
Adenosine 2A Receptor Activation Amplifies Ibrutinib Antiplatelet Effect; Implications in Chronic Lymphocytic Leukemia

 , ,
, ,
Simple Summary
Abstract
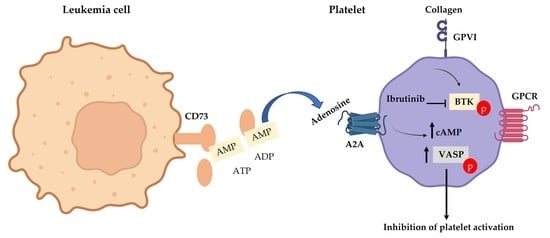
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Preparation of Human Platelets
2.3. Light Transmission Aggregometry (LTA)
2.4. Platelet Function Assay in Whole Blood
2.5. Platelet Activation and GPVI Expression Using Flow cytometry
2.6. Platelet Mepacrine Uptake and Release Assay Using Flow Cytometry
2.7. ELISA
2.8. Western Blot
2.9. Statistical Analysis
3. Results
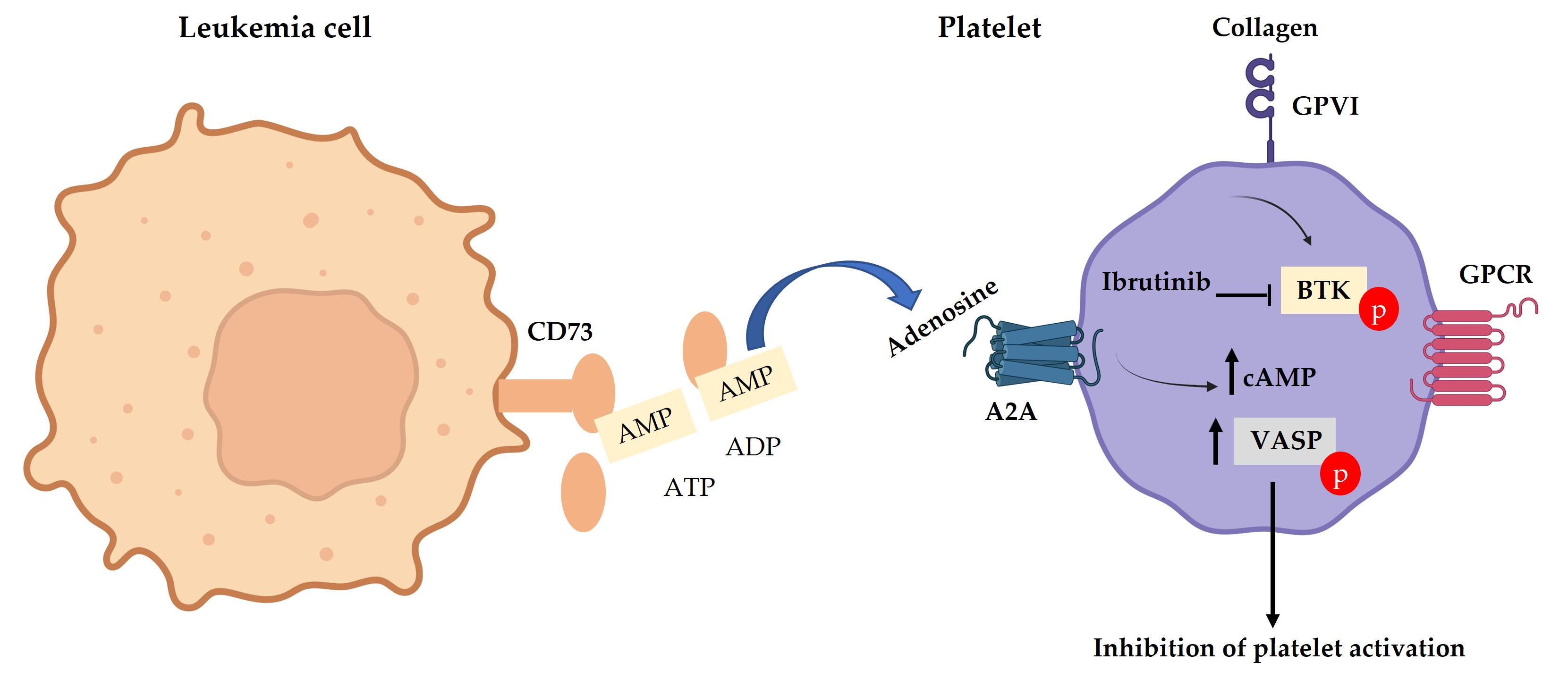
3.1. Low GPVI Expression and Collagen Response in CLL Patients
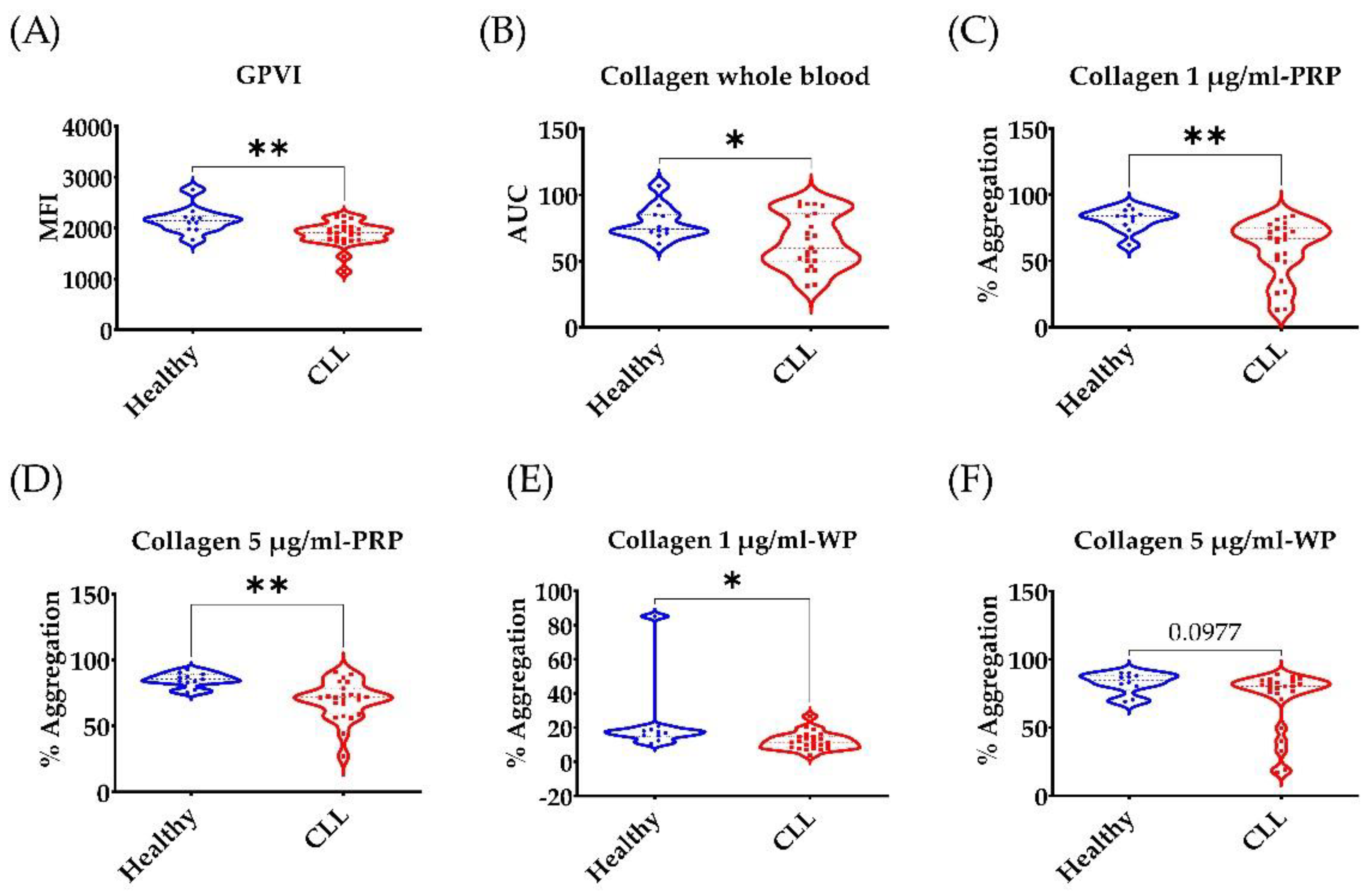
3.2. Broad Disruption of Platelet Response in CLL Patients
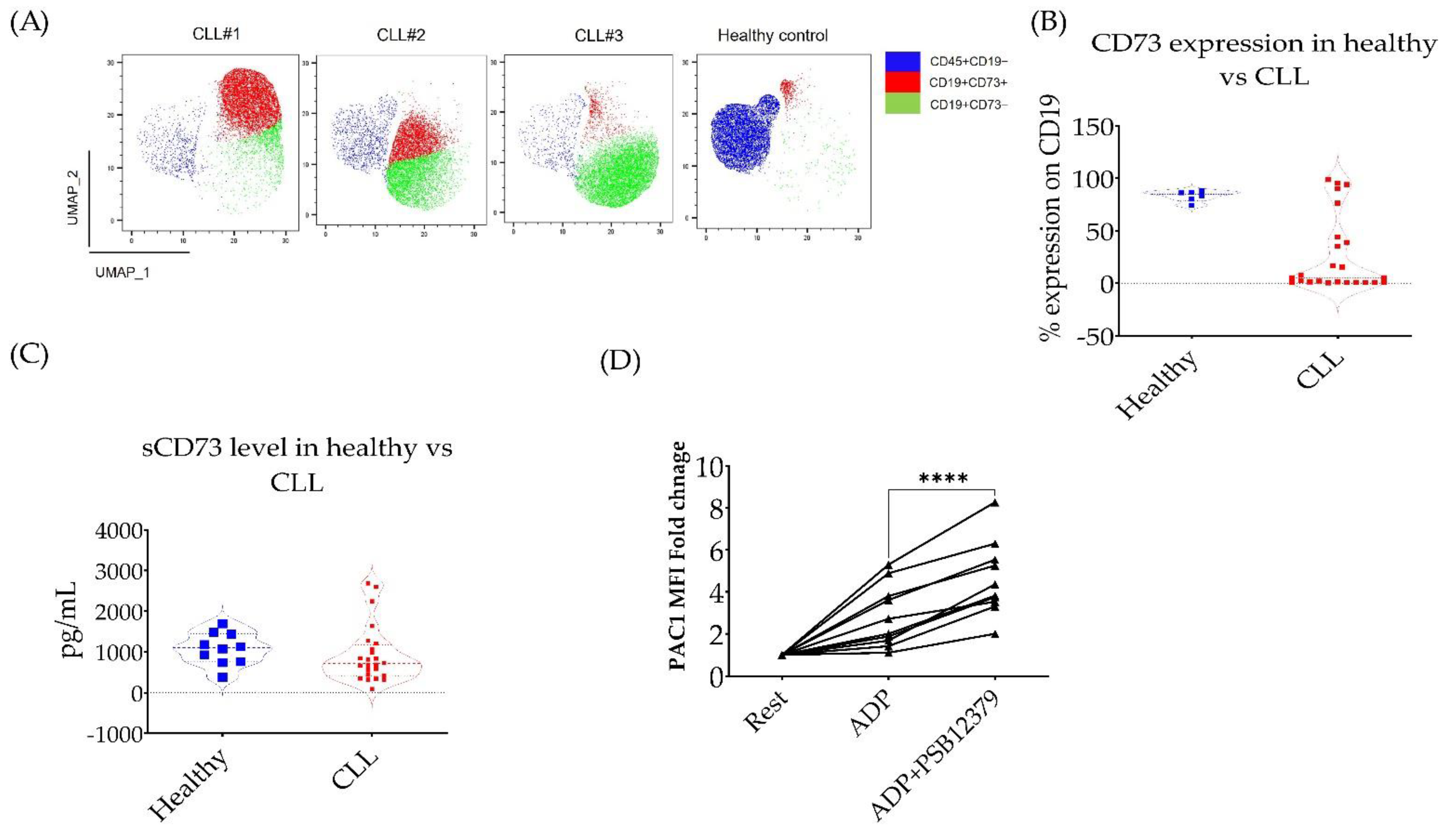
3.3. CD73 Contributes to Platelet Function in CLL Patients
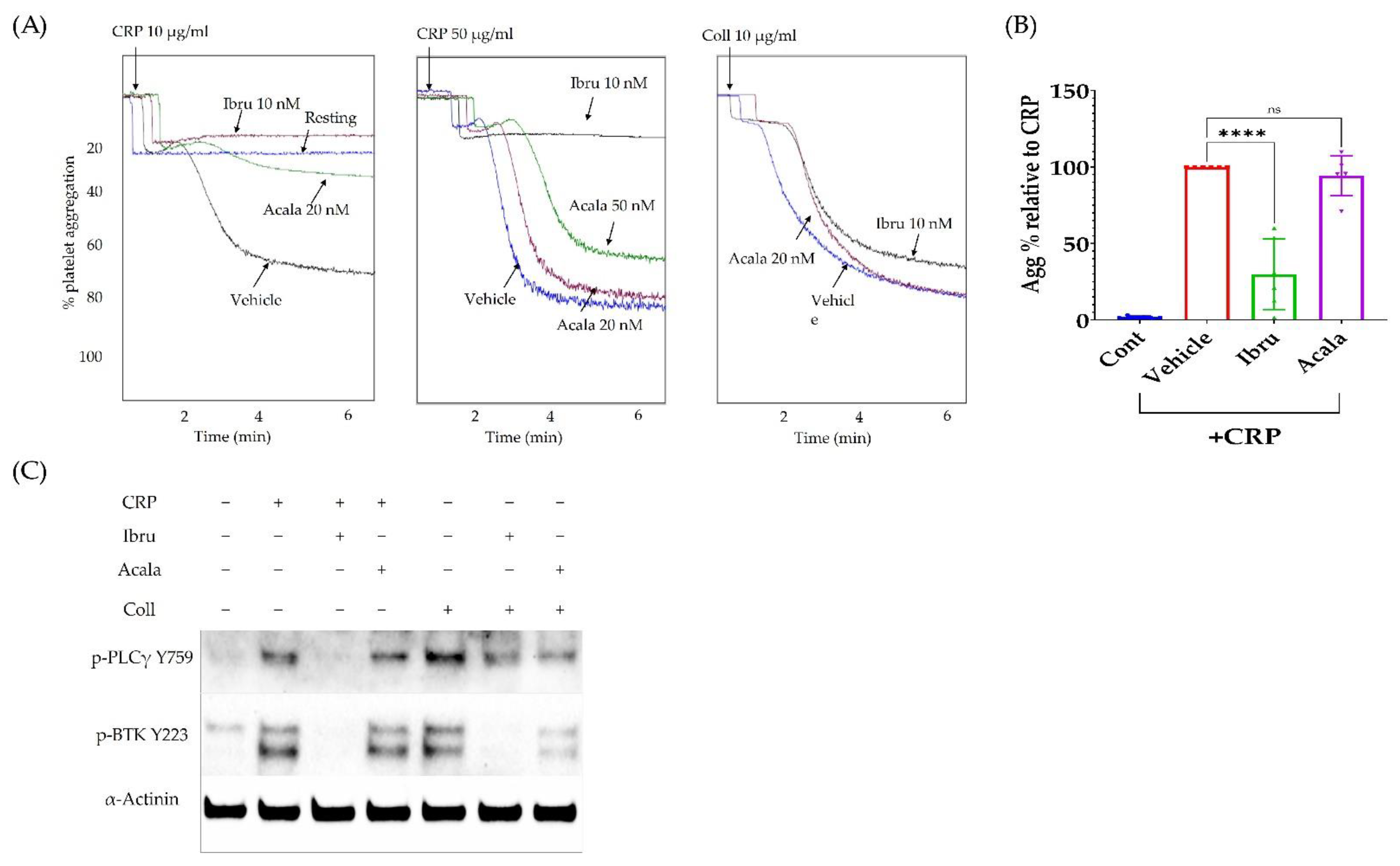
3.4. Ibrutinib Inhibits CRP but Not Collagen-Induced Platelet Aggregation
3.5. A2A Activation Amplifies the Antiplatelet Effect of Ibrutinib
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wiestner, A. Ibrutinib and Venetoclax—Doubling Down on CLL. N. Engl. J. Med. 2019, 380, 2169–2171. [Google Scholar] [CrossRef] [PubMed]
- Redaelli, A.; Laskin, B.L.; Stephens, J.M.; Botteman, M.F.; Pashos, C.L. The clinical and epidemiological burden of chronic lymphocytic leukaemia. Eur. J. Cancer Care 2004, 13, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Ma, J.; Guo, A.; Lu, P.; Leonard, J.P.; Coleman, M.; Liu, M.; Buggy, J.J.; Furman, R.R.; Wang, Y.L. BTK inhibition targets in vivo CLL proliferation through its effects on B-cell receptor signaling activity. Leukemia 2014, 28, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Chiorazzi, N.; Hatzi, K.; Albesiano, E. B-Cell Chronic Lymphocytic Leukemia, a Clonal Disease of B Lymphocytes with Receptors that Vary in Specificity for (Auto)antigens. Ann. N. Y. Acad. Sci. 2005, 1062, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Saba, H.I.; Mufti, G. Advances in Malignant Hematology; John Wiley & Sons, Incorporated: Hoboken, NJ, USA, 2011. [Google Scholar]
- Series, J.; Garcia, C.; Levade, M.; Viaud, J.; Sie, P.; Ysebaert, L.; Payrastre, B. Differences and similarities in the effects of ibrutinib and acalabrutinib on platelet functions. Haematologica 2019, 104, 2292–2299. [Google Scholar] [CrossRef]
- Byrd, J.C.; Harrington, B.; O’Brien, S.; Jones, J.A.; Schuh, A.; Devereux, S.; Chaves, J.; Wierda, W.G.; Awan, F.T.; Brown, J.R.; et al. Acalabrutinib (ACP-196) in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 323–332. [Google Scholar] [CrossRef]
- Dmitrieva, E.A.; Nikitin, E.A.; Ignatova, A.A.; Vorobyev, V.I.; Poletaev, A.V.; Seregina, E.A.; Voronin, K.A.; Polokhov, D.M.; Maschan, A.A.; Novichkova, G.A.; et al. Platelet function and bleeding in chronic lymphocytic leukemia and mantle cell lymphoma patients on ibrutinib. J. Thromb. Haemost. 2020, 18, 2672–2684. [Google Scholar] [CrossRef]
- Korycka-Wołowiec, A.; Wołowiec, D.; Robak, T. Pharmacodynamic considerations of small molecule targeted therapy for treating B-cell malignancies in the elderly. Expert Opin. Drug Metab. Toxicol. 2015, 11, 1371–1391. [Google Scholar] [CrossRef]
- Zhou, H.; Hu, P.; Yan, X.; Zhang, Y.; Shi, W. Ibrutinib in Chronic Lymphocytic Leukemia: Clinical Applications, Drug Resistance, and Prospects. OncoTargets Ther. 2020, 13, 4877–4892. [Google Scholar] [CrossRef]
- Deeks, E.D. Ibrutinib: A Review in Chronic Lymphocytic Leukaemia. Drugs 2017, 77, 225–236. [Google Scholar] [CrossRef]
- Shatzel, J.J.; Olson, S.R.; Tao, D.L.; McCarty, O.J.T.; Danilov, A.V.; DeLoughery, T.G. Ibrutinib-associated bleeding: Pathogenesis, management and risk reduction strategies. J. Thromb. Haemost. 2017, 15, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R.; Moslehi, J.; Ewer, M.S.; O’Brien, S.M.; Ghia, P.; Cymbalista, F.; Shanafelt, T.D.; Fraser, G.; Rule, S.; Coutre, S.E.; et al. Incidence of and risk factors for major haemorrhage in patients treated with ibrutinib: An integrated analysis. Br. J. Haematol. 2019, 184, 558–569. [Google Scholar] [CrossRef] [PubMed]
- Kamel, S.; Horton, L.; Ysebaert, L.; Levade, M.; Burbury, K.; Tan, S.; Cole-Sinclair, M.; Reynolds, J.; Filshie, R.; Schischka, S.; et al. Ibrutinib inhibits collagen-mediated but not ADP-mediated platelet aggregation. Leukemia 2015, 29, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Luu, S.; Gardiner, E.E.; Andrews, R.K. Bone Marrow Defects and Platelet Function: A Focus on MDS and CLL. Cancers 2018, 10, 147. [Google Scholar] [CrossRef] [PubMed]
- Nicolson, P.L.R.; Hughes, C.E.; Watson, S.; Nock, S.H.; Hardy, A.T.; Watson, C.N.; Montague, S.J.; Clifford, H.; Huissoon, A.P.; Malcor, J.D.; et al. Inhibition of Btk by Btk-specific concentrations of ibrutinib and acalabrutinib delays but does not block platelet aggregation mediated by glycoprotein VI. Haematologica 2018, 103, 2097–2108. [Google Scholar] [CrossRef]
- Atkinson, B.T.; Ellmeier, W.; Watson, S.P. Tec regulates platelet activation by GPVI in the absence of Btk. Blood 2003, 102, 3592–3599. [Google Scholar] [CrossRef]
- Rigg, R.A.; Aslan, J.E.; Healy, L.D.; Wallisch, M.; Thierheimer, M.L.; Loren, C.P.; Pang, J.; Hinds, M.T.; Gruber, A.; McCarty, O.J. Oral administration of Bruton’s tyrosine kinase inhibitors impairs GPVI-mediated platelet function. Am. J. Physiol. Cell Physiol. 2016, 310, C373–C380. [Google Scholar] [CrossRef]
- Lee, R.H.; Piatt, R.; Conley, P.B.; Bergmeier, W. Effects of ibrutinib treatment on murine platelet function during inflammation and in primary hemostasis. Haematologica 2017, 102, e89–e92. [Google Scholar] [CrossRef]
- Johnston-Cox, H.A.; Ravid, K. Adenosine and blood platelets. Purinergic Signal. 2011, 7, 357–365. [Google Scholar] [CrossRef]
- Chen, S.; Wainwright, D.A.; Wu, J.D.; Wan, Y.; Matei, D.E.; Zhang, Y.; Zhang, B. CD73: An emerging checkpoint for cancer immunotherapy. Immunotherapy 2019, 11, 983–997. [Google Scholar] [CrossRef]
- Valsecchi, R.; Coltella, N.; Belloni, D.; Ponente, M.; Ten Hacken, E.; Scielzo, C.; Scarfo, L.; Bertilaccio, M.T.; Brambilla, P.; Lenti, E.; et al. HIF-1alpha regulates the interaction of chronic lymphocytic leukemia cells with the tumor microenvironment. Blood 2016, 127, 1987–1997. [Google Scholar] [CrossRef] [PubMed]
- Serra, S.; Horenstein, A.L.; Vaisitti, T.; Brusa, D.; Rossi, D.; Laurenti, L.; D’Arena, G.; Coscia, M.; Tripodo, C.; Inghirami, G.; et al. CD73-generated extracellular adenosine in chronic lymphocytic leukemia creates local conditions counteracting drug-induced cell death. Blood 2011, 118, 6141–6152. [Google Scholar] [CrossRef] [PubMed]
- Chambers, A.M.; Matosevic, S. Immunometabolic Dysfunction of Natural Killer Cells Mediated by the Hypoxia-CD73 Axis in Solid Tumors. Front. Mol. Biosci. 2019, 6, 60. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, L.; Pacher, P.; Vizi, E.S.; Hasko, G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 2013, 19, 355–367. [Google Scholar] [CrossRef]
- Mazziotta, C.; Rotondo, J.C.; Lanzillotti, C.; Campione, G.; Martini, F.; Tognon, M. Cancer biology and molecular genetics of A3 adenosine receptor. Oncogene 2022, 41, 301–308. [Google Scholar] [CrossRef]
- Elaskalani, O.; Abdol Razak, N.B.; Metharom, P. Neutrophil extracellular traps induce aggregation of washed human platelets independently of extracellular DNA and histones. Cell Commun. Signal. 2018, 16, 24. [Google Scholar] [CrossRef]
- Carrim, N.; Walsh, T.G.; Consonni, A.; Torti, M.; Berndt, M.C.; Metharom, P. Role of focal adhesion tyrosine kinases in GPVI-dependent platelet activation and reactive oxygen species formation. PLoS ONE 2014, 9, e113679. [Google Scholar] [CrossRef]
- Toth, O.; Calatzis, A.; Penz, S.; Losonczy, H.; Siess, W. Multiple electrode aggregometry: A new device to measure platelet aggregation in whole blood. Thromb. Haemost. 2006, 96, 781–788. [Google Scholar]
- Gordon, N.; Thom, J.; Cole, C.; Baker, R. Rapid detection of hereditary and acquired platelet storage pool deficiency by flow cytometry. Br. J. Haematol. 1995, 89, 117–123. [Google Scholar] [CrossRef]
- Levade, M.; David, E.; Garcia, C.; Laurent, P.A.; Cadot, S.; Michallet, A.S.; Bordet, J.C.; Tam, C.; Sie, P.; Ysebaert, L.; et al. Ibrutinib treatment affects collagen and von Willebrand factor-dependent platelet functions. Blood 2014, 124, 3991–3995. [Google Scholar] [CrossRef]
- Bhattarai, S.; Freundlieb, M.; Pippel, J.; Meyer, A.; Abdelrahman, A.; Fiene, A.; Lee, S.Y.; Zimmermann, H.; Yegutkin, G.G.; Strater, N.; et al. alpha,beta-Methylene-ADP (AOPCP) Derivatives and Analogues: Development of Potent and Selective ecto-5’-Nucleotidase (CD73) Inhibitors. J. Med. Chem. 2015, 58, 6248–6263. [Google Scholar] [CrossRef] [PubMed]
- Mohebali, D.; Kaplan, D.; Carlisle, M.; Supiano, M.A.; Rondina, M.T. Alterations in platelet function during aging: Clinical correlations with thromboinflammatory disease in older adults. J. Am. Geriatr. Soc. 2014, 62, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Pulte, D.; Olson, K.E.; Broekman, M.J.; Islam, N.; Ballard, H.S.; Furman, R.R.; Olson, A.E.; Marcus, A.J. CD39 activity correlates with stage and inhibits platelet reactivity in chronic lymphocytic leukemia. J. Transl. Med. 2007, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Research CfDEa. Ibrutinib. In Clinical Pharmacology and Biopharmaceutics Review(s); 2013(205552Orig1s000):14; FDA: Silver Spring, MD, USA, 2013. Available online: https://www.accessdata.fda.gov (accessed on 22 November 2022).
- Research Cfdea. Acalabrutinib. In Multi-Discipline Review; 2017(210259Orig1s000):85; FDA: Silver Spring, MD, USA, 2017. Available online: https://www.accessdata.fda.gov (accessed on 22 November 2022).
- Marjoram, R.J.; Li, Z.; He, L.; Tollefsen, D.M.; Kunicki, T.J.; Dickeson, S.K.; Santoro, S.A.; Zutter, M.M. alpha2beta1 integrin, GPVI receptor, and common FcRgamma chain on mouse platelets mediate distinct responses to collagen in models of thrombosis. PLoS ONE 2014, 9, e114035. [Google Scholar] [CrossRef]
- Woyach, J.A.; Furman, R.R.; Liu, T.M.; Ozer, H.G.; Zapatka, M.; Ruppert, A.S.; Xue, L.; Li, D.H.; Steggerda, S.M.; Versele, M.; et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N. Engl. J. Med. 2014, 370, 2286–2294. [Google Scholar] [CrossRef] [PubMed]
- Honigberg, L.A.; Smith, A.M.; Sirisawad, M.; Verner, E.; Loury, D.; Chang, B.; Li, S.; Pan, Z.; Thamm, D.H.; Miller, R.A.; et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. USA 2010, 107, 13075–13080. [Google Scholar] [CrossRef] [PubMed]
- Crawford, J.J.; Johnson, A.R.; Misner, D.L.; Belmont, L.D.; Castanedo, G.; Choy, R.; Coraggio, M.; Dong, L.; Eigenbrot, C.; Erickson, R.; et al. Discovery of GDC-0853: A Potent, Selective, and Noncovalent Bruton’s Tyrosine Kinase Inhibitor in Early Clinical Development. J. Med. Chem. 2018, 61, 2227–2245. [Google Scholar] [CrossRef]
- Li, X.; Zuo, Y.; Tang, G.; Wang, Y.; Zhou, Y.; Wang, X.; Guo, T.; Xia, M.; Ding, N.; Pan, Z. Discovery of a series of 2,5-diaminopyrimidine covalent irreversible inhibitors of Bruton’s tyrosine kinase with in vivo antitumor activity. J. Med. Chem. 2014, 57, 5112–5128. [Google Scholar] [CrossRef]
- Kaptein, A.; de Bruin, G.; Hoek, M.E.; van de Kar, B.; de Jong, A.; Gulrajani, M.; Demont, D.; Covey, T.; Mittag, D.; Barf, T. Potency and Selectivity of BTK Inhibitors in Clinical Development for B-Cell Malignancies. Blood 2018, 132, 1871. [Google Scholar] [CrossRef]
- Cristalli, G.; Volpini, R.; Vittori, S.; Camaioni, E.; Monopoli, A.; Conti, A.; Dionisotti, S.; Zocchi, C.; Ongini, E. 2-Alkynyl derivatives of adenosine-5’-N-ethyluronamide: Selective A2 adenosine receptor agonists with potent inhibitory activity on platelet aggregation. J. Med. Chem. 1994, 37, 1720–1726. [Google Scholar] [CrossRef]
- Benz, P.M.; Laban, H.; Zink, J.; Gunther, L.; Walter, U.; Gambaryan, S.; Dib, K. Vasodilator-Stimulated Phosphoprotein (VASP)-dependent and -independent pathways regulate thrombin-induced activation of Rap1b in platelets. Cell Commun. Signal. 2016, 14, 21. [Google Scholar] [CrossRef] [PubMed]
- Durrant, T.N.; van den Bosch, M.T.; Hers, I. Integrin alphaIIbbeta3 outside-in signaling. Blood 2017, 130, 1607–1619. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, S.; Sato, I. Plasma adenosine levels and P-selectin expression on platelets in preeclampsia. Obstet. Gynecol. 2001, 98, 354–355. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Yoneyama, Y.; Suzuki, S.; Sawa, R.; Otsubo, Y.; Araki, T. Regulation of platelet aggregation in vitro by plasma adenosine in preeclampsia. Gynecol. Obstet. Investig. 2001, 51, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, Y.; Suzuki, S.; Sawa, R.; Otsubo, Y.; Power, G.G.; Araki, T. Plasma adenosine levels increase in women with normal pregnancies. Am. J. Obstet. Gynecol. 2000, 182, 1200–1203. [Google Scholar] [CrossRef]
- Bonello, L.; Laine, M.; Kipson, N.; Mancini, J.; Helal, O.; Fromonot, J.; Gariboldi, V.; Condo, J.; Thuny, F.; Frere, C.; et al. Ticagrelor increases adenosine plasma concentration in patients with an acute coronary syndrome. J. Am. Coll. Cardiol. 2014, 63, 872–877. [Google Scholar] [CrossRef]
- Kerbaul, F.; By, Y.; Gariboldi, V.; Mekkaoui, C.; Fesler, P.; Collart, F.; Brimioulle, S.; Jammes, Y.; Ruf, J.; Guieu, R. Acute pulmonary embolism decreases adenosine plasma levels in anesthetized pigs. ISRN Cardiol. 2011, 2011, 750301. [Google Scholar] [CrossRef][Green Version]
- Kamath, S.; Blann, A.D.; Caine, G.J.; Gurney, D.; Chin, B.S.; Lip, G.Y. Platelet P-selectin levels in relation to plasma soluble P-selectin and beta-thromboglobulin levels in atrial fibrillation. Stroke 2002, 33, 1237–1242. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Untreated CLL Cohort | Health Cohort | p-Value (CLL vs. Healthy) | |
|---|---|---|---|
| Number of participants | 23 | 11 | |
| Median Age, yr (range) | 69 (52–89) | 62 (57–69) | 0.0389 |
| WBC (×109/L), mean (range) | 40.82 (13–112.3) | 5.54 (4.1–7.9) | 0.0004 |
| Platelets (×109/L), mean (range) | 215.1 (128–327) | 231.5 (138–326) | 0.4247 |
| MPV (fL), mean (range) | 8.57 (7–10.2) | 9.18 (8.2–10.4) | 0.0813 |
| Lymphocytes (×109/L), mean (range) | 34.49 (10.7–103.9) | 1.63 (1–2.6) | 0.0006 |
| Lymphocytes %, mean (range) | 79.91 (61–94.1) | 29.47 (18.4–36.3) | <0.0001 |
| RBC (×1012/L), mean (range) | 4.72 (3.6–5.9) | 5.00 (4.3–5.6) | 0.1563 |
| Hb g/L, mean (range) | 137.0 (116–158) | 146.3 (127–169) | 0.0446 |
| HCT %, mean (range) | 41.89 (32.9–50.6) | 44.88 (38.6–51.1) | 0.0552 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elaskalani, O.; Gilmore, G.; Hagger, M.; Baker, R.I.; Metharom, P. Adenosine 2A Receptor Activation Amplifies Ibrutinib Antiplatelet Effect; Implications in Chronic Lymphocytic Leukemia. Cancers 2022, 14, 5750. https://doi.org/10.3390/cancers14235750
Elaskalani O, Gilmore G, Hagger M, Baker RI, Metharom P. Adenosine 2A Receptor Activation Amplifies Ibrutinib Antiplatelet Effect; Implications in Chronic Lymphocytic Leukemia. Cancers. 2022; 14(23):5750. https://doi.org/10.3390/cancers14235750
Chicago/Turabian StyleElaskalani, Omar, Grace Gilmore, Madison Hagger, Ross I. Baker, and Pat Metharom. 2022. "Adenosine 2A Receptor Activation Amplifies Ibrutinib Antiplatelet Effect; Implications in Chronic Lymphocytic Leukemia" Cancers 14, no. 23: 5750. https://doi.org/10.3390/cancers14235750
APA StyleElaskalani, O., Gilmore, G., Hagger, M., Baker, R. I., & Metharom, P. (2022). Adenosine 2A Receptor Activation Amplifies Ibrutinib Antiplatelet Effect; Implications in Chronic Lymphocytic Leukemia. Cancers, 14(23), 5750. https://doi.org/10.3390/cancers14235750