
SGLT-2 Inhibitors in Cancer Treatment—Mechanisms of Action and Emerging New Perspectives

 , , , ,
, , , ,  , and
, and
Abstract
Simple Summary
Abstract
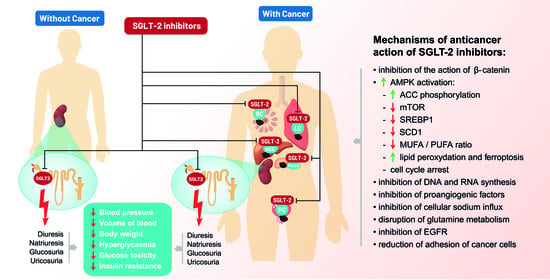
1. Introduction
2. SGLT-2 Inhibitors and Cancer in Clinical Studies
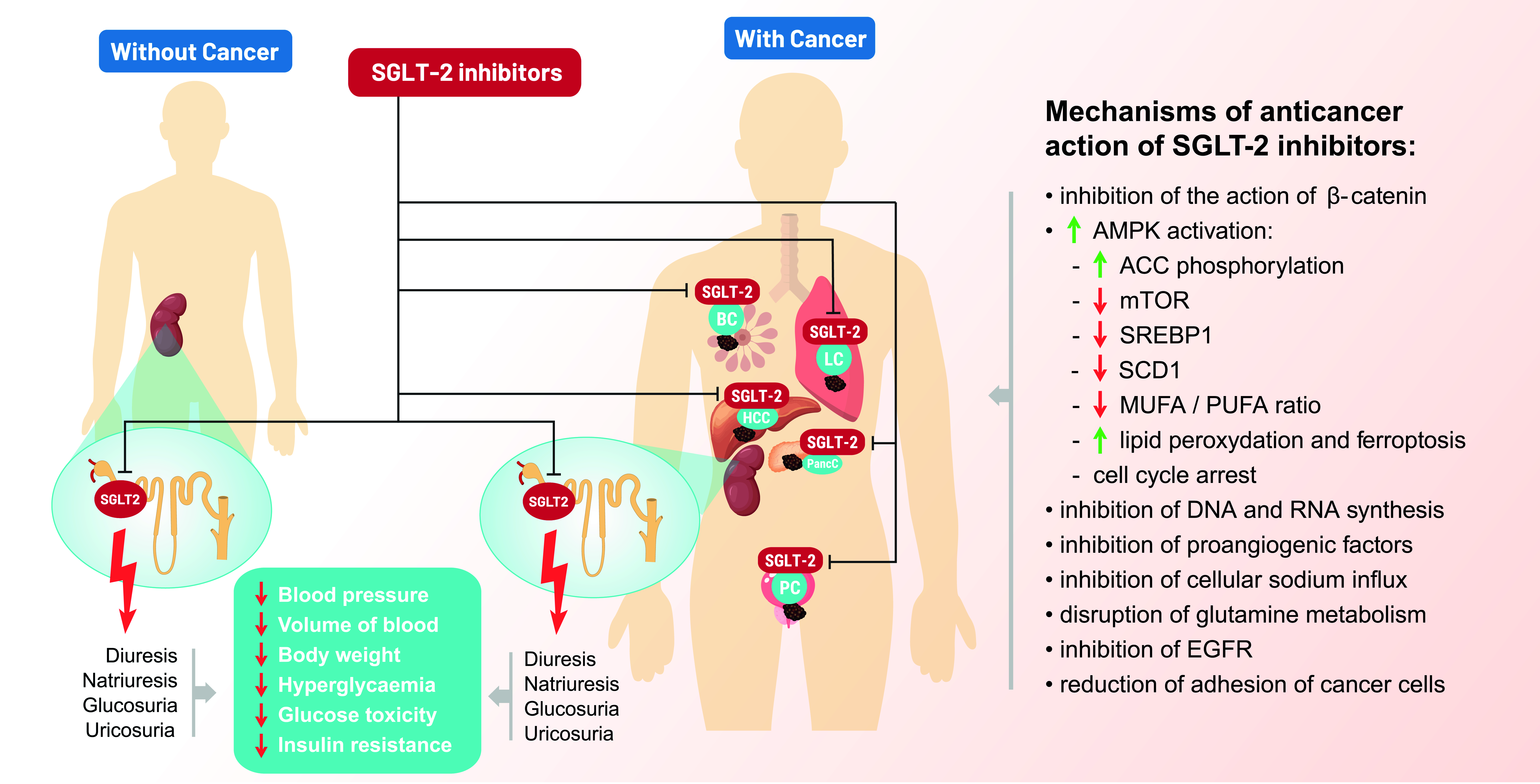
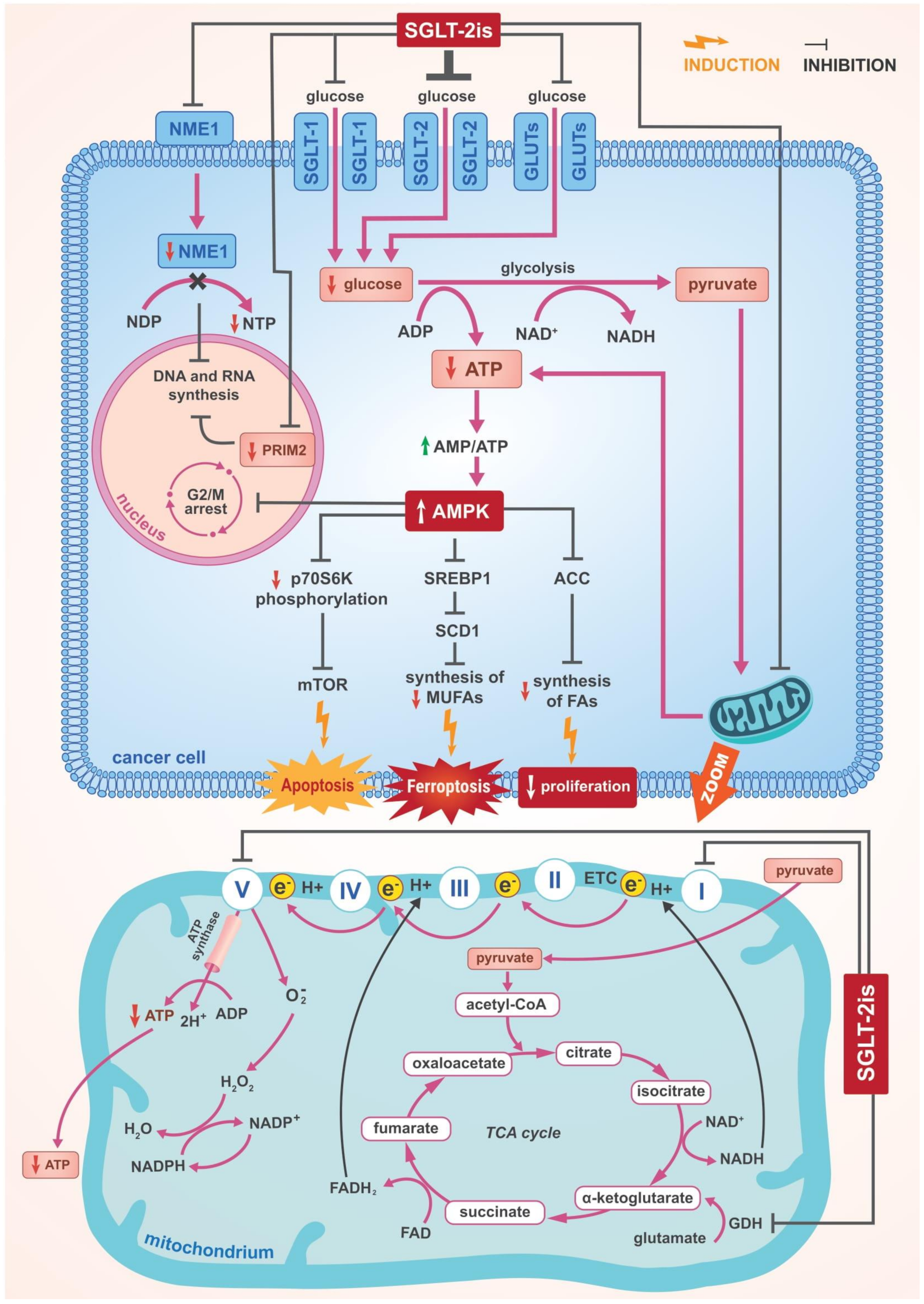
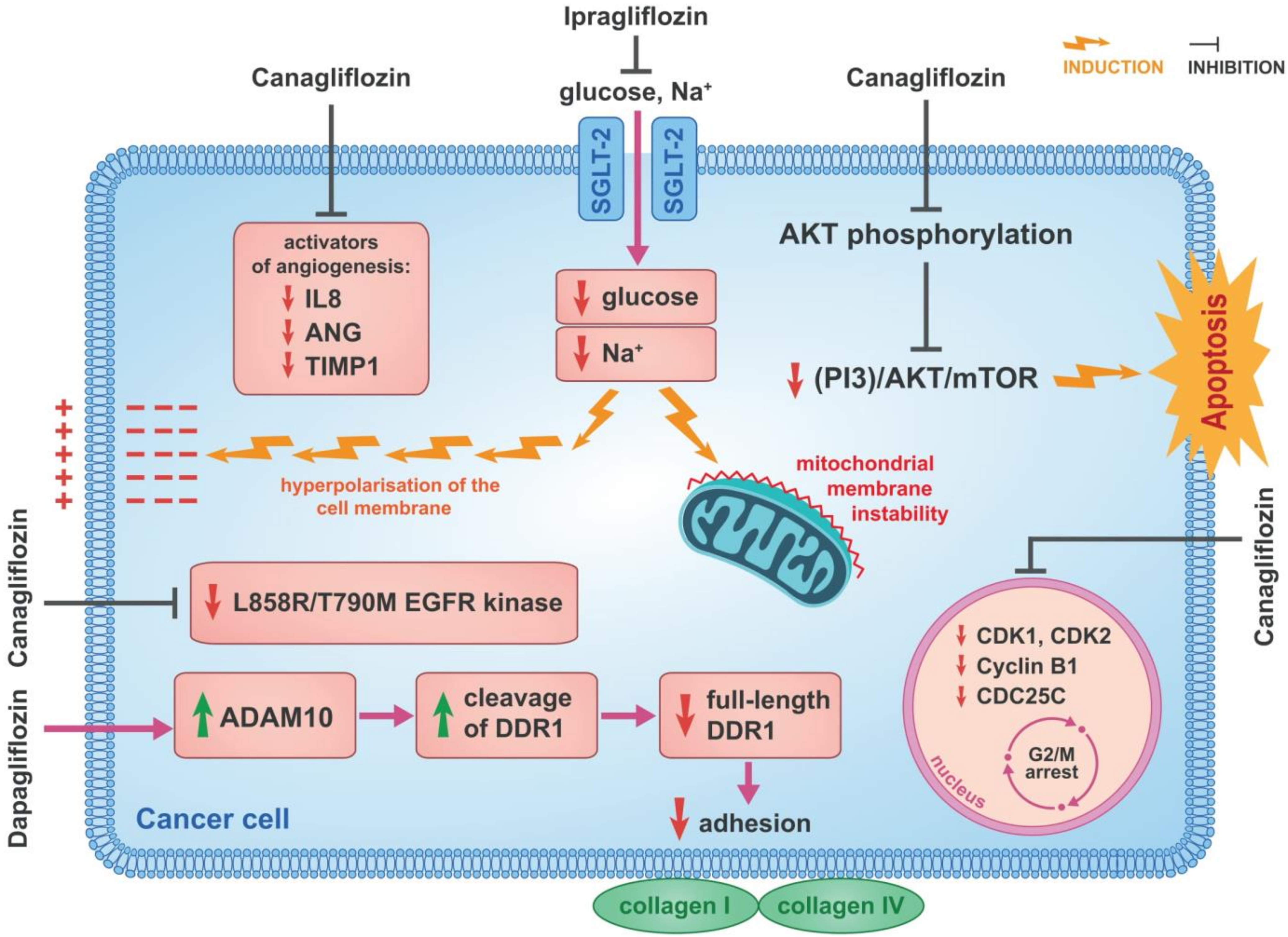
3. SGLT-2 Inhibitors—Anticancer Mechanisms of Action
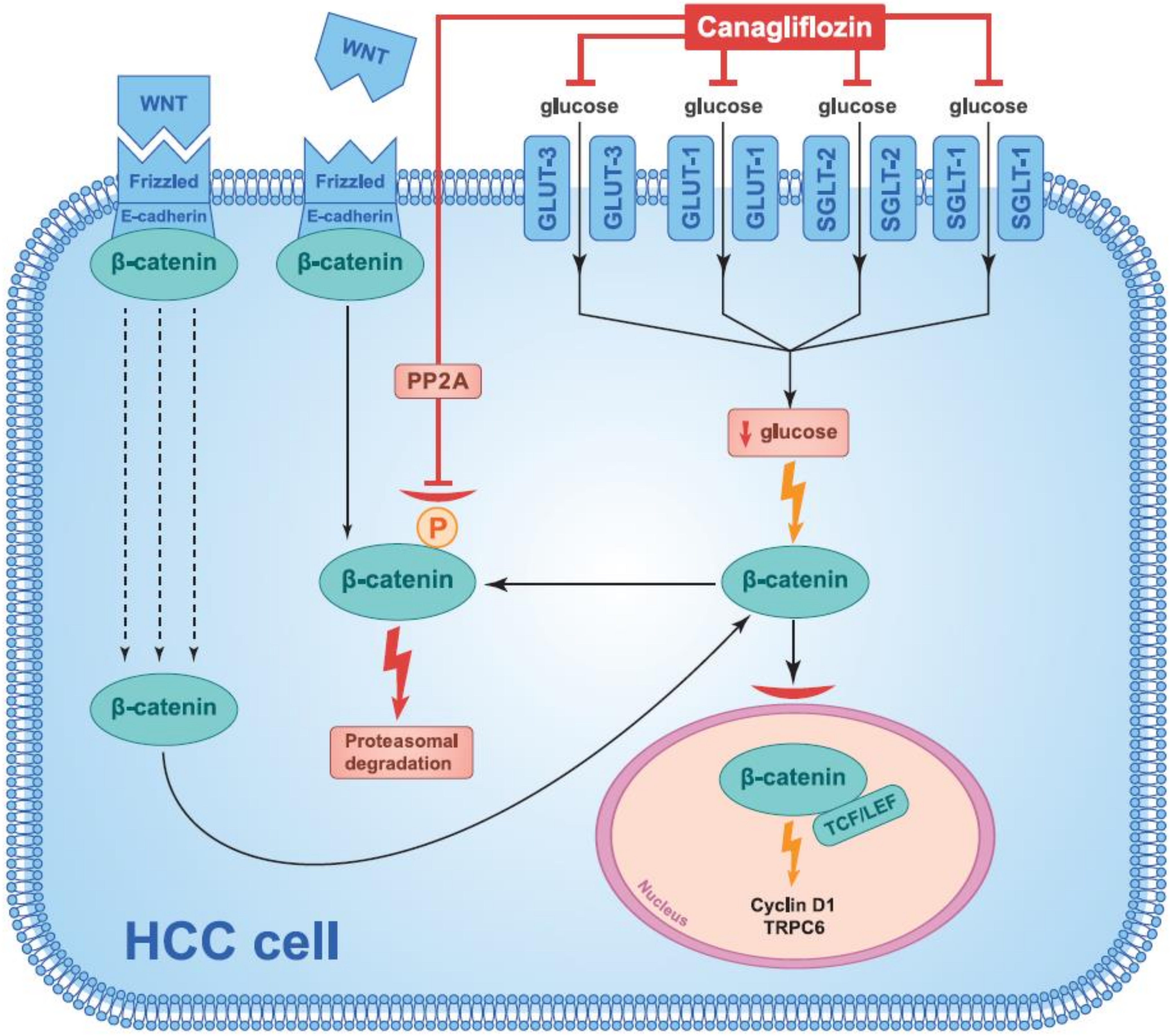
3.1. Inhibition of β-Catenin Action
3.2. The Role of AMPK Activation
3.2.1. The Inhibition of Complex I and α Subunit of ATP Synthase F1 in the Mitochondrial Electron Transport Chain
3.2.2. Suppression of SREBP1 and Further Consequences
3.2.3. Cell Cycle Arrest
3.2.4. Other Effects of AMPK Activation
3.3. Inhibition of DNA and RNA Synthesis
3.4. Cell Cycle and Proangiogenic Activities
3.5. Inhibition of Cellular Sodium Influx
3.6. Disruption of Glutamine Metabolism
3.7. Inhibition of EGFR
3.8. Reduction of Cancer Cell Adherence
4. SGLT-2 Inhibitors and Other Anticancer Drugs
5. Clinical Importance and Perspectives
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Komatsu, S.; Nomiyama, T.; Numata, T.; Kawanami, T.; Hamaguchi, Y.; Iwaya, C.; Horikawa, T.; Fujimura-Tanaka, Y.; Hamanoue, N.; Motonaga, R.; et al. SGLT2 inhibitor ipragliflozin attenuates breast cancer cell proliferation. Endocr. J. 2020, 67, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Scafoglio, C.; Hirayama, B.A.; Kepe, V.; Liu, J.; Ghezzi, C.; Satyamurthy, N.; Moatamed, N.A.; Huang, J.; Koepsell, H.; Barrio, J.R.; et al. Functional expression of sodium-glucose transporters in cancer. Proc. Natl. Acad. Sci. USA 2015, 112, E4111–E4119. [Google Scholar] [CrossRef] [PubMed]
- Shiba, K.; Tsuchiya, K.; Komiya, C.; Miyachi, Y.; Mori, K.; Shimazu, N.; Yamaguchi, S.; Ogasawara, N.; Katoh, M.; Itoh, M.; et al. Canagliflozin, an SGLT2 inhibitor, attenuates the development of hepatocellular carcinoma in a mouse model of human NASH. Sci. Rep. 2018, 8, 2362. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Okada, S.; Yamada, E.; Shimoda, Y.; Osaki, A.; Tagaya, Y.; Shibusawa, R.; Okada, J.; Yamada, M. Effect of dapagliflozin on colon cancer cell. Endocr. J. 2015, 62, 1133–1137. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Dai, Q.; Shi, W.; Zhai, S.; Song, Y.; Han, J. SGLT2 inhibitors and risk of cancer in type 2 diabetes: A systematic review and meta-analysis of randomised controlled trials. Diabetologia 2017, 60, 1862–1872. [Google Scholar] [CrossRef] [PubMed]
- Kaji, K.; Nishimura, N.; Seki, K.; Sato, S.; Saikawa, S.; Nakanishi, K.; Furukawa, M.; Kawaratani, H.; Kitade, M.; Moriya, K.; et al. Sodium glucose cotransporter 2 inhibitor canagliflozin attenuates liver cancer cell growth and angiogenic activity by inhibiting glucose uptake. Int. J. Cancer 2018, 142, 1712–1722. [Google Scholar] [CrossRef]
- Villani, L.A.; Smith, B.K.; Marcinko, K.; Ford, R.J.; Broadfield, L.A.; Green, A.E.; Houde, V.P.; Muti, P.; Tsakiridis, T.; Steinberg, G.R. The diabetes medication Canagliflozin reduces cancer cell proliferation by inhibiting mitochondrial complex-I supported respiration. Mol. Metab. 2016, 5, 1048–1056. [Google Scholar] [CrossRef]
- Obara, K.; Shirakami, Y.; Maruta, A.; Ideta, T.; Miyazaki, T.; Kochi, T.; Sakai, H.; Tanaka, T.; Seishima, M.; Shimizu, M. Preventive effects of the sodium glucose cotransporter 2 inhibitor tofogliflozin on diethylnitrosamine-induced liver tumorigenesis in obese and diabetic mice. Oncotarget 2017, 8, 58353–58363. [Google Scholar] [CrossRef]
- Kuang, H.; Liao, L.; Chen, H.; Kang, Q.; Shu, X.; Wang, Y. Therapeutic effect of sodium glucose co-transporter 2 inhibitor dapagliflozin on renal cell carcinoma. Med. Sci. Monit. 2017, 23, 3737–3745. [Google Scholar] [CrossRef]
- Scafoglio, C.R.; Villegas, B.; Abdelhady, G.; Bailey, S.T.; Liu, J.; Shirali, A.S.; Wallace, W.D.; Magyar, C.E.; Grogan, T.R.; Elashoff, D.; et al. Sodium-glucose transporter 2 is a diagnostic and therapeutic target for early-stage lung adenocarcinoma. Sci. Transl. Med. 2018, 10, eaat5933. [Google Scholar] [CrossRef]
- Nasiri, A.R.; Rodrigues, M.R.; Li, Z.; Leitner, B.P.; Perry, R.J. SGLT2 inhibition slows tumor growth in mice by reversing hyperinsulinemia. Cancer Metab. 2019, 7, 10. [Google Scholar] [CrossRef]
- Papadopoli, D.; Uchenunu, O.; Palia, R.; Chekkal, N.; Hulea, L.; Topisirovic, I.; Pollak, M.; St-Pierre, J. Perturbations of cancer cell metabolism by the antidiabetic drug canagliflozin. Neoplasia 2021, 23, 391–399. [Google Scholar] [CrossRef]
- Abdul-Ghani, M.; Stefano Del Prato, S.; Chilton, R.; DeFronzo, R.A. SGLT2 Inhibitors and Cardiovascular Risk: Lessons Learned From the EMPA-REG OUTCOME Study. Diabetes Care 2016, 39, 717–725. [Google Scholar] [CrossRef]
- Sanjay, K. Sodium-Glucose Cotransporter 2 (SGLT2) Inhibitors and cardiovascular Disease: A Systematic Review. Cardiol. Ther. 2016, 5, 161–168. [Google Scholar] [CrossRef]
- Tamargo, J. Sodium–glucose Cotransporter 2 Inhibitors in Heart Failure: Potential Mechanisms of Action, Adverse Effects and Future Developments. Eur. Cardiol. Rev. 2019, 14, 23–32. [Google Scholar] [CrossRef]
- Packer, M. Lessons learned from the DAPA-HF trial concerning the mechanisms of benefit of SGLT2 inhibitors on heart failure events in the context of other large-scale trials nearing completion. Cardiovasc. Diabetol. 2019, 18, 129. [Google Scholar] [CrossRef]
- Mahaffey, K.W.; Neal, B.; Perkovic, V.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Fabbrini, E.; Sun, T.; Li, Q.; et al. Canagliflozin for Primary and Secondary Prevention of Cardiovascular Events Results from the CANVAS Program (Canagliflozin Cardiovascular Assessment Study). Circulation 2018, 137, 323–334. [Google Scholar] [CrossRef]
- Rådholm, K.; Figtree, G.; Perkovic, V.; Solomon, S.D.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Barrett, T.D.; Shaw, W.; Desai, M.; et al. Canagliflozin and Heart Failure in Type 2 Diabetes Mellitus Results from the CANVAS Program. Circulation 2018, 138, 458–468. [Google Scholar] [CrossRef]
- Shah, S.R.; Najim, N.I.; Abbasi, Z.; Fatima, M.; Jangda, A.A.; Shahnawaz, W.; Shahid, M.; Shah, S.A. Canagliflozin and Cardiovascular disease—results of the CANVAS trial. J. Community Hosp. Intern. Med. Perspect. 2018, 8, 267–268. [Google Scholar] [CrossRef]
- Sezai, A.; Sekino, H.; Unosawa, S.; Taoka, M.; Osaka, S.; Tanaka, M. Canagliflozin for Japanese patients with chronic heart failure and type II. Cardiovasc. Diabetol. 2019, 18, 76. [Google Scholar] [CrossRef]
- Brown, E.; Heerspink, H.J.L.; Cuthbertson, D.J.; Wilding, J.P.H. SGLT2 inhibitors and GLP-1 receptor agonists: Established and emerging indications. Lancet 2021, 398, 262–276. [Google Scholar] [CrossRef] [PubMed]
- Cherney, D.; Dagogo-Jack, S.; McGuire, D.K.; Cosentino, F.; Pratley, R.; Shih, W.J.; Frederich, R.; Maldonado, M.; Liu, J.; Wang, S.; et al. Kidney outcomes using a sustained ≥40% decline in eGFR: A meta-analysis of SGLT2 inhibitor trials. Clin. Cardiol. 2021, 44, 1139–1143. [Google Scholar] [CrossRef] [PubMed]
- Heston, T.F.; Olson, A.H.; Randall, N.R. Canagliflozin lowers blood sugar, but does it also lower cardiovascular risk? Maybe not. Ann. Transl. Med. 2017, 5, 473. [Google Scholar] [CrossRef] [PubMed]
- Kondo, H.; Takahashi, N. Reduced hospitalization for heart failure using anti-diabetic drug dapagliflozin: Implications of DECLARE–TIMI 58 for the basic science community. Cardiovasc. Res. 2019, 115, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Butler, J.; Hamo, C.E.; Filippatos, G.; Pocock, S.J.; Bernstein, R.A.; Brueckmann, M.; Cheung, A.K.; George, J.T.; Green, J.B.; Januzzi, J.L.; et al. The potential role and rationale for treatmentof heart failure with sodium–glucose co-transporter 2 inhibitors. Eur. J. Heart Fail. 2017, 19, 1390–1400. [Google Scholar] [CrossRef]
- Maejima, Y. SGLT2 Inhibitors Play a Salutary Role in Heart Failure via Modulation of the Mitochondrial Function. Front. Cardiovasc. Med. 2019, 6, 186. [Google Scholar] [CrossRef]
- Dutka, M.; Bobiński, R.; Ulman-Włodarz, I.; Hajduga, M.; Bujok, J.; Pająk, C.; Ćwiertnia, M. Sodium glucose cotransporter 2 inhibitors: Mechanisms of action in heart failure. Heart Fail. Rev. 2021, 26, 603–622. [Google Scholar] [CrossRef]
- Nakano, D.; Kawaguchi, T.; Iwamoto, H.; Hayakawa, M.; Koga, H.; Torimura, T. Effects of canagliflozin on growth and metabolic reprograming in hepatocellular carcinoma cells: Multi-omnics analysis of metabolomics and absolute quantification proteomics (iMPAQT). PLoS ONE 2020, 15, e0232283. [Google Scholar] [CrossRef]
- Madunić, I.V.; Madunić, J.; Breljak, D.; Karaica, D.; Sabolić, I. Sodium-glucose cotransporters: New targets of cancer therapy? Arh. Hig. Rada Toksikol. 2018, 69, 278–285. [Google Scholar] [CrossRef]
- Vrhovac, I.; Breljak, D.; Sabolić, I. Glucose transporters in the mammalian blood cells. Period. Biol. 2014, 116, 131–138. [Google Scholar]
- Mueckler, M.; Thorens, B. The SLC2 (GLUT) family of membrane transporters. Mol. Asp. Med. 2013, 34, 121–138. [Google Scholar] [CrossRef]
- Wright, E. Glucose transport families SLC5 and SLC50. Mol. Asp. Med. 2013, 34, 183–196. [Google Scholar] [CrossRef]
- Wright, E.; Loo, D.; Hirayama, B. Biology of human sodium glucose transporters. Physiol. Rev. 2011, 91, 733–794. [Google Scholar] [CrossRef]
- Hediger, M.; Rhoads, D. Molecular physiology of sodiumglucose cotransporters. Physiol. Rev. 1994, 74, 993–1026. [Google Scholar] [CrossRef]
- Chao, E.C.; Henry, R.R. SGLT2 inhibition—A novel strategy for diabetes treatment. Nat. Rev. Drug Discov. 2010, 9, 551–559. [Google Scholar] [CrossRef]
- Bhartia, M.; Tahrani, A.A.; Barnett, A.H. SGLT-2 inhibitors in development for type 2 diabetes treatment. Rev. Diabet. Stud. 2011, 8, 348–354. [Google Scholar] [CrossRef][Green Version]
- Ferrannini, E.; Solini, A. SGLT2 inhibition in diabetes mellitus: Rationale and clinical prospects. Nat. Rev. Endocrinol. 2012, 8, 495–502. [Google Scholar] [CrossRef]
- Wright, E.M.; Turk, E. The sodium/glucose cotransport family SLC5. Pflug. Arch. 2004, 447, 510–518. [Google Scholar] [CrossRef]
- Perry, R.J.; Shulman, G.I. Sodium-glucose cotransporter-2 inhibitors: Understanding the mechanisms for therapeutic promise and persisting risks. J. Biol. Chem. 2020, 295, 14379–14390. [Google Scholar] [CrossRef]
- Sabolic, I.; Vrhovac, I.; Eror, D.B.; Gerasimova, M.; Rose, M.; Breljak, D.; Ljubojevic, M.; Brzica, H.; Sebastiani, A.; Thal, S.C.; et al. Expression of Na+-D-glucose cotransporter SGLT2 in rodents is kidneyspecific and exhibits sex and species differences. Am. J. Physiol.-Cell Physiol. 2012, 302, 1174–1188. [Google Scholar] [CrossRef] [PubMed]
- Vrhovac, I.; Balen Eror, D.; Klessen, D.; Burger, C.; Breljak, D.; Kraus, O.; Radović, N.; Jadrijević, S.; Aleksic, I.; Walles, T.; et al. Localizations of Na+-Dglucose cotransporters SGLT1 and SGLT2 in human kidney and of SGLT1 in human small intestine, liver, lung, and heart. Pflügers Arch. 2015, 467, 1881–1898. [Google Scholar] [CrossRef] [PubMed]
- Madunić, I.V.; Breljak, D.; Karaica, D.; Koepsell, H.; Sabolić, I. Expression profiling and immunolocalization of Na+-D-glucose-cotransporter 1 in mice employing knockout mice as specificity control indicate novel locations and differences between mice and rats. Pflügers Arch. 2017, 469, 1545–1565. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Williams, S.; Ho, S.; Loraine, H.; Hagan, D.; Whaley, J.M.; Feder, J.N. Quantitative PCR tissue expression profiling of the human SGLT2 gene and related family members. Diabetes Ther. 2010, 1, 57–92. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, Y. Expression of SGLT1 in human hearts and impairment of cardiac glucose uptake by phlorizin during ischemiareperfusion injury in mice. PLoS ONE 2015, 10, e0130605. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.M. Renal Na+-glucose cotransporters. Am. J. Physiol. Renal Physiol. 2001, 280, F10–F18. [Google Scholar] [CrossRef]
- Zhou, J.; Zhu, J.; Yu, S.J.; Ma, H.L.; Chen, J.; Ding, X.F.; Chen, G.; Liang, Y.; Zhang, Q. Sodium-glucose co-transporter-2 (SGLT-2) inhibition reduces glucose uptake to induce breast cancer cell growth arrest through AMPK/mTOR pathway. Biomed. Pharmacother. 2020, 132, 110821. [Google Scholar] [CrossRef]
- Ishikawa, N.; Oguri, T.; Isobe, T.; Fujitaka, K.; Kohno, N. SGLT gene expression in primary lung cancers and their metastatic lesions. Jpn. J. Cancer Res. 2001, 92, 874–879. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, X.; Liu, X.; Qi, P.; Wang, H.; Ma, Z.; Chai, Y. MicroRNA-296, a suppressor non-coding RNA, downregulates SGLT2 expression in lung cancer. Int. J. Oncol. 2019, 54, 199–208. [Google Scholar] [CrossRef]
- Billger, M.; Kirk, J.; Chang, J.; Bédard, A.; Attalla, B.; Haile, S.; Söderberg, M. A study in a rat initiation-promotion bladder tumour model demonstrated no promoter/progressor potential of dapagliflozin. Regul. Toxicol. Pharmacol. 2019, 103, 166–173. [Google Scholar] [CrossRef]
- Yamamoto, L.; Yamashita, S.; Nomiyama, T.; Kawanami, T.; Hamaguchi, Y.; Shigeoka, T.; Horikawa, T.; Tanaka, Y.; Yanase, T.; Kawanami, D.; et al. Sodium-glucose cotransporter 2 inhibitor canagliflozin attenuates lung cancer cell proliferation in vitro. Diabetol. Int. 2021, 12, 389–398. [Google Scholar] [CrossRef]
- Koepsell, H. The Na+ -D-glucose cotransporters SGLT1 and SGLT2 are targets for the treatment of diabetes and cancer. Pharmacol. Ther. 2017, 170, 148–165. [Google Scholar] [CrossRef]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner-La Rocca, H.P.; Choi, D.J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef]
- Yang, X.Q.; Xu, C.; Sun, Y.; Han, R.F. Diabetes mellitus increases the risk of bladder cancer: An updated meta-analysis. Asian Pac. J. Cancer Prev. 2013, 14, 2583–2589. [Google Scholar] [CrossRef][Green Version]
- Fang, H.; Yao, B.; Yan, Y.; Xu, H.; Liu, Y.; Tang, H.; Zhou, J.; Cao, L.; Wang, W.; Zhang, J.; et al. Diabetes mellitus increases the risk of bladder cancer: An updated meta-analysis of observational studies. Diabetes Technol. Ther. 2013, 15, 914–922. [Google Scholar] [CrossRef]
- Zhu, Z.; Wang, X.; Shen, Z.; Lu, Y.; Zhong, S.; Xu, C. Risk of bladder cancer in patients with diabetes mellitus: An updated meta-analysis of 36 observational studies. BMC Cancer 2013, 13, 310. [Google Scholar] [CrossRef]
- Tseng, C.H. Diabetes and risk of bladder cancer: A study using the National Health Insurance database in Taiwan. Diabetologia 2011, 54, 2009–2015. [Google Scholar] [CrossRef]
- Dąbrowski, M. Diabetes, Antidiabetic Medications and Cancer Risk in Type 2 Diabetes: Focus on SGLT-2 Inhibitors. Int. J. Mol. Sci. 2021, 22, 1680. [Google Scholar] [CrossRef]
- Shi, N.; Shi, Y.; Xu, J.; Si, Y.; Yang, T.; Zhang, M.; Ng, D.M.; Li, X.; Xie, F. SGLT-2i and Risk of Malignancy in Type 2 Diabetes: A Meta-Analysis of Randomized Controlled Trials. Front. Public Health 2021, 9, 668368. [Google Scholar] [CrossRef]
- Carstensen, B.; Jørgensen, M.E.; Friis, S. The epidemiology of diabetes and cancer. Curr. Diabetes Rep. 2014, 14, 535. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Carstensen, B.; Witte, D.; Bowker, S.L.; Lipscombe, L.; Renehan, A.G.; Diabetes and Cancer Research Consortium. Diabetes and cancer (1): Evaluating the temporal relationship between type 2 diabetes and cancer incidence. Diabetologia 2012, 55, 1607–1618. [Google Scholar] [CrossRef] [PubMed]
- Onitilo, A.A.; Engel, J.M.; Glurich, I.; Stankowski, R.V.; Williams, G.M.; Doi, S.A. Diabetes and cancer II: Role of diabetes medications and influence of shared risk factors. Cancer Causes Control 2012, 23, 991–1008. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.H. Pioglitazone and bladder cancer: A population-based study of Taiwanese. Diabetes Care 2012, 35, 278–280. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mamtani, R.; Haynes, K.; Bilker, W.B.; Vaughn, D.J.; Strom, B.L.; Glanz, K.; Lewis, J.D. Association Between Longer Therapy with Thiazolidinediones and Risk of Bladder Cancer: A Cohort Study. J. Natl. Cancer Inst. 2012, 104, 1411–1421. [Google Scholar] [CrossRef]
- Azoulay, L.; Yin, H.; Filion, K.B.; Assayag, J.; Majdan, A.; Pollak, M.N.; Suissa, S. The use of pioglitazone and the risk of bladder cancer in people with type 2 diabetes: Nested case-control study. BMJ 2012, 344, e3645. [Google Scholar] [CrossRef]
- Wei, L.; Macdonald, T.M.; Mackenzie, I.S. Pioglitazone and bladder cancer: A propensity score matched cohort study. Br. J. Clin. Pharmacol. 2013, 75, 254–259. [Google Scholar] [CrossRef]
- Tseng, C.H. A review on thiazolidinediones and bladder cancer in human studies. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2014, 32, 1–45. [Google Scholar] [CrossRef]
- Lewis, J.D.; Ferrara, A.; Peng, T.; Hedderson, M.; Bilker, W.B.; Quesenberry, C.P., Jr.; Vaughn, D.J.; Nessel, L.; Selby, J.; Strom, B.L. Risk of bladder cancer among diabetic patients treated with pioglitazone: Interim report of a longitudinal cohort study. Diabetes Care 2011, 34, 916–922. [Google Scholar] [CrossRef]
- Lin, H.W.; Tseng, C.H. A review on the relationship between SGLT2 inhibitors and cancer. Int. J. Endocrinol. 2014, 2014, 719578. [Google Scholar] [CrossRef]
- Suissa, M.; Yin, H.; Yu, O.H.Y.; Wong, S.M.; Azoulay, L. Sodium-Glucose Cotransporter 2 Inhibitors and the Short-term Risk of Breast Cancer Among Women with Type 2 Diabetes. Diabetes Care 2021, 44, e9–e11. [Google Scholar] [CrossRef]
- De Jonghe, S.; Proctor, J.; Vinken, P.; Feyen, B.; Wynant, I.; Marien, D.; Geys, H.; Mamidi, R.N.; Johnson, M.D. Carcinogenicity in rats of the SGLT2 inhibitor canagliflozin. Chem. Biol. Interact. 2014, 224, 1–12. [Google Scholar] [CrossRef]
- Taub, M.E.; Ludwig-Schwellinger, E.; Ishiguro, N.; Kishimoto, W.; Yu, H.; Wagner, K.; Tweedie, D. Sex-, Species-, and Tissue-Specific Metabolism of Empagliflozin in Male Mouse Kidney Forms an Unstable Hemiacetal Metabolite (M466/2) That Degrades to 4-Hydroxycrotonaldehyde, a Reactive and Cytotoxic Species. Chem. Res. Toxicol. 2015, 28, 103–115. [Google Scholar] [CrossRef]
- Prentice, D.E.; Meikle, A.W. A review of drug-induced Leydig cell hyperplasia and neoplasia in the rat and some comparisons with man. Hum. Exp. Toxicol. 1995, 14, 562–572. [Google Scholar] [CrossRef]
- Roe, F.J.C. Relevance for man of the effects of lactose, polyols and other carbohydrates on calcium metabolism seen in rats: A review. Hum. Toxicol. 1989, 8, 87–98. [Google Scholar] [CrossRef]
- Dicembrini, I.; Nreu, B.; Mannucci, E.; Monami, M. Sodium-glucose co-transporter-2 (SGLT-2) inhibitors and cancer: A meta-analysis of randomized controlled trials. Diabetes Obes. Metab. 2019, 21, 1871–1877. [Google Scholar] [CrossRef]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R.; et al. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef]
- Jones, D. Diabetes field cautiously upbeat despite possible setback for leading SGLT2 inhibitor. Nat. Rev. Drug Discov. 2011, 10, 645–646. [Google Scholar] [CrossRef]
- Pelletier, R.; Ng, K.; Alkabbani, W.; Labib, Y.; Mourad, N.; Gamble, J.M. The association of sodium-glucose cotransporter 2 inhibitors with cancer: An overview of quantitative systematic reviews. Endocrinol. Diabetes Metab. 2020, 23, e00145. [Google Scholar] [CrossRef]
- Ptaszynska, A.; Cohen, S.M.; Messing, E.M.; Reilly, T.P.; Johnsson, E.; Johnsson, K. Assessing Bladder Cancer Risk in Type 2 Diabetes Clinical Trials: The Dapagliflozin Drug Development Program as a ‘Case Study’. Diabetes Ther. 2015, 6, 357–375. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, D.L.; Chen, Z.Z.; Gou, B.F. Associations of body mass index with cancer incidence among populations, genders, and menopausal status: A systematic review and meta-analysis. Cancer Epidemiol. 2016, 42, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.W.; Zhao, L.G.; Yang, Y.; Ma, X.; Wang, Y.Y.; Xiang, Y.B. Obesity and risk of bladder cancer: A dose-response meta-analysis of 15 cohort studies. PLoS ONE 2015, 10, e0119313. [Google Scholar] [CrossRef] [PubMed]
- Renehan, A.G.; Tyson, M.; Egger, M.; Heller, R.F.; Zwahlen, M. Body-mass index and incidence of cancer: A systematic review and meta-analysis of prospective observational studies. Lancet 2008, 371, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Kinduryte Schorling, O.; Clark, D.; Zwiener, I.; Kaspers, S.; Lee, J.; Iliev, H. Pooled safety and tolerability analysis of empagliflozin in patients with type 2 diabetes mellitus. Adv. Ther. 2020, 37, 3463–3484. [Google Scholar] [CrossRef] [PubMed]
- Kohler, S.; Zeller, C.; Iliev, H.; Kaspers, S. Safety and Tolerability of Empagliflozin in Patients with Type 2 Diabetes: Pooled Analysis of Phase I-III Clinical Trials. Adv. Ther. 2017, 34, 1707–1726. [Google Scholar] [CrossRef]
- Benedetti, R.; Benincasa, G.; Glass, K.; Chianese, U.; Vietri, M.T.; Congi, R.; Altucci, L.; Napoli, C. Effects of novel SGLT2 inhibitors on cancer incidence in hyperglycemic patients: A meta-analysis of randomized clinical trials. Pharmacol. Res. 2022, 175, 106039. [Google Scholar] [CrossRef]
- Okada, J.; Yamada, E.; Saito, T.; Yokoo, H.; Osaki, A.; Shimoda, Y.; Ozawa, A.; Nakajima, Y.; Pessin, J.E.; Okada, S.; et al. Dapagliflozin Inhibits Cell Adhesion to Collagen I and IV and Increases Ectodomain Proteolytic Cleavage of DDR1 by Increasing ADAM10 Activity. Molecules 2020, 25, 495. [Google Scholar] [CrossRef]
- Hung, M.H.; Chen, Y.L.; Chen, L.J.; Chu, P.Y.; Hsieh, F.S.; Tsai, M.H.; Shih, C.T.; Chao, T.I.; Huang, C.Y.; Chen, K.F. Canagliflozin inhibits growth of hepatocellular carcinoma via blocking glucose-influx-induced β-catenin activation. Cell Death Dis. 2019, 10, 420. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell. Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef]
- Amann, T.; Maegdefrau, U.; Hartmann, A.; Agaimy, A.; Marienhagen, J.; Weiss, T.S.; Stoeltzing, O.; Warnecke, C.; Schölmerich, J.; Oefner, P.J.; et al. GLUT1 expression is increased in hepatocellular carcinoma and promotes tumorigenesis. Am. J. Pathol. 2009, 174, 1544–1552. [Google Scholar] [CrossRef]
- Younes, M.; Brown, R.W.; Mody, D.R.; Fernandez, L.; Laucirica, R. GLUT1 expression in human breast carcinoma: Correlation with known prognostic markers. Anticancer Res. 1995, 15, 2895–2898. [Google Scholar]
- Haber, R.S.; Weiser, K.R.; Pritsker, A.; Reder, I.; Burstein, D.E. GLUT1 glucose transporter expression in benign and malignant thyroid nodules. Thyroid 1997, 7, 363–367. [Google Scholar] [CrossRef]
- Haber, R.S.; Rathan, A.; Weiser, K.R.; Pritsker, A.; Itzkowitz, S.H.; Bodian, C.; Slater, G.; Weiss, A.; Burstein, D.E. GLUT1 glucose transporter expression in colorectal carcinoma: A marker for poor prognosis. Cancer 1998, 83, 34–40. [Google Scholar] [CrossRef]
- Tohma, T.; Okazumi, S.; Makino, H.; Cho, A.; Mochizuki, R.; Shuto, K.; Kudo, H.; Matsubara, K.; Gunji, H.; Matsubara, H.; et al. Overexpression of glucose transporter 1 in esophageal squamous cell carcinomas: A marker for poor prognosis. Dis. Esophagus 2005, 18, 185–189. [Google Scholar] [CrossRef]
- Thorens, B.; Sarkar, H.K.; Kaback, H.R.; Lodish, H.F. Cloning and functional expression in bacteria of a novel glucose transporter present in liver, intestine, kidney, and beta-pancreatic islet cells. Cell 1988, 55, 281–290. [Google Scholar] [CrossRef]
- Fukumoto, H.; Seino, S.; Imura, H.; Seino, Y.; Eddy, R.L.; Fukushima, Y.; Byers, M.G.; Shows, T.B.; Bell, G.I. Sequence, tissue distribution, and chromosomal localization of mRNA encoding a human glucose transporter-like protein. Proc. Natl. Acad. Sci. USA 1988, 85, 5434–5438. [Google Scholar] [CrossRef] [PubMed]
- Karim, S.; Adams, D.H.; Lalor, P.F. Hepatic expression and cellular distribution of the glucose transporter family. World J. Gastroenterol. 2012, 18, 6771–6781. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Jeon, H.M.; Ju, M.K.; Kim, C.H.; Yoon, G.; Han, S.I.; Park, H.G.; Kang, H.S. Wnt/snail signaling regulates cytochrome C oxidase and glucose metabolism. Cancer Res. 2012, 72, 3607–3617. [Google Scholar] [CrossRef] [PubMed]
- Pate, K.T.; Stringari, C.; Sprowl-Tanio, S.; Wang, K.; TeSlaa, T.; Hoverter, N.P.; McQuade, M.M.; Garner, C.; Digman, M.A.; Teitell, M.A.; et al. Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J. 2014, 33, 1454–1473. [Google Scholar] [CrossRef] [PubMed]
- Shibata, T.; Aburatani, H. Exploration of liver cancer genomes. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 340–349. [Google Scholar] [CrossRef]
- Reya, T.; Duncan, A.W.; Ailles, L.; Domen, J.; Scherer, D.C.; Willert, K.; Hintz, L.; Nusse, R.; Weissman, I.L. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature 2003, 423, 409–414. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2016, 36, 1461. [Google Scholar] [CrossRef]
- Osataphan, S.; Macchi, C.; Singhal, G.; Chimene-Weiss, J.; Sales, V.; Kozuka, C.; Dreyfuss, J.M.; Pan, H.; Tangcharoenpaisan, Y.; Morningstar, J.; et al. SGLT2 inhibition reprograms systemic metabolism via FGF21-dependent and -independent mechanisms. JCI Insight 2019, 4, e123130. [Google Scholar] [CrossRef]
- Yuan, H.; Han, Y.; Wang, X.; Li, N.; Liu, Q.; Yin, Y.; Wang, H.; Pan, L.; Li, L.; Song, K.; et al. SETD2 restricts prostate cancer metastasis by integrating EZH2 and AMPK signaling pathways. Cancer Cell 2020, 38, 350–365. [Google Scholar] [CrossRef]
- Leprivier, G.; Rotblat, B. How does mTOR sense glucose starvation? AMPK is the usual suspect. Cell Death Discov. 2020, 6, 27. [Google Scholar] [CrossRef]
- Steinberg, G.R.; Carling, D. AMP-activated protein kinase: The current landscape for drug development. Nat. Rev. Drug Discov. 2019, 18, 527–551. [Google Scholar] [CrossRef]
- Carling, D.; Mayer, F.V.; Sanders, M.J.; Gamblin, S.J. AMP-activated protein kinase: Nature’s energy sensor. Nat. Chem. Biol. 2011, 7, 512–518. [Google Scholar] [CrossRef]
- Hardie, D.G.; Carling, D. The AMP-activated protein kinase: Fuel gauge of the mammalian cell. Eur. J. Biochem. 1997, 246, 259–273. [Google Scholar] [CrossRef]
- Sanders, M.J.; Grondin, P.O.; Hegarty, B.D.; Snowden, M.A.; Carling, D. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem. J. 2007, 403, 139–148. [Google Scholar] [CrossRef]
- Xiao, B.; Sanders, M.J.; Underwood, E.; Heath, R.; Mayer, F.V.; Carmena, D.; Jing, C.; Walker, P.A.; Eccleston, J.F.; Haire, L.F.; et al. Structure of mammalian AMPK and its regulation by ADP. Nature 2011, 472, 230–233. [Google Scholar] [CrossRef]
- Ross, F.A.; Jensen, T.E.; Hardie, D.G. Differential regulation by AMP and ADP of AMPK complexes containing different gamma subunit isoforms. Biochem. J. 2016, 473, 189–199. [Google Scholar] [CrossRef]
- Lounis, M.A.; Bergeron, K.F.; Burhans, M.S.; Ntambi, J.M.; Mounier, C. Oleate activates SREBP-1 signaling activity in SCD1-deficient hepatocytes. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E710–E720. [Google Scholar] [CrossRef]
- Kahn, B.B.; Alquier, T.; Carling, D.; Hardie, D.G. AMP-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005, 1, 15–25. [Google Scholar] [CrossRef]
- Lally, J.S.V.; Ghoshal, S.; DePeralta, D.K.; Moaven, O.; Wei, L.; Masia, R.; Erstad, D.J.; Fujiwara, N.; Leong, V.; Houde, V.P.; et al. Inhibition of Acetyl-CoA Carboxylase by Phosphorylation or the Inhibitor ND-654 Suppresses Lipogenesis and Hepatocellular Carcinoma. Cell Metab. 2019, 29, 174–182. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, M.; Yao, X.; Fei, Y.; Lin, Z.; Li, Z.; Cai, K.; Zhao, Y.; Luo, Z. HCAR1/MCT1 regulates tumor ferroptosis through the lactate-mediated AMPK-SCD1 activity and its therapeutic implications. Cell Rep. 2020, 33, 108487. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology, and role in disease. Nat. Rev. Mol. Cell. Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Luis, G.; Godfroid, A.; Nishiumi, S.; Cimino, J.; Blacher, S.; Maquoi, E.; Wery, C.; Collignon, A.; Longuespée, R.; Montero-Ruiz, L.; et al. Tumor resistance to ferroptosis driven by Stearoyl-CoA Desaturase-1 (SCD1) in cancer cells and Fatty Acid Biding Protein-4 (FABP4) in tumor microenvironment promote tumor recurrence. Redox Biol. 2021, 43, 102006. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [PubMed]
- Magtanong, L.; Ko, P.J.; To, M.; Cao, J.Y.; Forcina, G.C.; Tarangelo, A.; Ward, C.C.; Cho, K.; Patti, G.J.; Nomura, D.K.; et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant Cell State. Cell Chem. Biol. 2019, 26, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Tesfay, L.; Paul, B.T.; Konstorum, A.; Deng, Z.; Cox, A.O.; Lee, J.; Furdui, C.M.; Hegde, P.; Torti, F.M.; Torti, S.V. Stearoyl-CoA desaturase 1 protects ovarian cancer cells from ferroptotic cell death. Cancer Res. 2019, 79, 5355–5366. [Google Scholar] [CrossRef]
- Fritz, V.; Benfodda, Z.; Rodier, G.; Henriquet, C.; Iborra, F.; Avancès, C.; Allory, Y.; de la Taille, A.; Culine, S.; Blancou, H.; et al. Abrogation of de novo lipogenesis by stearoyl-CoA desaturase 1 inhibition interferes with oncogenic signaling and blocks prostate cancer progression in mice. Mol. Cancer Ther. 2010, 9, 1740–1754. [Google Scholar] [CrossRef]
- Yan, H.; Li, Z.; Shen, Q.; Wang, Q.; Tian, J.; Jiang, Q.; Gao, L. Aberrant expression of cell cycle and material metabolism related genes contributes to hepatocellular carcinoma occurrence. Pathol. Res. Pract. 2017, 213, 316–321. [Google Scholar] [CrossRef]
- Liu, F.; Li, H.; Chang, H.; Wang, J.; Lu, J. Identification of hepatocellular carcinoma-associated hub genes and pathways by integrated microarray analysis. Tumori 2015, 101, 206–214. [Google Scholar] [CrossRef]
- Sanli, T.; Steinberg, G.R.; Singh, G.; Tsakiridis, T. AMP-activated protein kinase (AMPK) beyond metabolism: A novel genomic stress sensor participating in the DNA damage response pathway. Cancer Biol. Ther. 2014, 15, 156–169. [Google Scholar] [CrossRef]
- Lee, C.W.; Wong, L.L.; Tse, E.Y.; Liu, H.F.; Leong, V.Y.; Lee, J.M.; Hardie, D.G.; Ng, I.O.; Ching, Y.P. AMPK promotes p53 acetylation via phosphorylation and inactivation of SIRT1 in liver cancer cells. Cancer Res. 2012, 72, 4394–4404. [Google Scholar] [CrossRef]
- Kennedy, S.P.; O’Neill, M.; Cunningham, D.; Morris, P.G.; Toomey, S.; Blanco-Aparicio, C.; Martinez, S.; Pastor, J.; Eustace, A.J.; Hennessy, B.T. Preclinical evaluation of a novel triple-acting PIM/PI3K/mTOR inhibitor, IBL-302, in breast cancer. Oncogene 2020, 39, 3028–3040. [Google Scholar] [CrossRef]
- Xu, D.; Zhou, Y.; Xie, X.; He, L.; Ding, J.; Pang, S.; Shen, B.; Zhou, C. Inhibitory effects of canagliflozin on pancreatic cancer are mediated via the downregulation of glucose transporter-1 and lactate dehydrogenase A. Int. J. Oncol. 2020, 57, 1223–1233. [Google Scholar] [CrossRef]
- Xie, Z.; Wang, F.; Lin, L.; Duan, S.; Liu, X.; Li, X.; Li, T.; Xue, M.; Cheng, Y.; Ren, H.; et al. An SGLT2 inhibitor modulates SHH expression by activating AMPK to inhibit the migration and induce the apoptosis of cervical carcinoma cells. Cancer Lett. 2020, 495, 200–210. [Google Scholar] [CrossRef]
- Puts, G.S.; Leonard, M.K.; Pamidimukkala, N.V.; Snyder, D.E.; Kaetzel, D.M. Nuclear functions of NME proteins. Lab. Investig. 2018, 98, 211–218. [Google Scholar] [CrossRef]
- Hindupur, S.K.; Colombi, M.; Fuhs, S.R.; Matter, M.S.; Guri, Y.; Adam, K.; Cornu, M.; Piscuoglio, S.; Ng, C.; Betz, C.; et al. The protein histidine phosphatase LHPP is a tumour suppressor. Nature 2018, 555, 678–682. [Google Scholar] [CrossRef]
- Zerbe, L.K.; Kuchta, R.D. The p58 subunit of human DNA primase is important for primer initiation, elongation, and counting. Biochemistry 2002, 41, 4891–4900. [Google Scholar] [CrossRef]
- Chen, X.Y.; Li, D.F.; Han, J.C.; Wang, B.; Dong, Z.P.; Yu, L.N.; Pan, Z.H.; Qu, C.J.; Chen, Y.; Sun, S.; et al. Reprogramming induced by isoliquiritigenin diminishes melanoma cachexia through mTORC2-AKT-GSK3beta signaling. Oncotarget 2017, 8, 34565–34575. [Google Scholar] [CrossRef]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef]
- Agani, F.; Jiang, B.H. Oxygen-independent regulation of HIF-1: Novel involvement of PI3K/AKT/mTOR pathway in cancer. Curr. Cancer Drug Targets 2013, 13, 245–2451. [Google Scholar] [CrossRef]
- Polet, F.; Feron, O. Endothelial cell metabolism and tumour angiogenesis: Glucose and glutamine as essential fuels and lactate as the driving force. J. Intern. Med. 2013, 273, 156–165. [Google Scholar] [CrossRef]
- Cruys, B.; Wong, B.W.; Kuchnio, A.; Verdegem, D.; Cantelmo, A.R.; Conradi, L.C.; Vandekeere, S.; Bouché, A.; Cornelissen, I.; Vinckier, S.; et al. Glycolytic regulation of cell rearrangement in angiogenesis. Nat. Commun. 2016, 7, 12240. [Google Scholar] [CrossRef] [PubMed]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquière, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Benjamin, L.E. Tumorigenesis and the angiogenic switch. Nat. Rev. Cancer 2003, 3, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Moon, E.J.; Lee, S.W.; Kim, M.S.; Kim, K.W.; Kim, Y.J. Angiogenic activity of pyruvic acid in in vivo and in vitro angiogenesis models. Cancer Res. 2001, 61, 3290–3293. [Google Scholar] [PubMed]
- Jung, S.Y.; Song, H.S.; Park, S.Y.; Chung, S.H.; Kim, Y.J. Pyruvate promotes tumor angiogenesis through HIF-1-dependent PAI-1 expression. Int. J. Oncol. 2011, 38, 571–576. [Google Scholar] [CrossRef][Green Version]
- Kihira, Y.; Yamano, N.; Izawa-Ishizawa, Y.; Ishizawa, K.; Ikeda, Y.; Tsuchiya, K.; Tamaki, T.; Tomita, S. Basic fibroblast growth factor regulates glucose metabolism through glucose transporter 1 induced by hypoxia-inducible factor-1alpha in adipocytes. Int. J. Biochem. Cell Biol. 2011, 43, 1602–1611. [Google Scholar] [CrossRef]
- Torimura, T.; Ueno, T.; Kin, M.; Harada, R.; Taniguchi, E.; Nakamura, T.; Sakata, R.; Hashimoto, O.; Sakamoto, M.; Kumashiro, R.; et al. Overexpression of angiopoietin-1 and angiopoietin-2 in hepatocellular carcinoma. J. Hepatol. 2004, 40, 799–807. [Google Scholar] [CrossRef]
- Diaz-Sanchez, A.; Matilla, A.; Nuñez, O.; Lorente, R.; Fernandez, A.; Rincón, D.; Campos, R.; Bañares, R.; Clemente, G. Serum angiopoietin-2 level as a predictor of tumor invasiveness in patients with hepatocellular carcinoma. Scand. J. Gastroenterol. 2013, 48, 334–343. [Google Scholar] [CrossRef]
- Faillaci, F.; Marzi, L.; Critelli, R.; Milosa, F.; Schepis, F.; Turola, E.; Andreani, S.; Vandelli, G.; Bernabucci, V.; Lei, B.; et al. Liver Angiopoietin-2 Is a Key Predictor of De Novo or Recurrent Hepatocellular Cancer After Hepatitis C Virus Direct-Acting Antivirals. Hepatology 2018, 68, 1010–1024. [Google Scholar] [CrossRef]
- Qin, G.; Luo, M.; Chen, J.; Dang, Y.; Chen, G.; Li, L.; Zeng, J.; Lu, Y.; Yang, J. Reciprocal activation between MMP-8 and TGF-β1 stimulates EMT and malignant progression of hepatocellular carcinoma. Cancer Lett. 2016, 374, 85–95. [Google Scholar] [CrossRef]
- Lempinen, M.; Lyytinen, I.; Nordin, A.; Tervahartiala, T.; Mäkisalo, H.; Sorsa, T.; Isoniemi, H. Prognostic value of serum MMP-8, -9 and TIMP-1 in patients with hepatocellular carcinoma. Ann. Med. 2013, 45, 482–487. [Google Scholar] [CrossRef]
- Wei, T.; Zhang, L.N.; Lv, Y.; Ma, X.Y.; Zhi, L.; Liu, C.; Ma, F.; Zhang, X.F. Overexpression of platelet-derived growth factor receptor alpha promotes tumor progression and indicates poor prognosis in hepatocellular carcinoma. Oncotarget 2014, 5, 10307–10317. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Nakano, D.; Okamura, S.; Shimose, S.; Hayakawa, M.; Niizeki, T.; Koga, H.; Torimura, T. Spontaneous regression of hepatocellular carcinoma with reduction in angiogenesis-related cytokines after treatment with sodium-glucose cotransporter 2 inhibitor in a cirrhotic patient with diabetes mellitus. Hepatol. Res. 2019, 49, 479–486. [Google Scholar] [CrossRef]
- Mao, W.; Zhang, J.; Komer, H.; Jiang, Y.; Ying, S. The emerging role of voltage-gated sodium channels in tumor biology. Front. Oncol. 2019, 9, 124. [Google Scholar] [CrossRef]
- Liu, G.; Zhu, J.; Yu, M.; Cai, C.; Zhou, Y.; Yu, M.; Fu, Z.; Gong, Y.; Yang, B.; Li, Y.; et al. Glutamate dehydrogenase is a novel prognostic marker and predicts metastases in colorectal cancer patients. J. Transl. Med. 2015, 13, 144. [Google Scholar] [CrossRef]
- Di Conza, G.; Tsai, C.H.; Ho, P.C. Fifty shades of alpha-Ketoglutarate on cellular programming. Mol. Cell 2019, 76, 1–3. [Google Scholar] [CrossRef]
- Yang, C.; Ko, B.; Hensley, C.T.; Jiang, L.; Wasti, A.T.; Kim, J.; Sudderth, J.; Calvaruso, M.A.; Lumata, L.; Mitsche, M.; et al. Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol. Cell 2014, 56, 414–424. [Google Scholar] [CrossRef]
- Li, H.; Tong, C.W.; Leung, Y.; Wong, M.H.; To, K.K.; Leung, K.S. Identification of Clinically Approved Drugs Indacaterol and Canagliflozin for Repurposing to Treat Epidermal Growth Factor Tyrosine Kinase Inhibitor-Resistant Lung Cancer. Front. Oncol. 2017, 7, 288. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR Regulate Autophagy through Direct Phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Shibutani, S.T.; Saitoh, T.; Nowag, H.; Münz, C.; Yoshimori, T. Autophagy and Autophagy-Related Proteins in the Immune System. Nat. Immunol. 2015, 16, 1014–1024. [Google Scholar] [CrossRef]
- Sciarretta, S.; Maejima, Y.; Zablocki, D.; Sadoshima, J. The Role of Autophagy in the Heart. Annu. Rev. Physiol. 2018, 80, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Hutson, T.E.; Tomczak, P.; Michaelson, M.D.; Bukowski, R.M.; Rixe, O.; Oudard, S.; Negrier, S.; Szczylik, C.; Kim, S.T.; et al. Sunitinib versus Interferon Alfa in Metastatic Renal-Cell Carcinoma. N. Engl. J. Med. 2007, 356, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, P.; Kang, Y.K.; Rutkowski, P.; Schuette, J.; Rosen, L.S.; Seddon, B.; Yalcin, S.; Gelderblom, H.; Williams, C.C., Jr.; Fumagalli, E.; et al. Clinical Outcomes of Patients with Advanced Gastrointestinal Stromal Tumors: Safety and Efficacy in a Worldwide Treatment-Use Trial of Sunitinib. Cancer 2015, 121, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, G.; Autorino, R.; Bruni, G.; Cartenì, G.; Ricevuto, E.; Tudini, M.; Ficorella, C.; Romano, C.; Aieta, M.; Giordano, A.; et al. Cardiovascular Toxicity Following Sunitinib Therapy in Metastatic Renal Cell Carcinoma: A Multicenter Analysis. Ann. Oncol. 2009, 20, 1535–1542. [Google Scholar] [CrossRef] [PubMed]
- Narayan, V.; Keefe, S.; Haas, N.; Wang, L.; Puzanov, I.; Putt, M.; Catino, A.; Fang, J.; Agarwal, N.; Hyman, D.; et al. Prospective Evaluation of Sunitinib-Induced Cardiotoxicity in Patients with Metastatic Renal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 3601–3609. [Google Scholar] [CrossRef]
- Ren, C.; Sun, K.; Zhang, Y.; Hu, Y.; Hu, B.; Zhao, J.; He, Z.; Ding, R.; Wang, W.; Liang, C. Sodium-Glucose CoTransporter-2 Inhibitor Empagliflozin Ameliorates Sunitinib-Induced Cardiac Dysfunction via Regulation of AMPK-mTOR Signaling Pathway-Mediated Autophagy. Front. Pharmacol. 2021, 12, 664181. [Google Scholar] [CrossRef]
- Kerkela, R.; Woulfe, K.C.; Durand, J.B.; Vagnozzi, R.; Kramer, D.; Chu, T.F.; Beahm, C.; Chen, M.H.; Force, T. Sunitinib-induced Cardiotoxicity Is Mediated by Off-Target Inhibition of AMP-Activated Protein Kinase. Clin. Transl Sci. 2009, 2, 15–25. [Google Scholar] [CrossRef]
- Laderoute, K.; Calaoagan, J.M.; Madrid, P.B.; Klon, A.E.; Ehrlich, P.J. SU11248 (Sunitinib) Directly Inhibits the Activity of Mammalian 5′-AMP-Activated Protein Kinase (AMPK). Cancer Biol. Ther. 2010, 10, 68–76. [Google Scholar] [CrossRef]
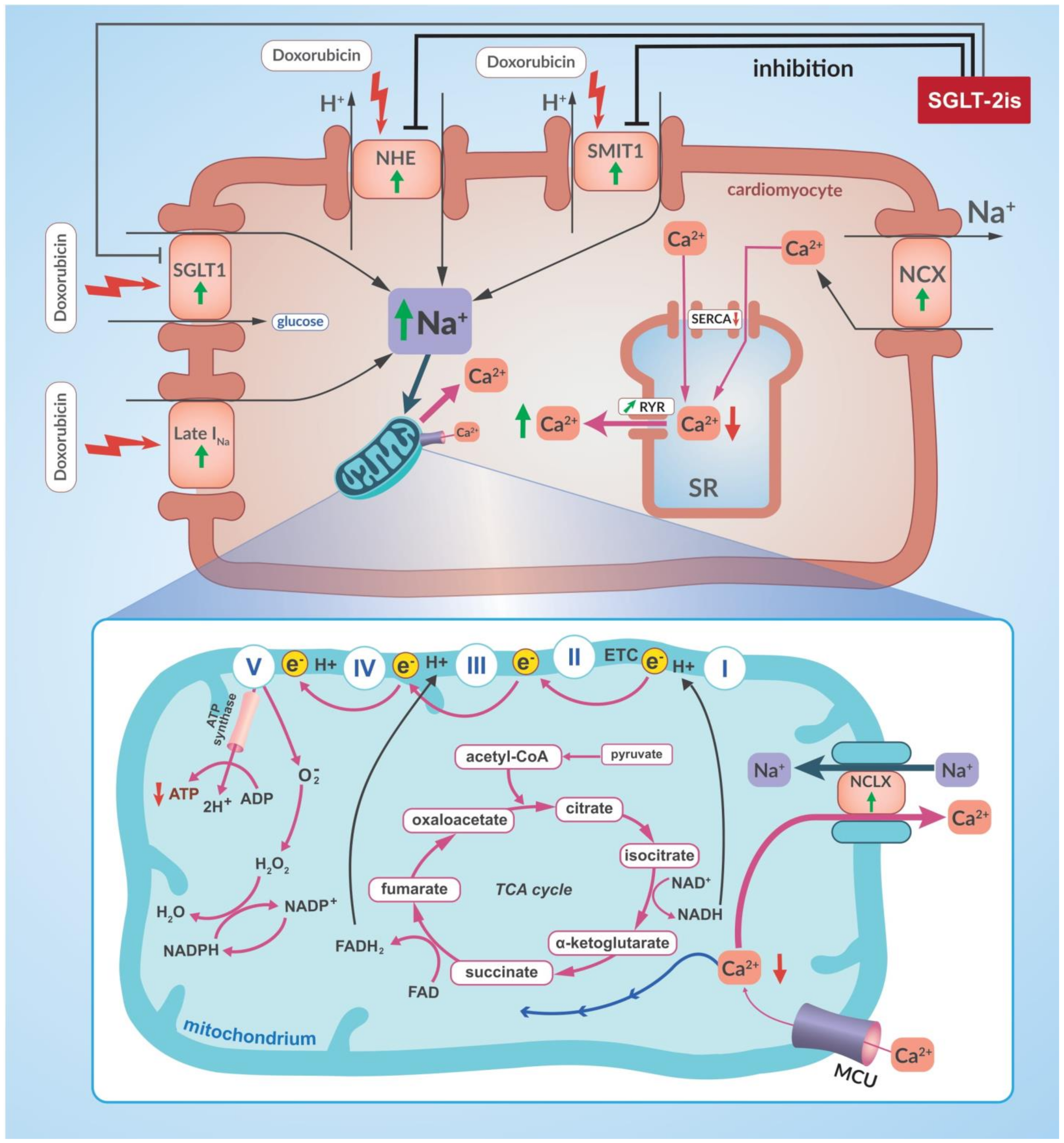
- Quagliariello, V.; De Laurentiis, M.; Rea, D.; Barbieri, A.; Monti, M.G.; Carbone, A.; Paccone, A.; Altucci, L.; Conte, M.; Canale, M.L.; et al. The SGLT-2 inhibitor empagliflozin improves myocardial strain, reduces cardiac fibrosis and pro-inflammatory cytokines in non-diabetic mice treated with doxorubicin. Cardiovasc. Diabetol. 2021, 20, 150. [Google Scholar] [CrossRef]
- Quagliariello, V.; Vecchione, R.; Coppola, C.; Di Cicco, C.; De Capua, A.; Piscopo, G.; Paciello, R.; Narciso, V.; Formisano, C.; Taglialatela-Scafati, O.; et al. Cardioprotective effects of nanoemulsions loaded with anti-inflammatory nutraceuticals against doxorubicin-induced cardiotoxicity. Nutrients 2018, 10, 1304. [Google Scholar] [CrossRef]
- Mele, D.; Tocchetti, C.G.; Pagliaro, P.; Madonna, R.; Novo, G.; Pepe, A.; Zito, C.; Maurea, N.; Spallarossa, P. Pathophysiology of anthracycline cardiotoxicity. J. Cardiovasc. Med. 2016, 17, S3–S11. [Google Scholar] [CrossRef]
- Bertero, E.; Roma, L.P.; Ameri, P.; Maack, C. Cardiac effects of SGLT2 inhibitors: The sodium hypothesis. Cardiovasc. Res. 2018, 114, 12–18. [Google Scholar] [CrossRef]
- Goerg, J.; Sommerfeld, M.; Greiner, B.; Lauer, D.; Seckin, Y.; Kulikov, A.; Ivkin, D.; Kintscher, U.; Okovityi, S.; Kaschina, E. Low-Dose Empagliflozin Improves Systolic Heart Function after Myocardial Infarction in Rats: Regulation of MMP9, NHE1, and SERCA2a. Int. J. Mol. Sci. 2021, 22, 5437. [Google Scholar] [CrossRef]
- Baartscheer, A.; Schumacher, C.A.; Wüst, R.C.; Fiolet, J.W.; Stienen, G.J.; Coronel, R.; Zuurbier, C.J. Empagliflozin decreases myocardial cytoplasmic Na. Diabetologia 2017, 60, 568–573. [Google Scholar] [CrossRef]
- Eliaa, S.G.; Al-Karmalawy, A.A.; Saleh, R.M.; Elshal, M.F. Empagliflozin and Doxorubicin Synergistically Inhibit the Survival of Triple-Negative Breast Cancer Cells via Interfering with the mTOR Pathway and Inhibition of Calmodulin: In Vitro and Molecular Docking Studies. ACS Pharmacol. Transl. Sci. 2020, 3, 1330–1338. [Google Scholar] [CrossRef]
- Zhong, J.; Sun, P.; Xu, N.; Liao, M.; Xu, C.; Ding, Y.; Cai, J.; Zhang, Y.; Xie, W. Canagliflozin inhibits p-gp function and early autophagy and improves the sensitivity to the antitumor effect of doxorubicin. Biochem. Pharmacol. 2020, 175, 113856. [Google Scholar] [CrossRef]
- Sahakian, N.; Cattieuw, L.; Ramillon-Cury, C.; Corroller, A.B.; Silvestre-Aillaud, P.; Béliard, S.; Valéro, R. SGLT2 inhibitors as potentially helpful drugs in PI3K inhibitor-induced diabetes: A case report. Clin. Diabetes Endocrinol. 2021, 7, 17. [Google Scholar] [CrossRef]
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef]
- Jump, C.; Abramson, V.; Wellons, M. Ketoacidosis with canagliflozin prescribed for phosphoinositide 3-kinase inhibitor–induced hyperglycemia: A case report. J. Investig. Med. High Impact Case Rep. 2017, 5, 2324709617725351. [Google Scholar] [CrossRef]
- Jump, D.B. Fatty acid regulation of hepatic lipid metabolism. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 115–120. [Google Scholar] [CrossRef]
- Angelopoulou, A.; Kolokithas-Ntoukas, A.; Papaioannou, L.; Kakazanis, Z.; Khoury, N.; Zoumpourlis, V.; Papatheodorou, S.; Kardamakis, D.; Bakandritsos, A.; Hatziantoniou, S.; et al. Canagliflozin-loaded magnetic nanoparticles as potential treatment of hypoxic tumors in combination with radiotherapy. Nanomedicine 2018, 13, 2435–2454. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Cancer Overview. Available online: https://www.who.int/health-topics/cancer#tab=tab_1 (accessed on 7 November 2022).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| First Author, Publication Year, Country | Study Design | Cancer Type | No. of Participants | Type of SGLT-2 Inhibitors | Comparator | Reported OR or IRR or HR or RR (95% CI) | Notes |
|---|---|---|---|---|---|---|---|
| Ptaszyńska, 2015, Poland [81] | Case study | All cancer types | 9339 | Dapagliflozin | Placebo or other active treatment | IRR 1.035 (0.724, 1.481), p = 0.849 | 21 phase 2b/3 clinical trials, duration 12–208 weeks |
| Tang, 2017, China [5] | Pairwaise meta-analysis | All cancer types | 34,569 | Canagliflozin Dapagliflozin Empagliflozin | Placebo or other active treatment | OR 1.14 (0.96, 1.36), p = 0.60 | 46 indenedent randomized controlled trials, mean duration 61 weeks |
| Dicembrini, 2019, Italy [76] | Meta-analysis | All cancer types | 48,185 | Canagliflozin Dapagliflozin Empagliflozin Ertugliflozin | Placebo or other active treatment | OR 0.98 (0.77, 1.24), p = 0.82 | 27 randomized controlled trials with duration at least 52 weeks, mean duration 84 weeks |
| Suissa, 2021, Canada [71] | Primary analysis | Breast cancer | 46,569 | Canagliflozin Dapagliflozin Empagliflozin | DPP-4 inhibitors | HR (95% CI) 1.0 (0.76, 1.30) | Median follow-up 2.6 years |
| Shi, 2021, China [60] | Meta-analysis | All cancer types | 88,973 | Canagliflozin Dapagliflozin Empagliflozin Ertugliflozin Tofogliflozin Bexagliflozin | Placebo or other active treatment | RR (95% CI) 1.05 (0.97, 1.14), p = 0.20 | 77 randomized controlled trials with duration from 10 to 416 weeks |
| Dąbrowski, 2021, Poland [59] | Meta-analysis | All cancer types | 66,568 | Canagliflozin Dapagliflozin Empagliflozin Ertugliflozin Sotagliflozin | Placebo | RR (95% CI) 1.11 (0.98, 1.26), p = 0.10 | 8 cardiovascular and renal randomized controlled trials |
| Benedetti, 2021, Italy [87] | Meta-analysis | All cancer types | 48,985 | Canagliflozin Dapagliflozin Empagliflozin Ertugliflozin | Placebo | RR (95% CI) 0.35 (0.33, 0.37), p = 0.00 | 20 randomized clinical trials, with a particularly reduced risk of cancer for dapagliflozin and ertugliflozin (RR 0.06, CI 0.06–0.07 and RR 0.22, CI 0.18–0.26, respectively) |
| Cancer Type | Type of SGLT-2 Inhibitor Drug | Mechanism of Action |
|---|---|---|
| Hepatocellular carcinoma (HCC) | Canagliflozin | Inhibition of the action of β-catenin |
| HCC, prostate cancer, lung cancer, liver cancer and breast cancer | Canagliflozin | Inhibition of complex I and subunit α of ATP synthase F1 in the mitochondrial electron transport chain |
| Breast cancer, HCC | Canagliflozin | Suppression of SREBP1 and SCD1 |
| HCC | Canagliflozin | Cell cycle arrest |
| Breast cancer, pancreatic cancer | Canagliflozin | Inhibition of mTOR |
| HCC | Canagliflozin | Inhibition of DNA and RNA synthesis |
| HCC | Canagliflozin | Induction of G2/M arrest and inhibition of proangiogenic factors |
| Breast cancer | Ipragliflozin | Inhibition of cellular sodium influx |
| Breast cancer | Canagliflozin | Disruption of glutamine metabolism |
| Lung cancer | Canagliflozin | Inhibition of L858R/T790M EGFR kinase |
| Human colon carcinoma | Dapagliflozin | Reduction in the adhesion of cancer cells |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dutka, M.; Bobiński, R.; Francuz, T.; Garczorz, W.; Zimmer, K.; Ilczak, T.; Ćwiertnia, M.; Hajduga, M.B. SGLT-2 Inhibitors in Cancer Treatment—Mechanisms of Action and Emerging New Perspectives. Cancers 2022, 14, 5811. https://doi.org/10.3390/cancers14235811
Dutka M, Bobiński R, Francuz T, Garczorz W, Zimmer K, Ilczak T, Ćwiertnia M, Hajduga MB. SGLT-2 Inhibitors in Cancer Treatment—Mechanisms of Action and Emerging New Perspectives. Cancers. 2022; 14(23):5811. https://doi.org/10.3390/cancers14235811
Chicago/Turabian StyleDutka, Mieczysław, Rafał Bobiński, Tomasz Francuz, Wojciech Garczorz, Karolina Zimmer, Tomasz Ilczak, Michał Ćwiertnia, and Maciej B. Hajduga. 2022. "SGLT-2 Inhibitors in Cancer Treatment—Mechanisms of Action and Emerging New Perspectives" Cancers 14, no. 23: 5811. https://doi.org/10.3390/cancers14235811
APA StyleDutka, M., Bobiński, R., Francuz, T., Garczorz, W., Zimmer, K., Ilczak, T., Ćwiertnia, M., & Hajduga, M. B. (2022). SGLT-2 Inhibitors in Cancer Treatment—Mechanisms of Action and Emerging New Perspectives. Cancers, 14(23), 5811. https://doi.org/10.3390/cancers14235811