

ATR Inhibitors in Platinum-Resistant Ovarian Cancer

Abstract
Simple Summary
Abstract
1. Introduction
2. What Is PROC?
3. Therapeutic Strategies for PROC
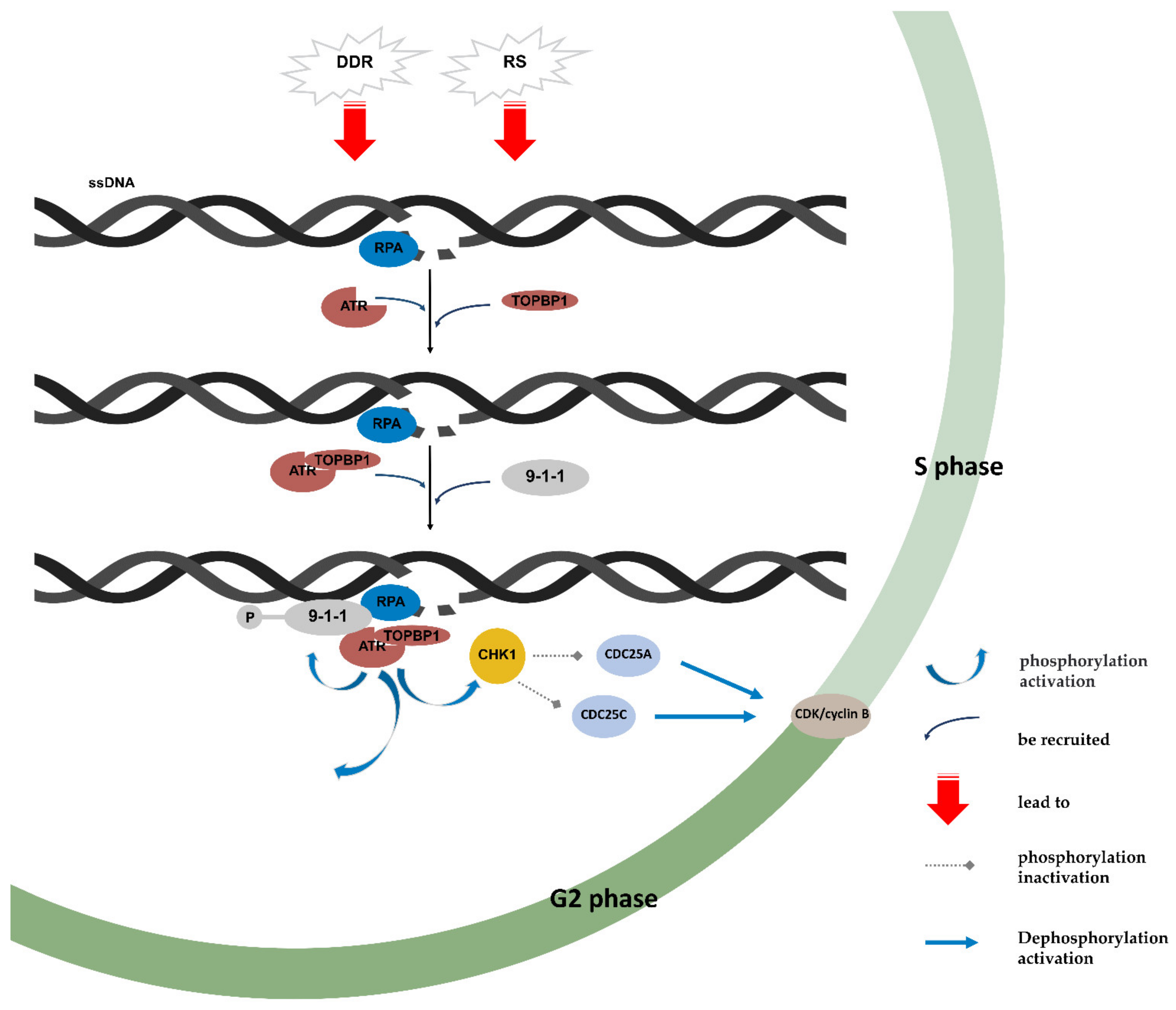
4. ATR/CHK1 Pathway
5. Preclinical Trials of ATRi
5.1. Combination with Radiotherapy
5.2. Combination with Chemotherapy
5.3. Combination with Immunotherapy
5.4. Combination with Drugs That Target DNA Damage Repair Pathways
5.4.1. ATRi and CHK1 Inhibitor, ATM/CHK2 Axis Inhibitor
5.4.2. ATRi and Poly (ADP-Ribose) Polymerase Inhibitors (PARPi)
5.4.3. ATRi and Topoisomerase Inhibitors
5.4.4. ATRi and WEE1 Inhibitors
6. Clinical Trials of ATRi
6.1. Berzosertib
6.2. Ceralasertib
6.3. M1774
7. Molecular Markers and Determinants of ATRi Sensitivity
8. Discussion
- (1)
- If lowering the dose and extending the duration of medication increase its efficacy, that remains an open question. ATRis inhibit the activation of ATR and its downstream molecules, leading to the restart of the cell cycle and the damage to HR. It should be remembered that long-term inhibition of the ATR/CHK1 pathway is more damaging to HR by increasing the transcription of E2F. This implies that long-term, low-dose treatment seems to be a better option. However, the results of models in vitro and clinical trials indicate that the toxicity of continuous administration appears to be intolerable. It could also be that the combination with chemotherapeutic drugs adds toxicity. It is worth trying to lower the dose or extend the treatment time in future experiments. Another option would be to pretreat with ATRis before using other chemotherapeutic agents or targeted agents.
- (2)
- What types of cancers should be selected in the design of in vitro experiments with ATRis? We found that ATRis also showed anticancer effects in chronic lymphocytic leukaemia, mantle cell lymphoma, non-small cell lung cancer, gastric cancer and other tumors [138,139,144,145]. Middleton et al. [140] speculated that tumor benefit from ATRis may be more dependent on tumor type. It is crucial to carefully select the types of cell lines used in studies. We hypothesize that cancers with a high RS or a high incidence rate of defects in DDR might be a good choice.
- (3)
- Is there an opportunity for ATR inhibitors to expand the indication for PARPis? As an important treatment option for OC, PARPis are now widely used in patients with HRD and PROC. As pointed out in our literature review, ATRis can overcome cells’ resistance to PARPis, enhancing the response to PARPis. This makes it possible for PARPis to perform better in PROC patients. However, it is necessary to test this in more cell lines with initial resistance to PARPis.
- (4)
- How effective are ATRis in other pathological types of OC? As described above, ATRis have a common chemotherapy sensitization effect in gynecological tumor cell lines, independent of BRCA status. As ATRi clinical studies are still mainly in phase II, almost all enrolled patients were advanced cancer patients who were resistant to platinum after multiline therapy. In addition, the efficacy of ATRis in patients with other types of OC is unclear due to the higher incidence of HGSOC. Patients with other types of OC may also benefit from clinical trials of ATRis.
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA A Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Fang, Y.; Chen, K.; Li, S.; Tang, S.; Ren, Y.; Cen, Y.; Fei, W.; Zhang, B.; Shen, Y.; et al. Single-Cell RNA Sequencing Reveals the Tissue Architecture in Human High-Grade Serous Ovarian Cancer. Clin. Cancer Res. 2022, 28, 3590–3602. [Google Scholar] [CrossRef]
- Peres, L.C.; Cushing-Haugen, K.L.; Köbel, M.; Harris, H.R.; Berchuck, A.; Rossing, M.A.; Schildkraut, J.M.; Doherty, J.A. Invasive Epithelial Ovarian Cancer Survival by Histotype and Disease Stage. J. Natl. Cancer Inst. 2019, 111, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.K.; Alvarez, R.D.; Bakkum-Gamez, J.N.; Barroilhet, L.; Behbakht, K.; Berchuck, A.; Chen, L.; Cristea, M.; DeRosa, M.; Eisenhauer, E.L.; et al. Ovarian Cancer, Version 1.2022, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Comprehencive Cancer Netw. 2022, 19, 191–226. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.; Tinker, A.V.; Friedlander, M. “Platinum resistant” ovarian cancer: What is it, who to treat and how to measure benefit? Gynecol. Oncol. 2014, 133, 624–631. [Google Scholar] [CrossRef]
- Markman, M.; Rothman, R.; Hakes, T.; Reichman, B.; Hoskins, W.; Rubin, S.; Jones, W.; Almadrones, L.; Lewis, J.L. Second-line platinum therapy in patients with ovarian cancer previously treated with cisplatin. J. Clin. Oncol. 1991, 9, 389–393. [Google Scholar] [CrossRef]
- Blackledge, G.; Lawton, F.; Redman, C.; Kelly, K. Response of patients in phase II studies of chemotherapy in ovarian cancer: Implications for patient treatment and the design of phase II trials. Br. J. Cancer 1989, 59, 650–653. [Google Scholar] [CrossRef] [PubMed]
- Gore, M.; Fryatt, I.; Wiltshaw, E.; Dawson, T. Treatment of relapsed carcinoma of the ovary with cisplatin or carboplatin following initial treatment with these compounds. Gynecol. Oncol. 1990, 36, 207–211. [Google Scholar] [CrossRef]
- Nishino, M.; Jagannathan, J.P.; Ramaiya, N.H.; Van den Abbeele, A.D. Revised RECIST Guideline Version 1.1: What Oncologists Want to Know and What Radiologists Need to Know. Am. J. Roentgenol. 2010, 195, 281–289. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Therasse, P.; Arbuck, S.G.; Eisenhauer, E.A.; Wanders, J.; Kaplan, R.S.; Rubinstein, L.; Verweij, J.; Van Glabbeke, M.; Van Oosterom, A.T.; Christian, M.C.; et al. New Guidelines to Evaluate the Response to Treatment in Solid Tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J. Natl. Cancer Inst. 2000, 92, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Rustin, G.J.S.; Vergote, I.; Eisenhauer, E.; Pujade-Lauraine, E.; Quinn, M.; Thigpen, T.; Du Bois, A.; Kristensen, G.; Jakobsen, A.; Sagae, S.; et al. Definitions for Response and Progression in Ovarian Cancer Clinical Trials Incorporating RECIST 1.1 and CA 125 Agreed by the Gynecological Cancer Intergroup (GCIG). Int. J. Gynecol. Cancer 2011, 21, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Ciccarelli, R.B.; Solomon, M.J.; Varshavsky, A.; Lippard, S.J. In vivo effects of cis- and trans-diamminedichloroplatinum(II) on SV40 chromosomes: Differential repair, DNA-protein crosslinking, and inhibition of replication. Biochemistry 1985, 24, 7533–7540. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2011, 31, 1869–1883. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Hilpert, F.; Weber, B.; Reuss, A.; Poveda, A.; Kristensen, G.; Sorio, R.; Vergote, I.; Witteveen, P.; Bamias, A.; et al. Bevacizumab Combined With Chemotherapy for Platinum-Resistant Recurrent Ovarian Cancer: The AURELIA Open-Label Randomized Phase III Trial. J. Clin. Oncol. 2014, 32, 1302–1308. [Google Scholar] [CrossRef] [PubMed]
- Bamias, A.; Gibbs, E.; Lee, C.K.; Davies, L.; Dimopoulos, M.; Zagouri, F.; Veillard, A.-S.; Kosse, J.; Santaballa, A.; Mirza, M.R.; et al. Bevacizumab with or after chemotherapy for platinum-resistant recurrent ovarian cancer: Exploratory analyses of the AURELIA trial. Ann. Oncol. 2017, 28, 1842–1848. [Google Scholar] [CrossRef]
- Hall, M.; Gourley, C.; McNeish, I.; Ledermann, J.; Gore, M.; Jayson, G.; Perren, T.; Rustin, G.; Kaye, S. Targeted anti-vascular therapies for ovarian cancer: Current evidence. Br. J. Cancer 2013, 108, 250–258. [Google Scholar] [CrossRef]
- Gelmon, K.A.; Tischkowitz, M.; Mackay, H.; Swenerton, K.; Robidoux, A.; Tonkin, K.; Hirte, H.; Huntsman, D.; Clemons, M.; Gilks, B.; et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: A phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011, 12, 852–861. [Google Scholar] [CrossRef]
- Kristeleit, R.; Shapiro, G.I.; Burris, H.A.; Oza, A.M.; LoRusso, P.; Patel, M.R.; Domchek, S.M.; Balmaña, J.; Drew, Y.; Chen, L.-M.; et al. A Phase I–II Study of the Oral PARP Inhibitor Rucaparib in Patients with Germline BRCA1/2-Mutated Ovarian Carcinoma or Other Solid Tumors. Clin. Cancer Res. 2017, 23, 4095–4106. [Google Scholar] [CrossRef]
- Bronger, H. Immunology and Immune Checkpoint Inhibition in Ovarian Cancer—Current Aspects. Geburtshilfe Frauenheilkd. 2021, 81, 1128–1144. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, M.; Anchora, L.P.; Tortorella, L.; Fanfani, F.; Gallotta, V.; Pacciani, M.; Scambia, G.; Fagotti, A. Secondary cytoreductive surgery in patients with isolated platinum-resistant recurrent ovarian cancer: A retrospective analysis. Gynecol. Oncol. 2014, 134, 257–261. [Google Scholar] [CrossRef]
- Musella, A.; Marchetti, C.; Palaia, I.; Perniola, G.; Giorgini, M.; Lecce, F.; Vertechy, L.; Iadarola, R.; De Felice, F.; Monti, M.; et al. Secondary Cytoreduction in Platinum-Resistant Recurrent Ovarian Cancer: A Single-Institution Experience. Ann. Surg. Oncol. 2015, 22, 4211–4216. [Google Scholar] [CrossRef]
- Petrillo, M.; Sozzi, G.; Dessole, M.; Capobianco, G.; Dessole, S.; Madonia, M.; Cherchi, P.L.; Paoletti, A.M.; Scambia, G.; Chiantera, V. The role of surgery in platinum-resistant ovarian cancer: A call to the scientific community. Semin. Cancer Biol. 2021, 77, 194–202. [Google Scholar] [CrossRef]
- Herrera, F.G.; Irving, M.; Kandalaft, L.E.; Coukos, G. Rational combinations of immunotherapy with radiotherapy in ovarian cancer. Lancet Oncol. 2019, 20, e417–e433. [Google Scholar] [CrossRef] [PubMed]
- Falzone, L.; Scandurra, G.; Lombardo, V.; Gattuso, G.; Lavoro, A.; Distefano, A.B.; Scibilia, G.; Scollo, P. A multidisciplinary approach remains the best strategy to improve and strengthen the management of ovarian cancer (Review). Int. J. Oncol. 2021, 59, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Cimprich, K.A.; Shin, T.B.; Keith, C.T.; Schreiber, S.L. cDNA cloning and gene mapping of a candidate human cell cycle checkpoint protein. Proc. Natl. Acad. Sci. USA 1996, 93, 2850–2855. [Google Scholar] [CrossRef]
- Hu, S.; Hui, Z.; Duan, J.; Garrido, C.; Xie, T.; Ye, X.Y. Discovery of small-molecule ATR inhibitors for potential cancer treatment: A patent review from 2014 to present. Expert Opin. Ther. Pat. 2022, 32, 401–421. [Google Scholar] [CrossRef]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, A.; Hall, S.; Curtin, N.; Drew, Y. Targeting ATR as Cancer Therapy: A new era for synthetic lethality and synergistic combinations? Pharmacol. Ther. 2019, 207, 107450. [Google Scholar] [CrossRef]
- Nguyen, B.; Sokoloski, J.; Galletto, R.; Elson, E.L.; Wold, M.S.; Lohman, T.M. Diffusion of Human Replication Protein A along Single-Stranded DNA. J. Mol. Biol. 2014, 426, 3246–3261. [Google Scholar] [CrossRef]
- Treuner, K.; Ramsperger, U.; Knippers, R. Replication Protein A Induces the Unwinding of Long Double-stranded DNA Regions. J. Mol. Biol. 1996, 259, 104–112. [Google Scholar] [CrossRef]
- Zou, L.; Elledge, S.J. Sensing DNA Damage Through ATRIP Recognition of RPA-ssDNA Complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Lu, T.; Wang, X.; Zheng, H.; Wang, L.-E.; Wei, Q.; Hittelman, W.N.; Li, L. Disruption of the Rad9/Rad1/Hus1 (9–1–1) complex leads to checkpoint signaling and replication defects. Oncogene 2004, 23, 5586–5593. [Google Scholar] [CrossRef]
- Bass, T.E.; Cortez, D. Quantitative phosphoproteomics reveals mitotic function of the ATR activator ETAA1. J. Cell Biol. 2019, 218, 1235–1249. [Google Scholar] [CrossRef]
- Saldivar, J.C.; Hamperl, S.; Bocek, M.J.; Chung, M.; Bass, T.E.; Cisneros-Soberanis, F.; Samejima, K.; Xie, L.; Paulson, J.R.; Earnshaw, W.C.; et al. An intrinsic S/G 2 checkpoint enforced by ATR. Science 2018, 361, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Dent, P.; Jelinek, T.; Morrison, D.K.; Weber, M.J.; Sturgill, T.W. Reversal of Raf-1 Activation by Purified and Membrane-Associated Protein Phosphatases. Science 1995, 268, 1902–1906. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.-Y.; Graves, P.R.; Thoma, R.S.; Wu, Z.; Shaw, A.S.; Piwnica-Worms, H. Mitotic and G 2 Checkpoint Control: Regulation of 14-3-3 Protein Binding by Phosphorylation of Cdc25C on Serine-216. Science 1997, 277, 1501–1505. [Google Scholar] [CrossRef]
- Sørensen, C.S.; Syljuåsen, R.G.; Falck, J.; Schroeder, T.; Rönnstrand, L.; Khanna, K.K.; Zhou, B.-B.; Bartek, J.; Lukas, J. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell 2003, 3, 247–258. [Google Scholar] [CrossRef]
- Sørensen, C.S.; Syljuåsen, R.G. Safeguarding genome integrity: The checkpoint kinases ATR, CHK1 and WEE1 restrain CDK activity during normal DNA replication. Nucleic Acids Res. 2011, 40, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, H.; Powell, S.N.; Iliakis, G.; Wang, Y. ATR affecting cell radiosensitivity is dependent on homologous recombination repair but independent of nonhomologous end joining. Cancer Res. 2004, 64, 7139–7143. [Google Scholar] [CrossRef]
- Brown, A.D.; Sager, B.W.; Gorthi, A.; Tonapi, S.S.; Brown, E.J.; Bishop, A.J.R. ATR suppresses endogenous DNA damage and allows completion of homologous recombination repair. PLoS ONE 2014, 9, e91222. [Google Scholar] [CrossRef] [PubMed]
- Tibbetts, R.S.; Cortez, D.; Brumbaugh, K.M.; Scully, R.; Livingston, D.; Elledge, S.J.; Abraham, R.T. Functional interactions between BRCA1 and the checkpoint kinase ATR during genotoxic stress. Genes Dev. 2000, 14, 2989–3002. [Google Scholar] [CrossRef] [PubMed]
- Ahlskog, J.K.; Larsen, B.D.; Achanta, K.; Sørensen, C.S. ATM/ATR -mediated phosphorylation of PALB 2 promotes RAD 51 function. EMBO Rep. 2016, 17, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, C.; Hansen, L.T.; Dziegielewski, J.; Syljuåsen, R.G.; Lundin, C.; Bartek, J.; Helleday, T. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat. Cell Biol. 2005, 7, 195–201. [Google Scholar] [CrossRef]
- Bahassi, E.M.; Ovesen, J.L.; Riesenberg, A.L.; Bernstein, W.Z.; Hasty, P.E.; Stambrook, P.J. The checkpoint kinases Chk1 and Chk2 regulate the functional associations between hBRCA2 and Rad51 in response to DNA damage. Oncogene 2008, 27, 3977–3985. [Google Scholar] [CrossRef]
- Dibitetto, D.; Sims, J.R.; Ascenção, C.F.R.; Feng, K.; Kim, D.; Oberly, S.; Freire, R.; Smolka, M.B. Intrinsic ATR signaling shapes DNA end resection and suppresses toxic DNA-PKcs signaling. NAR Cancer 2020, 2, zcaa006. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Huang, T.-T.; Horibata, S.; Lee, J.-M. Cell cycle checkpoints and beyond: Exploiting the ATR/CHK1/WEE1 pathway for the treatment of PARP inhibitor–resistant cancer. Pharmacol. Res. 2022, 178, 106162. [Google Scholar] [CrossRef]
- Kim, D.; Liu, Y.; Oberly, S.; Freire, R.; Smolka, M.B. ATR-mediated proteome remodeling is a major determinant of homologous recombination capacity in cancer cells. Nucleic Acids Res. 2018, 46, 8311–8325. [Google Scholar] [CrossRef]
- Nishida, H.; Tatewaki, N.; Nakajima, Y.; Magara, T.; Ko, K.M.; Hamamori, Y.; Konishi, T. Inhibition of ATR protein kinase activity by schisandrin B in DNA damage response. Nucleic Acids Res. 2009, 37, 5678–5689. [Google Scholar] [CrossRef]
- Fokas, E.; Prevo, R.; Pollard, J.R.; Reaper, P.M.; Charlton, P.A.; Cornelissen, B.; Vallis, K.A.; Hammond, E.M.; Olcina, M.M.; Gillies McKenna, W.; et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 2012, 3, e441. [Google Scholar] [CrossRef]
- Teng, P.-N.; Bateman, N.W.; Darcy, K.M.; Hamilton, C.A.; Maxwell, G.L.; Bakkenist, C.J.; Conrads, T.P. Pharmacologic inhibition of ATR and ATM offers clinically important distinctions to enhancing platinum or radiation response in ovarian, endometrial, and cervical cancer cells. Gynecol. Oncol. 2015, 136, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Reaper, P.M.; Griffiths, M.R.; Long, J.M.; Charrier, J.-D.; MacCormick, S.; Charlton, P.A.; Golec, J.M.C.; Pollard, J.R. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. 2011, 7, 428–430. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.B.; Newsome, D.; Wang, Y.; Boucher, D.M.; Eustace, B.; Gu, Y.; Hare, B.; Johnson, M.A.; Li, H.; Milton, S.; et al. Potentiation of tumor responses to DNA damaging therapy by the selective ATR inhibitor VX-970. Oncotarget 2014, 5, 5674–5685. [Google Scholar] [CrossRef] [PubMed]
- Huntoon, C.J.; Flatten, K.S.; Hendrickson, A.E.W.; Huehls, A.M.; Sutor, S.L.; Kaufmann, S.H.; Karnitz, L.M. ATR Inhibition Broadly Sensitizes Ovarian Cancer Cells to Chemotherapy Independent of BRCA Status. Cancer Res. 2013, 73, 3683–3691. [Google Scholar] [CrossRef]
- Nie, S.; Wan, Y.; Wang, H.; Liu, J.; Yang, J.; Sun, R.; Meng, H.; Ma, X.; Jiang, Y.; Cheng, W. CXCL2-mediated ATR/CHK1 signaling pathway and platinum resistance in epithelial ovarian cancer. J. Ovarian Res. 2021, 14, 1–11. [Google Scholar] [CrossRef]
- Wallez, Y.; Dunlop, C.R.; Johnson, T.I.; Koh, S.-B.; Fornari, C.; Yates, J.W.; Fernández, S.B.D.Q.; Lau, A.; Richards, F.M.; Jodrell, D.I. The ATR Inhibitor AZD6738 Synergizes with Gemcitabine In Vitro and In Vivo to Induce Pancreatic Ductal Adenocarcinoma Regression. Mol. Cancer Ther. 2018, 17, 1670–1682. [Google Scholar] [CrossRef]
- Ashworth, A. ATR Inhibitors and Paclitaxel in Melanoma. Clin. Cancer Res. 2021, 27, 4667–4668. [Google Scholar] [CrossRef]
- Cui, J.; Dean, D.; Hornicek, F.J.; Pollock, R.E.; Hoffman, R.M.; Duan, Z. ATR inhibition sensitizes liposarcoma to doxorubicin by increasing DNA damage. Am. J. Cancer Res. 2022, 12, 1577–1592. [Google Scholar]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef]
- Sun, L.-L.; Yang, R.-Y.; Li, C.-W.; Chen, M.-K.; Shao, B.; Hsu, J.-M.; Chan, L.-C.; Yang, Y.; Hsu, J.L.; Lai, Y.-J.; et al. Inhibition of ATR downregulates PD-L1 and sensitizes tumor cells to T cell-mediated killing. Am. J. Cancer Res. 2018, 8, 1307–1316. [Google Scholar]
- Patin, E.C.; Dillon, M.T.; Nenclares, P.; Grove, L.; Soliman, H.; Leslie, I.; Northcote, D.; Bozhanova, G.; Crespo-Rodriguez, E.; Baldock, H.; et al. Harnessing radiotherapy-induced NK-cell activity by combining DNA damage–response inhibition and immune checkpoint blockade. J. Immunother. Cancer 2022, 10, e004306. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Pan, W.; Xing, Y.; Xiao, Y.; Chen, J.; Xu, Z. Recent advances in DDR (DNA damage response) inhibitors for cancer therapy. Eur. J. Med. Chem. 2022, 230, 114109. [Google Scholar] [CrossRef] [PubMed]
- Massey, A.J. Inhibition of ATR-dependent feedback activation of Chk1 sensitises cancer cells to Chk1 inhibitor monotherapy. Cancer Lett. 2016, 383, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Mittica, G.; Ghisoni, E.; Giannone, G.; Genta, S.; Aglietta, M.; Sapino, A.; Valabrega, G. PARP Inhibitors in Ovarian Cancer. Recent Patents Anti-Cancer Drug Discov. 2018, 13, 392–410. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Giudice, E.; Gentile, M.; Salutari, V.; Ricci, C.; Musacchio, L.; Carbone, M.V.; Ghizzoni, V.; Camarda, F.; Tronconi, F.; Nero, C.; et al. PARP Inhibitors Resistance: Mechanisms and Perspectives. Cancers 2022, 14, 1420. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; George, E.; Ragland, R.L.; Rafail, S.; Zhang, R.; Krepler, C.; Morgan, M.A.; Herlyn, M.; Brown, E.J.; Simpkins, F. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clin. Cancer Res. 2017, 23, 3097–3108. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Xu, H.; George, E.; Hallberg, D.; Kumar, S.; Jagannathan, V.; Medvedev, S.; Kinose, Y.; Devins, K.; Verma, P.; et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat. Commun. 2020, 11, 3726. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.C.; Yap, T.A.; Boss, D.S.; Carden, C.P.; Mergui-Roelvink, M.; Gourley, C.; De Greve, J.; Lubinski, J.; Shanley, S.; Messiou, C.; et al. Poly(ADP)-Ribose Polymerase Inhibition: Frequent Durable Responses in BRCA Carrier Ovarian Cancer Correlating With Platinum-Free Interval. J. Clin. Oncol. 2010, 28, 2512–2519. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Wu, Q.; Luan, S.; Yin, Z.; He, C.; Yin, L.; Zou, Y.; Yuan, Z.; Li, L.; Song, X.; et al. A comprehensive review of topoisomerase inhibitors as anticancer agents in the past decade. Eur. J. Med. Chem. 2019, 171, 129–168. [Google Scholar] [CrossRef] [PubMed]
- Jossé, R.; Martin, S.E.; Guha, R.; Ormanoglu, P.; Pfister, T.D.; Reaper, P.M.; Barnes, C.S.; Jones, J.; Charlton, P.; Pollard, J.R.; et al. ATR Inhibitors VE-821 and VX-970 Sensitize Cancer Cells to Topoisomerase I Inhibitors by Disabling DNA Replication Initiation and Fork Elongation Responses. Cancer Res 2014, 74, 6968–6979. [Google Scholar] [CrossRef]
- Hur, J.; Ghosh, M.; Kim, T.H.; Park, N.; Pandey, K.; Cho, Y.B.; Hong, S.D.; Katuwal, N.B.; Kang, M.; An, H.J.; et al. Synergism of AZD6738, an ATR Inhibitor, in Combination with Belotecan, a Camptothecin Analogue, in Chemotherapy-Resistant Ovarian Cancer. Int. J. Mol. Sci. 2021, 22, 1223. [Google Scholar] [CrossRef]
- Kong, A.; Mehanna, H. WEE1 Inhibitor: Clinical Development. Curr. Oncol. Rep. 2021, 23, 1–8. [Google Scholar] [CrossRef]
- Rødland, G.E.; Hauge, S.; Hasvold, G.; Bay, L.T.E.; Raabe, T.T.H.; Joel, M.; Syljuåsen, R.G. Differential Effects of Combined ATR/WEE1 Inhibition in Cancer Cells. Cancers 2021, 13, 3790. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Kang, X.; Zhang, X.; Xie, W.; Su, Y.; Liu, X.; Guo, L.; Guo, E.; Li, F.; Hu, D.; et al. WEE1 inhibitor and ataxia telangiectasia and RAD3-related inhibitor trigger stimulator of interferon gene-dependent immune response and enhance tumor treatment efficacy through programmed death-ligand 1 blockade. Cancer Sci. 2021, 112, 4444–4456. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; George, E.; Kinose, Y.; Kim, H.; Shah, J.B.; Peake, J.D.; Ferman, B.; Medvedev, S.; Murtha, T.; Barger, C.J.; et al. CCNE1 copy number is a biomarker for response to combination WEE1-ATR inhibition in ovarian and endometrial cancer models. Cell Rep. Med. 2021, 2, 100394. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Redon, C.E.; Sciuto, L.; Padiernos, E.; Ji, J.; Lee, M.-J.; Yuno, L.M.A.; Lee, S.; Zhang, Y.; Tran, L.; et al. Phase I Study of ATR Inhibitor M6620 in Combination With Topotecan in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2018, 36, 1594–1602. [Google Scholar] [CrossRef]
- Middleton, M.R.; Dean, E.; Evans, T.R.J.; Shapiro, G.I.; Pollard, J.; Hendriks, B.S.; Falk, M.; Diaz-Padilla, I.; Plummer, R. Phase 1 study of the ATR inhibitor berzosertib (formerly M6620, VX-970) combined with gemcitabine ± cisplatin in patients with advanced solid tumours. Br. J. Cancer 2021, 125, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, G.I.; Wesolowski, R.; Devoe, C.; Lord, S.; Pollard, J.; Hendriks, B.S.; Falk, M.; Diaz-Padilla, I.; Plummer, R.; Yap, T.A. Phase 1 study of the ATR inhibitor berzosertib in combination with cisplatin in patients with advanced solid tumours. Br. J. Cancer 2021, 125, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; O’Carrigan, B.; Penney, M.S.; Lim, J.S.; Brown, J.S.; Luken, M.J.D.M.; Tunariu, N.; Perez-Lopez, R.; Rodrigues, D.N.; Riisnaes, R.; et al. Phase I Trial of First-in-Class ATR Inhibitor M6620 (VX-970) as Monotherapy or in Combination with Carboplatin in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2020, 38, 3195–3204. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Cheng, S.-C.; Hendrickson, A.E.W.; Penson, R.T.; Schumer, S.T.; Doyle, L.A.; Lee, E.K.; Kohn, E.C.; Duska, L.R.; Crispens, M.A.; et al. Berzosertib plus gemcitabine versus gemcitabine alone in platinum-resistant high-grade serous ovarian cancer: A multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 957–968. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.; Hendrickson, A.W.; Penson, R.; Doyle, A.; Kohn, E.; Duska, L.; Crispens, M.; Olawaiye, A.; Winer, I.; Barroilhet, L.; et al. Randomized phase II (RP2) study of ATR inhibitor M6620 in combination with gemcitabine versus gemcitabine alone in platinum-resistant high grade serous ovarian cancer (HGSOC). Ann. Oncol. 2019, 30, v897. [Google Scholar] [CrossRef]
- Sabat, C.; Ginestet, C.; Chassagnon, G. Gemcitabine and nab-paclitaxel induced interstitial pneumonia. Diagn. Interv. Imaging 2021, 102, 763–764. [Google Scholar] [CrossRef]
- Banerjee, S.; Vergotte, I.; Colombo, N.; Barve, M.; Grisham, R.; Mehr, K.; Falk, M.; Beier, F.; Hennessy, M.; Schroeder, A.; et al. Randomized, phase Ib/II study of M6620 + avelumab + carboplatin vs standard care (sc) in patients (pts) with platinum-sensitive poly (ADP-ribose) polymerase inhibitor-(PARPi)-resistant ovarian cancer. Ann. Oncol. 2019, 30, v431–v432. [Google Scholar] [CrossRef]
- Jones, C.D.; Blades, K.; Foote, K.M.; Guichard, S.M.; Jewsbury, P.J.; McGuire, T.; Nissink, J.W.; Odedra, R.; Tam, K.; Thommes, P.; et al. Abstract 2348: Discovery of AZD6738, a potent and selective inhibitor with the potential to test the clinical efficacy of ATR kinase inhibition in cancer patients. Cancer Res. 2013, 73, 2348. [Google Scholar] [CrossRef]
- Brunner, A.M.; Liu, Y.; Mendez, L.M.; Garcia, J.S.; Amrein, P.C.; Neuberg, D.S.; Dean, E.; Smith, S.; Stone, R.M.; Fathi, A.T.; et al. Inhibition of ATR with AZD6738 (Ceralasertib) for the Treatment of Progressive or Relapsed Myelodysplastic Syndromes and Chronic Myelomonocytic Leukemia: Safety and Preliminary Activity from a Phase Ib/II Study. Blood 2021, 138, 1521. [Google Scholar] [CrossRef]
- Mahdi, H.; Hafez, N.; Doroshow, D.; Sohal, D.; Keedy, V.; Do, K.T.; LoRusso, P.; Jürgensmeier, J.; Avedissian, M.; Sklar, J.; et al. Ceralasertib-Mediated ATR Inhibition Combined with Olaparib in Advanced Cancers Harboring DNA Damage Response and Repair Alterations (Olaparib Combinations). JCO Precis. Oncol. 2021, 5, 1432–1442. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Smith, S.A.; Mortimer, P.; Loembé, A.-B.; Cho, H.; Kim, K.-M.; Smith, C.; Willis, S.; Irurzun-Arana, I.; Berges, A.; et al. Phase I Study of Ceralasertib (AZD6738), a Novel DNA Damage Repair Agent, in Combination with Weekly Paclitaxel in Refractory Cancer. Clin. Cancer Res. 2021, 27, 4700–4709. [Google Scholar] [CrossRef]
- Yap, T.A.; Krebs, M.G.; Postel-Vinay, S.; El-Khouiery, A.; Soria, J.-C.; Lopez, J.; Berges, A.; Cheung, S.Y.A.; Irurzun-Arana, I.; Goldwin, A.; et al. Ceralasertib (AZD6738), an Oral ATR Kinase Inhibitor, in Combination with Carboplatin in Patients with Advanced Solid Tumors: A Phase I Study. Clin. Cancer Res. 2021, 27, 5213–5224. [Google Scholar] [CrossRef]
- Shah, P.D.; Wethington, S.L.; Pagan, C.; Latif, N.; Tanyi, J.; Martin, L.P.; Morgan, M.; Burger, R.A.; Haggerty, A.; Zarrin, H.; et al. Combination ATR and PARP Inhibitor (CAPRI): A phase 2 study of ceralasertib plus olaparib in patients with recurrent, platinum-resistant epithelial ovarian cancer. Gynecol. Oncol. 2021, 163, 246–253. [Google Scholar] [CrossRef]
- Kim, R.; Kwon, M.; An, M.; Kim, S.; Smith, S.; Loembé, A.; Mortimer, P.; Armenia, J.; Lukashchuk, N.; Shah, N.; et al. Phase II study of ceralasertib (AZD6738) in combination with durvalumab in patients with advanced/metastatic melanoma who have failed prior anti-PD-1 therapy. Ann. Oncol. 2021, 33, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.; Kim, G.; Kim, R.; Kim, K.-T.; Kim, S.T.; Smith, S.; Mortimer, P.G.S.; Hong, J.Y.; Loembé, A.-B.; Irurzun-Arana, I.; et al. Phase II study of ceralasertib (AZD6738) in combination with durvalumab in patients with advanced gastric cancer. J. Immunother. Cancer 2022, 10, e005041. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.; Tolcher, A.; Plummer, R.; Mukker, J.; Enderlin, M.; Hicking, C.; Locatelli, G.; Szucs, Z.; Gounaris, I.; de Bono, J. 457MO A phase I study of ATR inhibitor M1774 in patients with solid tumours (DDRiver Solid Tumours 301): Part A1 results. Ann. Oncol. 2022, 33, S747–S748. [Google Scholar] [CrossRef]
- Kurz, L.; Miklyaeva, A.; Skowron, M.A.; Overbeck, N.; Poschmann, G.; Becker, T.; Eul, K.; Kurz, T.; Schönberger, S.; Calaminus, G.; et al. ARID1A Regulates Transcription and the Epigenetic Landscape via POLE and DMAP1 While ARID1A Deficiency or Pharmacological Inhibition Sensitizes Germ Cell Tumor Cells to ATR Inhibition. Cancers 2020, 12, 905. [Google Scholar] [CrossRef]
- Williamson, C.T.; Miller, R.; Pemberton, H.N.; Jones, S.E.; Campbell, J.; Konde, A.; Badham, N.; Rafiq, R.; Brough, R.; Gulati, A.; et al. ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nat. Commun. 2016, 7, 13837. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Eberhart, C.G.; Pratilas, C.A.; Blakeley, J.O.; Davis, C.; Stojanova, M.; Reilly, K.; Meeker, A.K.; Heaphy, C.M.; Rodriguez, F.J. Therapeutic Vulnerability to ATR Inhibition in Concurrent NF1 and ATRX-Deficient/ALT-Positive High-Grade Solid Tumors. Cancers 2022, 14, 3015. [Google Scholar] [CrossRef]
- Gupta, M.; Concepcion, C.P.; Fahey, C.G.; Keshishian, H.; Bhutkar, A.; Brainson, C.F.; Sanchez-Rivera, F.J.; Pessina, P.; Kim, J.Y.; Simoneau, A.; et al. BRG1 Loss Predisposes Lung Cancers to Replicative Stress and ATR Dependency. Cancer Res. 2020, 80, 3841–3854. [Google Scholar] [CrossRef]
- Kurashima, K.; Kashiwagi, H.; Shimomura, I.; Suzuki, A.; Takeshita, F.; Mazevet, M.; Harata, M.; Yamashita, T.; Yamamoto, Y.; Kohno, T.; et al. SMARCA4 deficiency-associated heterochromatin induces intrinsic DNA replication stress and susceptibility to ATR inhibition in lung adenocarcinoma. NAR Cancer 2020, 2, zcaa005. [Google Scholar] [CrossRef] [PubMed]
- Sethy, R.; Rakesh, R.; Patne, K.; Arya, V.; Sharma, T.; Haokip, D.T.; Kumari, R.; Muthuswami, R. Regulation of ATM and ATR by SMARCAL1 and BRG1. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 1076–1092. [Google Scholar] [CrossRef]
- Schleicher, E.M.; Dhoonmoon, A.; Jackson, L.M.; Clements, K.E.; Stump, C.L.; Nicolae, C.M.; Moldovan, G.-L. Dual genome-wide CRISPR knockout and CRISPR activation screens identify mechanisms that regulate the resistance to multiple ATR inhibitors. PLoS Genet. 2020, 16, e1009176. [Google Scholar] [CrossRef]
- Klattenhoff, A.W.; Thakur, M.; Chu, C.S.; Ray, D.; Habib, S.L.; Kidane, D. Loss of NEIL3 DNA glycosylase markedly increases replication associated double strand breaks and enhances sensitivity to ATR inhibitor in glioblastoma cells. Oncotarget 2017, 8, 112942–112958. [Google Scholar] [CrossRef] [PubMed]
- Hustedt, N.; Álvarez-Quilón, A.; McEwan, A.; Yuan, J.Y.; Cho, T.; Koob, L.; Hart, T.; Durocher, D. A consensus set of genetic vulnerabilities to ATR inhibition. Open Biol. 2019, 9, 190156. [Google Scholar] [CrossRef] [PubMed]
- Job, A.; Tatura, M.; Schäfer, C.; Lutz, V.; Schneider, H.; Lankat-Buttgereit, B.; Zielinski, A.; Borgmann, K.; Bauer, C.; Gress, T.M.; et al. The POLD1R689W variant increases the sensitivity of colorectal cancer cells to ATR and CHK1 inhibitors. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hocke, S.; Guo, Y.; Job, A.; Orth, M.; Ziesch, A.; Lauber, K.; De Toni, E.N.; Gress, T.M.; Herbst, A.; Göke, B.; et al. A synthetic lethal screen identifies ATR-inhibition as a novel therapeutic approach for POLD1-deficient cancers. Oncotarget 2016, 7, 7080–7095. [Google Scholar] [CrossRef] [PubMed]
- Job, A.; Schmitt, L.-M.; von Wenserski, L.; Lankat-Buttgereit, B.; Gress, T.M.; Buchholz, M.; Gallmeier, E. Inactivation of PRIM1 Function Sensitizes Cancer Cells to ATR and CHK1 Inhibitors. Neoplasia 2018, 20, 1135–1143. [Google Scholar] [CrossRef]
- Jiang, H.-G.; Chen, P.; Su, J.-Y.; Wu, M.; Qian, H.; Wang, Y.; Li, J. Knockdown of REV3 synergizes with ATR inhibition to promote apoptosis induced by cisplatin in lung cancer cells. J. Cell. Physiol. 2017, 232, 3433–3443. [Google Scholar] [CrossRef]
- Wang, C.; Wang, G.; Feng, X.; Shepherd, P.; Zhang, J.; Tang, M.; Chen, Z.; Srivastava, M.; McLaughlin, M.E.; Navone, N.M.; et al. Genome-wide CRISPR screens reveal synthetic lethality of RNASEH2 deficiency and ATR inhibition. Oncogene 2019, 38, 2451–2463. [Google Scholar] [CrossRef]
- Gilad, O.; Nabet, B.Y.; Ragland, R.L.; Schoppy, D.W.; Smith, K.D.; Durham, A.C.; Brown, E.J. Combining ATR Suppression with Oncogenic Ras Synergistically Increases Genomic Instability, Causing Synthetic Lethality or Tumorigenesis in a Dosage-Dependent Manner. Cancer Res 2010, 70, 9693–9702. [Google Scholar] [CrossRef]
- Gralewska, P.; Gajek, A.; Rybaczek, D.; Marczak, A.; Rogalska, A. The Influence of PARP, ATR, CHK1 Inhibitors on Premature Mitotic Entry and Genomic Instability in High-Grade Serous BRCAMUT and BRCAWT Ovarian Cancer Cells. Cells 2022, 11, 1889. [Google Scholar] [CrossRef]
- Schoonen, P.M.; Kok, Y.P.; Wierenga, E.; Bakker, B.; Foijer, F.; Spierings, D.C.J.; Van Vugt, M.A.T.M. Premature mitotic entry induced by ATR inhibition potentiates olaparib inhibition-mediated genomic instability, inflammatory signaling, and cytotoxicity in BRCA2-deficient cancer cells. Mol. Oncol. 2019, 13, 2422–2440. [Google Scholar] [CrossRef]
- Dhoonmoon, A.; Schleicher, E.M.; Clements, K.E.; Nicolae, C.M.; Moldovan, G.-L. Genome-wide CRISPR synthetic lethality screen identifies a role for the ADP-ribosyltransferase PARP14 in DNA replication dynamics controlled by ATR. Nucleic Acids Res. 2020, 48, 7252–7264. [Google Scholar] [CrossRef]
- Shigechi, T.; Tomida, J.; Sato, K.; Kobayashi, M.; Eykelenboom, J.K.; Pessina, F.; Zhang, Y.; Uchida, E.; Ishiai, M.; Lowndes, N.F.; et al. ATR–ATRIP Kinase Complex Triggers Activation of the Fanconi Anemia DNA Repair Pathway. Cancer Res. 2012, 72, 1149–1156. [Google Scholar] [CrossRef] [PubMed]
- Schwab, R.A.; Blackford, A.N.; Niedzwiedz, W. ATR activation and replication fork restart are defective in FANCM-deficient cells. EMBO J. 2010, 29, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Fan, X.; Dueva, R.; Soni, A.; Iliakis, G. Radiation-dose-dependent functional synergisms between ATM, ATR and DNA-PKcs in checkpoint control and resection in G2-phase. Sci. Rep. 2019, 9, 1–19. [Google Scholar] [CrossRef]
- Mohni, K.N.; Kavanaugh, G.M.; Cortez, D. ATR Pathway Inhibition Is Synthetically Lethal in Cancer Cells with ERCC1 Deficiency. Cancer Res. 2014, 74, 2835–2845. [Google Scholar] [CrossRef]
- Al-Subhi, N.; Ali, R.; Abdel-Fatah, T.; Moseley, P.M.; Chan, S.Y.T.; Green, A.R.; Ellis, I.O.; Rakha, E.A.; Madhusudan, S. Targeting ataxia telangiectasia-mutated- and Rad3-related kinase (ATR) in PTEN-deficient breast cancers for personalized therapy. Breast Cancer Res. Treat. 2018, 169, 277–286. [Google Scholar] [CrossRef]
- Jackson, C.B.; Noorbakhsh, S.I.; Sundaram, R.K.; Kalathil, A.N.; Ganesa, S.; Jia, L.; Breslin, H.; Burgenske, D.M.; Gilad, O.; Sarkaria, J.N.; et al. Temozolomide Sensitizes MGMT-Deficient Tumor Cells to ATR Inhibitors. Cancer Res. 2019, 79, 4331–4338. [Google Scholar] [CrossRef]
- El Touny, L.H.; Hose, C.; Connelly, J.; Harris, E.; Monks, A.; Dull, A.B.; Wilsker, D.F.; Hollingshead, M.G.; Gottholm-Ahalt, M.; Alcoser, S.Y.; et al. ATR inhibition reverses the resistance of homologous recombination deficient MGMTlow/MMRproficient cancer cells to temozolomide. Oncotarget 2021, 12, 2114–2130. [Google Scholar] [CrossRef]
- Ali, R.; Alblihy, A.; Toss, M.S.; Algethami, M.; Al Sunni, R.; Green, A.R.; Rakha, E.A.; Madhusudan, S. XRCC1 deficient triple negative breast cancers are sensitive to ATR, ATM and Wee1 inhibitor either alone or in combination with olaparib. Ther. Adv. Med. Oncol. 2020, 12, 1758835920974201. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Abdel-Fatah, T.; Perry, C.; Moseley, P.; Albarakati, N.; Mohan, V.; Seedhouse, C.; Chan, S.; Madhusudan, S. Ataxia Telangiectasia Mutated and Rad3 Related (ATR) Protein Kinase Inhibition Is Synthetically Lethal in XRCC1 Deficient Ovarian Cancer Cells. PLoS ONE 2013, 8, e57098. [Google Scholar] [CrossRef] [PubMed]
- Buisson, R.; Lawrence, M.S.; Benes, C.H.; Zou, L. APOBEC3A and APOBEC3B Activities Render Cancer Cells Susceptible to ATR Inhibition. Cancer Res 2017, 77, 4567–4578. [Google Scholar] [CrossRef]
- Lloyd, R.L.; Urban, V.; Muñoz-Martínez, F.; Ayestaran, I.; Thomas, J.C.; de Renty, C.; O’Connor, M.J.; Forment, J.V.; Galanty, Y.; Jackson, S.P. Loss of Cyclin C or CDK8 provides ATR inhibitor resistance by suppressing transcription-associated replication stress. Nucleic Acids Res. 2021, 49, 8665–8683. [Google Scholar] [CrossRef] [PubMed]
- King, D.; Southgate, H.E.D.; Roetschke, S.; Gravells, P.; Fields, L.; Watson, J.B.; Chen, L.; Chapman, D.; Harrison, D.; Yeomanson, D.; et al. Increased Replication Stress Determines ATR Inhibitor Sensitivity in Neuroblastoma Cells. Cancers 2021, 13, 6215. [Google Scholar] [CrossRef] [PubMed]
- Toh, M.; Ngeow, J. Homologous Recombination Deficiency: Cancer Predispositions and Treatment Implications. Oncologist 2021, 26, e1526–e1537. [Google Scholar] [CrossRef]
- Qiu, Z.; Fa, P.; Liu, T.; Prasad, C.B.; Ma, S.; Hong, Z.; Chan, E.R.; Wang, H.; Li, Z.; He, K.; et al. A Genome-Wide Pooled shRNA Screen Identifies PPP2R2A as a Predictive Biomarker for the Response to ATR and CHK1 Inhibitors. Cancer Res. 2020, 80, 3305–3318. [Google Scholar] [CrossRef]
- Jo, U.; Murai, Y.; Chakka, S.; Chen, L.; Cheng, K.; Murai, J.; Saha, L.K.; Jenkins, L.M.M.; Pommier, Y. SLFN11 promotes CDT1 degradation by CUL4 in response to replicative DNA damage, while its absence leads to synthetic lethality with ATR/CHK1 inhibitors. Proc. Natl. Acad. Sci. USA 2021, 118, e2015654118. [Google Scholar] [CrossRef] [PubMed]
- Kundu, K.; Cardnell, R.J.; Zhang, B.; Shen, L.; Stewart, C.A.; Ramkumar, K.; Cargill, K.R.; Wang, J.; Gay, C.M.; Byers, L.A. SLFN11 biomarker status predicts response to lurbinectedin as a single agent and in combination with ATR inhibition in small cell lung cancer. Transl. Lung Cancer Res. 2021, 10, 4095–4105. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Graves, J.D.; Lin, F.-T.; Lin, W.-C. Overexpression of TopBP1, a canonical ATR/Chk1 activator, paradoxically hinders ATR/Chk1 activation in cancer. J. Biol. Chem. 2021, 296, 100382. [Google Scholar] [CrossRef]
- Mordes, D.A.; Glick, G.G.; Zhao, R.; Cortez, D. TopBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes Dev. 2008, 22, 1478–1489. [Google Scholar] [CrossRef] [PubMed]
- Komura, K.; Yoshikawa, Y.; Shimamura, T.; Chakraborty, G.; Gerke, T.; Hinohara, K.; Chadalavada, K.; Jeong, S.H.; Armenia, J.; Du, S.-Y.; et al. ATR inhibition controls aggressive prostate tumors deficient in Y-linked histone demethylase KDM5D. J. Clin. Investig. 2018, 128, 2979–2995. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; He, J.; Li, Y.; Lv, S.; Cui, H. NUSAP1 potentiates chemoresistance in glioblastoma through its SAP domain to stabilize ATR. Signal Transduct. Target. Ther. 2020, 5, 1–11. [Google Scholar] [CrossRef]
- Smith, K.D.; Fu, M.A.; Brown, E.J. Tim–Tipin dysfunction creates an indispensible reliance on the ATR–Chk1 pathway for continued DNA synthesis. J. Cell Biol. 2009, 187, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Abu-Odeh, M.; Hereema, N.A.; Aqeilan, R.I. WWOX modulates the ATR-mediated DNA damage checkpoint response. Oncotarget 2015, 7, 4344–4355. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ledermann, J.A.; Drew, Y.; Kristeleit, R.S. Homologous recombination deficiency and ovarian cancer. Eur. J. Cancer 2016, 60, 49–58. [Google Scholar] [CrossRef]
- Sancar, A.; Lindsey-Boltz, L.A.; Ünsal-Kaçmaz, K.; Linn, S. Molecular Mechanisms of Mammalian DNA Repair and the DNA Damage Checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef]
- Gaillard, H.; Garcia-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, A.; Zenke, F.T.; Curtin, N.J.; Drew, Y. The Role of ATR Inhibitors in Ovarian Cancer: Investigating Predictive Biomarkers of Response. Cells 2022, 11, 2361. [Google Scholar] [CrossRef] [PubMed]
- Menezes, D.L.; Holt, J.; Tang, Y.; Feng, J.; Barsanti, P.; Pan, Y.; Ghoddusi, M.; Zhang, W.; Thomas, G.; Holash, J.; et al. A Synthetic Lethal Screen Reveals Enhanced Sensitivity to ATR Inhibitor Treatment in Mantle Cell Lymphoma with ATM Loss-of-Function. Mol. Cancer Res. 2015, 13, 120–129. [Google Scholar] [CrossRef]
- Min, A.; Im, S.-A.; Jang, H.; Kim, S.; Lee, M.; Kim, D.K.; Yang, Y.; Kim, H.-J.; Lee, K.-H.; Kim, J.W.; et al. AZD6738, A Novel Oral Inhibitor of ATR, Induces Synthetic Lethality with ATM Deficiency in Gastric Cancer Cells. Mol. Cancer Ther. 2017, 16, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Middleton, F.K.; Pollard, J.R.; Curtin, N.J. The Impact of p53 Dysfunction in ATR Inhibitor Cytotoxicity and Chemo- and Radiosensitisation. Cancers 2018, 10, 275. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Kipps, T.; Kurzrock, R. ATM Mutations in Cancer: Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 1781–1791. [Google Scholar] [CrossRef] [PubMed]
- Heeke, A.L.; Pishvaian, M.J.; Lynce, F.; Xiu, J.; Brody, J.R.; Chen, W.-J.; Baker, T.M.; Marshall, J.L.; Isaacs, C. Prevalence of Homologous Recombination–Related Gene Mutations Across Multiple Cancer Types. JCO Precis. Oncol. 2018, 2018, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kwok, M.; Davies, N.; Agathanggelou, A.; Smith, E.; Petermann, E.; Yates, E.; Brown, J.; Lau, A.; Stankovic, T. Synthetic lethality in chronic lymphocytic leukaemia with DNA damage response defects by targeting the ATR pathway. Lancet 2015, 385, S58. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.; Knittel, G.; Welcker, D.; Yang, T.-P.; George, J.; Nowak, M.; Leeser, U.; Büttner, R.; Perner, S.; Peifer, M.; et al. ATM Deficiency Is Associated with Sensitivity to PARP1- and ATR Inhibitors in Lung Adenocarcinoma. Cancer Res. 2017, 77, 3040–3056. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| ATRi | Intervention | Main ID | Phase | Status | Condition or Disease |
|---|---|---|---|---|---|
| ART0380 | ART0380; ART0380 + Gemcitabine; ART0380 + Irinotecan | NCT04657068 | I/II | Recruiting | OC, advanced cancer, metastatic cancer, primary peritoneal cancer, and fallopian tube cancer with DDR genes |
| ATRN-119 | ATRN-119 | NCT04905914 | I/II | Recruiting | Advanced solid tumor with DDR genes |
| Berzosertib (VE-822, M6620, VX-970) | Berzosertib | NCT03718091 | II | Completed | Solid tumor, leiomyosarcoma, and osteosarcoma with HR mutations |
| Berzosertib + Avelumab | NCT04266912 | I/II | Recruiting | DDR-deficient metastatic or unresectable solid tumors with DDR genes | |
| Berzosertib + Carboplatin | EUCTR2013-005100-34-GB | I | Not recruiting | Advanced-stage solid tumors with DDR genes | |
| Berzosertib + Carboplatin + Avelumab | NCT03704467 | I | Completed | PARPi-resistant OC with BRCA 1/2 mutation | |
| Berzosertib + Carboplatin + Avelumab | EUCTR2018-001534-17-BE | Ib/II | Not recruiting | PARPi-resistant recurrent ovarian, primary [eritoneal, or fallopian tube cancer | |
| Berzosertib + Carboplatin + Paclitaxel | NCT03309150 | I | Active | Advanced-stage solid tumors | |
| Berzosertib + Carboplatin + Gemcitabine hydrochloride | NCT02627443 | I | Active | Platinu- sensitive recurrent and metastatic ovarian, primary peritoneal, or fallopian tube cancer | |
| Berzosertib + Cisplatin + Gemcitabine; Berzosertib + Cisplatin + Etoposide; Berzosertib + Irinotecan; Berzosertib+Gemcitabine; Berzosertib + Cisplatin; Berzosertib + Carboplatin | NCT02157792 | I | Completed | Advanced-stage solid tumors | |
| Berzosertib+ Chemotherapy | EUCTR2012-003126-25-GB | I | Not recruiting | Advanced Solid Tumors | |
| Berzosertib + Gemcitabine hydrochloride | NCT02595892 | II | Completed | Platinum-resistant recurrent ovarian, primary peritoneal, or fallopian tube cancer | |
| Berzosertib + Irinotecan Hydrochloride | NCT02595931 | I | Recruiting | Solid tumors that are metastatic or cannot be removed by surgery with DDR genes | |
| Berzosertib+Lurbinectedin | NCT04802174 | I/II | Recruiting | Advanced solid tumors, SCLCS, mall cell cancers, and high-grade neuroendocrine cancers | |
| Berzosertib + Topotecan | NCT05246111 | I | Recruiting | Advanced solid tumor | |
| Berzosertib + Veliparib + Cisplatin | NCT02723864 | I | Completed | Refractory solid tumors | |
| Ceralasertib (AZD6738) | Ceralasertib | NCT04564027 | II | Recruiting | Advanced solid tumours with deleterious ATM mutation |
| Ceralasertib | EUCTR2020-002529-27-FR | IIa | Authorised | Advanced cancer whose tumours contain molecular alterations | |
| Ceralasertib; Ceralasertib+ Carboplatin; Ceralasertib + Olaparib; Ceralasertib+ Durvalumab | NCT02264678 | I/II | Recruiting | Platinum sensitive OC with BRCA mutant or RAD51C/D mutant or HRD positive status, head and neck SCC, ATM proficiency/deficiency NSCLC, and gastric or breast cancer | |
| Ceralasertib; Ceralasertib + Olaparib; Adavosertib + Olaparib | NCT03579316 | II | Recruiting | Recurrent ovarian, primary peritoneal, or fallopian tube cancer | |
| Ceralasertib; Ceralasertib + Olaparib | EUCTR2019-003791-39-GB | II | Authorised | platinum-sensitive epithelial ovarian cancer | |
| Ceralasertib+ Durvalumab | KCT0003806 | II | Not recruiting | Metastatic solid tumor | |
| Ceralasertib+ Durvalumab | CTR20221743 | I | Not recruiting | Advanced solid tumors | |
| Ceralasertib+ Gemcitabine | NCT03669601 | I | Recruiting | Advanced or metastatic solid tumour | |
| Ceralasertib+ Gemcitabine | EUCTR2017-003935-12-GB | I | Authorised | Advanced or metastatic solid tumour | |
| Ceralasertib + Olaparib | NCT02576444 | II | Active | Cancer, including HGSC harboring DDR, and repair alterations | |
| Ceralasertib + Olaparib | NCT03462342 | II | Recruiting | Recurrent OC | |
| Ceralasertib + Olaparib | NCT03878095 | II | Recruiting | Malignant solid neoplasm, refractory cholangiocarcinoma, or refractory malignant solid neoplasm with IDH1 and IDH2 mutant | |
| Ceralasertib monotherapy; Ceralasertib + Olaparib | NCT04065269 | II | Recruiting | Gynaecological cancers | |
| Ceralasertib + Olaparib; Olaparib monotherapy | NCT04239014 | II | Withdrawn | OC | |
| Ceralasertib + Paclitaxel | NCT02630199 | I | Completed | Refractory cancer | |
| Ceralasertib + Paclitaxel | KCT0003403 | I | Recruiting | Advanced or metastatic solid tumour | |
| Ceralasertib + Palliative Radiotherapy | NCT02223923 | I | Unknown | Solid-tumour refractory to conventional treatment | |
| Elimusertib (BAY1895344) | Elimusertib | NCT03188965 | II | Active | Advanced solid tumor and lymphomas with DDR defects |
| Elimusertib | NCT05071209 | I/II | Recruiting | Relapsed or refractory solid tumors | |
| Elimusertib + Cisplatin; Elimusertib + Cisplatin + Gemcitabine | NCT04491942 | I | Recruiting | Advanced solid tumors with emphasis on urothelial cancer | |
| Elimusertib + Copanlisib | NCT05010096 | Ib | Withdrawn | Advanced solid tumors with at least one DRR-related gene mutation | |
| Elimusertib+ Gemcitabine | NCT04616534 | I | Recruiting | Advanced solid tumors, advanced pancreatic and OC, and advanced solid tumors | |
| Elimusertib +Niraparib | NCT04267939 | Ib | Recruiting | Recurrent EOC, fallopian tube, or primary peritoneal cancer, and recurrent advanced solid tumors | |
| Elimusertib+ Pembrolizumab | NCT04095273 | I | Active | Advanced solid tumor with putative biomarkers of DDR deficiency | |
| M1774 | M1774; M1774 + Niraparib | NCT04170153 | II | Recruiting | Metastatic or locally advanced unresectable solid tumors |
| M1774+ DDR inhibitor; M1774 + Immune Checkpoint Inhibitor | NCT05396833 | I | Recruiting | Metastatic or locally advanced unresectable solid tumors | |
| M4344 (VX803) | M4344; M4344 + Carboplatin | NCT02278250 | I | Completed | Advanced solid tumors |
| M4344 monotherapy; M4344 + Niraparib | NCT04655183 | I/II | Withdrawn | Advanced solid tumors, breast cancer | |
| M4344 + Niraparib | NCT04149145 | I | Not recruiting | PARPi-resistant recurrent OC | |
| RP-3500 | RP-3500; RP-3500 + Talazoparib + Gemcitabine | NCT04497116 | I/II | Recruiting | Advanced solid tumors |
| RP-3500; RP-3500 + RP-6306 | NCT04855656 | I | Recruiting | Advanced solid tumors | |
| RP-3500 + Niraparib and/or Olaparib | NCT04972110 | I/II | Recruiting | Advanced solid tumors, adult |
| Regulatory Mechanism | Patient Selection |
|---|---|
| Blocked DNA synthesis and accumulation of damaged DNA | ARID1A-deficient [93,94]; ATRX-Deficient [95]; BRG1 Loss (SMARCA4-deficient) [96,97,98]; LUC7L3-deficient [99]; NEIL3-deficient [100]; POLE3/POLE4-deficient [101]; POLD1-deficient [102,103]; PRIM1-deficient [104]; REV3-deficient [105]; RNASEH2-deficient [106]; RAS-transformed [107]. |
| Impaired DNA damage repair (DDR) | HRD [108] (AXL [109]; BRCA [108,110], RAD51 [111], PARP14 [111]; FANCM [112,113]; NEIL3-deficient); CCNE1 amplification [74]; DNA-PKcs-deficient [114]; ERCC1-deficient [115]; LIAS-deficient [99]; MED12 and PTEN-deficient [116]; MGMT-deficient [117,118]; XRCC1-deficient [119,120]. |
| Stalling of replication fork progression | APOBEC3B reduced [121]; BRG1 Loss (SMARCA4-deficient); FANCM-deficient; Loss of Cyclin C and CDK [122]; MYCN amplification [123]; PBRM1-defective [124]; PPP2R2A-deficient [125]; RAD51 reduced [111]; REV3-deficient; SLFN11-deficient [126,127]; TopBP1-deficient [128,129]. |
| Regulation of the cell cycle | ARID1A-deficient; ATM-deficient; CCNE1 amplification; DNA-PKcs-deficient; FANCM-deficient; KDM5D-defective [130]; NUSAP1-deficient [131]; Tim-Tipin-deficient [132]; WWOX-deficient [133]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Wang, T.; Fei, X.; Zhang, M. ATR Inhibitors in Platinum-Resistant Ovarian Cancer. Cancers 2022, 14, 5902. https://doi.org/10.3390/cancers14235902
Li S, Wang T, Fei X, Zhang M. ATR Inhibitors in Platinum-Resistant Ovarian Cancer. Cancers. 2022; 14(23):5902. https://doi.org/10.3390/cancers14235902
Chicago/Turabian StyleLi, Siyu, Tao Wang, Xichang Fei, and Mingjun Zhang. 2022. "ATR Inhibitors in Platinum-Resistant Ovarian Cancer" Cancers 14, no. 23: 5902. https://doi.org/10.3390/cancers14235902
APA StyleLi, S., Wang, T., Fei, X., & Zhang, M. (2022). ATR Inhibitors in Platinum-Resistant Ovarian Cancer. Cancers, 14(23), 5902. https://doi.org/10.3390/cancers14235902