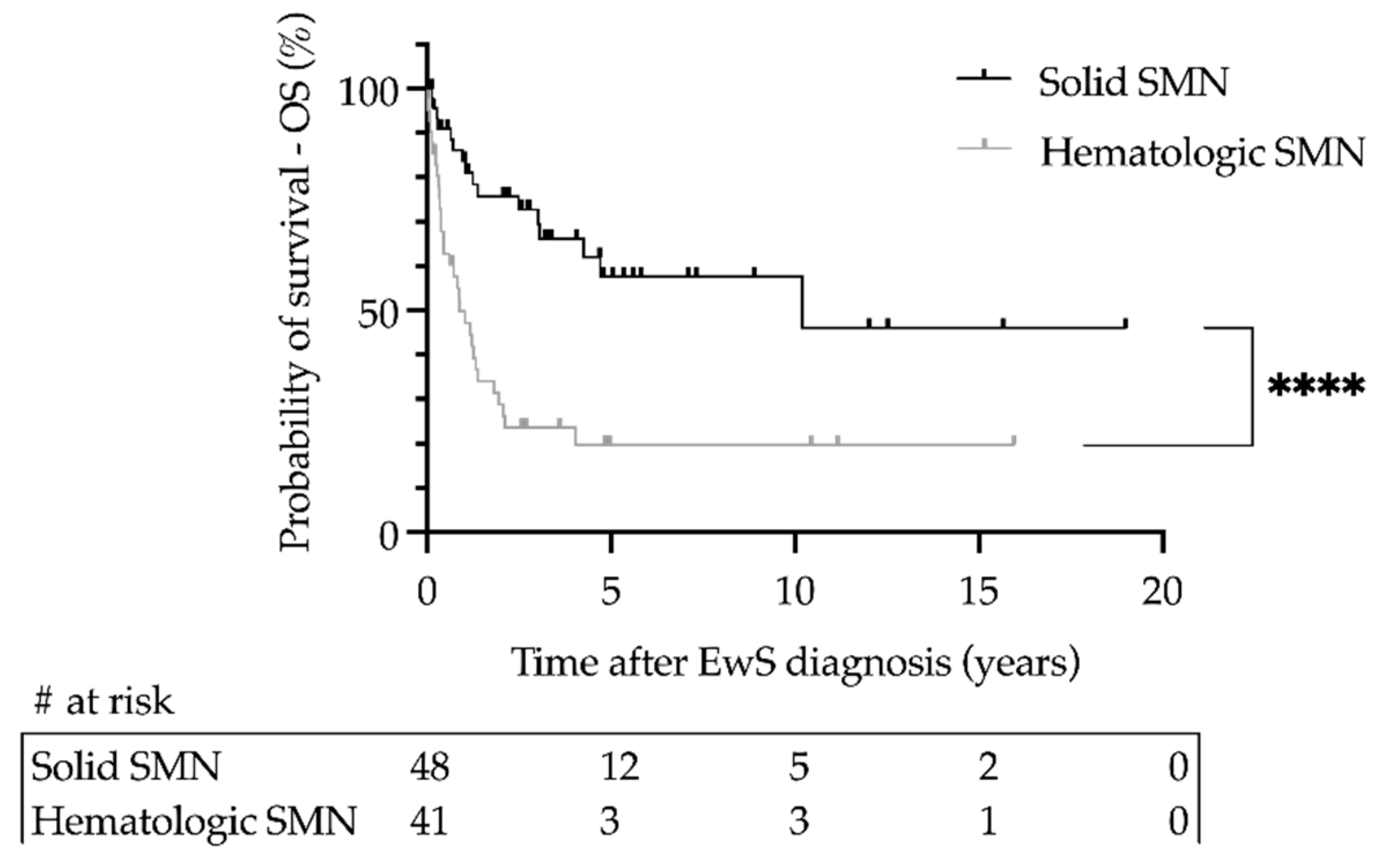
3.3. Cumulative Incidences and Outcome of Secondary Malignant Neoplasms
3.4. Literature Search
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grunewald, T.G.P.; Cidre-Aranaz, F.; Surdez, D.; Tomazou, E.M.; de Alava, E.; Kovar, H.; Sorensen, P.H.; Delattre, O.; Dirksen, U. Ewing sarcoma. Nat. Rev. Dis. Prim. 2018, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Worch, J.; Cyrus, J.; Goldsby, R.; Matthay, K.K.; Neuhaus, J.; DuBois, S.G. Racial differences in the incidence of mesenchymal tumors associated with EWSR1 translocation. Cancer Epidemiol. Biomark. Prev. 2011, 20, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Jawad, M.U.; Cheung, M.C.; Min, E.S.; Schneiderbauer, M.M.; Koniaris, L.G.; Scully, S.P. Ewing sarcoma demonstrates racial disparities in incidence-related and sex-related differences in outcome: An analysis of 1631 cases from the SEER database, 1973–2005. Cancer 2009, 115, 3526–3536. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, N.; Hawkins, D.S.; Dirksen, U.; Lewis, I.J.; Ferrari, S.; Le Deley, M.-C.; Kovar, H.; Grimer, R.; Whelan, J.; Claude, L.; et al. Ewing Sarcoma: Current Management and Future Approaches through Collaboration. J. Clin. Oncol. 2015, 33, 3036–3046. [Google Scholar] [CrossRef]
- Delattre, O.; Zucman, J.; Plougastel, B.; Desmaze, C.; Melot, T.; Peter, M.; Kovar, H.; Joubert, I.; de Jong, P.; Rouleau, G.; et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 1992, 359, 162–165. [Google Scholar] [CrossRef]
- Zucman, J.; Melot, T.; Desmaze, C.; Ghysdael, J.; Plougastel, B.; Peter, M.; Zucker, J.M.; Triche, T.J.; Sheer, D.; Turc-Carel, C.; et al. Combinatorial generation of variable fusion proteins in the Ewing family of tumours. EMBO J. 1993, 12, 4481–4487. [Google Scholar] [CrossRef]
- Aurias, A.; Rimbaut, C.; Buffe, D.; Zucker, J.M.; Mazabraud, A. Translocation involving chromosome 22 in Ewing’s sarcoma. A cytogenetic study of four fresh tumors. Cancer Genet. Cytogenet. 1984, 12, 21–25. [Google Scholar] [CrossRef]
- Kreyer, J.; Ranft, A.; Timmermann, B.; Juergens, H.; Jung, S.; Wiebe, K.; Boelling, T.; Schuck, A.; Vieth, V.; Streitbuerger, A.; et al. Impact of the Interdisciplinary Tumor Board of the Cooperative Ewing Sarcoma Study Group on local therapy and overall survival of Ewing sarcoma patients after induction therapy. Pediatr. Blood Cancer 2018, 65, e27384. [Google Scholar] [CrossRef]
- Dirksen, U. Efficacy of add-on treosulfan and melphalan high-dose therapy in patients with high-risk metastatic Ewing sarcoma: Report from the International Ewing 2008R3 trial. J. Clin. Oncol. 2020, 38, 11501. [Google Scholar] [CrossRef]
- Pappo, A.S.; Dirksen, U. Rhabdomyosarcoma, Ewing Sarcoma, and Other Round Cell Sarcomas. J. Clin. Oncol. 2017, 36, 168–179. [Google Scholar] [CrossRef]
- Ladenstein, R.; Pötschger, U.; Le Deley, M.-C.; Whelan, J.; Paulussen, M.; Oberlin, O.; Van Den Berg, H.; Dirksen, U.; Hjorth, L.; Michon, J.; et al. Primary disseminated multifocal Ewing sarcoma: Results of the Euro-EWING 99 trial. J. Clin. Oncol. 2010, 28, 3284–3291. [Google Scholar] [CrossRef] [PubMed]
- Caruso, J.; Shulman, D.S.; DuBois, S.G. Second malignancies in patients treated for Ewing sarcoma: A systematic review. Pediatr. Blood Cancer 2019, 66, e27938. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.K.; Helenowski, I.; Hijiya, N. Secondary malignancies in pediatric cancer survivors: Perspectives and review of the literature. Int. J. Cancer 2014, 135, 1764–1773. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Sklar, C. Second cancers in survivors of childhood cancer. Nat. Rev. Cancer 2002, 2, 124–132. [Google Scholar] [CrossRef]
- Hijiya, N.; Ness, K.K.; Ribeiro, R.C.; Hudson, M.M. Acute leukemia as a secondary malignancy in children and adolescents: Current findings and issues. Cancer 2009, 115, 23–35. [Google Scholar] [CrossRef]
- Pui, C.H.; Ribeiro, R.C.; Hancock, M.L.; Rivera, G.K.; Evans, W.E.; Raimondi, S.C.; Head, D.R.; Behm, F.G.; Mahmoud, M.H.; Sandlund, J.T.; et al. Acute myeloid leukemia in children treated with epipodophyllotoxins for acute lymphoblastic leukemia. N. Engl. J. Med. 1991, 325, 1682–1687. [Google Scholar] [CrossRef] [PubMed]
- Heyn, R.; Khan, F.; Ensign, L.G.; Donaldson, S.S.; Ruymann, F.; Smith, M.A.; Vietti, T.; Maurer, H.M. Acute myeloid leukemia in patients treated for rhabdomyosarcoma with cyclophosphamide and low-dose etoposide on Intergroup Rhabdomyosarcoma Study III: An interim report. Med. Pediatr. Oncol. 1994, 23, 99–106. [Google Scholar] [CrossRef]
- Bhatia, S.; Krailo, M.D.; Chen, Z.; Burden, L.; Askin, F.B.; Dickman, P.S.; Grier, H.E.; Link, M.P.; Meyers, P.; Perlman, E.; et al. Therapy-related myelodysplasia and acute myeloid leukemia after Ewing sarcoma and primitive neuroectodermal tumor of bone: A report from the Children’s Oncology Group. Blood 2007, 109, 46–51. [Google Scholar] [CrossRef]
- Travis, L.B.; Demark Wahnefried, W.; Allan, J.M.; Wood, M.E.; Ng, A.K. Aetiology, genetics and prevention of secondary neoplasms in adult cancer survivors. Nat. Rev. Clin. Oncol. 2013, 10, 289–301. [Google Scholar] [CrossRef]
- Morton, L.M.; Onel, K.; Curtis, R.E.; Hungate, E.A.; Armstrong, G.T. The rising incidence of second cancers: Patterns of occurrence and identification of risk factors for children and adults. Am. Soc. Clin. Oncol. Educ. Book 2014, 34, e57–e67. [Google Scholar] [CrossRef]
- Turcotte, L.M.; Neglia, J.P.; Reulen, R.C.; Ronckers, C.M.; van Leeuwen, F.E.; Morton, L.M.; Hodgson, D.C.; Yasui, Y.; Oeffinger, K.C.; Henderson, T.O. Risk, Risk Factors, and Surveillance of Subsequent Malignant Neoplasms in Survivors of Childhood Cancer: A Review. J. Clin Oncol. 2018, 36, 2145–2152. [Google Scholar] [CrossRef] [PubMed]
- Dunst, J.; Ahrens, S.; Paulussen, M.; Rube, C.; Winkelmann, W.; Zoubek, A.; Harms, D.; Jurgens, H. Second malignancies after treatment for Ewing’s sarcoma: A report of the CESS-studies. Int. J. Radiat. Oncol. Biol. Phys. 1998, 42, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, F.; Rubino, C.; Guerin, S.; Diallo, I.; Samand, A.; Hawkins, M.; Oberlin, O.; Lefkopoulos, D.; De Vathaire, F. Risk of a second malignant neoplasm after cancer in childhood treated with radiotherapy: Correlation with the integral dose restricted to the irradiated fields. Int. J. Radiat. Oncol. Biol. Phys. 2008, 70, 908–915. [Google Scholar] [CrossRef] [PubMed]
- Kuttesch, J.F.; Wexler, L.H.; Marcus, R.B.; Fairclough, D.; Weaver-McClure, L.; White, M.; Mao, L.; Delaney, T.F.; Pratt, C.B.; Horowitz, M.E.; et al. Second malignancies after Ewing’s sarcoma: Radiation dose-dependency of secondary sarcomas. J. Clin. Oncol. 1996, 14, 2818–2825. [Google Scholar] [CrossRef] [PubMed]
- Tucker, M.A.; D’Angio, G.J.; Boice, J.D., Jr.; Strong, L.C.; Li, F.P.; Stovall, M.; Stone, B.J.; Green, D.M.; Lombardi, F.; Newton, W.; et al. Bone sarcomas linked to radiotherapy and chemotherapy in children. N. Engl. J. Med. 1987, 317, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Schuck, A.; Ahrens, S.; Paulussen, M.; Kuhlen, M.; Könemann, S.; Rübe, C.; Winkelmann, W.; Kotz, R.; Dunst, J.; Willich, N.; et al. Local therapy in localized Ewing tumors: Results of 1058 patients treated in the CESS 81, CESS 86, and EICESS 92 trials. Int. J. Radiat. Oncol. Biol. Phys. 2003, 55, 168–177. [Google Scholar] [CrossRef]
- Paulussen, M.; Ahrens, S.; Lehnert, M.; Taeger, D.; Hense, H.W.; Wagner, A.; Dunst, J.; Harms, D.; Reiter, A.; Henze, G.; et al. Second malignancies after ewing tumor treatment in 690 patients from a cooperative German/Austrian/Dutch study. Ann. Oncol. 2001, 12, 1619–1630. [Google Scholar] [CrossRef]
- Paulussen, M.; Craft, A.W.; Lewis, I.; Hackshaw, A.; Douglas, C.; Dunst, J.; Schuck, A.; Winkelmann, W.; Köhler, G.; Poremba, C.; et al. Results of the EICESS-92 Study: Two randomized trials of Ewing’s sarcoma treatment--cyclophosphamide compared with ifosfamide in standard-risk patients and assessment of benefit of etoposide added to standard treatment in high-risk patients. J. Clin. Oncol. 2008, 26, 4385–4393. [Google Scholar] [CrossRef]
- Le Deley, M.-C.; Paulussen, M.; Lewis, I.; Brennan, B.; Ranft, A.; Whelan, J.; Le Teuff, G.; Michon, J.; Ladenstein, R.; Marec-Bérard, P.; et al. Cyclophosphamide compared with ifosfamide in consolidation treatment of standard-risk Ewing sarcoma: Results of the randomized noninferiority Euro-EWING99-R1 trial. J. Clin. Oncol. 2014, 32, 2440–2448. [Google Scholar] [CrossRef]
- Whelan, J.; Le Deley, M.-C.; Dirksen, U.; Le Teuff, G.; Brennan, B.; Gaspar, N.; Hawkins, D.S.; Amler, S.; Bauer, S.; Bielack, S.; et al. High-Dose Chemotherapy and Blood Autologous Stem-Cell Rescue Compared with Standard Chemotherapy in Localized High-Risk Ewing Sarcoma: Results of Euro-E.W.I.N.G. 99 and Ewing-2008. J. Clin. Oncol. 2018, 36, 3110–3119. [Google Scholar] [CrossRef]
- Dirksen, U.; Brennan, B.; Le Deley, M.-C.; Cozic, N.; Van Den Berg, H.; Bhadri, V.; Brichard, B.; Claude, L.; Craft, A.; Amler, S.; et al. High-Dose Chemotherapy Compared with Standard Chemotherapy and Lung Radiation in Ewing Sarcoma with Pulmonary Metastases: Results of the European Ewing Tumour Working Initiative of National Groups, 99 Trial and EWING 2008. J. Clin. Oncol. 2019, 37, 3192–3202. [Google Scholar] [CrossRef] [PubMed]
- Juergens, C.; Weston, C.; Lewis, I.; Whelan, J.; Paulussen, M.; Oberlin, O.; Michon, J.; Zoubek, A.; Juergens, H.; Craft, A. Safety assessment of intensive induction with vincristine, ifosfamide, doxorubicin, and etoposide (VIDE) in the treatment of Ewing tumors in the EURO-E.W.I.N.G. 99 clinical trial. Pediatr. Blood Cancer 2006, 47, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, M.M.; Wilson, L.M.; Burton, H.S.; Potok, M.H.; Winter, D.L.; Marsden, H.B.; Stovall, M.A. Radiotherapy, alkylating agents, and risk of bone cancer after childhood cancer. J. Natl. Cancer Inst. 1996, 88, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, J.P.; Goodman, P.; Leisenring, W.; Ness, K.K.; Meyers, P.A.; Wolden, S.L.; Smith, S.M.; Stovall, M.; Hammond, S.; Robison, L.L.; et al. Long-term survivors of childhood Ewing sarcoma: Report from the childhood cancer survivor study. J. Natl. Cancer Inst. 2010, 102, 1272–1283. [Google Scholar] [CrossRef] [PubMed]
- Navid, F.; Billups, C.; Liu, T.; Krasin, M.J.; Rodriguez-Galindo, C. Second cancers in patients with the Ewing sarcoma family of tumours. Eur. J. Cancer 2008, 44, 983–991. [Google Scholar] [CrossRef]
- Longhi, A.; Ferrari, S.; Tamburini, A.; Luksch, R.; Fagioli, F.; Bacci, G.; Ferrari, C. Late effects of chemotherapy and radiotherapy in osteosarcoma and Ewing sarcoma patients: The Italian Sarcoma Group Experience (1983–2006). Cancer 2012, 118, 5050–5059. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.N.; Chastain, K.; Chou, J.F.; Moskowitz, C.S.; Adsuar, R.; Wexler, L.H.; Chou, A.J.; DeRosa, A.; Candela, J.; Magnan, H.; et al. Morbidity and mortality after treatment of Ewing sarcoma: A single-institution experience. Pediatr. Blood Cancer 2017, 64, e26562. [Google Scholar] [CrossRef]
- Kaatsch, P.; Grabow, D.; Spix, C. German Childhood Cancer Registry-Annual Report 2017 (1980–2016); Institute of Medical Biostatistics, Epidemiology and Informatics (IMBEI) at the University Medical Center of the Johannes Gutenberg University Mainz: Mainz, Germany, 2018; Volume 2018. [Google Scholar]
- Erdmann, F.K.P.; Grabow, D.; Spix, C. German Childhood Cancer Registry-Annual Report 2019 (1980–2018); Institute of Medical Biostatistics, Epidemiology and Informatics (IMBEI) at the University Medical Center of the Johannes Gutenberg University Mainz: Mainz, Germany, 2020; Volume 2019. [Google Scholar]
- Friedman, D.L.; Whitton, J.; Leisenring, W.; Mertens, A.C.; Hammond, S.; Stovall, M.; Donaldson, S.S.; Meadows, A.T.; Robison, L.L.; Neglia, J.P. Subsequent neoplasms in 5-year survivors of childhood cancer: The Childhood Cancer Survivor Study. J. Natl. Cancer Inst. 2010, 102, 1083–1095. [Google Scholar] [CrossRef]
- Leavey, P.J.; Laack, N.N.; Krailo, M.D.; Buxton, A.; Randall, R.L.; DuBois, S.G.; Reed, D.R.; Grier, H.E.; Hawkins, D.S.; Pawel, B.; et al. Phase III Trial Adding Vincristine-Topotecan-Cyclophosphamide to the Initial Treatment of Patients With Nonmetastatic Ewing Sarcoma: A Children’s Oncology Group Report. J. Clin. Oncol. 2021, 39, 4029–4038. [Google Scholar] [CrossRef]
- Sultan, I.; Rihani, R.; Hazin, R.; Rodriguez-Galindo, C. Second malignancies in patients with Ewing Sarcoma Family of Tumors: A population-based study. Acta Oncol. 2010, 49, 237–244. [Google Scholar] [CrossRef]
- Neglia, J.P.; Friedman, D.L.; Yasui, Y.; Mertens, A.C.; Hammond, S.; Stovall, M.; Donaldson, S.S.; Meadows, A.T.; Robison, L.L. Second malignant neoplasms in five-year survivors of childhood cancer: Childhood cancer survivor study. J. Natl. Cancer Inst. 2001, 93, 618–629. [Google Scholar] [CrossRef] [PubMed]
- Kenney, L.B.; Yasui, Y.; Inskip, P.D.; Hammond, S.; Neglia, J.P.; Mertens, A.C.; Meadows, A.T.; Friedman, D.; Robison, L.L.; Diller, L. Breast cancer after childhood cancer: A report from the Childhood Cancer Survivor Study. Ann. Intern. Med. 2004, 141, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Oeffinger, K.C.; Mertens, A.C.; Sklar, C.A.; Kawashima, T.; Hudson, M.M.; Meadows, A.T.; Friedman, D.L.; Marina, N.; Hobbie, W.; Kadan-Lottick, N.S.; et al. Chronic health conditions in adult survivors of childhood cancer. N. Engl. J. Med. 2006, 355, 1572–1582. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Robison, L.L.; Oberlin, O.; Greenberg, M.; Bunin, G.; Fossati-Bellani, F.; Meadows, A.T. Breast cancer and other second neoplasms after childhood Hodgkin’s disease. N. Engl. J. Med. 1996, 334, 745–751. [Google Scholar] [CrossRef]
- Schellong, G.; Riepenhausen, M.; Ehlert, K.; Bramswig, J.; Dorffel, W.; Schmutzler, R.K.; Rhiem, K.; Bick, U. Breast cancer in young women after treatment for Hodgkin’s disease during childhood or adolescence–an observational study with up to 33-year follow-up. Dtsch. Arztebl Int. 2014, 111, 3–9. [Google Scholar]
- Rodriguez-Galindo, C.; Poquette, C.A.; Marina, N.M.; Head, D.R.; Cain, A.; Meyer, W.H.; Santana, V.M.; Pappo, A.S. Hematologic abnormalities and acute myeloid leukemia in children and adolescents administered intensified chemotherapy for the Ewing sarcoma family of tumors. J. Pediatr. Hematol. Oncol. 2000, 22, 321–329. [Google Scholar] [CrossRef]
- Paulides, M.; Kremers, A.; Stöhr, W.; Bielack, S.; Jürgens, H.; Treuner, J.; Beck, J.; Langer, T.; German Late Effects Working Group in the Society of Pediatric Oncology and Haematology (GPOH). Prospective longitudinal evaluation of doxorubicin-induced cardiomyopathy in sarcoma patients: A report of the late effects surveillance system (LESS). Pediatr. Blood Cancer 2006, 46, 489–495. [Google Scholar] [CrossRef]
- Schiffman, J.D.; Wright, J. Ewing’s Sarcoma and Second Malignancies. Sarcoma 2011, 2011, 736841. [Google Scholar] [CrossRef]
- Kaatsch, P.; Reinisch, I.; Spix, C.; Berthold, F.; Janka-Schaub, G.; Mergenthaler, A.; Michaelis, J.; Blettner, M. Case-control study on the therapy of childhood cancer and the occurrence of second malignant neoplasms in Germany. Cancer Causes Control 2009, 20, 965–980. [Google Scholar] [CrossRef]
- Constine, L.S.; Ronckers, C.M.; Hua, C.H.; Olch, A.; Kremer, L.C.M.; Jackson, A.; Bentzen, S.M. Pediatric Normal Tissue Effects in the Clinic (PENTEC): An International Collaboration to Analyse Normal Tissue Radiation Dose-Volume Response Relationships for Paediatric Cancer Patients. Clin. Oncol. 2019, 31, 199–207. [Google Scholar] [CrossRef]
- Sheppard, D.G.; Libshitz, H.I. Post-radiation sarcomas: A review of the clinical and imaging features in 63 cases. Clin. Radiol. 2001, 56, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Granowetter, L.; Womer, R.; Devidas, M.; Krailo, M.; Wang, C.; Bernstein, M.; Marina, N.; Leavey, P.; Gebhardt, M.; Healey, J.; et al. Dose-intensified compared with standard chemotherapy for nonmetastatic Ewing sarcoma family of tumors: A Children’s Oncology Group Study. J. Clin. Oncol. 2009, 27, 2536–2541. [Google Scholar] [CrossRef]
- Mertens, A.C.; Liu, Q.; Neglia, J.P.; Wasilewski, K.; Leisenring, W.; Armstrong, G.T.; Robison, L.L.; Yasui, Y. Cause-specific late mortality among 5-year survivors of childhood cancer: The Childhood Cancer Survivor Study. J. Natl. Cancer Inst. 2008, 100, 1368–1379. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.H. Epipodophyllotoxin-related acute myeloid leukaemia. Lancet 1991, 338, 1468. [Google Scholar] [CrossRef] [PubMed]
- Relling, M.V.; Boyett, J.M.; Blanco, J.G.; Raimondi, S.; Behm, F.G.; Sandlund, J.T.; Rivera, G.K.; Kun, L.E.; Evans, W.E.; Pui, C.H. Granulocyte colony-stimulating factor and the risk of secondary myeloid malignancy after etoposide treatment. Blood 2003, 101, 3862–3867. [Google Scholar] [CrossRef][Green Version]
- Teepen, J.C.; van Leeuwen, F.E.; Tissing, W.J.; van Dulmen-den Broeder, E.; van den Heuvel-Eibrink, M.M.; van der Pal, H.J.; Loonen, J.J.; Bresters, D.; Versluys, B.; Neggers, S.; et al. Long-Term Risk of Subsequent Malignant Neoplasms After Treatment of Childhood Cancer in the DCOG LATER Study Cohort: Role of Chemotherapy. J. Clin. Oncol. 2017, 35, 2288–2298. [Google Scholar] [CrossRef]
- Henderson, T.O.; Rajaraman, P.; Stovall, M.; Constine, L.S.; Olive, A.; Smith, S.A.; Mertens, A.; Meadows, A.; Neglia, J.; Hammond, S.; et al. Risk factors associated with secondary sarcomas in childhood cancer survivors: A report from the childhood cancer survivor study. Int. J. Radiat. Oncol. Biol. Phys. 2012, 84, 224–230. [Google Scholar] [CrossRef]
- Jurgens, H.; Dirksen, U. Ewing sarcoma treatment. Eur. J. Cancer 2011, 47 (Suppl. S3), S366–S367. [Google Scholar] [CrossRef]
- Whelan, J.; Hackshaw, A.; McTiernan, A.; Grimer, R.; Spooner, D.; Bate, J.; Ranft, A.; Paulussen, M.; Juergens, H.; Craft, A.; et al. Survival is influenced by approaches to local treatment of Ewing sarcoma within an international randomised controlled trial: Analysis of EICESS-92. Clin. Sarcoma Res. 2018, 8, 6. [Google Scholar] [CrossRef]
- Li, X.; Li, W.; Mo, W.; Yang, Z. Acute lymphoblastic leukemia arising after treatment of Ewing sarcoma was misdiagnosed as bone marrow metastasis of Ewing sarcoma: A case report. Medicine 2018, 97, e9644. [Google Scholar] [CrossRef]
- Gröbner, S.N.; Project, I.P.-S.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; et al. Author Correction: The landscape of genomic alterations across childhood cancers. Nature 2018, 559, E10. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| EwS Trial | CESS 81 | CESS 86 | EICESS 92 | EURO E.W.I.N.G. 99 | Ewing 2008 | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Number of Cycles | 12 | 12 | 14 | 14 | 14 | 8 | 8 | 14 | 14 | 8 | 14 | 15 | ||||||||||
| Risk Strata | - | SR | HR | SR | HR | R1 = SR | R2 = HR | R3 = VHR | R1 = SR | R2 = HR | R3 = VHR | |||||||||||
| Regimen | VACA | VACA | VAIA | VAIA + VACA | VAIA | VAIA | EVAIA | VIDE + VAC ♀ | VIDE + VAI ♂ | VIDE + VAI | VIDE + VAI + BU/MEL | VIDE + VAI + ME/ME | VIDE + VAI + TREO/MEL | VIDE + VAI + BU/MEL | VIDE + VAI | VIDE + VAC ♀ | VIDE + VAI ♂ | VIDE + VAI | VIDE + VAI + BU/MEL | VIDE + VAC | VIDE + VAC + TREO/MEL | |
| Chemotherapeutic Agent and Dose | V (mg/m2) | 24 | 24 | 24 | 21 | 21 | 10.5 | 10.5 | 21 | 10.5 | 21 | |||||||||||
| A (mg/m2) | 6 | 6 | 6 | 10.5 | 12 | 1.5 | 1.5 | 12 | 1.5 | 12 | ||||||||||||
| C (g/m2) | 14.4 | 14.4 | - | 12 | - | 10.5 | - | - | - | - | 12 | - | - | - | 12 | |||||||
| I (g/m2) | - | - | 72 | 24 | 84 | 60 | 102 | 60 | 60 | 54 | 102 | 60 | 54 | |||||||||
| D (mg/m2) | 480 | 480 | 480 | 420 | 360 | 360 | 360 | |||||||||||||||
| E (g/m2) | - | - | - | - | 6.3 | 2.7 | 3.15 | 2.7 | 2.7 | |||||||||||||
| BU (mg/m2) | - | - | - | - | - | 600 | - | - | 600 | - | - | - | - | 600 | - | - | ||||||
| MEL (mg/m2) | - | - | - | - | - | 140 | 140 | - | - | - | - | 140 | - | 140 | ||||||||
| TREO (g/m2) | - | - | - | - | - | - | 36 | - | - | - | - | - | - | - | 36 | |||||||
| Irradiation Dose (Gy) | Preoperative | - | - | 45 | 54.4 | 54.4 | 54.4 | |||||||||||||||
| Definitive | 46–60 | 45–60 ↑ | 55 | 44.8–54.5 ↑ | 44.8–54.5 ↑ | 45–54 ↑ | ||||||||||||||||
| Postoperative | 36 | 45 | 45 | 44.8–54.4 | 44.8–54.4 | 45–54 | ||||||||||||||||
| Attributable Distribution of Primary EwS Patients at Diagnosis | Number of Patients with SMNs (n, %) | Median Observation Time from Primary EwS Diagnosis to SMNs (Years) |
|---|---|---|
| EwS trial (n = 96) | ||
| CESS 81 | 2 (of 184), 1.1% | 21.7 |
| CESS 86 | 16 (of 490), 3.3% | 11.9 |
| EICESS 92 | 21 (of 875), 2.4% | 6 |
| EURO E.W.I.N.G. 99 | 36 (of 1548), 2.3% | 4.9 |
| Ewing 2008 | 21 (of 1421), 1.5% | 2.3 |
| Sex (%) (n = 96) | ||
| Male | 45 (46.9%) | |
| Female | 51 (53.1%) | |
| Metastases (n = 96) | ||
| Yes | 31 (32.3%) | |
| No | 65 (67.7%) | |
| Age (n = 96) | ||
| median (range) | 14.4 (2.4–68.6) years | |
| Localization (n = 96) | ||
| Cranium | 5 (5.2%) | |
| Hand/foot | 6 (6.3%) | |
| Upper limb | 9 (9.4%) | |
| Lower limb | 21 (21.9%) | |
| Axial skeleton | 29 (30.2%) | |
| Pelvis | 26 (27%) |
| EwS Trial | Type of SMNs | Number of Patients with SMNs (n, %) |
|---|---|---|
| Across trials (n = 96) | Solid | 53 (55.2%) |
| Hematologic | 43 (44.8%) | |
| CESS 81 (n = 2) | Solid | 2 (100%) |
| Osteosarcoma | 0 | |
| Other sarcoma | 0 | |
| Carcinoma | 2 | |
| Other | 0 | |
| Hematologic | 0 (0%) | |
| Leukemia, lymphoma | 0 | |
| Myelodysplastic syndrome | 0 | |
| CESS 86 (n = 16) | Solid | 11 (73.3%) |
| Osteosarcoma | 4 | |
| Other sarcoma | 3 | |
| Carcinoma | 4 | |
| Other | 0 | |
| Hematologic | 4 (26.7%) | |
| Leukemia, lymphoma | 3 | |
| Myelodysplastic syndrome | 1 | |
| EICESS 92 (n = 21) | Solid | 12 (54.5%) |
| Osteosarcoma | 4 | |
| Other sarcoma | 2 | |
| Carcinoma | 5 | |
| Other | 1 | |
| Hematologic | 10 (45.5%) | |
| Leukemia, lymphoma | 5 | |
| Myelodysplastic syndrome | 5 | |
| Euro E.W.I.N.G. 99 (n = 36) | Solid | 18 (50%) |
| Osteosarcoma | 7 | |
| Other sarcoma | 2 | |
| Carcinoma | 7 | |
| Other | 2 | |
| Hematologic | 18 (50%) | |
| Leukemia, lymphoma | 9 | |
| Myelodysplastic syndrome | 9 | |
| Ewing 2008 (n = 21) | Solid | 9 (42.9%) |
| Osteosarcoma | 0 | |
| Other sarcoma | 2 | |
| Carcinoma | 5 | |
| Other | 2 | |
| Hematologic | 12 (57.1%) | |
| Leukemia, lymphoma | 6 | |
| Myelodysplastic syndrome | 6 |
| Study Details | EwS Characteristics at Diagnosis | Characteristics of Secondary Malignant Neoplasms | Comments | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Author (Publication Year) | Reported Time Period (Time to Publication) (Years) | Cohort | Cohort Size (Patients) | Median Follow-Up (Range) (Years) | Median Age (Range) (Years) | Metastases (%) | Females (%) | Tumor Volume > 100 mL (%) | Number | Solid (%) | Predominant Type (%) | CI (%/Years) | Latency (Range) (Years) | Risk Factors | |
| Hawkins et al. (1996) [33] | 1940–1983 (13) | EwS survivors | 207 | 7.1 | N A | N A | N A | N A | N A | N A | N A | 5.4/20 | N A | Sarcoma: CTX (alkylating agents, dose-dependent) Sarcoma: RTX (4/5 tumors in RTX field) | Selective description of secondary bone cancer after childhood cancer |
| Ginsberg et al. (2010) [34] | 1970–1986 (24) | EwS | 403 | Alive: 23.0 (16–33) Deceased: 11.2 (5–28) * | 13.5 (6–20) | N A | N A | N A | 36 | 94.5 | BC (36) | 9/25 | 14.5 (4–32) | Solid: RTX (p = 0.28) BC: WLI | NMSC excluded |
| Kuttesch et al. (1996) [24] | 1963–1990 (6) | EwS | 266 | 9.5 | 14.2 (4.2–28; 90% < 21) | N A | 56.25 | N A | 16 | 87.5 | Sarcoma (62.5) | 9.2/20 | 7.6 (3.5–25.7) | All SMN: RTX (>48 Gy) (p = 0.043) Sarcoma: RTX (100% in RTX field) (p = 0.002) | Combination of actinomycin D and RTX reduce risk |
| Dunst et al. (1998) [22] | 1981–1991 (7) | EwS | 674 | 5.1 | 13.25 (8–21) | 25 | 87.5 | 50 | 8 | 37.5 | AML (50) | 4.7/15 | 6 (1.5–11.4) | Sarcoma: RTX (100% with RTX) | Selection bias for RTX |
| Navid et al. (2008) [35] | 1979–2004 (4) | EwS family of tumors | 237 | N A | 8.3 | 50 | N A | 12 | 33.3 | MDS/leukemia (66.6) | 4.7/10 | 3.3 (1.4–19.6) | Hematologic: CTX (alkylating agents, topoisomerase-II inhibitors, dose-dependent) All SMN: Localized stage (p = 0.036) Earlier treatment protocol (p = 0.001) | ||
| Longhi et al. (2012) [36] | 1983–2006 (6) | Localized EwS, <40 years | 581 | 7.2 | 16.36 (6–39) | 0 | N A | N A | 15 | 80 | OS (40) | 5/25 | 7 (1–21.1) Hematological: 3.1 Solid: 7.8 | Female sex | |
| Friedmann et al. (2017) [37] | 1974–2012 (5) | EwS, <40 years | 300 | 7.8 | MDS/AML: 17.4 (5–32) Solid: 14.6 (6–24) | 30 | 30 | 30 | 15 | 60 | MDS/AML (60) | 15/25 | 10.9 (0.9–27.7) MDS/AML: 3.2 (0.9–4.6) Solid: 21.3 (10.5–27.7) | Hematologic: CTX (alkylating agents, topoisomerase-II inhibitors, dose-dependent) | NMSC and melanoma excluded |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaiser, I.; Kauertz, K.; Zöllner, S.K.; Hartmann, W.; Langer, T.; Jürgens, H.; Ranft, A.; Dirksen, U. Secondary Malignancies after Ewing Sarcoma—Epidemiological and Clinical Analysis of an International Trial Registry. Cancers 2022, 14, 5920. https://doi.org/10.3390/cancers14235920
Kaiser I, Kauertz K, Zöllner SK, Hartmann W, Langer T, Jürgens H, Ranft A, Dirksen U. Secondary Malignancies after Ewing Sarcoma—Epidemiological and Clinical Analysis of an International Trial Registry. Cancers. 2022; 14(23):5920. https://doi.org/10.3390/cancers14235920
Chicago/Turabian StyleKaiser, Isabelle, Katja Kauertz, Stefan K. Zöllner, Wolfgang Hartmann, Thorsten Langer, Heribert Jürgens, Andreas Ranft, and Uta Dirksen. 2022. "Secondary Malignancies after Ewing Sarcoma—Epidemiological and Clinical Analysis of an International Trial Registry" Cancers 14, no. 23: 5920. https://doi.org/10.3390/cancers14235920
APA StyleKaiser, I., Kauertz, K., Zöllner, S. K., Hartmann, W., Langer, T., Jürgens, H., Ranft, A., & Dirksen, U. (2022). Secondary Malignancies after Ewing Sarcoma—Epidemiological and Clinical Analysis of an International Trial Registry. Cancers, 14(23), 5920. https://doi.org/10.3390/cancers14235920