
RAD51 Is Implicated in DNA Damage, Chemoresistance and Immune Dysregulation in Solid Tumors

Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Bioinformatics Data Sources
2.2. Cell Types
2.3. Cell Viability and Apoptosis
2.4. Modulation of RAD51 Expression
2.5. Detection of DNA Breaks and End Resection with Different DNA Breaking Agents, Inhibitor or Gene Modulation
2.6. Strand Exchange Assay
2.7. Evaluation of RAD51 Inhibitor, Alone or in the Presence of a DNA Breaking Agent, In Vivo
2.8. Impact on Expression Profile
2.9. Investigating Genomic Instability
2.10. Statistics and Reproducibility
3. Results
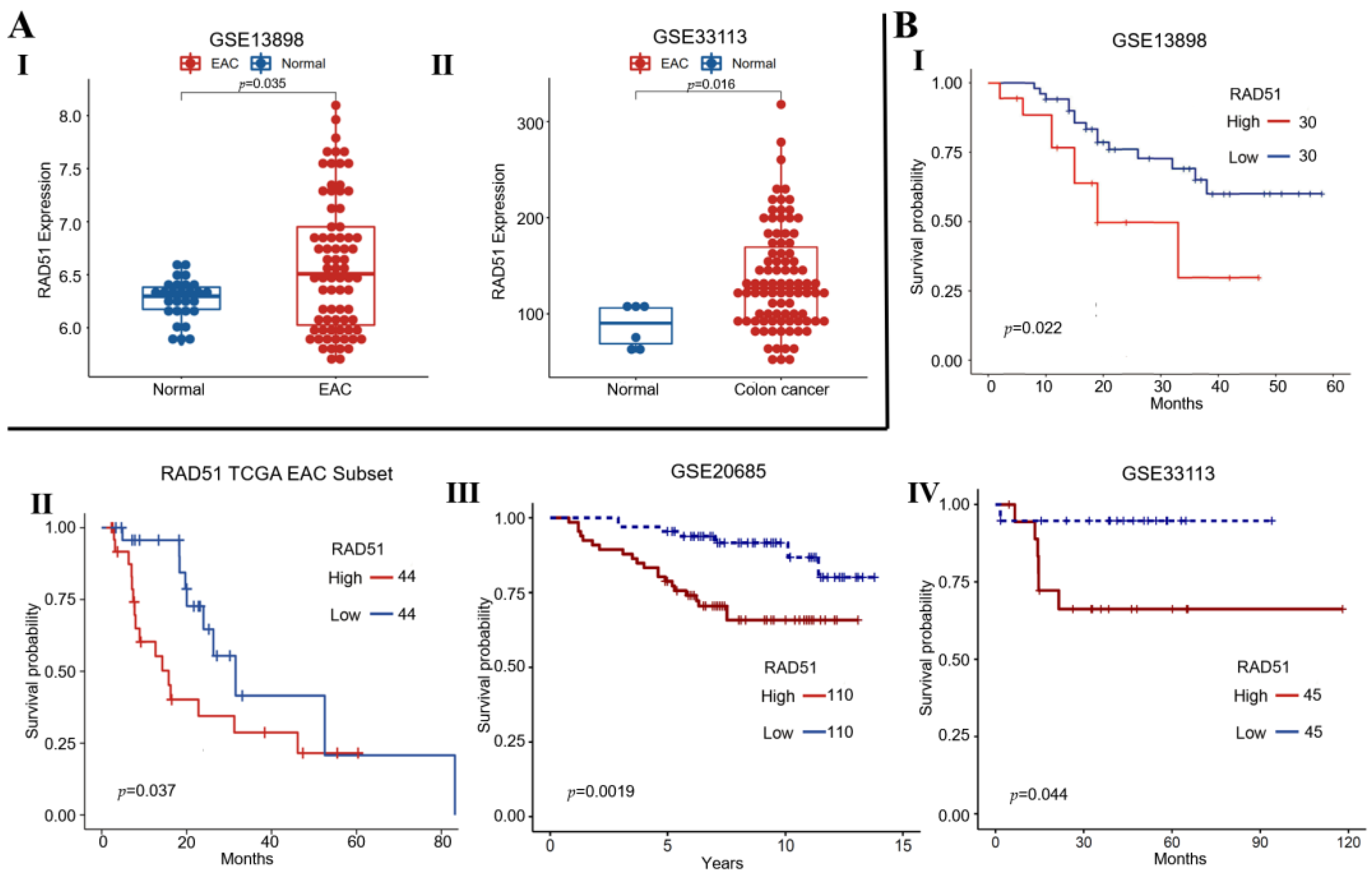
3.1. Elevated RAD51 Expression Correlates with Poor Survival
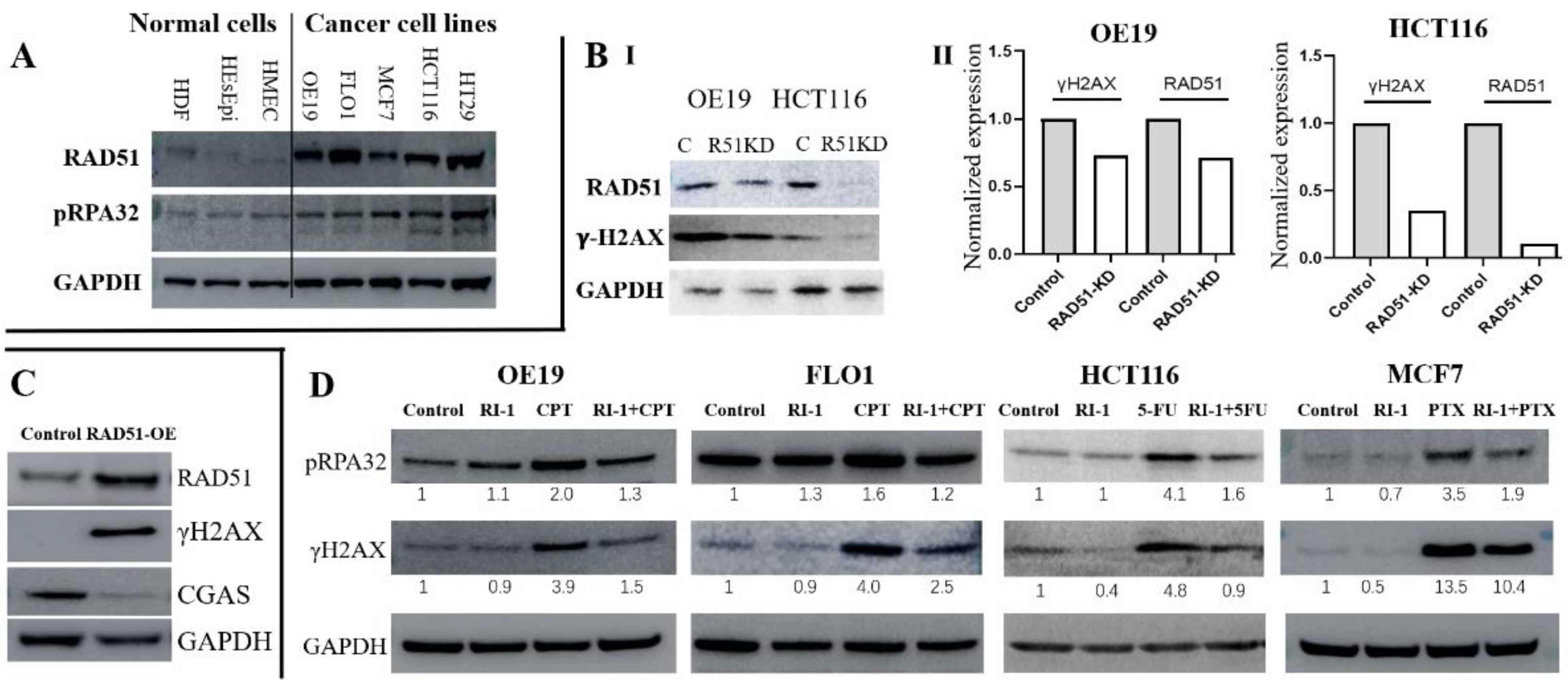
3.2. Elevated RAD51 Expression Contributes DNA Breaks in Solid Tumors
3.3. RAD51 Inhibitor Significantly Reduces DNA Breaking Agent-Induced Genomic Instability in Cancer Cell Lines
3.4. RAD51 Inhibitor Potentiates Cytotoxicity of DNA Breaking Agent in Cancer Cell Lines
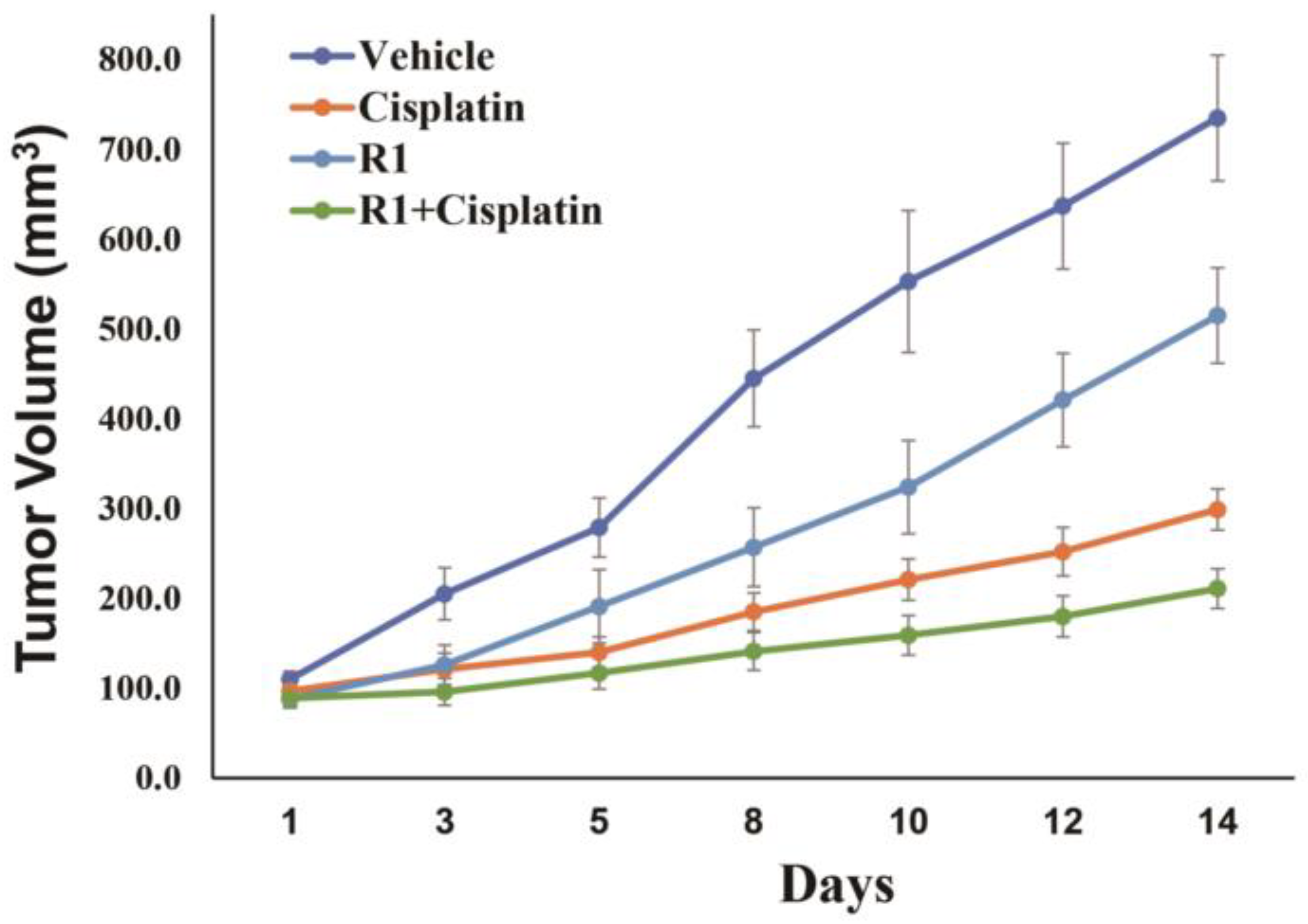
3.5. RAD51 Inhibitor Increases Sensitivity of DNA Breaking Agent in EAC Cells In Vivo
3.6. Association of RAD51 with Immune Dysregulation
3.7. Impact of Elevated RAD51 on Cell Cycle
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Solomon, E.; Borrow, J.; Goddard, A.D. Chromosome Aberrations and Cancer. Science 1991, 254, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Heyer, W.D.; Ehmsen, K.T.; Liu, J. Regulation of Homologous Recombination in Eukaryotes. Annu. Rev. Genet. 2010, 44, 113–139. [Google Scholar] [CrossRef] [PubMed]
- Moynahan, M.E.; Jasin, M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 2010, 11, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Huszno, J.; Kołosza, Z.; Grzybowska, E. BRCA1 mutation in breast cancer patients: Analysis of prognostic factors and survival. Oncol. Lett. 2019, 17, 1986–1995. [Google Scholar] [CrossRef]
- Cousineau, I.; Abaji, C.; Belmaaza, A. BRCA1 Regulates RAD51 Function in Response to DNA Damage and Suppresses Spontaneous Sister Chromatid Replication Slippage: Implications for Sister Chromatid Cohesion, Genome Stability, and Carcinogenesis. Cancer Res. 2005, 65, 11384–11391. [Google Scholar] [CrossRef]
- Guirouilh-Barbat, J.; Lambert, S.; Bertrand, P.; Lopez, B.S. Is homologous recombination really an error-free process? Front. Genet. 2014, 5, 175. [Google Scholar] [CrossRef]
- Pal, J.; Nanjappa, P.; Kumar, S.; Shi, J.; Buon, L.; Munshi, N.C.; Shammas, M.A. Impact of RAD51C-mediated Homologous Recombination on Genomic Integrity in Barrett’s Adenocarcinoma Cells. J. Gastroenterol. Hepatol. Res. 2017, 6, 2286–2295. [Google Scholar] [CrossRef]
- Pal, J.; Bertheau, R.; Buon, L.; Qazi, A.; Batchu, R.B.; Bandyopadhyay, S.; Ali-Fehmi, R.; Beer, D.G.; Weaver, D.W.; Reis, R.J.S.; et al. Genomic evolution in Barrett’s adenocarcinoma cells: Critical roles of elevated hsRAD51, homologous recombination and Alu sequences in the genome. Oncogene 2011, 30, 3585–3598. [Google Scholar] [CrossRef]
- Shammas, M.A.; Reis, R.J.S.; Koley, H.; Batchu, R.B.; Li, C.; Munshi, N.C. Dysfunctional homologous recombination mediates genomic instability and progression in myeloma. Blood 2009, 113, 2290–2297. [Google Scholar] [CrossRef]
- Lu, R.; Pal, J.; Buon, L.; Nanjappa, P.; Shi, J.; Fulciniti, M.; Tai, Y.-T.; Guo, L.; Yu, M.; Gryaznov, S.; et al. Targeting homologous recombination and telomerase in Barrett’s adenocarcinoma: Impact on telomere maintenance, genomic instability and tumor growth. Oncogene 2014, 33, 1495–1505. [Google Scholar] [CrossRef] [PubMed]
- Bärlund, M.; Monni, O.; Kononen, J.; Cornelison, R.; Torhorst, J.; Sauter, G.; Olli-P, K.; Kallioniemi, A. Multiple genes at 17q23 undergo amplification and overexpression in breast cancer. Cancer Res. 2000, 60, 5340–5344. [Google Scholar] [PubMed]
- Hansen, L.T.; Lundin, C.; Helleday, T.; Poulsen, H.S.; Sorensen, C.; Petersen, L.N.; Spang-Thomsen, M. DNA repair rate and etoposide (VP16) resistance of tumor cell subpopulations derived from a single human small cell lung cancer. Lung Cancer 2003, 40, 157–164. [Google Scholar] [CrossRef]
- Maacke, H.; Jost, K.; Opitz, S.; Miska, S.; Yuan, Y.; Hasselbach, L.; Lüttges, J.; Kalthoff, H.; Stürzbecher, H.-W. DNA repair and recombination factor Rad51 is over-expressed in human pancreatic adenocarcinoma. Oncogene 2000, 19, 2791–2795. [Google Scholar] [CrossRef]
- Qiao, G.B.; Wu, Y.L.; Yang, X.N.; Zhong, W.-Z.; Xie, D.; Guan, X.-Y.; Fischer, D.; Kolberg, H.-C.; Kruger, S.; Stuerzbecher, H.-W. High-level expression of Rad51 is an independent prognostic marker of survival in non-small-cell lung cancer patients. Br. J. Cancer 2005, 93, 137–143. [Google Scholar] [CrossRef]
- Nandi, B.; Talluri, S.; Kumar, S.; Yenumula, C.; Gold, J.S.; Prabhala, R.; Munshi, N.C.; Shammas, M.A. The roles of homologous recombination and the immune system in the genomic evolution of cancer. J. Transl. Sci. 2019, 5, 10–15761. [Google Scholar] [CrossRef]
- Aguilera, A.; Gomez-González, B. Genome instability: A mechanistic view of its causes and consequences. Nat. Rev. Genet. 2008, 9, 204–217. [Google Scholar] [CrossRef]
- Wright, W.D.; Shah, S.S.; Heyer, W.D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef]
- Argueso, J.L.; Westmoreland, J.; Mieczkowski, P.A.; Gawel, M.; Petes, T.D.; Resnick, M.A. Double-strand breaks associated with repetitive DNA can reshape the genome. Proc. Natl. Acad. Sci. USA 2008, 105, 11845–11850. [Google Scholar] [CrossRef]
- Nakad, R.; Schumacher, B. DNA damage response and immune defense: Links and mechanisms. Front. Genet. 2016, 7, 147. [Google Scholar] [CrossRef]
- Pateras, I.S.; Havaki, S.; Nikitopoulou, X.; Vougas, K.; Townsend, P.A.; Panayiotidis, M.I.; Georgakilas, A.G.; Gorgoulis, V.G. The DNA damage response and immune signaling alliance: Is it good or bad? Nature decides when and where. Pharmacol. Ther. 2015, 154, 36–56. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Brann, T.W.; Zhou, M.; Yang, J.; Oguariri, R.M.; Lidie, K.B.; Imamichi, H.; Huang, D.-W.; Lempicki, R.A.; Baseler, M.W.; et al. Cutting edge: Ku70 is a novel cytosolic DNA sensor that induces type III rather than type I IFN. J. Immunol. 2011, 186, 4541–4545. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yuan, B.; Bao, M.; Lu, N.; Kim, T.; Liu, Y.-J. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat. Immunol. 2011, 12, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, B.J.; Mansur, D.S.; Peters, N.E.; Ren, H.; Smith, G.L. DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. Elife 2012, 1, e00047. [Google Scholar] [CrossRef]
- Kondo, T.; Kobayashi, J.; Saitoh, T.; Maruyama, K.; Ishii, K.J.; Barber, G.N.; Komatsu, K.; Akira, S.; Kawai, T. DNA damage sensor MRE11 recognizes cytosolic double-stranded DNA and induces type I interferon by regulating STING trafficking. Proc. Natl. Acad. Sci. USA 2013, 110, 2969–2974. [Google Scholar] [CrossRef]
- Volcic, M.; Karl, S.; Baumann, B.; Salles, D.; Daniel, P.; Fulda, S.; Wiesmüller, L. NF-ΰB regulates DNA double-strand break repair in conjunction with BRCA1-CtIP complexes. Nucleic Acids Res. 2012, 40, 181–195. [Google Scholar] [CrossRef]
- Kumar, S.; Buon, L.; Talluri, S.; Roncador, M.; Liao, C.; Zhao, J.; Shi, J.; Chakraborty, C.; Gonzalez, G.; Tai, Y.T.; et al. Integrated genomics and comprehensive validation reveal drivers of genomic evolution in esophageal adenocarcinoma. Commun. Biol. 2021, 4, 617. [Google Scholar] [CrossRef]
- Gravel, S.; Chapman, J.R.; Magill, C.; Jackson, S.P. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008, 22, 2767–2772. [Google Scholar] [CrossRef]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef]
- Huang, F.; Motlekar, N.A.; Burgwin, C.M.; Napper, A.D.; Diamond, S.L.; Mazin, A.V. Identification of Specific Inhibitors of Human RAD51 Recombinase Using High-Throughput Screening. ACS Chem. Biol. 2011, 6, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Balmus, G.; A Karp, N.; Ng, B.L.; Jackson, S.P.; Adams, D.J.; McIntyre, R.E. A high-throughput in vivo micronucleus assay for genome instability screening in mice. Nat. Protoc. 2015, 10, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Katyal, S.; Lee, Y.; Nitiss, K.C.; Downing, S.M.; Li, Y.; Shimada, M.; Zhao, J.; Russell, H.R.; Petrini, J.H.J.; Nitiss, J.L.; et al. Aberrant topoisomerase-1 DNA lesions are pathogenic in neurodegenerative genome instability syndromes. Nat. Neurosci. 2014, 17, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Talluri, S.; Pal, J.; Yuan, X.; Lu, R.; Nanjappa, P.; Samur, M.K.; Munshi, N.C.; Shammas, M.A. Role of apurinic/apyrimidinic nucleases in the regulation of homologous recombination in myeloma: Mechanisms and translational significance. Blood Cancer J. 2018, 8, 92. [Google Scholar] [CrossRef]
- Liao, C.; Zhao, J.; Kumar, S.; Chakraborty, C.; Talluri, S.; Munshi, N.C.; Shammas, M.A. RAD51 Inhibitor Reverses Etoposide-Induced Genomic Toxicity and Instability in Esophageal Adenocarcinoma Cells. Arch. Clin. Toxicol. (Middlet) 2020, 2, 3–9. [Google Scholar] [CrossRef]
- Xia, S.J.; Shammas, M.A.; Shmookler Reis, R.J. Elevated recombination in immortal human cells is mediated by HsRAD51 recombinase. Mol. Cell. Biol. 1997, 17, 7151–7158. [Google Scholar] [CrossRef]
- Tanaka, K.; Hiramoto, T.; Fukuda, T.; Miyagawa, K. A Novel Human Rad54 Homologue, Rad54B, Associates with Rad51. J. Biol. Chem. 2000, 275, 26316–26321. [Google Scholar] [CrossRef]
- Yasuhara, T.; Suzuki, T.; Katsura, M.; Miyagawa, K. Rad54B serves as a scaffold in the DNA damage response that limits checkpoint strength. Nat. Commun. 2014, 5, 5426. [Google Scholar] [CrossRef]
- Kumar, S.; Zhao, J.; Talluri, S.; Buon, L.; Mu, S.; Potluri, B.; Liao, C.; Shi, J.; Chakraborty, C.; Gonzalez, G.B.; et al. Apurinic/apyrimidinic nuclease 1 drives genomic evolution contributing to chemoresistance and tumorigenesis in solid tumor. bioRxiv 2022. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Marker Type | Marker | Correlation Coefficient | p Value |
|---|---|---|---|
| Inflammatory | GMCSF | 0.32 | 0.0004 |
| Inflammatory | IFNγ | 0.62 | 4.4 × 10−14 |
| Inflammatory | IL6 | 0.38 | 2.6 × 10−5 |
| Inflammatory | IL1β | 0.4 | 7.1 × 10−6 |
| Inflammatory | IL2 | 0.6 | 8.6 × 10−13 |
| Inflammatory | TNFα | 0.37 | 4.2 × 10−5 |
| Inflammatory | IL13 | 0.52 | 1.9 × 10−9 |
| T cell | OX40 | 0.6 | 7.9 × 10−13 |
| T cell | OX40L | 0.63 | 1.5 × 10−14 |
| T cell | TIGIT | 0.55 | 1.8 × 10−10 |
| T cell | PD-L1 | 0.55 | 1.7 × 10−10 |
| T cell | PD1 | 0.71 | 2.2 × 10−16 |
| T cell | TIM3 | 0.63 | 2.1 × 10−14 |
| B lymphocyte | HLADRA | 0.62 | 9.2 × 10−14 |
| T, NK and B cells | CD27 | 0.51 | 3.1 × 10−9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, C.; Talluri, S.; Zhao, J.; Mu, S.; Kumar, S.; Shi, J.; Buon, L.; Munshi, N.C.; Shammas, M.A. RAD51 Is Implicated in DNA Damage, Chemoresistance and Immune Dysregulation in Solid Tumors. Cancers 2022, 14, 5697. https://doi.org/10.3390/cancers14225697
Liao C, Talluri S, Zhao J, Mu S, Kumar S, Shi J, Buon L, Munshi NC, Shammas MA. RAD51 Is Implicated in DNA Damage, Chemoresistance and Immune Dysregulation in Solid Tumors. Cancers. 2022; 14(22):5697. https://doi.org/10.3390/cancers14225697
Chicago/Turabian StyleLiao, Chengcheng, Srikanth Talluri, Jiangning Zhao, Shidai Mu, Subodh Kumar, Jialan Shi, Leutz Buon, Nikhil C. Munshi, and Masood A. Shammas. 2022. "RAD51 Is Implicated in DNA Damage, Chemoresistance and Immune Dysregulation in Solid Tumors" Cancers 14, no. 22: 5697. https://doi.org/10.3390/cancers14225697
APA StyleLiao, C., Talluri, S., Zhao, J., Mu, S., Kumar, S., Shi, J., Buon, L., Munshi, N. C., & Shammas, M. A. (2022). RAD51 Is Implicated in DNA Damage, Chemoresistance and Immune Dysregulation in Solid Tumors. Cancers, 14(22), 5697. https://doi.org/10.3390/cancers14225697