

NPFFR2 Contributes to the Malignancy of Hepatocellular Carcinoma Development by Activating RhoA/YAP Signaling
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. HCC Patient Tissues
2.2. TCGA Data Analysis
2.3. RT–PCR and Real–Time PCR
2.4. Statistical Analysis
2.5. Cell Culture and Reagents
2.6. siRNAs, Plasmids, and Transfection
2.7. Lentivirus Infection
2.8. Invasion Assay
2.9. Migration Assay
2.10. Colony Formation and Soft Agar Assays
2.11. Western Blotting and RhoA Assay
2.12. Immunofluorescence
3. Results
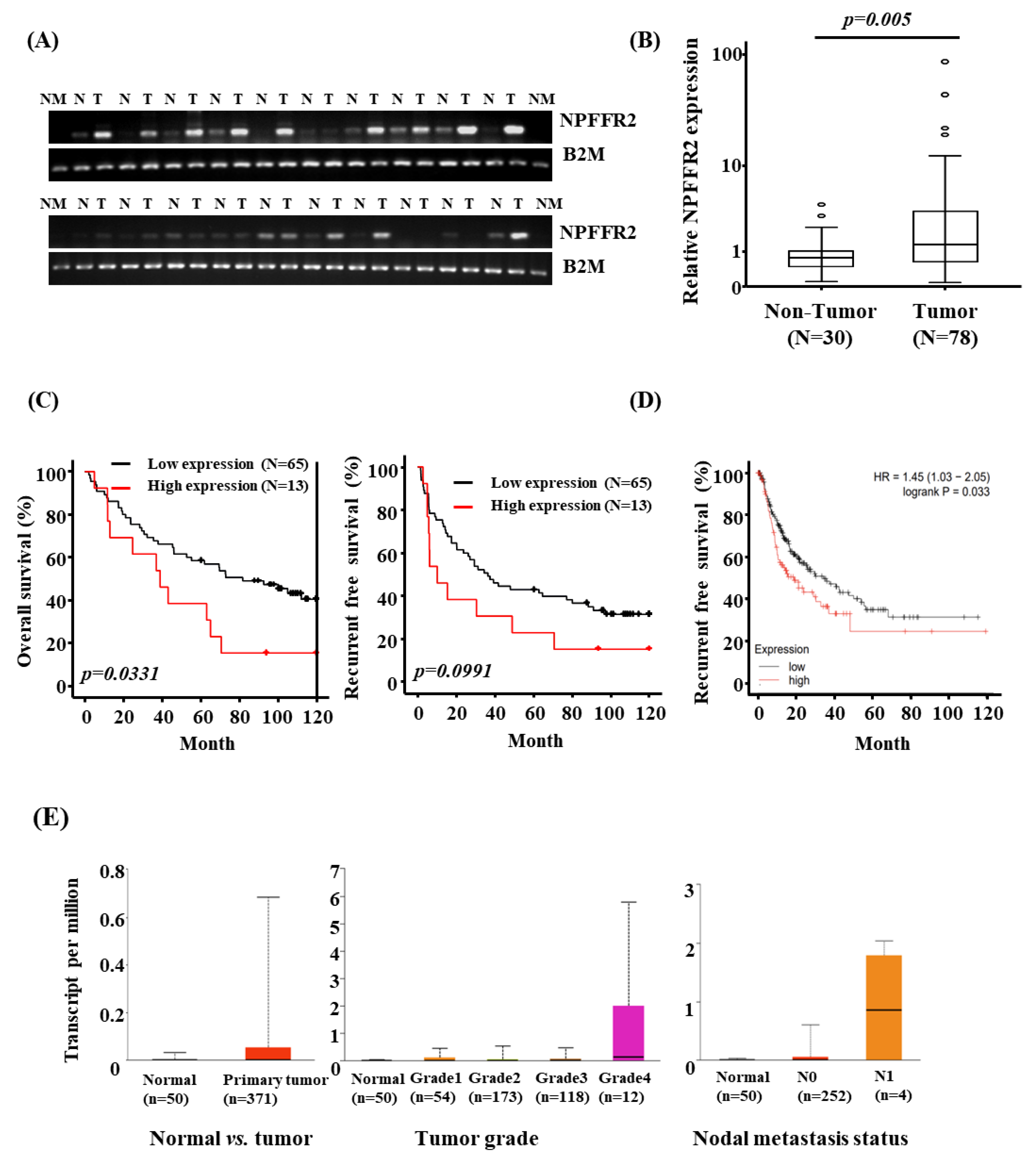
3.1. NPFFR2 Is Overexpressed in HCC Tissues and the Expression Is Related to Poor Prognosis
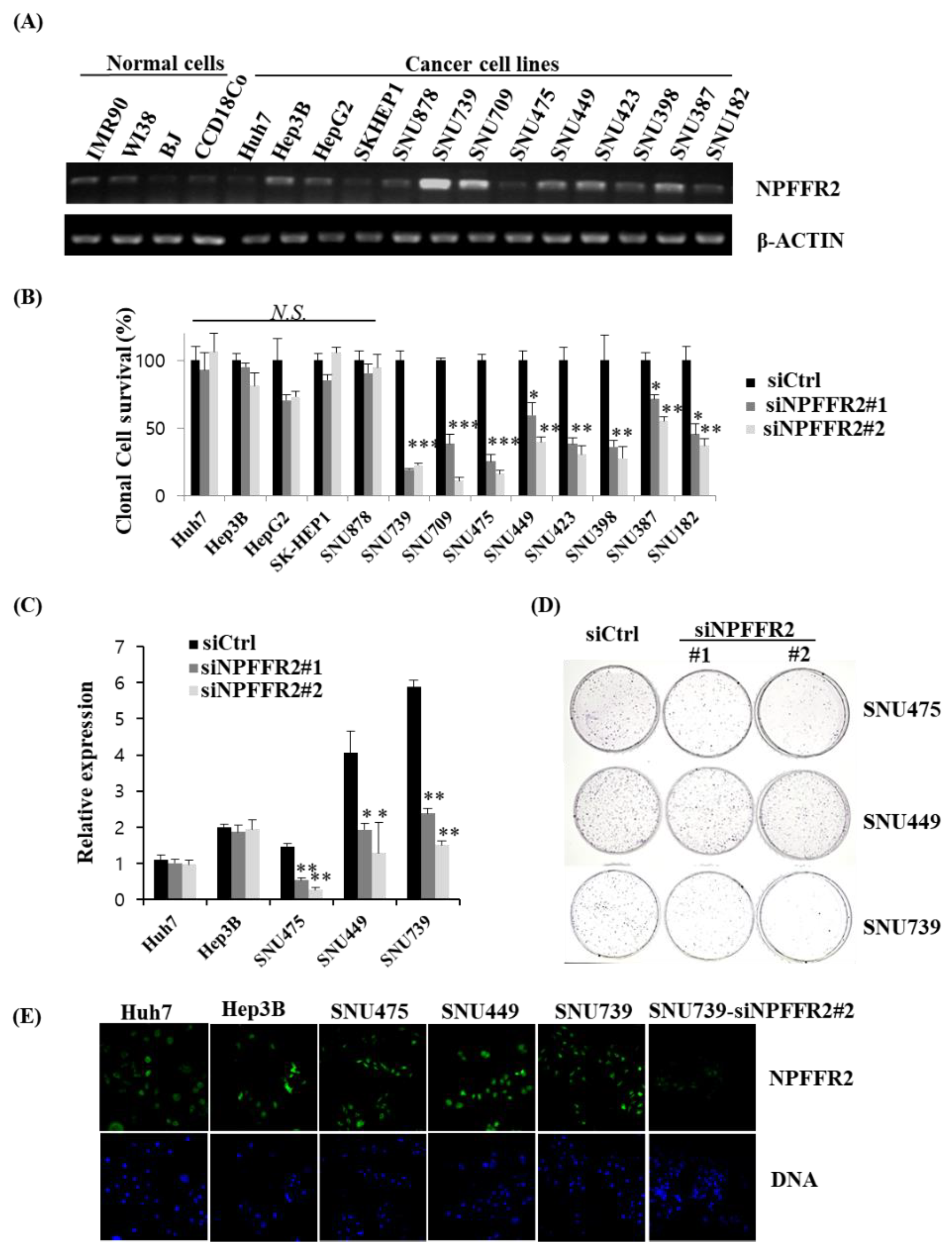
3.2. The Expression Levels of NPFFR2 Is Closely Associated with the Survival and Proliferation of HCC Cells
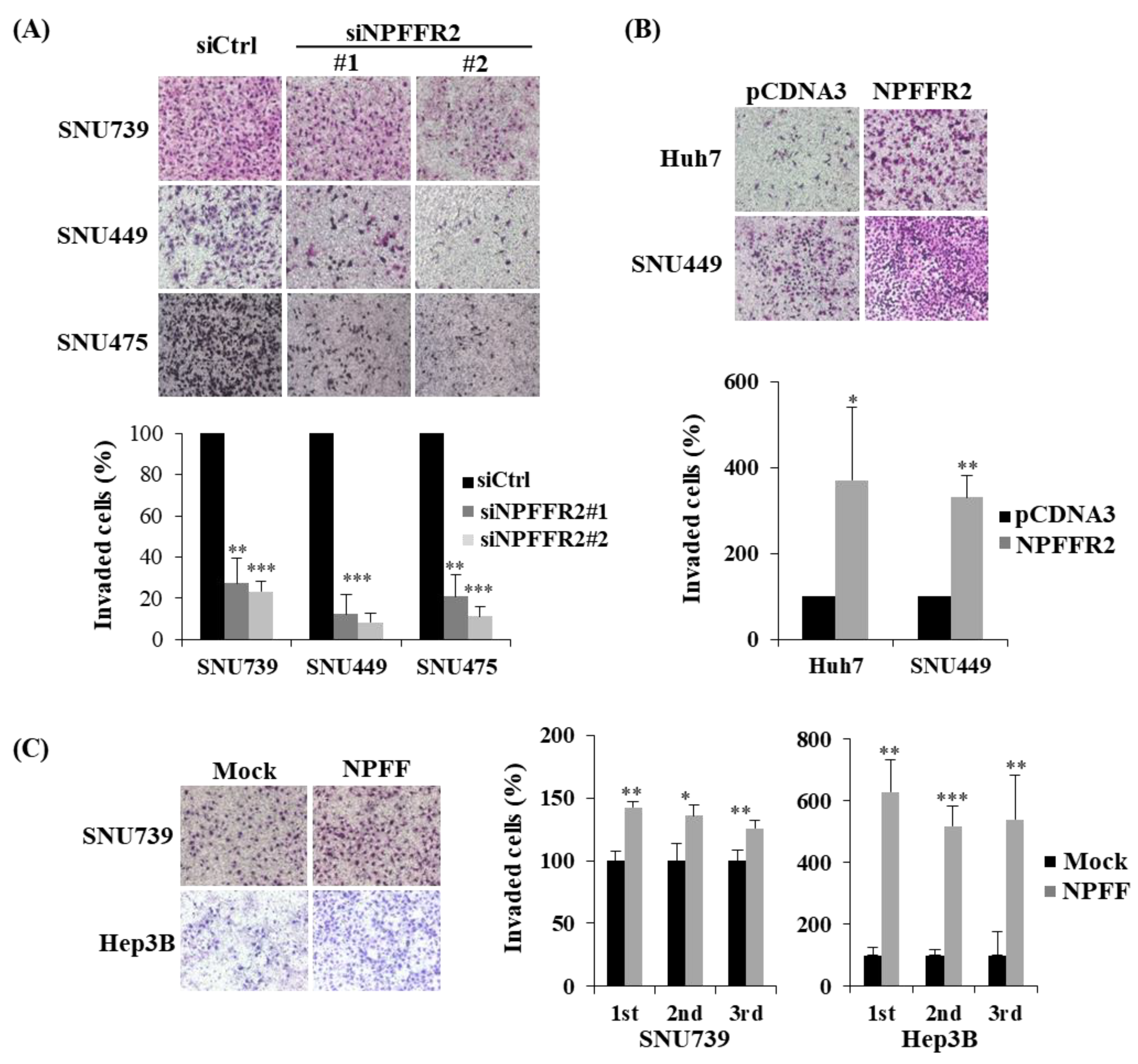
3.3. NPFFR2 Increases Invasiveness and Migration in HCC Cancer Cells
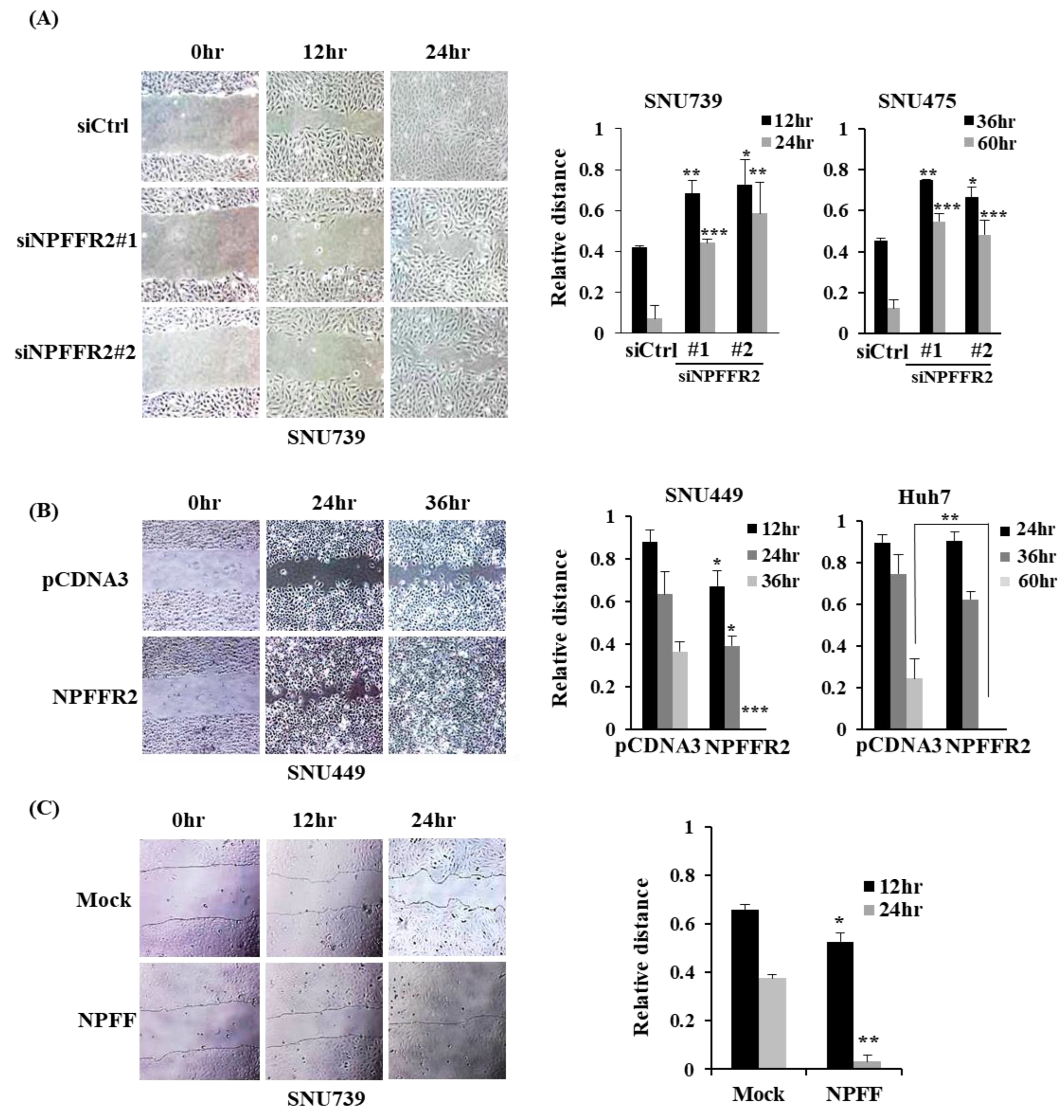
3.4. NPFFR2 Increases Migration in HCC Cancer Cells
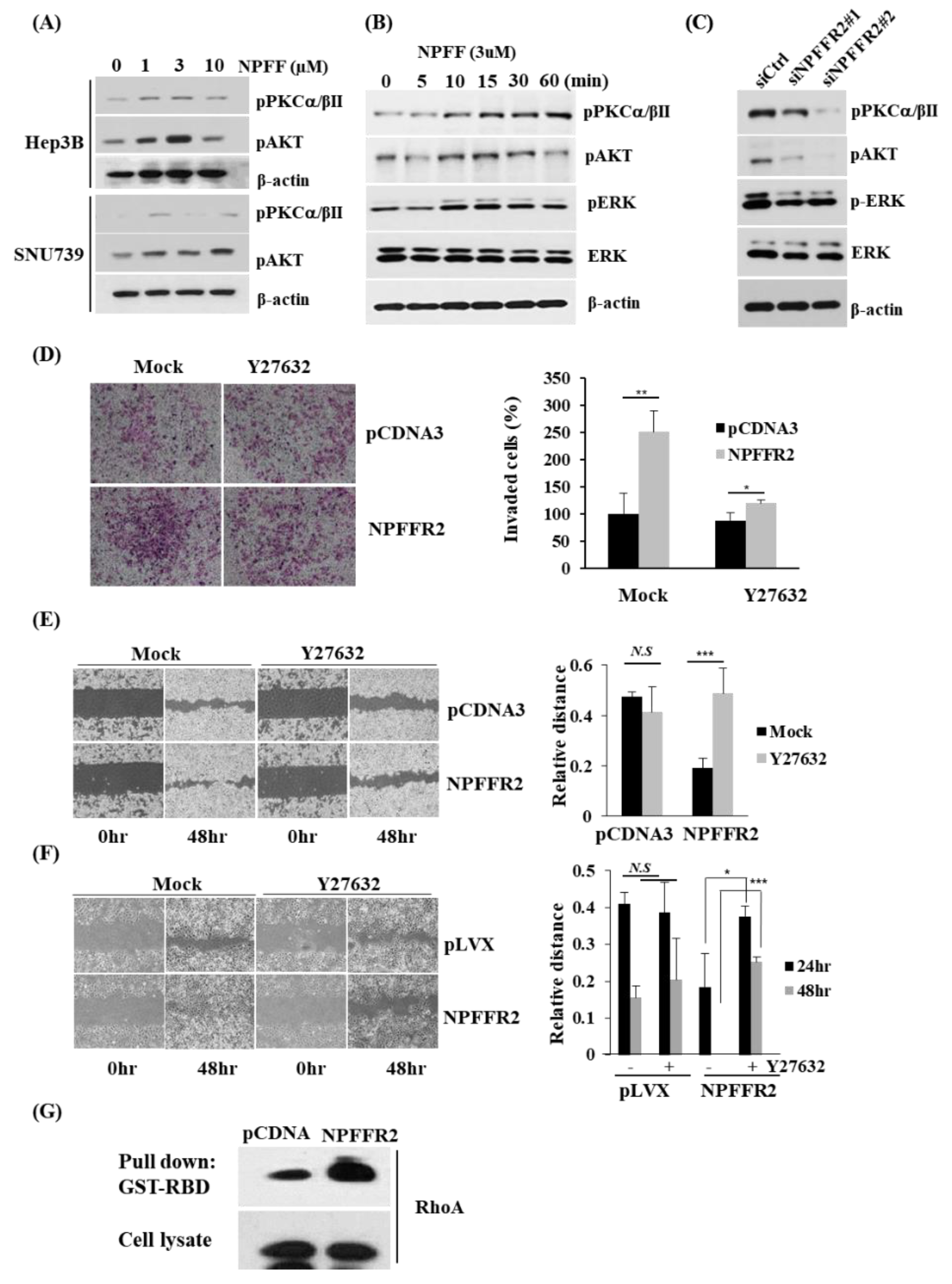
3.5. NPFFR2 Increases Invasiveness in Cancer Cells through RhoA Signaling
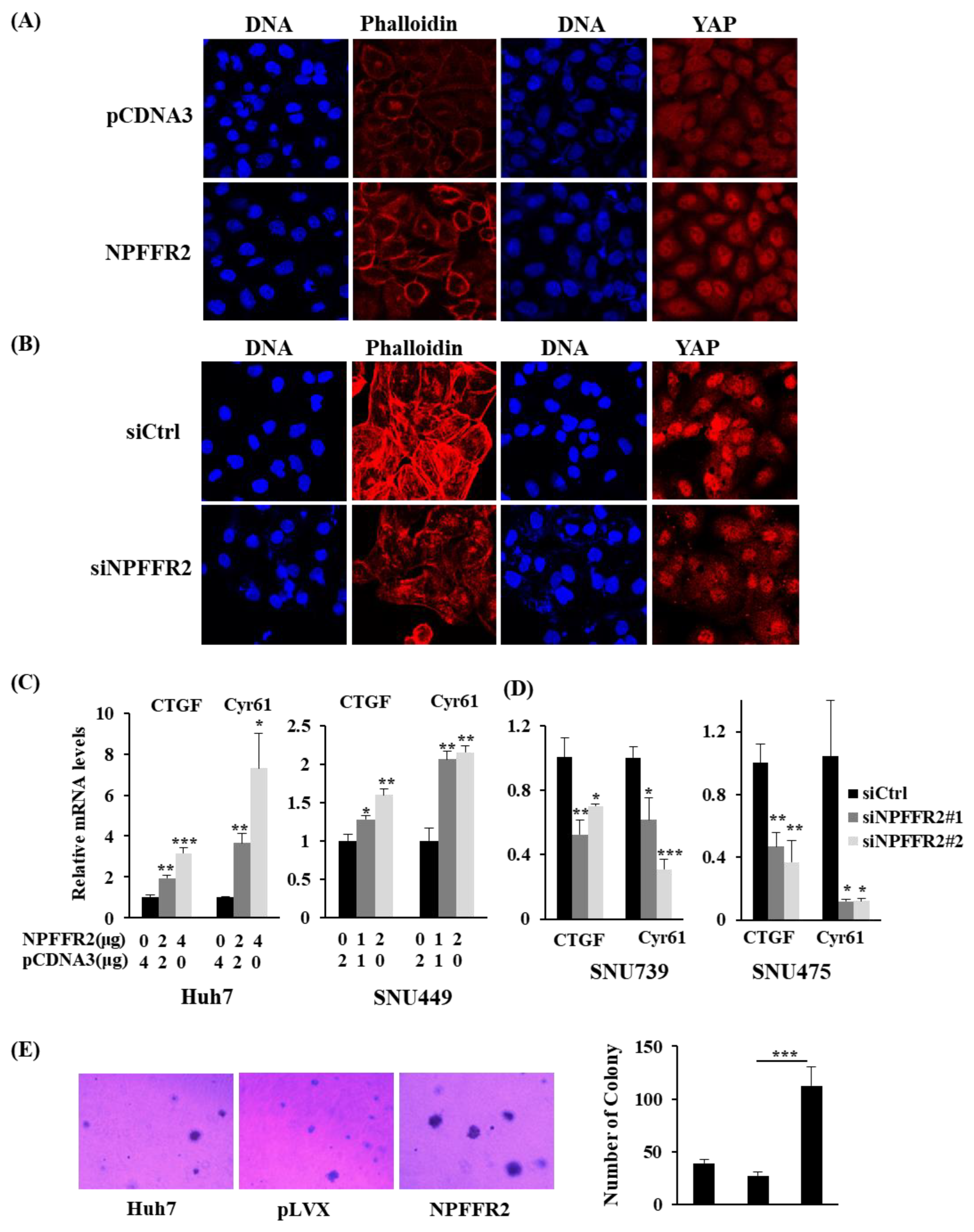
3.6. Regulation of RhoA by NPFFR2 Affects the Formation of F–Actin and the Activity of YAP
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rask-Andersen, M.; Masuram, S.; Schiöth, H.B. The Druggable Genome: Evaluation of Drug Targets in Clinical Trials Suggests Major Shifts in Molecular Class and Indication. Ann. Rev. Pharmacol. Toxicol. 2014, 54, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR Drug Discovery: New Agents, Targets and Indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef]
- Congreve, M.; de Graaf, C.; Swain, N.A.; Tate, C.G. Impact of GPCR Structures on Drug Discovery. Cell 2020, 181, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; Fitzhugh, W.; et al. Initial Sequencing and Analysis of the Human Genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.Y.T.; Fratta, W.; Majane, E.A.; Costa, E. Isolation, Sequencing, Synthesis, and Pharmacological Characterization of Two Brain Neuropeptides That Modulate the Action of Morphine. Proc. Natl. Acad. Sci. USA 1985, 82, 7757–7761. [Google Scholar] [CrossRef] [PubMed]
- Gouardères, C.; Jhamandas, K.; Sutak, M.; Zajac, J.M. Role of Opioid Receptors in the Spinal Antinociceptive Effects of Neuropeptide FF Analogues. Br. J. Pharmacol. 1996, 117, 493. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.T.; Liu, H.L.; Day, Y.J.; Chang, C.C.; Hsu, P.H.; Chen, J.C. Activation of NPFFR2 Leads to Hyperalgesia through the Spinal Inflammatory Mediator CGRP in Mice. Exp. Neurol. 2017, 291, 62–73. [Google Scholar] [CrossRef]
- Malin, D.H.; Lake, J.R.; Fowler, D.E.; Hammond, M.V.; Brown, S.L.; Leyva, J.E.; Prasco, P.E.; Dougherty, T.M. FMRF-NH2-like Mammalian Peptide Precipitates Opiate-Withdrawal Syndrome in the Rat. Peptides 1990, 11, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.T.; Liu, T.Y.; Yang, C.Y.; Yu, Y.L.; Chen, T.C.; Day, Y.J.; Chang, C.C.; Huang, G.J.; Chen, J.C. Chronic Activation of NPFFR2 Stimulates the Stress-Related Depressive Behaviors through HPA Axis Modulation. Psychoneuroendocrinology 2016, 71, 73–85. [Google Scholar] [CrossRef]
- Zhang, L.; Ip, C.K.; Lee, I.C.J.; Qi, Y.; Reed, F.; Karl, T.; Low, J.K.; Enriquez, R.F.; Lee, N.J.; Baldock, P.A.; et al. Diet-Induced Adaptive Thermogenesis Requires Neuropeptide FF Receptor-2 Signalling. Nat. Commun. 2018, 9, 4722. [Google Scholar] [CrossRef]
- Gutierrez, S.; Boada, M.D. Neuropeptide-Induced Modulation of Carcinogenesis in a Metastatic Breast Cancer Cell Line (MDA-MB-231LUC+). Cancer Cell Int. 2018, 18, 216. [Google Scholar] [CrossRef] [PubMed]
- Castro, T.A.; Cohen, M.C.; Rameshwar, P. The Expression of Neurokinin-1 and Preprotachykinin-1 in Breast Cancer Cells Depends on the Relative Degree of Invasive and Metastatic Potential. Clin. Exp. Metastasis 2006, 22, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Rao, G.; Patel, P.S.; Idler, S.P.; Maloof, P.; Gascon, P.; Potian, J.A.; Rameshwar, P. Facilitating Role of Preprotachykinin-I Gene in the Integration of Breast Cancer Cells within the Stromal Compartment of the Bone MarrowA Model of Early Cancer Progression. Cancer Res. 2004, 64, 2874–2881. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Singh, A.S.; Caplan, A.; Corcoran, K.E.; Fernandez, J.S.; Preziosi, M.; Rameshwar, P. Oncogenic and Metastatic Properties of Preprotachykinin-I and Neurokinin-1 Genes. Vascul. Pharmacol. 2006, 45, 235–242. [Google Scholar] [CrossRef]
- Moody, T.W.; Ramos-Alvarez, I.; Jensen, R.T. Neuropeptide G Protein-Coupled Receptors as Oncotargets. Front. Endocrinol. 2018, 9, 345. [Google Scholar] [CrossRef]
- Li, J.; Tian, Y.; Wu, A. Neuropeptide Y Receptors: A Promising Target for Cancer Imaging and Therapy. Regen. Biomater. 2015, 2, 215. [Google Scholar] [CrossRef]
- Sayiner, M.; Golabi, P.; Younossi, Z.M. Disease Burden of Hepatocellular Carcinoma: A Global Perspective. Dig. Dis. Sci. 2019, 64, 910–917. [Google Scholar] [CrossRef]
- Miller, K.D.; Nogueira, L.; Mariotto, A.B.; Rowland, J.H.; Yabroff, K.R.; Alfano, C.M.; Jemal, A.; Kramer, J.L.; Siegel, R.L. Cancer Treatment and Survivorship Statistics, 2019. CA Cancer J. Clin. 2019, 69, 363–385. [Google Scholar] [CrossRef]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular Carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- Prieto, J.; Melero, I.; Sangro, B. Immunological Landscape and Immunotherapy of Hepatocellular Carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 681–700. [Google Scholar] [CrossRef]
- Haider, M.; Elsayed, I.; Ahmed, I.S.; Fares, A.R. In Situ—Forming Microparticles for Controlled Release of Rivastigmine: In Vitro Optimization and In Vivo Evaluation. Pharmaceuticals 2021, 14, 66. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Yang, B.; Liao, Z.Y. Current Progress and Prospect of Immune Checkpoint Inhibitors in Hepatocellular Carcinoma (Review). Oncol. Lett. 2020, 20, 45. [Google Scholar] [CrossRef]
- Menyhárt, O.; Nagy, Á.; Gyórffy, B. Determining Consistent Prognostic Biomarkers of Overall Survival and Vascular Invasion in Hepatocellular Carcinoma. R. Soc. Open Sci. 2018, 5, 181006. [Google Scholar] [CrossRef] [PubMed]
- Park, E.R.; Kim, S.B.; Lee, J.S.; Kim, Y.H.; Lee, D.H.; Cho, E.H.; Park, S.H.; Han, C.J.; Kim, B.Y.; Choi, D.W.; et al. The Mitochondrial Hinge Protein, UQCRH, Is a Novel Prognostic Factor for Hepatocellular Carcinoma. Cancer Med. 2017, 6, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Rouméas, L.; Humbert, J.P.; Schneider, S.; Doebelin, C.; Bertin, I.; Schmitt, M.; Bourguignon, J.J.; Simonin, F.; Bihel, F. Effects of Systematic N-Terminus Deletions and Benzoylations of Endogenous RF-Amide Peptides on NPFF1R, NPFF2R, GPR10, GPR54 and GPR103. Peptides 2015, 71, 156–161. [Google Scholar] [CrossRef]
- Bonini, J.A.; Jones, K.A.; Adham, N.; Forray, C.; Artymyshyn, R.; Durkin, M.M.; Smith, K.E.; Tamm, J.A.; Boteju, L.W.; Lakhlani, P.P.; et al. Identification and Characterization of Two G Protein-Coupled Receptors for Neuropeptide FF. J. Biol. Chem. 2000, 275, 39324–39331. [Google Scholar] [CrossRef]
- Mollereau, C.; Mazarguil, H.; Zajac, J.M.; Roumy, M. Neuropeptide FF (NPFF) Analogs Functionally Antagonize Opioid Activities in NPFF2 Receptor-Transfected SH-SY5Y Neuroblastoma Cells. Mol. Pharmacol. 2005, 67, 965–975. [Google Scholar] [CrossRef] [PubMed]
- Seasholtz, T.M.; Majumdar, M.; Brown, J.H. Rho as a Mediator of G Protein-Coupled Receptor Signaling. Mol. Pharmacol. 1999, 55, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J.; Hall, A. The Small GTP-Binding Protein Rho Regulates the Assembly of Focal Adhesions and Actin Stress Fibers in Response to Growth Factors. Cell 1992, 70, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H.; et al. Regulation of the Hippo-YAP Pathway by G-Protein-Coupled Receptor Signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef]
- Dai, X.; She, P.; Chi, F.; Feng, Y.; Liu, H.; Jin, D.; Zhao, Y.; Guo, X.; Jiang, D.; Guan, K.L.; et al. Phosphorylation of Angiomotin by Lats1/2 Kinases Inhibits F-Actin Binding, Cell Migration, and Angiogenesis. J. Biol. Chem. 2013, 288, 34041–34051. [Google Scholar] [CrossRef]
- Overholtzer, M.; Zhang, J.; Smolen, G.A.; Muir, B.; Li, W.; Sgroi, D.C.; Deng, C.X.; Brugge, J.S.; Haber, D.A. Transforming Properties of YAP, a Candidate Oncogene on the Chromosome 11q22 Amplicon. Proc. Natl. Acad. Sci. USA 2006, 103, 12405–12410. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhang, J.; You, X.; Liu, Q.; Du, Y.; Gao, Y.; Shan, C.; Kong, G.; Wang, Y.; Yang, X.; et al. Hepatitis B Virus X Protein Modulates Oncogene Yes-Associated Protein by CREB to Promote Growth of Hepatoma Cells. Hepatology 2012, 56, 2051–2059. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.Z.; Yao, T.J.; Lee, N.P.Y.; Ng, I.O.L.; Chan, Y.T.; Zender, L.; Lowe, S.W.; Poon, R.T.P.; Luk, J.M. Yes-Associated Protein Is an Independent Prognostic Marker in Hepatocellular Carcinoma. Cancer 2009, 115, 4576–4585. [Google Scholar] [CrossRef]
- Nguyen, C.D.K.; Yi, C. YAP/TAZ Signaling and Resistance to Cancer Therapy. Trends Cancer 2019, 5, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Han, S.K.; Zylka, M.J.; Simon, M.I.; Anderson, D.J. A Diverse Family of GPCRs Expressed in Specific Subsets of Nociceptive Sensory Neurons. Cell 2001, 106, 619–632. [Google Scholar] [CrossRef]
- Elhabazi, K.; Humbert, J.P.; Bertin, I.; Schmitt, M.; Bihel, F.; Bourguignon, J.J.; Bucher, B.; Becker, J.A.J.; Sorg, T.; Meziane, H.; et al. Endogenous Mammalian RF-Amide Peptides, Including PrRP, Kisspeptin and 26RFa, Modulate Nociception and Morphine Analgesia via NPFF Receptors. Neuropharmacology 2013, 75, 164–171. [Google Scholar] [CrossRef]
- Lin, Y.T.; Huang, Y.L.; Tsai, S.C.; Chen, J.C. Ablation of NPFFR2 in Mice Reduces Response to Single Prolonged Stress Model. Cells 2020, 9, 2479. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, Y.; Jung, W.; Kim, M.-Y.; Shin, D.; Kim, G.H.; Kim, C.H.; Park, S.-H.; Cho, E.-H.; Choi, D.W.; Han, C.J.; et al. NPFFR2 Contributes to the Malignancy of Hepatocellular Carcinoma Development by Activating RhoA/YAP Signaling. Cancers 2022, 14, 5850. https://doi.org/10.3390/cancers14235850
Shin Y, Jung W, Kim M-Y, Shin D, Kim GH, Kim CH, Park S-H, Cho E-H, Choi DW, Han CJ, et al. NPFFR2 Contributes to the Malignancy of Hepatocellular Carcinoma Development by Activating RhoA/YAP Signaling. Cancers. 2022; 14(23):5850. https://doi.org/10.3390/cancers14235850
Chicago/Turabian StyleShin, Yuna, Wonhee Jung, Mi-Yeon Kim, Dongjo Shin, Geun Hee Kim, Chun Ho Kim, Sun-Hoo Park, Eung-Ho Cho, Dong Wook Choi, Chul Ju Han, and et al. 2022. "NPFFR2 Contributes to the Malignancy of Hepatocellular Carcinoma Development by Activating RhoA/YAP Signaling" Cancers 14, no. 23: 5850. https://doi.org/10.3390/cancers14235850
APA StyleShin, Y., Jung, W., Kim, M.-Y., Shin, D., Kim, G. H., Kim, C. H., Park, S.-H., Cho, E.-H., Choi, D. W., Han, C. J., Lee, K. H., Kim, S.-B., & Shin, H. J. (2022). NPFFR2 Contributes to the Malignancy of Hepatocellular Carcinoma Development by Activating RhoA/YAP Signaling. Cancers, 14(23), 5850. https://doi.org/10.3390/cancers14235850