
MicroRNAs and Drug Resistance in Non-Small Cell Lung Cancer: Where Are We Now and Where Are We Going



Abstract
Simple Summary
Abstract
1. Introduction
2. Relevant Sections
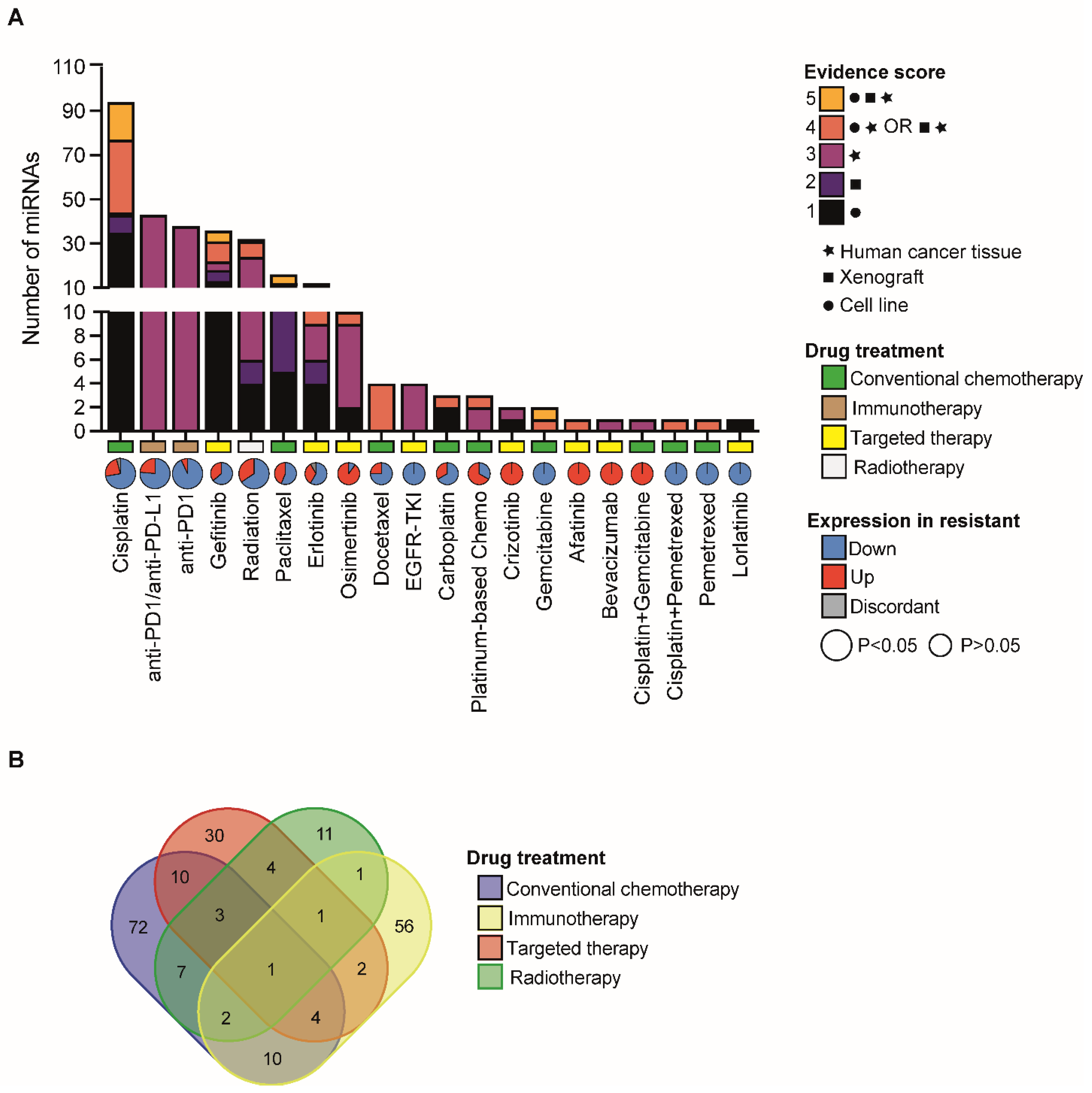
2.1. Annotation of miRNAs with a Functional Role in NSCLC Therapy Resistance
2.2. The Landscape of miRNAs Which Regulate Cisplatin Resistance in NSCLC
- −
- Cisplatin import/export: regulation of cisplatin import/export is an important mechanism of resistance to cisplatin. MiR-369-3p overexpression promoted cisplatin resistance by direct regulation of the expression of the SLC35F5 gene, a nucleotide sugar transporter involved in drug uptake [20]. In addition, miR-495-3p regulates cisplatin resistance by modulating the expression of ATP7A, a copper transporter that regulate cisplatin efflux from the cells [21].
- −
- DNA damage repair: upon entering the cells, cisplatin becomes activated by the displacement of chloride atoms by water molecules. Active cisplatin exerts its cytotoxic function mainly by generating monoadducts and crosslinks at the level of the DNA. Therefore, genetic and epigenetic alterations of genes involved in DNA damage response represent a major mechanism to cope with cisplatin-induced cell death. In line with this, miR-92a-3p regulates cisplatin resistance by directly binding to the 3′ untranslated region (UTR) of RAD21 mRNA, a member of the cohesin complex that can promote DNA repair at the G2 phase of the cell cycle [22]. Yet, miR-17-5p, which belongs to the same cluster of miR-92a-3p (e.g., miR-17-92 cluster), can protect cancer cells from cisplatin-induced apoptosis by regulating CDKN1A, a cell cycle inhibitor that blocks DNA synthesis by G1 arrest and whose levels are increased upon accumulation of DNA damage due to activation of ATM and ATR and consequent TP53 stabilization [22].
- −
- miRNA sponges: long non-coding RNAs (lncRNAs) are transcripts longer than 200 nucleotides that do not code for functional proteins. Since lncRNAs can contain MREs in their sequence, they can sequester miRNAs and impair their activity on target genes, therefore, acting as competitive endogenous RNAs or miRNA ‘sponges’. Over the last years, a high number of reports described lncRNAs that regulate cisplatin resistance by sponging specific miRNAs. For example, NORAD is a lncRNA upregulated by DNA damage and was found involved in cisplatin resistance by regulating SOX4 expression by sponging miR-129-1-3p [23]. Similarly, circular RNA (circRNA) Circ-PRMT5 promoted the resistance to cisplatin by competing with the binding of miR-4458 to REV3L [24], a catalytic subunit of DNA polymerase implicated in the tolerance of DNA adduct through translesion synthesis.
- −
- Pro-survival and apoptotic signaling pathways: when cells are exposed to platinum, both pro-survival and apoptotic signaling pathways are activated and compete for the final fate of the cells. A number of miRNAs have been associated with the direct or indirect regulation of apoptotic proteins or survival signaling pathways. MiR-103a-3p induces ERK signaling in NSCLC cells by targeting NF1 expression, a key negative regulator of the Ras signaling pathway [25]. In an independent study, Wang et al., found that miR-103a-3p could be released in an exosome (nanosized extracellular vesicles actively released by a variety of cells) from cancer-associated fibroblast and can induce resistance of NSCLC cells via direct regulation of BAK1, a pro-apoptotic BCL-2 family member [26]. Yet, miR-29c-3p affects cisplatin resistance by regulating PI3K/AKT signaling pathways due to binding to 3′UTR of AKT2 [27]. Similarly, miR-126-5p overexpression increases cisplatin sensitivity by inhibiting the PTEN/PI3K/AKT signaling pathway, an effect partly induced by the direct regulation of the metalloprotease ADAM9 [28]. In addition, miR-539-5p increases the sensitivity of cisplatin-resistant cells via the inactivation of the P13K/AKT/mTOR signaling pathway by targeting the protein kinase DCLK1 [29]. Interestingly, the increase in DCLK1 expression and cisplatin resistance was found to be also mediated by lncRNA SNHG1-dependent sponging of miR-330-5p [30]. Several independent studies associated also miR-21 upregulation with increased resistance to Cisplatin [31,32,33,34,35,36,37], mostly due to a direct regulation of PTEN expression [32,33,35,36]. Furthermore, miR-21 expression was reported to be regulated by KRAS wild-type or mutant [37], a major driver mutation in NSCLC.
- −
- Reactive oxygen species: besides DNA damage, activated cisplatin is also a potent inducer of reactive oxygen species, which induces cell death. In this scenario, miR-495-3p overexpression was reported to modulate cisplatin resistance through direct inhibition of NRF2 [38], a transcription factor that regulates the expression of important NADPH-generating enzymes and redox proteins crucial for protecting the cells from oxidative stress.
- −
- Epithelial-to-mesenchymal transition (EMT): EMT contributes to resistance to several therapeutic agents, including cisplatin [39]. In line with this, miR-128-3p upregulation drives chemoresistance and was associated with the overactivation of Wnt/beta-catenin and TGF-beta (TGF-β) pathways and consequent acquisition of mesenchymal and stem-like features [40]. Likewise, miR-181b-5p regulates the TGF-β pathway by direct inhibition of TGFBR1 expression, thus modulating EMT and sensitivity to cisplatin [41]. An independent study found that miR-181b-5p suppresses stem cell properties in tumor cells and enhances sensitivity to cisplatin treatment by directly targeting NOTCH [42].
- −
- Autophagy: autophagy is a crucial process that allows the recycling of important cellular components in response to stress conditions such as those induced by cisplatin treatment. Indeed, regulation of autophagy has been widely associated with cisplatin resistance phenotype. Rescue of miR-1-3p increases the sensitivity of cisplatin resistance cells by inhibiting ATG3, a key autophagic protein [43]. Furthermore, exosomal transfer of miR-425-3p was found to increase autophagic flux and chemoresistance by inhibiting AKT1 in the targeted NSCLC cells [44].
2.3. Beyond Targeting NSCLC Cells: The Role of miRNA in Regulating Immune Response to Cisplatin Treatment
2.4. miRNA Regulation of Platinum-Based Therapy Response
2.5. miRNAs Which Modulate Response to EGFR Inhibitors
2.6. miRNAs Associated to Resistance to Radiotherapy
2.7. Overview of the Experimental Strategies to Investigate the Functional Role of miRNA in NSCLC Therapy Response: Advantages and Limitations
2.7.1. Cell Lines
2.7.2. Animal Models
2.7.3. Human Cancer Tissue
3. Conclusions and Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Miller, K.D.; Nogueira, L.; Devasia, T.; Mariotto, A.B.; Yabroff, K.R.; Jemal, A.; Kramer, J.; Siegel, R.L. Cancer Treatment and Survivorship Statistics, 2022. CA Cancer J. Clin. 2022, 72, 409–436. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Fillmore, C.M.; Hammerman, P.S.; Kim, C.F.; Wong, K.-K. Non-Small-Cell Lung Cancers: A Heterogeneous Set of Diseases. Nat. Rev. Cancer 2014, 14, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Lim, Z.-F.; Ma, P.C. Emerging Insights of Tumor Heterogeneity and Drug Resistance Mechanisms in Lung Cancer Targeted Therapy. J. Hematol. Oncol. 2019, 12, 134. [Google Scholar] [CrossRef] [PubMed]
- Yanaihara, N.; Caplen, N.; Bowman, E.; Seike, M.; Kumamoto, K.; Yi, M.; Stephens, R.M.; Okamoto, A.; Yokota, J.; Tanaka, T.; et al. Unique MicroRNA Molecular Profiles in Lung Cancer Diagnosis and Prognosis. Cancer Cell 2006, 9, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research, N. Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Dama, E.; Melocchi, V.; Colangelo, T.; Cuttano, R.; Bianchi, F. Deciphering the Molecular Profile of Lung Cancer: New Strategies for the Early Detection and Prognostic Stratification. J. Clin. Med. 2019, 8, 108. [Google Scholar] [CrossRef]
- Si, W.; Shen, J.; Zheng, H.; Fan, W. The Role and Mechanisms of Action of MicroRNAs in Cancer Drug Resistance. Clin. Epigenet. 2019, 11, 25. [Google Scholar] [CrossRef]
- Treiber, T.; Treiber, N.; Meister, G. Regulation of MicroRNA Biogenesis and Its Crosstalk with Other Cellular Pathways. Nat. Rev. Mol. Cell Biol. 2019, 20, 5–20. [Google Scholar] [CrossRef]
- Dragomir, M.P.; Knutsen, E.; Calin, G.A. Classical and Noncanonical Functions of MiRNAs in Cancers. Trends Genet. 2022, 38, 379–394. [Google Scholar] [CrossRef]
- Chevillet, J.R.; Kang, Q.; Ruf, I.K.; Briggs, H.A.; Vojtech, L.N.; Hughes, S.M.; Cheng, H.H.; Arroyo, J.D.; Meredith, E.K.; Gallichotte, E.N.; et al. Quantitative and Stoichiometric Analysis of the MicroRNA Content of Exosomes. Proc. Natl. Acad. Sci. USA 2014, 111, 14888–14893. [Google Scholar] [CrossRef]
- Turchinovich, A.; Weiz, L.; Langheinz, A.; Burwinkel, B. Characterization of Extracellular Circulating MicroRNA. Nucleic Acids Res. 2011, 39, 7223–7233. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, J.D.; Chevillet, J.R.; Kroh, E.M.; Ruf, I.K.; Pritchard, C.C.; Gibson, D.F.; Mitchell, P.S.; Bennett, C.F.; Pogosova-Agadjanyan, E.L.; Stirewalt, D.L.; et al. Argonaute2 Complexes Carry a Population of Circulating MicroRNAs Independent of Vesicles in Human Plasma. Proc. Natl. Acad. Sci. USA 2011, 108, 5003–5008. [Google Scholar] [CrossRef] [PubMed]
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. MicroRNAs Are Transported in Plasma and Delivered to Recipient Cells by High-Density Lipoproteins. Nat. Cell Biol. 2011, 13, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Pardini, B.; Calin, G.A. MicroRNAs and Long Non-Coding RNAs and Their Hormone-Like Activities in Cancer. Cancers 2019, 11, 378. [Google Scholar] [CrossRef] [PubMed]
- Kelland, L. The Resurgence of Platinum-Based Cancer Chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef]
- Kumar, M.S.; Lu, J.; Mercer, K.L.; Golub, T.R.; Jacks, T. Impaired MicroRNA Processing Enhances Cellular Transformation and Tumorigenesis. Nat. Genet. 2007, 39, 673–677. [Google Scholar] [CrossRef]
- Yuan, M.; Huang, L.-L.; Chen, J.-H.; Wu, J.; Xu, Q. The Emerging Treatment Landscape of Targeted Therapy in Non-Small-Cell Lung Cancer. Signal Transduct. Target. Ther. 2019, 4, 61. [Google Scholar] [CrossRef]
- Rottenberg, S.; Disler, C.; Perego, P. The Rediscovery of Platinum-Based Cancer Therapy. Nat. Rev. Cancer 2021, 21, 37–50. [Google Scholar] [CrossRef]
- Huang, D.; Savage, S.R.; Calinawan, A.P.; Lin, C.; Zhang, B.; Wang, P.; Starr, T.K.; Birrer, M.J.; Paulovich, A.G. A Highly Annotated Database of Genes Associated with Platinum Resistance in Cancer. Oncogene 2021, 40, 6395–6405. [Google Scholar] [CrossRef]
- Hao, G.-J.; Ding, Y.-H.; Wen, H.; Li, X.-F.; Zhang, W.; Su, H.-Y.; Liu, D.-M.; Xie, N.-L. Attenuation of Deregulated MiR-369-3p Expression Sensitizes Non-Small Cell Lung Cancer Cells to Cisplatin via Modulation of the Nucleotide Sugar Transporter SLC35F5. Biochem. Biophys. Res. Commun. 2017, 488, 501–508. [Google Scholar] [CrossRef]
- Song, L.; Li, Y.; Li, W.; Wu, S.; Li, Z. MiR-495 Enhances the Sensitivity of Non-Small Cell Lung Cancer Cells to Platinum by Modulation of Copper-Transporting P-Type Adenosine Triphosphatase A (ATP7A). J. Cell. Biochem. 2014, 115, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Fu, W.; Liao, H.; Dai, L.; Jiang, Z.; Pan, Y.; Huang, H.; Mo, Y.; Li, S.; Yang, G.; et al. The Regulatory and Predictive Functions of MiR-17 and MiR-92 Families on Cisplatin Resistance of Non-Small Cell Lung Cancer. BMC Cancer 2015, 15, 731. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Xing, S.; Peng, A.; Yu, Z. NORAD Accelerates Chemo-Resistance of Non-Small-Cell Lung Cancer via Targeting at MiR-129-1-3p/SOX4 Axis. Biosci. Rep. 2020, 40, BSR20193489. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.; Ye, L.; Zhao, D.; Zhao, D.; Chen, Q. Circular RNA PRMT5 Confers Cisplatin-Resistance via MiR-4458/REV3L Axis in Non-Small-Cell Lung Cancer. Cell Biol. Int. 2020, 44, 2416–2426. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Yang, J.; Yang, S. MicroRNA-103a-3p Potentiates Chemoresistance to Cisplatin in Non-Small Cell Lung Carcinoma by Targeting Neurofibromatosis 1. Exp. Ther. Med. 2020, 19, 1797–1805. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Huang, H.; Wang, L.; Liu, Y.; Wang, M.; Zhao, S.; Lu, G.; Kang, X. Cancer-Associated Fibroblasts Secreted MiR-103a-3p Suppresses Apoptosis and Promotes Cisplatin Resistance in Non-Small Cell Lung Cancer. Aging 2021, 13, 14456–14468. [Google Scholar] [CrossRef]
- Sun, D.-M.; Tang, B.-F.; Li, Z.-X.; Guo, H.-B.; Cheng, J.-L.; Song, P.-P.; Zhao, X. MiR-29c Reduces the Cisplatin Resistance of Non-Small Cell Lung Cancer Cells by Negatively Regulating the PI3K/Akt Pathway. Sci. Rep. 2018, 8, 8007. [Google Scholar] [CrossRef]
- Liu, B.; Wang, R.; Liu, H. Mir-126-5p Promotes Cisplatin Sensitivity of Non-Small-Cell Lung Cancer by Inhibiting ADAM9. BioMed Res. Int. 2021, 2021, 6622342. [Google Scholar] [CrossRef]
- Deng, H.; Qianqian, G.; Ting, J.; Aimin, Y. MiR-539 Enhances Chemosensitivity to Cisplatin in Non-Small Cell Lung Cancer by Targeting DCLK1. Biomed. Pharmacother. 2018, 106, 1072–1081. [Google Scholar] [CrossRef]
- Ge, P.; Cao, L.; Zheng, M.; Yao, Y.; Wang, W.; Chen, X. LncRNA SNHG1 Contributes to the Cisplatin Resistance and Progression of NSCLC via MiR-330-5p/DCLK1 Axis. Exp. Mol. Pathol. 2021, 120, 104633. [Google Scholar] [CrossRef]
- Wei, J.; Gao, W.; Zhu, C.-J.; Liu, Y.-Q.; Mei, Z.; Cheng, T.; Shu, Y.-Q. Identification of Plasma MicroRNA-21 as a Biomarker for Early Detection and Chemosensitivity of Non-Small Cell Lung Cancer. Chin. J. Cancer 2011, 30, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Lu, X.; Liu, L.; Xu, J.; Feng, D.; Shu, Y. MiRNA-21: A Biomarker Predictive for Platinum-Based Adjuvant Chemotherapy Response in Patients with Non-Small Cell Lung Cancer. Cancer Biol. Ther. 2012, 13, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-L.; Wang, H.; Liu, J.; Wang, Z.-X. MicroRNA-21 (MiR-21) Expression Promotes Growth, Metastasis, and Chemo- or Radioresistance in Non-Small Cell Lung Cancer Cells by Targeting PTEN. Mol. Cell. Biochem. 2013, 372, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Huang, Y.; Chen, D.; He, J.; Zhu, W.; Zhang, Y.; Liu, X. Downregulation of MiR-21 Increases Cisplatin Sensitivity of Non-Small-Cell Lung Cancer. Cancer Genet. 2014, 207, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Fang, S.; Di, Y.; Ying, W.; Tan, Y.; Gu, W. Modulation of NF-ΚB/MiR-21/PTEN Pathway Sensitizes Non-Small Cell Lung Cancer to Cisplatin. PLoS ONE 2015, 10, e0121547. [Google Scholar] [CrossRef]
- Cao, L.; Chen, J.; Ou, B.; Liu, C.; Zou, Y.; Chen, Q. GAS5 Knockdown Reduces the Chemo-Sensitivity of Non-Small Cell Lung Cancer (NSCLC) Cell to Cisplatin (DDP) through Regulating MiR-21/PTEN Axis. Biomed. Pharmacother. 2017, 93, 570–579. [Google Scholar] [CrossRef]
- Shi, L.; Middleton, J.; Jeon, Y.-J.; Magee, P.; Veneziano, D.; Laganà, A.; Leong, H.-S.; Sahoo, S.; Fassan, M.; Booton, R.; et al. KRAS Induces Lung Tumorigenesis through MicroRNAs Modulation. Cell Death Dis. 2018, 9, 219. [Google Scholar] [CrossRef]
- Li, C.; Fan, K.; Qu, Y.; Zhai, W.; Huang, A.; Sun, X.; Xing, S. Deregulation of UCA1 Expression May Be Involved in the Development of Chemoresistance to Cisplatin in the Treatment of Non-Small-Cell Lung Cancer via Regulating the Signaling Pathway of MicroRNA-495/NRF2. J. Cell. Physiol. 2020, 235, 3721–3730. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and Drug Resistance: The Mechanistic Link and Clinical Implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef]
- Cai, J.; Guan, H.; Fang, L.; Yang, Y.; Zhu, X.; Yuan, J.; Wu, J.; Li, M. MicroRNA-374a Activates Wnt/Beta-Catenin Signaling to Promote Breast Cancer Metastasis. J. Clin. Investig. 2013, 123, 566–579. [Google Scholar] [CrossRef]
- Wang, X.; Chen, X.; Meng, Q.; Jing, H.; Lu, H.; Yang, Y.; Cai, L.; Zhao, Y. MiR-181b Regulates Cisplatin Chemosensitivity and Metastasis by Targeting TGFβR1/Smad Signaling Pathway in NSCLC. Sci. Rep. 2015, 5, 17618. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Meng, Q.; Qiao, W.; Ma, R.; Ju, W.; Hu, J.; Lu, H.; Cui, J.; Jin, Z.; Zhao, Y.; et al. MiR-181b/Notch2 Overcome Chemoresistance by Regulating Cancer Stem Cell-like Properties in NSCLC. Stem Cell Res. Ther. 2018, 9, 327. [Google Scholar] [CrossRef] [PubMed]
- Hua, L.; Zhu, G.; Wei, J. MicroRNA-1 Overexpression Increases Chemosensitivity of Non-Small Cell Lung Cancer Cells by Inhibiting Autophagy Related 3-Mediated Autophagy. Cell Biol. Int. 2018, 42, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yuwen, D.; Chen, J.; Zheng, B.; Gao, J.; Fan, M.; Xue, W.; Wang, Y.; Li, W.; Shu, Y.; et al. Exosomal Transfer Of Cisplatin-Induced MiR-425-3p Confers Cisplatin Resistance In NSCLC Through Activating Autophagy. Int. J. Nanomed. 2019, 14, 8121–8132. [Google Scholar] [CrossRef]
- Rébé, C.; Demontoux, L.; Pilot, T.; Ghiringhelli, F. Platinum Derivatives Effects on Anticancer Immune Response. Biomolecules 2019, 10, 13. [Google Scholar] [CrossRef]
- Hato, S.V.; Khong, A.; de Vries, I.J.M.; Lesterhuis, W.J. Molecular Pathways: The Immunogenic Effects of Platinum-Based Chemotherapeutics. Clin. Cancer Res. 2014, 20, 2831–2837. [Google Scholar] [CrossRef]
- Fujita, Y.; Yagishita, S.; Hagiwara, K.; Yoshioka, Y.; Kosaka, N.; Takeshita, F.; Fujiwara, T.; Tsuta, K.; Nokihara, H.; Tamura, T.; et al. The Clinical Relevance of the MiR-197/CKS1B/STAT3-Mediated PD-L1 Network in Chemoresistant Non-Small-Cell Lung Cancer. Mol. Ther. 2015, 23, 717–727. [Google Scholar] [CrossRef]
- Parra, E.R.; Villalobos, P.; Behrens, C.; Jiang, M.; Pataer, A.; Swisher, S.G.; William, W.N.; Zhang, J.; Lee, J.; Cascone, T.; et al. Effect of Neoadjuvant Chemotherapy on the Immune Microenvironment in Non–Small Cell Lung Carcinomas as Determined by Multiplex Immunofluorescence and Image Analysis Approaches. J. Immunother. Cancer 2018, 6, 48. [Google Scholar] [CrossRef]
- Zhang, P.; Ma, Y.; Lv, C.; Huang, M.; Li, M.; Dong, B.; Liu, X.; An, G.; Zhang, W.; Zhang, J.; et al. Upregulation of Programmed Cell Death Ligand 1 Promotes Resistance Response in Non-small-cell Lung Cancer Patients Treated with Neo-adjuvant Chemotherapy. Cancer Sci. 2016, 107, 1563–1571. [Google Scholar] [CrossRef]
- Shin, J.; Chung, J.-H.; Kim, S.H.; Lee, K.S.; Suh, K.J.; Lee, J.Y.; Kim, J.-W.; Lee, J.-O.; Kim, J.-W.; Kim, Y.-J.; et al. Effect of Platinum-Based Chemotherapy on PD-L1 Expression on Tumor Cells in Non-Small Cell Lung Cancer. Cancer Res. Treat. 2019, 51, 1086–1097. [Google Scholar] [CrossRef]
- Guo, L.; Song, P.; Xue, X.; Guo, C.; Han, L.; Fang, Q.; Ying, J.; Gao, S.; Li, W. Variation of Programmed Death Ligand 1 Expression After Platinum-Based Neoadjuvant Chemotherapy in Lung Cancer. J. Immunother. 2019, 42, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Fournel, L.; Wu, Z.; Stadler, N.; Damotte, D.; Lococo, F.; Boulle, G.; Ségal-Bendirdjian, E.; Bobbio, A.; Icard, P.; Trédaniel, J.; et al. Cisplatin Increases PD-L1 Expression and Optimizes Immune Check-Point Blockade in Non-Small Cell Lung Cancer. Cancer Lett. 2019, 464, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Wakita, D.; Iwai, T.; Harada, S.; Suzuki, M.; Yamamoto, K.; Sugimoto, M. Cisplatin Augments Antitumor T-Cell Responses Leading to a Potent Therapeutic Effect in Combination With PD-L1 Blockade. Anticancer Res. 2019, 39, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Li, H.; Chen, T.; Ren, H.; Shi, P.; Chen, M. LncRNA MALAT1 Depressed Chemo-Sensitivity of NSCLC Cells through Directly Functioning on MiR-197-3p/P120 Catenin Axis. Mol. Cells 2019, 42, 270–283. [Google Scholar] [CrossRef]
- Yin, J.; Zhao, J.; Hu, W.; Yang, G.; Yu, H.; Wang, R.; Wang, L.; Zhang, G.; Fu, W.; Dai, L.; et al. Disturbance of the Let-7/LIN28 Double-Negative Feedback Loop Is Associated with Radio- and Chemo-Resistance in Non-Small Cell Lung Cancer. PLoS ONE 2017, 12, e0172787. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, J.; Zhu, R.; Zhang, J.; Zhang, Y. DLX6-AS1 Activated by H3K4me1 Enhanced Secondary Cisplatin Resistance of Lung Squamous Cell Carcinoma through Modulating MiR-181a-5p/MiR-382-5p/CELF1 Axis. Sci. Rep. 2021, 11, 21014. [Google Scholar] [CrossRef]
- Wang, C.; Wang, S.; Ma, F.; Zhang, W. MiRNA-328 Overexpression Confers Cisplatin Resistance in Non-small Cell Lung Cancer via Targeting of PTEN. Mol. Med. Rep. 2018, 18, 4563–4570. [Google Scholar] [CrossRef]
- Jiang, Z.; Yin, J.; Fu, W.; Mo, Y.; Pan, Y.; Dai, L.; Huang, H.; Li, S.; Zhao, J. MiRNA 17 Family Regulates Cisplatin-Resistant and Metastasis by Targeting TGFbetaR2 in NSCLC. PLoS ONE 2014, 9, e94639. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Li, B.T.; Chaft, J.E.; Kris, M.G. Chemotherapy Remains an Essential Element of Personalized Care for Persons with Lung Cancers. Ann. Oncol. 2016, 27, 1829–1835. [Google Scholar] [CrossRef]
- Lin, X.; Lai, X.; Feng, W.; Yu, X.; Gu, Q.; Zheng, X. MiR-30a Sensitized Lung Cancer against Neoadjuvant Chemotherapy by Depressing Autophagy. Jpn. J. Clin. Oncol. 2021, 51, 675–684. [Google Scholar] [CrossRef]
- Xu, X.; Jin, S.; Ma, Y.; Fan, Z.; Yan, Z.; Li, W.; Song, Q.; You, W.; Lyu, Z.; Song, Y.; et al. MiR-30a-5p Enhances Paclitaxel Sensitivity in Non-Small Cell Lung Cancer through Targeting BCL-2 Expression. J. Mol. Med. 2017, 95, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Fang, L.; Huang, Y.; Li, R.; Xu, X.; Hu, Z.; Zhang, L.; Yang, Y.; Zhu, X.; Zhang, H.; et al. Simultaneous Overactivation of Wnt/β-Catenin and TGFβ Signalling by MiR-128-3p Confers Chemoresistance-Associated Metastasis in NSCLC. Nat. Commun. 2017, 8, 15870. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhou, X.; Chen, Y.; Huang, Y.; He, J.; Luo, H. MiR-186-5p Targeting SIX1 Inhibits Cisplatin Resistance in Non-Small-Cell Lung Cancer Cells (NSCLCs). Neoplasma 2020, 67, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Zhang, Z.; Sun, L.; Fang, Y.; Xu, X.; Zhou, G. MiR-186 Regulates Chemo-Sensitivity to Paclitaxel via Targeting MAPT in Non-Small Cell Lung Cancer (NSCLC). Mol. Biosyst. 2016, 12, 3417–3424. [Google Scholar] [CrossRef]
- Chatterjee, A.; Chattopadhyay, D.; Chakrabarti, G. MiR-17-5p Downregulation Contributes to Paclitaxel Resistance of Lung Cancer Cells through Altering Beclin1 Expression. PLoS ONE 2014, 9, e95716. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Chattopadhyay, D.; Chakrabarti, G. MiR-16 Targets Bcl-2 in Paclitaxel-Resistant Lung Cancer Cells and Overexpression of MiR-16 along with MiR-17 Causes Unprecedented Sensitivity by Simultaneously Modulating Autophagy and Apoptosis. Cell. Signal. 2015, 27, 189–203. [Google Scholar] [CrossRef]
- Yang, L.-Z.; Lei, C.-C.; Zhao, Y.-P.; Sun, H.-W.; Yu, Q.-H.; Yang, E.-J.; Zhan, X. MicroRNA-34c-3p Target Inhibiting NOTCH1 Suppresses Chemosensitivity and Metastasis of Non-Small Cell Lung Cancer. J. Int. Med. Res. 2020, 48, 300060520904847. [Google Scholar] [CrossRef]
- Fu, W.-F.; Chen, W.-B.; Dai, L.; Yang, G.-P.; Jiang, Z.-Y.; Pan, L.; Zhao, J.; Chen, G. Inhibition of MiR-141 Reverses Cisplatin Resistance in Non-Small Cell Lung Cancer Cells via Upregulation of Programmed Cell Death Protein 4. Eur. Rev. Med. Pharmacol. Sci 2016, 20, 2565–2572. [Google Scholar]
- Wang, D.; Ma, J.; Ji, X.; Xu, F.; Wei, Y. MiR-141 Regulation of EIF4E Expression Affects Docetaxel Chemoresistance of Non-Small Cell Lung Cancer. Oncol. Rep. 2017, 37, 608–616. [Google Scholar] [CrossRef]
- Hao, G.-J.; Hao, H.-J.; Ding, Y.-H.; Wen, H.; Li, X.-F.; Wang, Q.-R.; Zhang, B.-B. Suppression of EIF4G2 by MiR-379 Potentiates the Cisplatin Chemosensitivity in Nonsmall Cell Lung Cancer Cells. FEBS Lett. 2017, 591, 636–645. [Google Scholar] [CrossRef]
- Zheng, S.; Wang, C.; Yan, H.; Du, Y. Blocking Hsa_circ_0074027 Suppressed Non-Small Cell Lung Cancer Chemoresistance via the MiR-379-5p/IGF1 Axis. Bioengineered 2021, 12, 8347–8357. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.-H.; Yang, J.C.-H.; Mok, T.S.; Loong, H.H. Overview of Current Systemic Management of EGFR-Mutant NSCLC. Ann. Oncol. 2018, 29, i3–i9. [Google Scholar] [CrossRef] [PubMed]
- Morin, M.J. From Oncogene to Drug: Development of Small Molecule Tyrosine Kinase Inhibitors as Anti-Tumor and Anti-Angiogenic Agents. Oncogene 2000, 19, 6574–6583. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Yang, J.C.-H.; Yamamoto, N.; O’Byrne, K.; Hirsh, V.; Mok, T.; Geater, S.L.; Orlov, S.; Tsai, C.-M.; Boyer, M.; et al. Phase III Study of Afatinib or Cisplatin plus Pemetrexed in Patients with Metastatic Lung Adenocarcinoma with EGFR Mutations. J. Clin. Oncol. 2013, 31, 3327–3334. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, Y.-L.; Chen, G.; Feng, J.; Liu, X.-Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus Chemotherapy as First-Line Treatment for Patients with Advanced EGFR Mutation-Positive Non-Small-Cell Lung Cancer (OPTIMAL, CTONG-0802): A Multicentre, Open-Label, Randomised, Phase 3 Study. Lancet Oncol. 2011, 12, 735–742. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.-L.; Thongprasert, S.; Yang, C.-H.; Chu, D.-T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or Carboplatin-Paclitaxel in Pulmonary Adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Cooper, A.J.; Sequist, L.V.; Lin, J.J. Third-Generation EGFR and ALK Inhibitors: Mechanisms of Resistance and Management. Nat. Rev. Clin. Oncol. 2022, 19, 499–514. [Google Scholar] [CrossRef]
- Passaro, A.; Jänne, P.A.; Mok, T.; Peters, S. Overcoming Therapy Resistance in EGFR-Mutant Lung Cancer. Nat. Cancer 2021, 2, 377–391. [Google Scholar] [CrossRef]
- Cao, X.; Lai, S.; Hu, F.; Li, G.; Wang, G.; Luo, X.; Fu, X.; Hu, J. MiR-19a Contributes to Gefitinib Resistance and Epithelial Mesenchymal Transition in Non-Small Cell Lung Cancer Cells by Targeting c-Met. Sci. Rep. 2017, 7, 2939. [Google Scholar] [CrossRef]
- Shen, H.; Zhu, F.; Liu, J.; Xu, T.; Pei, D.; Wang, R.; Qian, Y.; Li, Q.; Wang, L.; Shi, Z.; et al. Alteration in Mir-21/PTEN Expression Modulates Gefitinib Resistance in Non-Small Cell Lung Cancer. PLoS ONE 2014, 9, e103305. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, A.; Capula, M.; Minari, R.; Mazzaschi, G.; Gregori, A.; El Hassouni, B.; Papini, F.; Bordi, P.; Verzè, M.; Avan, A.; et al. Dynamic Evaluation of Circulating MiRNA Profile in EGFR-Mutated NSCLC Patients Treated with EGFR-TKIs. Cells 2021, 10, 1520. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Cui, J.; Guo, X.-T.; Cao, X.; Li, Q. Increased Expression of MiR-641 Contributes to Erlotinib Resistance in Non-Small-Cell Lung Cancer Cells by Targeting NF1. Cancer Med. 2018, 7, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Guo, Z.; Li, Y. Long Non-Coding RNA Prostate Cancer-Associated Transcript 6 Inhibited Gefitinib Sensitivity of Non-Small Cell Lung Cancer by Serving as a Competing Endogenous RNA of MiR-326 to up-Regulate Interferon-Alpha Receptor 2. Bioengineered 2022, 13, 3785–3796. [Google Scholar] [CrossRef]
- Shemesh, M.; Lochte, S.; Piehler, J.; Schreiber, G. IFNAR1 and IFNAR2 Play Distinct Roles in Initiating Type I Interferon–Induced JAK-STAT Signaling and Activating STATs. Sci. Signal. 2021, 14, eabe4627. [Google Scholar] [CrossRef]
- Gong, K.; Guo, G.; Panchani, N.; Bender, M.E.; Gerber, D.E.; Minna, J.D.; Fattah, F.; Gao, B.; Peyton, M.; Kernstine, K.; et al. EGFR Inhibition Triggers an Adaptive Response by Co-Opting Antiviral Signaling Pathways in Lung Cancer. Nat. Cancer 2020, 1, 394–409. [Google Scholar] [CrossRef]
- Ge, P.; Cao, L.; Chen, X.; Jing, R.; Yue, W. MiR-762 Activation Confers Acquired Resistance to Gefitinib in Non-Small Cell Lung Cancer. BMC Cancer 2019, 19, 1203. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, W.; Chang, H.; Han, Z.; Yu, X.; Zhang, T. Reciprocal Regulation of MiR-206 and IL-6/STAT3 Pathway Mediates IL6-Induced Gefitinib Resistance in EGFR-Mutant Lung Cancer Cells. J. Cell. Mol. Med. 2019, 23, 7331–7341. [Google Scholar] [CrossRef]
- Wu, K.; Li, J.; Qi, Y.; Zhang, C.; Zhu, D.; Liu, D.; Zhao, S. SNHG14 Confers Gefitinib Resistance in Non-Small Cell Lung Cancer by up-Regulating ABCB1 via Sponging MiR-206-3p. Biomed. Pharmacother. 2019, 116, 108995. [Google Scholar] [CrossRef]
- Jiao, D.; Jiang, C.; Zhu, L.; Zheng, J.; Liu, X.; Liu, X.; Chen, J.; Tang, X.; Chen, Q. MiR-1/133a and MiR-206/133b Clusters Overcome HGF Induced Gefitinib Resistance in Non-Small Cell Lung Cancers with EGFR Sensitive Mutations. J. Drug Target. 2021, 29, 1111–1117. [Google Scholar] [CrossRef]
- Liu, Y.-N.; Tsai, M.-F.; Wu, S.-G.; Chang, T.-H.; Tsai, T.-H.; Gow, C.-H.; Wang, H.-Y.; Shih, J.-Y. MiR-146b-5p Enhances the Sensitivity of NSCLC to EGFR Tyrosine Kinase Inhibitors by Regulating the IRAK1/NF-ΚB Pathway. Mol. Ther. Nucleic Acids 2020, 22, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Hu, W.; Pan, L.; Fu, W.; Dai, L.; Jiang, Z.; Zhang, F.; Zhao, J. Let-7 and MiR-17 Promote Self-renewal and Drive Gefitinib Resistance in Non-small Cell Lung Cancer. Oncol. Rep. 2019, 42, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; He, L.; Ma, J.; Zhang, J.; Wang, L.; Wang, J. The Relationship between MiR-17-5p, MiR-92a, and Let-7b Expression with Non-Small Cell Lung Cancer Targeted Drug Resistance. J. BUON 2017, 22, 454–461. [Google Scholar] [PubMed]
- Zhang, W.; Lin, J.; Wang, P.; Sun, J. MiR-17-5p down-Regulation Contributes to Erlotinib Resistance in Non-Small Cell Lung Cancer Cells. J. Drug Target. 2017, 25, 125–131. [Google Scholar] [CrossRef]
- Chen, R.; Qian, Z.; Xu, X.; Zhang, C.; Niu, Y.; Wang, Z.; Sun, J.; Zhang, X.; Yu, Y. Exosomes-Transmitted MiR-7 Reverses Gefitinib Resistance by Targeting YAP in Non-Small-Cell Lung Cancer. Pharmacol. Res. 2021, 165, 105442. [Google Scholar] [CrossRef]
- Zhou, G.; Zhang, F.; Guo, Y.; Huang, J.; Xie, Y.; Yue, S.; Chen, M.; Jiang, H.; Li, M. MiR-200c Enhances Sensitivity of Drug-Resistant Non-Small Cell Lung Cancer to Gefitinib by Suppression of PI3K/Akt Signaling Pathway and Inhibites Cell Migration via Targeting ZEB1. Biomed. Pharmacother. 2017, 85, 113–119. [Google Scholar] [CrossRef]
- Lin, C.-C.; Wu, C.-Y.; Tseng, J.T.; Hung, C.-H.; Wu, S.-Y.; Huang, Y.-T.; Chang, W.-Y.; Su, P.-L.; Su, W.-C. Extracellular Vesicle MiR-200c Enhances Gefitinib Sensitivity in Heterogeneous EGFR-Mutant NSCLC. Biomedicines 2021, 9, 243. [Google Scholar] [CrossRef]
- Hu, F.-Y.; Cao, X.-N.; Xu, Q.-Z.; Deng, Y.; Lai, S.-Y.; Ma, J.; Hu, J.-B. MiR-124 Modulates Gefitinib Resistance through SNAI2 and STAT3 in Non-Small Cell Lung Cancer. J. Huazhong Univ. Sci. Technolog. Med. Sci. 2016, 36, 839–845. [Google Scholar] [CrossRef]
- Yu, F.; Liu, J.-B.; Wu, Z.-J.; Xie, W.-T.; Zhong, X.-J.; Hou, L.-K.; Wu, W.; Lu, H.-M.; Jiang, X.-H.; Jiang, J.-J.; et al. Tumor Suppressive MicroRNA-124a Inhibits Stemness and Enhances Gefitinib Sensitivity of Non-Small Cell Lung Cancer Cells by Targeting Ubiquitin-Specific Protease 14. Cancer Lett. 2018, 427, 74–84. [Google Scholar] [CrossRef]
- Fan, D.; Yang, Y.; Zhang, W. A Novel Circ_MACF1/MiR-942-5p/TGFBR2 Axis Regulates the Functional Behaviors and Drug Sensitivity in Gefitinib-Resistant Non-Small Cell Lung Cancer Cells. BMC Pulm. Med. 2022, 22, 27. [Google Scholar] [CrossRef]
- Zhang, W.; Dong, Y.-Z.; Du, X.; Peng, X.-N.; Shen, Q.-M. MiRNA-153-3p Promotes Gefitinib-Sensitivity in Non-Small Cell Lung Cancer by Inhibiting ATG5 Expression and Autophagy. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 2444–2452. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Lin, J.; Lin, D.; Zou, C.; Kurata, J.; Lin, R.; He, Z.; Su, Y. Down-Regulation of MiR-214 Reverses Erlotinib Resistance in Non-Small-Cell Lung Cancer through up-Regulating LHX6 Expression. Sci. Rep. 2017, 7, 781. [Google Scholar] [CrossRef] [PubMed]
- Vadla, G.P.; Daghat, B.; Patterson, N.; Ahmad, V.; Perez, G.; Garcia, A.; Manjunath, Y.; Kaifi, J.T.; Li, G.; Chabu, C.Y. Combining Plasma Extracellular Vesicle Let-7b-5p, MiR-184 and Circulating MiR-22-3p Levels for NSCLC Diagnosis and Drug Resistance Prediction. Sci. Rep. 2022, 12, 6693. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, C.; Wang, Z.; Liu, J.; Sun, W.; Shen, K.; Lv, Y.; Zhu, S.; Zhan, P.; Lv, T.; et al. Elevated Exosome-Derived MiRNAs Predict Osimertinib Resistance in Non-Small Cell Lung Cancer. Cancer Cell. Int. 2021, 21, 428. [Google Scholar] [CrossRef]
- Delaney, G.P.; Barton, M.B. Evidence-Based Estimates of the Demand for Radiotherapy. Clin. Oncol. 2015, 27, 70–76. [Google Scholar] [CrossRef]
- Hayman, T.J.; Glazer, P.M. Regulation of the Cell-Intrinsic DNA Damage Response by the Innate Immune Machinery. Int. J. Mol. Sci. 2021, 22, 12761. [Google Scholar] [CrossRef]
- Castle, K.D.; Kirsch, D.G. Establishing the Impact of Vascular Damage on Tumor Response to High-Dose Radiation Therapy. Cancer Res. 2019, 79, 5685–5692. [Google Scholar] [CrossRef]
- Chen, X.; Xu, Y.; Jiang, L.; Tan, Q. MiRNA-218-5p Increases Cell Sensitivity by Inhibiting PRKDC Activity in Radiation-Resistant Lung Carcinoma Cells. Thorac. Cancer 2021, 12, 1549–1557. [Google Scholar] [CrossRef]
- Wang, X.-C.; Du, L.-Q.; Tian, L.-L.; Wu, H.-L.; Jiang, X.-Y.; Zhang, H.; Li, D.-G.; Wang, Y.-Y.; Wu, H.-Y.; She, Y.; et al. Expression and Function of MiRNA in Postoperative Radiotherapy Sensitive and Resistant Patients of Non-Small Cell Lung Cancer. Lung Cancer 2011, 72, 92–99. [Google Scholar] [CrossRef]
- Li, L.; Liu, H.; Du, L.; Xi, P.; Wang, Q.; Li, Y.; Liu, D. MiR-449a Suppresses LDHA-Mediated Glycolysis to Enhance the Sensitivity of Non-Small Cell Lung Cancer Cells to Ionizing Radiation. Oncol. Res. 2018, 26, 547–556. [Google Scholar] [CrossRef]
- Jin, Y.; Su, Z.; Sheng, H.; Li, K.; Yang, B.; Li, S. Circ_0086720 Knockdown Strengthens the Radiosensitivity of Non-Small Cell Lung Cancer via Mediating the MiR-375/SPIN1 Axis. Neoplasma 2021, 68, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Qin, P.; Li, Y.; Liu, J.; Wang, N. Knockdown of LINC00473 Promotes Radiosensitivity of Non-Small Cell Lung Cancer Cells via Sponging MiR-513a-3p. Free Radic. Res. 2020, 54, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ren, P.; Zhang, Y.; Gong, B.; Yu, D.; Sun, X. Long Non-coding RNA GAS5 Increases the Radiosensitivity of A549 Cells through Interaction with the MiR-21/PTEN/Akt Axis. Oncol. Rep. 2020, 43, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.-P.; He, C.-Y.; Zhu, Z.-T. Role of MicroRNA-21 in Radiosensitivity in Non-Small Cell Lung Cancer Cells by Targeting PDCD4 Gene. Oncotarget 2017, 8, 23675–23689. [Google Scholar] [CrossRef]
- Jiang, S.; Wang, R.; Yan, H.; Jin, L.; Dou, X.; Chen, D. MicroRNA-21 Modulates Radiation Resistance through Upregulation of Hypoxia-Inducible Factor-1α-Promoted Glycolysis in Non-Small Cell Lung Cancer Cells. Mol. Med. Rep. 2016, 13, 4101–4107. [Google Scholar] [CrossRef]
- Ma, Y.; Xia, H.; Liu, Y.; Li, M. Silencing MiR-21 Sensitizes Non-Small Cell Lung Cancer A549 Cells to Ionizing Radiation through Inhibition of PI3K/Akt. Biomed Res. Int. 2014, 2014, 617868. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Zhang, Z.-B.; Zhao, J.; Tan, X.-G.; Luo, J.-C. Overexpression of MiRNA-21 Promotes Radiation-Resistance of Non-Small Cell Lung Cancer. Radiat. Oncol. 2013, 8, 146. [Google Scholar] [CrossRef]
- He, Z.; Liu, Y.; Xiao, B.; Qian, X. MiR-25 Modulates NSCLC Cell Radio-Sensitivity through Directly Inhibiting BTG2 Expression. Biochem. Biophys. Res. Commun. 2015, 457, 235–241. [Google Scholar] [CrossRef]
- McDermott, M.; Eustace, A.J.; Busschots, S.; Breen, L.; Crown, J.; Clynes, M.; O’Donovan, N.; Stordal, B. In Vitro Development of Chemotherapy and Targeted Therapy Drug-Resistant Cancer Cell Lines: A Practical Guide with Case Studies. Front. Oncol. 2014, 4, 40. [Google Scholar] [CrossRef]
- Hafner, M.; Niepel, M.; Chung, M.; Sorger, P.K. Growth Rate Inhibition Metrics Correct for Confounders in Measuring Sensitivity to Cancer Drugs. Nat. Methods 2016, 13, 521–527. [Google Scholar] [CrossRef]
- Pribluda, A.; de la Cruz, C.C.; Jackson, E.L. Intratumoral Heterogeneity: From Diversity Comes Resistance. Clin. Cancer Res. 2015, 21, 2916–2923. [Google Scholar] [CrossRef] [PubMed]
- Guarize, J.; Bianchi, F.; Marino, E.; Belloni, E.; Vecchi, M.; Donghi, S.; Lo Iacono, G.; Casadio, C.; Cuttano, R.; Barberis, M.; et al. MicroRNA Expression Profile in Primary Lung Cancer Cells Lines Obtained by Endobronchial Ultrasound Transbronchial Needle Aspiration. J. Thorac. Dis. 2018, 10, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Roscilli, G.; De Vitis, C.; Ferrara, F.F.; Noto, A.; Cherubini, E.; Ricci, A.; Mariotta, S.; Giarnieri, E.; Giovagnoli, M.R.; Torrisi, M.R.; et al. Human Lung Adenocarcinoma Cell Cultures Derived from Malignant Pleural Effusions as Model System to Predict Patients Chemosensitivity. J. Transl. Med. 2016, 14, 61. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Krawczyk, E.; Suprynowicz, F.A.; Palechor-Ceron, N.; Yuan, H.; Dakic, A.; Simic, V.; Zheng, Y.-L.; Sripadhan, P.; Chen, C.; et al. Conditional Reprogramming and Long-Term Expansion of Normal and Tumor Cells from Human Biospecimens. Nat. Protoc. 2017, 12, 439–451. [Google Scholar] [CrossRef]
- Kodack, D.P.; Farago, A.F.; Dastur, A.; Held, M.A.; Dardaei, L.; Friboulet, L.; von Flotow, F.; Damon, L.J.; Lee, D.; Parks, M.; et al. Primary Patient-Derived Cancer Cells and Their Potential for Personalized Cancer Patient Care. Cell. Rep. 2017, 21, 3298–3309. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia Enables Predictive Modelling of Anticancer Drug Sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Drost, J.; Clevers, H. Organoids in Cancer Research. Nat. Rev. Cancer 2018, 18, 407–418. [Google Scholar] [CrossRef]
- Khan, A.A.; Betel, D.; Miller, M.L.; Sander, C.; Leslie, C.S.; Marks, D.S. Transfection of Small RNAs Globally Perturbs Gene Regulation by Endogenous MicroRNAs. Nat. Biotechnol. 2009, 27, 549–555. [Google Scholar] [CrossRef]
- Thomson, D.W.; Bracken, C.P.; Goodall, G.J. Experimental Strategies for MicroRNA Target Identification. Nucleic Acids Res. 2011, 39, 6845–6853. [Google Scholar] [CrossRef]
- Krützfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of MicroRNAs in Vivo with “Antagomirs”. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef]
- Ebert, M.S.; Sharp, P.A. MicroRNA Sponges: Progress and Possibilities. RNA 2010, 16, 2043–2050. [Google Scholar] [CrossRef] [PubMed]
- Aquino-Jarquin, G. Emerging Role of CRISPR/Cas9 Technology for MicroRNAs Editing in Cancer Research. Cancer Res. 2017, 77, 6812–6817. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Ajay, S.S.; Yook, J.I.; Kim, H.S.; Hong, S.H.; Kim, N.H.; Dhanasekaran, S.M.; Chinnaiyan, A.M.; Athey, B.D. New Class of MicroRNA Targets Containing Simultaneous 5′-UTR and 3′-UTR Interaction Sites. Genome Res. 2009, 19, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Hausser, J.; Syed, A.P.; Bilen, B.; Zavolan, M. Analysis of CDS-Located MiRNA Target Sites Suggests That They Can Effectively Inhibit Translation. Genome Res. 2013, 23, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Mayr, C.; Bartel, D.P. Widespread Shortening of 3′UTRs by Alternative Cleavage and Polyadenylation Activates Oncogenes in Cancer Cells. Cell 2009, 138, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Messina, A.; Langlet, F.; Chachlaki, K.; Roa, J.; Rasika, S.; Jouy, N.; Gallet, S.; Gaytan, F.; Parkash, J.; Tena-Sempere, M.; et al. A MicroRNA Switch Regulates the Rise in Hypothalamic GnRH Production before Puberty. Nat. Neurosci. 2016, 19, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Gengenbacher, N.; Singhal, M.; Augustin, H.G. Preclinical Mouse Solid Tumour Models: Status Quo, Challenges and Perspectives. Nat. Rev. Cancer 2017, 17, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Rottenberg, S.; Borst, P. Drug Resistance in the Mouse Cancer Clinic. Drug Resist. Updat. 2012, 15, 81–89. [Google Scholar] [CrossRef]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinská, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Mælandsmo, G.M.; et al. Patient-Derived Xenograft Models: An Emerging Platform for Translational Cancer Research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef]
- Gómez-Cuadrado, L.; Tracey, N.; Ma, R.; Qian, B.; Brunton, V.G. Mouse Models of Metastasis: Progress and Prospects. Dis. Model. Mech. 2017, 10, 1061–1074. [Google Scholar] [CrossRef]
- Romani, P.; Nirchio, N.; Arboit, M.; Barbieri, V.; Tosi, A.; Michielin, F.; Shibuya, S.; Benoist, T.; Wu, D.; Hindmarch, C.C.T.; et al. Mitochondrial Fission Links ECM Mechanotransduction to Metabolic Redox Homeostasis and Metastatic Chemotherapy Resistance. Nat. Cell Biol. 2022, 24, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Sanmamed, M.F.; Chester, C.; Melero, I.; Kohrt, H. Defining the Optimal Murine Models to Investigate Immune Checkpoint Blockers and Their Combination with Other Immunotherapies. Ann. Oncol. 2016, 27, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- de Rie, D.; Abugessaisa, I.; Alam, T.; Arner, E.; Arner, P.; Ashoor, H.; Åström, G.; Babina, M.; Bertin, N.; Burroughs, A.M.; et al. An Integrated Expression Atlas of MiRNAs and Their Promoters in Human and Mouse. Nat. Biotechnol. 2017, 35, 872–878. [Google Scholar] [CrossRef] [PubMed]
- De La Rochere, P.; Guil-Luna, S.; Decaudin, D.; Azar, G.; Sidhu, S.S.; Piaggio, E. Humanized Mice for the Study of Immuno-Oncology. Trends Immunol. 2018, 39, 748–763. [Google Scholar] [CrossRef] [PubMed]
- Montani, F.; Marzi, M.J.; Dezi, F.; Dama, E.; Carletti, R.M.; Bonizzi, G.; Bertolotti, R.; Bellomi, M.; Rampinelli, C.; Maisonneuve, P.; et al. MiR-Test: A Blood Test for Lung Cancer Early Detection. J. Natl. Cancer Inst. 2015, 107, djv063. [Google Scholar] [CrossRef]
- Dama, E.; Melocchi, V.; Mazzarelli, F.; Colangelo, T.; Cuttano, R.; Candia, L.D.; Ferretti, G.M.; Taurchini, M.; Graziano, P.; Bianchi, F. Non-Coding RNAs as Prognostic Biomarkers: A Mirna Signature Specific for Aggressive Early-Stage Lung Adenocarcinomas. Non-Coding RNA 2020, 6, 48. [Google Scholar] [CrossRef]
- Salgia, R.; Kulkarni, P. The Genetic/Non-Genetic Duality of Drug “Resistance” in Cancer. Trends Cancer 2018, 4, 110–118. [Google Scholar] [CrossRef]
- de Bruin, E.C.; McGranahan, N.; Mitter, R.; Salm, M.; Wedge, D.C.; Yates, L.; Jamal-Hanjani, M.; Shafi, S.; Murugaesu, N.; Rowan, A.J.; et al. Spatial and Temporal Diversity in Genomic Instability Processes Defines Lung Cancer Evolution. Science 2014, 346, 251–256. [Google Scholar] [CrossRef]
- Zhang, J.; Fujimoto, J.; Wedge, D.C.; Song, X.; Seth, S.; Chow, C.W.; Cao, Y.; Gumbs, C.; Gold, K.A.; Kalhor, N.; et al. Intratumor Heterogeneity in Localized Lung Adenocarcinomas Delineated by Multiregion Sequencing. Science 2014, 346, 256–259. [Google Scholar] [CrossRef]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef]
- Aran, D.; Sirota, M.; Butte, A.J. Systematic Pan-Cancer Analysis of Tumour Purity. Nat. Commun. 2015, 6, 8971. [Google Scholar] [CrossRef]
- Barkley, D.; Moncada, R.; Pour, M.; Liberman, D.A.; Dryg, I.; Werba, G.; Wang, W.; Baron, M.; Rao, A.; Xia, B.; et al. Cancer Cell States Recur across Tumor Types and Form Specific Interactions with the Tumor Microenvironment. Nat. Genet. 2022, 54, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Cazet, A.S.; Hui, M.N.; Elsworth, B.L.; Wu, S.Z.; Roden, D.; Chan, C.-L.; Skhinas, J.N.; Collot, R.; Yang, J.; Harvey, K.; et al. Targeting Stromal Remodeling and Cancer Stem Cell Plasticity Overcomes Chemoresistance in Triple Negative Breast Cancer. Nat. Commun. 2018, 9, 2897. [Google Scholar] [CrossRef] [PubMed]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem Cell-Associated Heterogeneity in Glioblastoma Results from Intrinsic Tumor Plasticity Shaped by the Microenvironment. Nat. Commun. 2019, 10, 1787. [Google Scholar] [CrossRef] [PubMed]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e24. [Google Scholar] [CrossRef]
- Hirsch, F.R.; Scagliotti, G.V.; Mulshine, J.L.; Kwon, R.; Curran, W.J.; Wu, Y.-L.; Paz-Ares, L. Lung Cancer: Current Therapies and New Targeted Treatments. Lancet 2017, 389, 299–311. [Google Scholar] [CrossRef]
- Esposito, M.; Ganesan, S.; Kang, Y. Emerging Strategies for Treating Metastasis. Nat. Cancer 2021, 2, 258–270. [Google Scholar] [CrossRef]
- Biswas, A.K.; Han, S.; Tai, Y.; Ma, W.; Coker, C.; Quinn, S.A.; Shakri, A.R.; Zhong, T.J.; Scholze, H.; Lagos, G.G.; et al. Targeting S100A9-ALDH1A1-Retinoic Acid Signaling to Suppress Brain Relapse in EGFR-Mutant Lung Cancer. Cancer Discov. 2022, 12, 1002–1021. [Google Scholar] [CrossRef]
- Rinaldi, G.; Pranzini, E.; Van Elsen, J.; Broekaert, D.; Funk, C.M.; Planque, M.; Doglioni, G.; Altea-Manzano, P.; Rossi, M.; Geldhof, V.; et al. In Vivo Evidence for Serine Biosynthesis-Defined Sensitivity of Lung Metastasis, but Not of Primary Breast Tumors, to MTORC1 Inhibition. Mol. Cell 2021, 81, 386–397.e7. [Google Scholar] [CrossRef]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The Biology and Management of Non-Small Cell Lung Cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef]
- Shukuya, T.; Ghai, V.; Amann, J.M.; Okimoto, T.; Shilo, K.; Kim, T.-K.; Wang, K.; Carbone, D.P. Circulating MicroRNAs and Extracellular Vesicle-Containing MicroRNAs as Response Biomarkers of Anti-Programmed Cell Death Protein 1 or Programmed Death-Ligand 1 Therapy in NSCLC. J. Thorac. Oncol. 2020, 15, 1773–1781. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.-X.; Yu, R.; Wu, X.; Wu, S.-Y.; Pi, C.; Chen, Z.-H.; Zhang, X.-C.; Gao, C.-Y.; Shao, Y.W.; Liu, L.; et al. Correlation of Plasma Exosomal MicroRNAs with the Efficacy of Immunotherapy in EGFR/ALK Wild-Type Advanced Non-Small Cell Lung Cancer. J. Immunother. Cancer 2020, 8, e000376. [Google Scholar] [CrossRef]
- Boeri, M.; Milione, M.; Proto, C.; Signorelli, D.; Lo Russo, G.; Galeone, C.; Verri, C.; Mensah, M.; Centonze, G.; Martinetti, A.; et al. Circulating MiRNAs and PD-L1 Tumor Expression Are Associated with Survival in Advanced NSCLC Patients Treated with Immunotherapy: A Prospective Study. Clin. Cancer Res. 2019, 25, 2166–2173. [Google Scholar] [CrossRef]
- Fan, J.; Yin, Z.; Xu, J.; Wu, F.; Huang, Q.; Yang, L.; Jin, Y.; Yang, G. Circulating MicroRNAs Predict the Response to Anti-PD-1 Therapy in Non-Small Cell Lung Cancer. Genomics 2020, 112, 2063–2071. [Google Scholar] [CrossRef] [PubMed]
- Halvorsen, A.R.; Sandhu, V.; Sprauten, M.; Flote, V.G.; Kure, E.H.; Brustugun, O.T.; Helland, Å. Circulating MicroRNAs Associated with Prolonged Overall Survival in Lung Cancer Patients Treated with Nivolumab. Acta Oncol. 2018, 57, 1225–1231. [Google Scholar] [CrossRef] [PubMed]
- Rajakumar, T.; Horos, R.; Jehn, J.; Schenz, J.; Muley, T.; Pelea, O.; Hofmann, S.; Kittner, P.; Kahraman, M.; Heuvelman, M.; et al. A Blood-Based MiRNA Signature with Prognostic Value for Overall Survival in Advanced Stage Non-Small Cell Lung Cancer Treated with Immunotherapy. NPJ Precis. Oncol. 2022, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- Chaft, J.E.; Shyr, Y.; Sepesi, B.; Forde, P.M. Preoperative and Postoperative Systemic Therapy for Operable Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2022, 40, 546–555. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Common Pathway/Signalling | Drug | miRNA | Reference (PMID) |
|---|---|---|---|
| Autophagy | Cisplatin | miR-1-3p | 29851226 |
| Cisplatin | miR-425-3p | 31632022 | |
| Gefitinib | miR-153-3p | 30964170 | |
| Cell cycle and DNA repair | Cisplatin | miR-92a-3p | 26482648 |
| Cisplatin | miR-4458 | 32808744 | |
| Radiotherapy | miR-218-5p | 33759399 | |
| Cisplatin | miR-17-5p | 26482648 | |
| Radiotherapy | miR-25-3p | 25576360 | |
| Drug transport | Cisplatin | miR-369-3p | 28511796 |
| Cisplatin | miR-495-3p | 24038379 | |
| Gefitinib | miR-206 | 31121484 | |
| EMT and/or Stem-like properties | Cisplatin | miR-128-3p | 28627514 |
| Cisplatin | miR-181b-5p | 26620926, 30470250 | |
| Gefitinib | miR-200c-3p | 27930974 | |
| Gefitinib | miR-124-3p | 29702194, 27924500 | |
| Gefitinib | Let-7 | 31233201 | |
| Gefitinib | miR-17-5p | 31233201 | |
| MET signalling | Gefitinib | miR-19a | 28592790 |
| Gefitinib | miR-206 | 33955799 | |
| Metabolism | Radiotherapy | MiR-449a | 28800787 |
| Radiotherapy | miR-21-5p | 27035555 | |
| PI3K/AKT signalling | Cisplatin | miR-29c-3p | 29789623 |
| Cisplatin | miR-126-5p | 34055989 | |
| Cisplatin | miR-539-5p | 30119173 | |
| Cisplatin | miR-21-5p | 22237007, 22956424, 28686971 | |
| Gefitinib | miR-21-5p | 25058005 | |
| Gefitinib | miR-206 | 33955799 | |
| Osimertinib | miR-184 | 35461372 | |
| Osimertinib | miR-22-3p | 35461372 | |
| Radiotherapy | miR-126-3p | 20728239 | |
| Gefitinib | miR-200c-3p | 27930974 | |
| Radiotherapy | miR-21-5p | 32020207, 24804226, 22956424 | |
| RAS signalling | Cisplatin | miR-103a-3p | 32104235 |
| Erlotinib | miR-641 | 29493886 | |
| STAT signalling | Gefitinib | miR-326 | 35081872 |
| Gefitinib | miR-762 | 25597412 | |
| Gefitinib | miR-206 | 31507089 | |
| Cisplatin | miR-197-3p | 25597412 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuttano, R.; Afanga, M.K.; Bianchi, F. MicroRNAs and Drug Resistance in Non-Small Cell Lung Cancer: Where Are We Now and Where Are We Going. Cancers 2022, 14, 5731. https://doi.org/10.3390/cancers14235731
Cuttano R, Afanga MK, Bianchi F. MicroRNAs and Drug Resistance in Non-Small Cell Lung Cancer: Where Are We Now and Where Are We Going. Cancers. 2022; 14(23):5731. https://doi.org/10.3390/cancers14235731
Chicago/Turabian StyleCuttano, Roberto, Miriam Kuku Afanga, and Fabrizio Bianchi. 2022. "MicroRNAs and Drug Resistance in Non-Small Cell Lung Cancer: Where Are We Now and Where Are We Going" Cancers 14, no. 23: 5731. https://doi.org/10.3390/cancers14235731
APA StyleCuttano, R., Afanga, M. K., & Bianchi, F. (2022). MicroRNAs and Drug Resistance in Non-Small Cell Lung Cancer: Where Are We Now and Where Are We Going. Cancers, 14(23), 5731. https://doi.org/10.3390/cancers14235731