
Targeting Leukemia-Initiating Cells and Leukemic Niches: The Next Therapy Station for T-Cell Acute Lymphoblastic Leukemia?

Simple Summary
Abstract
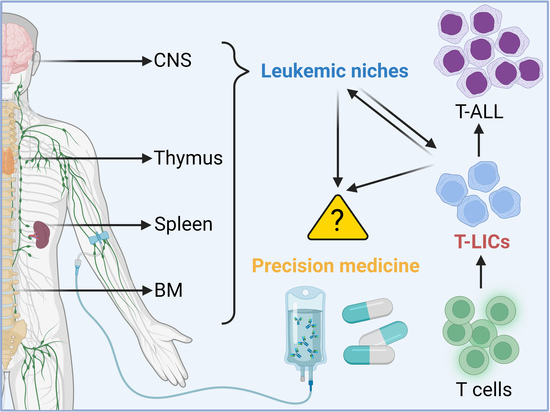
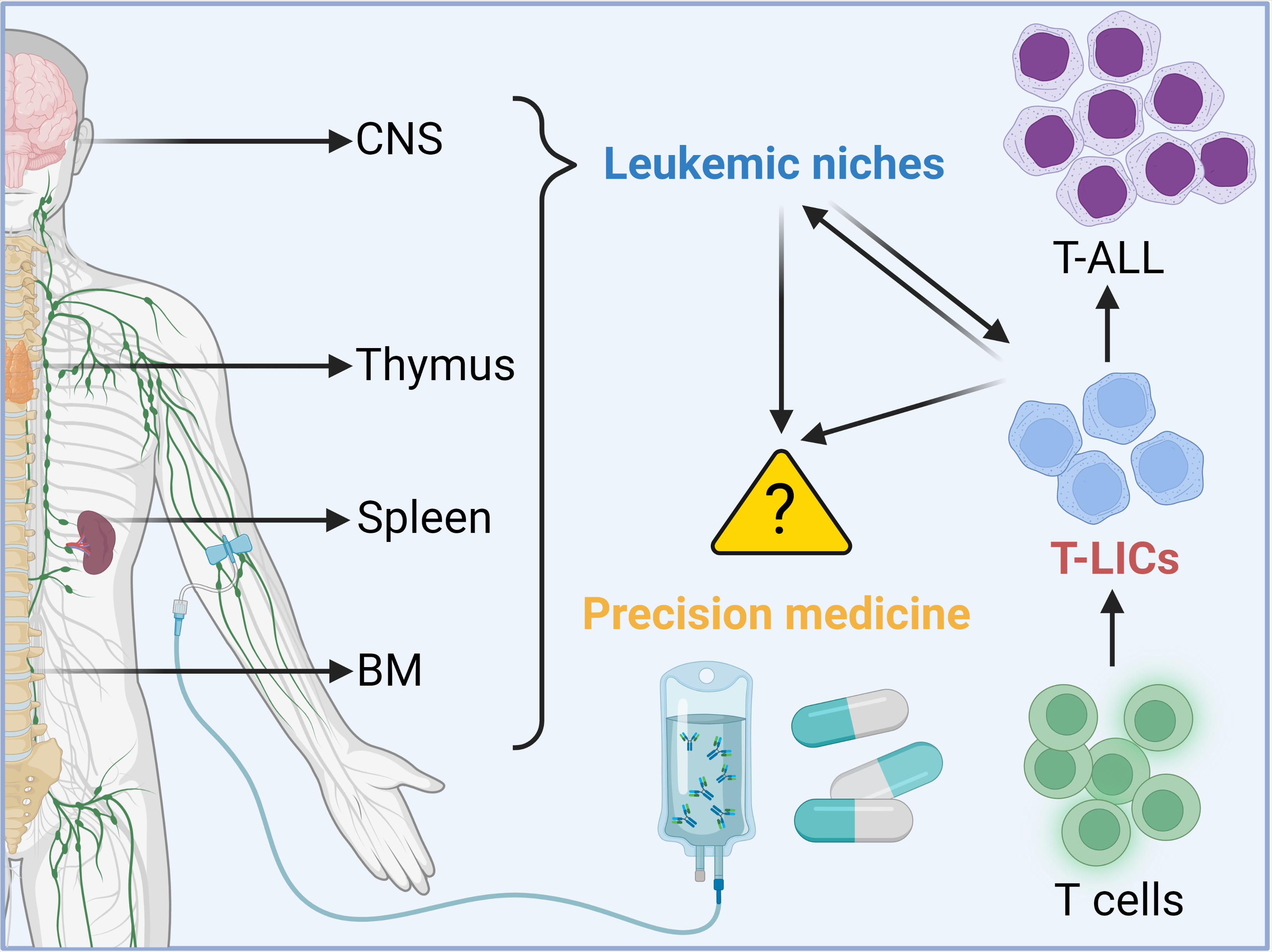
1. Introduction
2. LICs in T-ALL
2.1. T-LICs in Mouse Models
2.2. T-LICs in Zebrafish Models
2.3. Therapies Targeting T-LICs
3. Leukemic Niches in T-ALL
3.1. Effects of Leukemic Niches on T-ALL Cells
3.1.1. CXC Chemokine Ligand 12 (CXCL12)/CXC Chemokine Receptor 4 (CXCR4) Signaling

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Treatment | Clinical Trials | Ref. | |||
|---|---|---|---|---|---|---|
| Age | Phase | NCT No. | Status | |||
| CXCR4 | BL-8040 + nelarabine | ≥18 y | 2 | NCT02763384 | Recruiting | - |
| MEK | selumetinib + dexamethasone | Child, adult, older adult | 1/2 | NCT03705507 | Recruiting | [76] |
| NOTCH1 | MK0752 | ≥12 m | 1 | NCT00100152 | Terminated | - |
| PF-03084014 | ≥16 y | 1 | NCT00878189 | Completed | [77,78] | |
| RO4929097 + dexamethasone | 1–21 y | 1 | NCT01088763 | Terminated | - | |
| BMS-906024 + dexamethasone | ≥18 y | 1 | NCT01363817 | Completed | - | |
| LY3039478 + dexamethasone | ≥2 y | 1/2 | NCT02518113 | Completed | [79] | |
| BCL2 | venetoclax + chemotherapy | Child, adult, older adult | 2 | NCT00501826 | Recruiting | - |
| venetoclax + navitoclax + chemotherapy | ≥4 y | 1 | NCT03181126 | Completed | [80] | |
| venetoclax + chemotherapy | ≤25 y | 1 | NCT03236857 | Recruiting | [81] | |
| venetoclax + vincristine liposomal | ≥18 y | 1/2 | NCT03504644 | Suspended | - | |
| venetoclax + low-intensity chemotherapy | ≥18 y | 1/2 | NCT03808610 | Recruiting | - | |
| venetoclax + navitoclax | ≥18 y | 1b/2 | NCT05054465 | Not yet recruiting | - | |
| venetoclax + azacitidine | 15–65 y | 2 | NCT05149378 | Recruiting | - | |
| venetoclax + navitoclax + chemotherapy | 4–30 y | 1/2 | NCT05192889 | Not yet recruiting | - | |
| venetoclax + ponatinib + mini-hyper CVD † | ≥18 y | 2 | NCT05268003 | Recruiting | - | |
| venetoclax + azacitidine | ≥15 y | 2 | NCT05376111 | Recruiting | - | |
| PI3K/mTOR | everolimus + hyper-CVAD †† | ≥10 y | 1/2 | NCT00968253 | Completed | - |
| everolimus + reinduction chemotherapy | 18 m–21 y | 1 | NCT01523977 | Completed | - | |
| temsirolimus + chemotherapy | 1–21 y | 1 | NCT01614197 | Completed | - | |
| BEZ235 | ≥18 y | 1 | NCT01756118 | Uk | [82] | |
| sapanisertib | ≥18 y | 2 | NCT02484430 | Active, not recruiting | - | |
| everolimus + chemotherapy | 2–29 y | 1 | NCT03328104 | Recruiting | - | |
| CDK4/6 | palbociclib + sorafenib, decitabine or dexamethasone | ≥15 y | 1 | NCT03132454 | Recruiting | - |
| palbociclib + chemotherapy | ≤21 y | 1 | NCT03515200 | Terminated | - | |
| ribociclib + everolimus + dexamethasone | 1–30 y | 1 | NCT03740334 | Active, not recruiting | - | |
| palbociclib + chemotherapy | 12 m–31 y | 1 | NCT03792256 | Active, not recruiting | - | |
| CD38 | isatuximab + chemotherapy | ≥16 y | 2 | NCT02999633 | Terminated | [83] |
| daratumumab + chemotherapy | 1–30 y | 2 | NCT03384654 | Active, not recruiting | - | |
| isatuximab + chemotherapy | 28 d–17 y | 2 | NCT03860844 | Recruiting | - | |
| daratumumab | ≤39 y | 2 | NCT04972942 | Not yet recruiting | - | |
| daratumumab + hyaluronidase | ≥18 y | 2 | NCT05289687 | Recruiting | - | |
| CD38-CD3 | XmAb18968 (bsAb) | ≥18 y | 1 | NCT05038644 | Recruiting | - |
| CD52 | alemtuzumab + chemotherapy | ≥15 y | 1/2 | NCT00061945 | Completed | - |
| alemtuzumab ± methotrexate, mercaptopurine | ≤30 y | 2 | NCT00089349 | Completed | [84] | |
| alemtuzumab ± cladribine | ≥18 y | 2 | NCT00199030 | Completed | - | |
| alemtuzumab + pentostatin | ≥18 y | 2 | NCT00453193 | Terminated | [85] | |
3.1.2. Insulin-like Growth Factor 1 (IGF1)/IGF1 Receptor (IGF1R) Signaling
3.1.3. Interleukin (IL7)/IL7 Receptor (IL7R) Signaling
3.1.4. CC Chemokine Ligand 19 (CCL19)/CC Chemokine Receptor 7 (CCR7) Signaling
3.2. Microenvironmental Alterations
3.2.1. BM Microenvironment
3.2.2. Thymic Microenvironment
3.2.3. Splenic Microenvironment
4. Preclinically- and Clinically-Evaluated Precision Medicine for T-ALL
4.1. Agents Targeting Aberrant Pathways
4.1.1. NOTCH1 Signaling
4.1.2. BCL2 Signaling
4.1.3. JAK-STAT Signaling
4.1.4. PI3K-AKT-mTOR Signaling
4.1.5. CDK4/6-Mediated Signaling
4.1.6. Other Signaling Pathways
4.2. Antibody-Based Therapy
4.2.1. CD38 mAbs
4.2.2. CD52 mAbs
4.2.3. IL7Rα mAbs
5. Challenges and Future Perspectives
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADCC | antibody-dependent cell mediated cytotoxicity |
| AKT | AKT serine/threonine kinase |
| AML | acute myelocytic leukemia |
| ATL | adult T-cell leukemia |
| B-ALL | B-cell acute lymphoblastic leukemia |
| BCL2 | B-cell lymphoma 2 |
| BM | bone marrow |
| CAR | chimeric antigen receptor |
| CCL19 | CC chemokine ligand 19 |
| CCR7 | CC chemokine receptor7 |
| CDC | complement-dependent cytotoxicity |
| CDK4/6 | cyclin dependent kinase 4/6 |
| CDKis | CDK4/6 inhibitors |
| CNS | central nervous system |
| CR | complete remissions |
| CSF | cerebrospinal fluid |
| CXCL12 | CXC chemokine ligand 12 |
| CXCR4 | CXC chemokine receptor 4 |
| DCs | dendritic cells |
| DLL4 | delta-like canonical Notch ligand 4 |
| DN | CD4−CD8− double-negative |
| DNMTs | DNA methyltransferases |
| DP | CD4+CD8+ double-positive |
| EHMT2 | euchromatic histone lysine methyltransferase 2 |
| ERK | extracellular regulated protein kinase |
| ETV6 | ETS variant transcription factor 6 |
| EZH2 | enhancer of zeste homolog 2 |
| FL | fetal liver |
| GSIs | γ-secretase inhibitors |
| H3K9 | histone 3 lysine 9 |
| HDACs | histone deacetylases |
| HIF1α | hypoxia inducible factor 1α |
| HPCs | hematopoietic progenitor cells |
| HSCs | hematopoietic stem cells |
| HSCT | hematopoietic stem-cell transplantation |
| IGF1(R) | insulin-like growth factor 1 (receptor) |
| IGFBP7 | insulin-like growth factor binding protein 7 |
| IL7(R) | interleukin 7 (receptor) |
| JAK | Janus kinase |
| KARS (Kras) | KRAS proto-oncogene |
| KLF4 | Krüppel-like factor 4 |
| LIC(s) | leukemia-initiating cell(s) |
| LMO1/2 (Lmo1/2) | LIM domain only 1/2 |
| LN | lymph node |
| LSCs | leukemia stem cells |
| mAbs | monoclonal antibodies |
| MAPK | mitogen-activated protein kinase |
| MBD2 | methyl-CpG binding domain protein 2 |
| MEK | MAPK kinase |
| mTOR(C1) | mechanistic target of rapamycin kinase (complex 1) |
| MYC (Myc) | MYC proto-oncogene |
| NOTCH1 | notch receptor 1 |
| NRR | negative regulatory region |
| PDX | patient-derived tumor xenograft |
| PHF6 | plant homeodomain factor 6 |
| PI3K | phosphatidylinositol 3-kinase |
| PKCθ | protein kinase C theta |
| PTEN (Pten) | phosphatase and tensin homolog |
| R/R | relapsed/refractory |
| ROS | reactive oxygen species |
| RUNX | runt-related transcription factor |
| SDF-1 | stromal cell derived factor-1 |
| STAT | signal transducer and activator of transcription |
| TAL1 (Tal1) | T-cell acute lymphocytic 1 (TAL) basic helix-loop-helix (bHLH) transcription factor 1 |
| T-ALL | T-cell acute lymphoblastic leukemia |
| TECs | thymic epithelial cells |
| TKI(s) | tyrosine kinase inhibitor(s) |
| T-LICs | T-ALL LICs |
| T-PLL | T-cell prolymphocytic leukemia |
| ZAP70 | zeta-chain-associated protein kinase 70 |
References
- Grunenberg, A.; Sala, E.; Kapp-Schwoerer, S.; Viardot, A. Pharmacotherapeutic management of T-cell acute lymphoblastic leukemia in adults: An update of the literature. Expert Opin. Pharm. 2022, 23, 561–571. [Google Scholar] [CrossRef]
- Fattizzo, B.; Rosa, J.; Giannotta, J.A.; Baldini, L.; Fracchiolla, N.S. The Physiopathology of T- Cell Acute Lymphoblastic Leukemia: Focus on Molecular Aspects. Front. Oncol. 2020, 10, 273. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.H.; Bertulfo, F.C.; Sanda, T. Leukemia-Initiating Cells in T-Cell Acute Lymphoblastic Leukemia. Front. Oncol. 2017, 7, 218. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322. [Google Scholar] [CrossRef]
- Kelliher, M.A.; Seldin, D.C.; Leder, P. Tal-1 induces T cell acute lymphoblastic leukemia accelerated by casein kinase IIalpha. EMBO J. 1996, 15, 5160–5166. [Google Scholar] [CrossRef]
- Aplan, P.D.; Jones, C.A.; Chervinsky, D.S.; Zhao, X.; Ellsworth, M.; Wu, C.; McGuire, E.A.; Gross, K.W. An scl gene product lacking the transactivation domain induces bony abnormalities and cooperates with LMO1 to generate T-cell malignancies in transgenic mice. EMBO J. 1997, 16, 2408–2419. [Google Scholar] [CrossRef]
- Larson, R.C.; Fisch, P.; Larson, T.A.; Lavenir, I.; Langford, T.; King, G.; Rabbitts, T.H. T cell tumours of disparate phenotype in mice transgenic for Rbtn-2. Oncogene 1994, 9, 3675–3681. [Google Scholar]
- Tremblay, M.; Tremblay, C.S.; Herblot, S.; Aplan, P.D.; Hebert, J.; Perreault, C.; Hoang, T. Modeling T-cell acute lymphoblastic leukemia induced by the SCL and LMO1 oncogenes. Genes Dev. 2010, 24, 1093–1105. [Google Scholar] [CrossRef] [PubMed]
- Tatarek, J.; Cullion, K.; Ashworth, T.; Gerstein, R.; Aster, J.C.; Kelliher, M.A. Notch1 inhibition targets the leukemia-initiating cells in a Tal1/Lmo2 mouse model of T-ALL. Blood 2011, 118, 1579–1590. [Google Scholar] [CrossRef] [PubMed]
- Chiang, M.Y.; Xu, L.; Shestova, O.; Histen, G.; L’Heureux, S.; Romany, C.; Childs, M.E.; Gimotty, P.A.; Aster, J.C.; Pear, W.S. Leukemia-associated NOTCH1 alleles are weak tumor initiators but accelerate K-ras-initiated leukemia. J. Clin. Investig. 2008, 118, 3181–3194. [Google Scholar] [CrossRef] [PubMed]
- Giambra, V.; Jenkins, C.R.; Wang, H.; Lam, S.H.; Shevchuk, O.O.; Nemirovsky, O.; Wai, C.; Gusscott, S.; Chiang, M.Y.; Aster, J.C.; et al. NOTCH1 promotes T cell leukemia-initiating activity by RUNX-mediated regulation of PKC-theta and reactive oxygen species. Nat. Med. 2012, 18, 1693–1698. [Google Scholar] [CrossRef]
- Chiang, M.Y.; Shestova, O.; Xu, L.; Aster, J.C.; Pear, W.S. Divergent effects of supraphysiologic Notch signals on leukemia stem cells and hematopoietic stem cells. Blood 2013, 121, 905–917. [Google Scholar] [CrossRef]
- King, B.; Trimarchi, T.; Reavie, L.; Xu, L.; Mullenders, J.; Ntziachristos, P.; Aranda-Orgilles, B.; Perez-Garcia, A.; Shi, J.; Vakoc, C.; et al. The ubiquitin ligase FBXW7 modulates leukemia-initiating cell activity by regulating MYC stability. Cell 2013, 153, 1552–1566. [Google Scholar] [CrossRef]
- Jena, N.; Sheng, J.; Hu, J.K.; Li, W.; Zhou, W.; Lee, G.; Tsichlis, N.; Pathak, A.; Brown, N.; Deshpande, A.; et al. CDK6-mediated repression of CD25 is required for induction and maintenance of Notch1-induced T-cell acute lymphoblastic leukemia. Leukemia 2016, 30, 1033–1043. [Google Scholar] [CrossRef]
- Kong, G.; Du, J.; Liu, Y.; Meline, B.; Chang, Y.I.; Ranheim, E.A.; Wang, J.; Zhang, J. Notch1 gene mutations target KRAS G12D-expressing CD8+ cells and contribute to their leukemogenic transformation. J. Biol. Chem. 2013, 288, 18219–18227. [Google Scholar] [CrossRef]
- Guo, W.; Lasky, J.L.; Chang, C.J.; Mosessian, S.; Lewis, X.; Xiao, Y.; Yeh, J.E.; Chen, J.Y.; Iruela-Arispe, M.L.; Varella-Garcia, M.; et al. Multi-genetic events collaboratively contribute to Pten-null leukaemia stem-cell formation. Nature 2008, 453, 529–533. [Google Scholar] [CrossRef]
- Schubbert, S.; Cardenas, A.; Chen, H.; Garcia, C.; Guo, W.; Bradner, J.; Wu, H. Targeting the MYC and PI3K pathways eliminates leukemia-initiating cells in T-cell acute lymphoblastic leukemia. Cancer Res. 2014, 74, 7048–7059. [Google Scholar] [CrossRef]
- Aifantis, I.; Raetz, E.; Buonamici, S. Molecular pathogenesis of T-cell leukaemia and lymphoma. Nat. Rev. Immunol. 2008, 8, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Grabher, C.; von Boehmer, H.; Look, A.T. Notch 1 activation in the molecular pathogenesis of T-cell acute lymphoblastic leukaemia. Nat. Rev. Cancer 2006, 6, 347–359. [Google Scholar] [CrossRef]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris, J.P.T.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef]
- Armstrong, F.; Brunet de la Grange, P.; Gerby, B.; Rouyez, M.C.; Calvo, J.; Fontenay, M.; Boissel, N.; Dombret, H.; Baruchel, A.; Landman-Parker, J.; et al. NOTCH is a key regulator of human T-cell acute leukemia initiating cell activity. Blood 2009, 113, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- Gerby, B.; Tremblay, C.S.; Tremblay, M.; Rojas-Sutterlin, S.; Herblot, S.; Hebert, J.; Sauvageau, G.; Lemieux, S.; Lecuyer, E.; Veiga, D.F.; et al. SCL, LMO1 and Notch1 reprogram thymocytes into self-renewing cells. PLoS Genet. 2014, 10, e1004768. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Peydro, M.; Fuentes, P.; Mosquera, M.; Garcia-Leon, M.J.; Alcain, J.; Rodriguez, A.; Garcia de Miguel, P.; Menendez, P.; Weijer, K.; Spits, H.; et al. The NOTCH1/CD44 axis drives pathogenesis in a T cell acute lymphoblastic leukemia model. J. Clin. Investig. 2018, 128, 2802–2818. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, R.; Hayashi, A.; Utsunomiya, A.; Nukada, Y.; Fukui, R.; Itoh, K.; Tezuka, K.; Ohashi, K.; Mizuno, K.; Sakamoto, M.; et al. Alteration of phosphatidylinositol 3-kinase cascade in the multilobulated nuclear formation of adult T cell leukemia/lymphoma (ATLL). Proc. Natl. Acad. Sci. USA 2005, 102, 15213–15218. [Google Scholar] [CrossRef]
- Gutierrez, A.; Sanda, T.; Grebliunaite, R.; Carracedo, A.; Salmena, L.; Ahn, Y.; Dahlberg, S.; Neuberg, D.; Moreau, L.A.; Winter, S.S.; et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood 2009, 114, 647–650. [Google Scholar] [CrossRef]
- Larson Gedman, A.; Chen, Q.; Kugel Desmoulin, S.; Ge, Y.; LaFiura, K.; Haska, C.L.; Cherian, C.; Devidas, M.; Linda, S.B.; Taub, J.W.; et al. The impact of NOTCH1, FBW7 and PTEN mutations on prognosis and downstream signaling in pediatric T-cell acute lymphoblastic leukemia: A report from the Children’s Oncology Group. Leukemia 2009, 23, 1417–1425. [Google Scholar] [CrossRef]
- Palomero, T.; Sulis, M.L.; Cortina, M.; Real, P.J.; Barnes, K.; Ciofani, M.; Caparros, E.; Buteau, J.; Brown, K.; Perkins, S.L.; et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat. Med. 2007, 13, 1203–1210. [Google Scholar] [CrossRef]
- Gachet, S.; Genesca, E.; Passaro, D.; Irigoyen, M.; Alcalde, H.; Clemenson, C.; Poglio, S.; Pflumio, F.; Janin, A.; Lasgi, C.; et al. Leukemia-initiating cell activity requires calcineurin in T-cell acute lymphoblastic leukemia. Leukemia 2013, 27, 2289–2300. [Google Scholar] [CrossRef] [PubMed]
- Giambra, V.; Jenkins, C.E.; Lam, S.H.; Hoofd, C.; Belmonte, M.; Wang, X.; Gusscott, S.; Gracias, D.; Weng, A.P. Leukemia stem cells in T-ALL require active Hif1α and Wnt signaling. Blood 2015, 125, 3917–3927. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Park, C.S.; Suppipat, K.; Mistretta, T.A.; Puppi, M.; Horton, T.M.; Rabin, K.; Gray, N.S.; Meijerink, J.P.P.; Lacorazza, H.D. Inactivation of KLF4 promotes T-cell acute lymphoblastic leukemia and activates the MAP2K7 pathway. Leukemia 2017, 31, 1314–1324. [Google Scholar] [CrossRef]
- Wendorff, A.A.; Quinn, S.A.; Rashkovan, M.; Madubata, C.J.; Ambesi-Impiombato, A.; Litzow, M.R.; Tallman, M.S.; Paietta, E.; Paganin, M.; Basso, G.; et al. Phf6 Loss Enhances HSC Self-Renewal Driving Tumor Initiation and Leukemia Stem Cell Activity in T-ALL. Cancer Discov. 2019, 9, 436–451. [Google Scholar] [CrossRef]
- Tremblay, C.S.; Chiu, S.K.; Saw, J.; McCalmont, H.; Litalien, V.; Boyle, J.; Sonderegger, S.E.; Chau, N.; Evans, K.; Cerruti, L.; et al. Small molecule inhibition of Dynamin-dependent endocytosis targets multiple niche signals and impairs leukemia stem cells. Nat. Commun. 2020, 11, 6211. [Google Scholar] [CrossRef] [PubMed]
- Cox, C.V.; Martin, H.M.; Kearns, P.R.; Virgo, P.; Evely, R.S.; Blair, A. Characterization of a progenitor cell population in childhood T-cell acute lymphoblastic leukemia. Blood 2007, 109, 674–682. [Google Scholar] [CrossRef]
- Chiu, P.P.; Jiang, H.; Dick, J.E. Leukemia-initiating cells in human T-lymphoblastic leukemia exhibit glucocorticoid resistance. Blood 2010, 116, 5268–5279. [Google Scholar] [CrossRef]
- Risueno, R.M.; Campbell, C.J.; Dingwall, S.; Levadoux-Martin, M.; Leber, B.; Xenocostas, A.; Bhatia, M. Identification of T-lymphocytic leukemia-initiating stem cells residing in a small subset of patients with acute myeloid leukemic disease. Blood 2011, 117, 7112–7120. [Google Scholar] [CrossRef]
- Gerby, B.; Clappier, E.; Armstrong, F.; Deswarte, C.; Calvo, J.; Poglio, S.; Soulier, J.; Boissel, N.; Leblanc, T.; Baruchel, A.; et al. Expression of CD34 and CD7 on human T-cell acute lymphoblastic leukemia discriminates functionally heterogeneous cell populations. Leukemia 2011, 25, 1249–1258. [Google Scholar] [CrossRef]
- Frazer, J.K.; Meeker, N.D.; Rudner, L.; Bradley, D.F.; Smith, A.C.; Demarest, B.; Joshi, D.; Locke, E.E.; Hutchinson, S.A.; Tripp, S.; et al. Heritable T-cell malignancy models established in a zebrafish phenotypic screen. Leukemia 2009, 23, 1825–1835. [Google Scholar] [CrossRef]
- Smith, A.C.; Raimondi, A.R.; Salthouse, C.D.; Ignatius, M.S.; Blackburn, J.S.; Mizgirev, I.V.; Storer, N.Y.; de Jong, J.L.; Chen, A.T.; Zhou, Y.; et al. High-throughput cell transplantation establishes that tumor-initiating cells are abundant in zebrafish T-cell acute lymphoblastic leukemia. Blood 2010, 115, 3296–3303. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, J.S.; Liu, S.; Wilder, J.L.; Dobrinski, K.P.; Lobbardi, R.; Moore, F.E.; Martinez, S.A.; Chen, E.Y.; Lee, C.; Langenau, D.M. Clonal evolution enhances leukemia-propagating cell frequency in T cell acute lymphoblastic leukemia through Akt/mTORC1 pathway activation. Cancer Cell 2014, 25, 366–378. [Google Scholar] [CrossRef] [PubMed]
- Simioni, C.; Neri, L.M.; Tabellini, G.; Ricci, F.; Bressanin, D.; Chiarini, F.; Evangelisti, C.; Cani, A.; Tazzari, P.L.; Melchionda, F.; et al. Cytotoxic activity of the novel Akt inhibitor, MK-2206, in T-cell acute lymphoblastic leukemia. Leukemia 2012, 26, 2336–2342. [Google Scholar] [CrossRef] [PubMed]
- Roderick, J.E.; Tesell, J.; Shultz, L.D.; Brehm, M.A.; Greiner, D.L.; Harris, M.H.; Silverman, L.B.; Sallan, S.E.; Gutierrez, A.; Look, A.T.; et al. c-Myc inhibition prevents leukemia initiation in mice and impairs the growth of relapsed and induction failure pediatric T-ALL cells. Blood 2014, 123, 1040–1050. [Google Scholar] [CrossRef]
- Medyouf, H.; Alcalde, H.; Berthier, C.; Guillemin, M.C.; dos Santos, N.R.; Janin, A.; Decaudin, D.; de The, H.; Ghysdael, J. Targeting calcineurin activation as a therapeutic strategy for T-cell acute lymphoblastic leukemia. Nat. Med. 2007, 13, 736–741. [Google Scholar] [CrossRef]
- Piya, S.; Yang, Y.; Bhattacharya, S.; Sharma, P.; Ma, H.; Mu, H.; He, H.; Ruvolo, V.; Baran, N.; Davis, R.E.; et al. Targeting the NOTCH1-MYC-CD44 axis in leukemia-initiating cells in T-ALL. Leukemia 2022, 36, 1261–1273. [Google Scholar] [CrossRef]
- Grimaldi, C.; Chiarini, F.; Tabellini, G.; Ricci, F.; Tazzari, P.L.; Battistelli, M.; Falcieri, E.; Bortul, R.; Melchionda, F.; Iacobucci, I.; et al. AMP-dependent kinase/mammalian target of rapamycin complex 1 signaling in T-cell acute lymphoblastic leukemia: Therapeutic implications. Leukemia 2012, 26, 91–100. [Google Scholar] [CrossRef]
- Diamanti, P.; Cox, C.V.; Moppett, J.P.; Blair, A. Parthenolide eliminates leukemia-initiating cell populations and improves survival in xenografts of childhood acute lymphoblastic leukemia. Blood 2013, 121, 1384–1393. [Google Scholar] [CrossRef]
- Passaro, D.; Quang, C.T.; Ghysdael, J. Microenvironmental cues for T-cell acute lymphoblastic leukemia development. Immunol. Rev. 2016, 271, 156–172. [Google Scholar] [CrossRef]
- Passaro, D. Myeloid cells hold the master key for T-ALL spread. Blood 2020, 136, 1799–1800. [Google Scholar] [CrossRef]
- Calvo, J.; Fahy, L.; Uzan, B.; Pflumio, F. Desperately seeking a home marrow niche for T-cell acute lymphoblastic leukaemia. Adv. Biol. Regul. 2019, 74, 100640. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Ferrer, S.; Bonnet, D.; Steensma, D.P.; Hasserjian, R.P.; Ghobrial, I.M.; Gribben, J.G.; Andreeff, M.; Krause, D.S. Bone marrow niches in haematological malignancies. Nat. Rev. Cancer 2020, 20, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.Y.; Yang, X.; Feng, W.L.; Liao, J.F.; Wang, L.N.; Feng, L.; Lin, Y.M.; Ren, Q.; Zheng, G.G. Organ-specific microenvironment modifies diverse functional and phenotypic characteristics of leukemia-associated macrophages in mouse T cell acute lymphoblastic leukemia. J. Immunol. 2015, 194, 2919–2929. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Shi, Y.; Pang, Y.; Dong, F.; Cheng, H.; Hao, S.; Xu, J.; Zhu, X.; Yuan, W.; Cheng, T.; et al. Notch1-induced T cell leukemia can be potentiated by microenvironmental cues in the spleen. J. Hematol. Oncol. 2014, 7, 71. [Google Scholar] [CrossRef]
- Xiong, H.; Mancini, M.; Gobert, M.; Shen, S.; Furtado, G.C.; Lira, S.A.; Parkhurst, C.N.; Garambois, V.; Brengues, M.; Tadokoro, C.E.; et al. Spleen plays a major role in DLL4-driven acute T-cell lymphoblastic leukemia. Theranostics 2021, 11, 1594–1608. [Google Scholar] [CrossRef]
- Hawkins, E.D.; Duarte, D.; Akinduro, O.; Khorshed, R.A.; Passaro, D.; Nowicka, M.; Straszkowski, L.; Scott, M.K.; Rothery, S.; Ruivo, N.; et al. T-cell acute leukaemia exhibits dynamic interactions with bone marrow microenvironments. Nature 2016, 538, 518–522. [Google Scholar] [CrossRef]
- Wang, W.; Zimmerman, G.; Huang, X.; Yu, S.; Myers, J.; Wang, Y.; Moreton, S.; Nthale, J.; Awadallah, A.; Beck, R.; et al. Aberrant Notch Signaling in the Bone Marrow Microenvironment of Acute Lymphoid Leukemia Suppresses Osteoblast-Mediated Support of Hematopoietic Niche Function. Cancer Res. 2016, 76, 1641–1652. [Google Scholar] [CrossRef]
- Georgievski, A.; Michel, A.; Thomas, C.; Mlamla, Z.; Pais de Barros, J.P.; Lemaire-Ewing, S.; Garrido, C.; Quere, R. Acute lymphoblastic leukemia-derived extracellular vesicles affect quiescence of hematopoietic stem and progenitor cells. Cell Death Dis. 2022, 13, 337. [Google Scholar] [CrossRef]
- Ghezzo, M.N.; Fernandes, M.T.; Pacheco-Leyva, I.; Rodrigues, P.M.; Machado, R.S.; Araujo, M.A.S.; Kalathur, R.K.; Futschik, M.E.; Alves, N.L.; Dos Santos, N.R. FoxN1-dependent thymic epithelial cells promote T-cell leukemia development. Carcinogenesis 2018, 39, 1463–1476. [Google Scholar] [CrossRef]
- Fernandes, M.T.; Ghezzo, M.N.; Silveira, A.B.; Kalathur, R.K.; Povoa, V.; Ribeiro, A.R.; Brandalise, S.R.; Dejardin, E.; Alves, N.L.; Ghysdael, J.; et al. Lymphotoxin-beta receptor in microenvironmental cells promotes the development of T-cell acute lymphoblastic leukaemia with cortical/mature immunophenotype. Br. J. Haematol. 2015, 171, 736–751. [Google Scholar] [CrossRef]
- Di Grande, A.; Peirs, S.; Donovan, P.D.; Van Trimpont, M.; Morscio, J.; Lintermans, B.; Reunes, L.; Vandamme, N.; Goossens, S.; Nguyen, H.A.; et al. The spleen as a sanctuary site for residual leukemic cells following ABT-199 monotherapy in ETP-ALL. Blood Adv. 2021, 5, 1963–1976. [Google Scholar] [CrossRef]
- de Bock, C.E.; Cools, J. T-ALL: Home Is where the CXCL12 Is. Cancer Cell 2015, 27, 745–746. [Google Scholar] [CrossRef]
- Crane, G.M.; Jeffery, E.; Morrison, S.J. Adult haematopoietic stem cell niches. Nat. Rev. Immunol. 2017, 17, 573–590. [Google Scholar] [CrossRef]
- Su, L.; Hu, Z.; Yang, Y.G. Role of CXCR4 in the progression and therapy of acute leukaemia. Cell Prolif. 2021, 54, e13076. [Google Scholar] [CrossRef] [PubMed]
- Mehrpouri, M. The contributory roles of the CXCL12/CXCR4/CXCR7 axis in normal and malignant hematopoiesis: A possible therapeutic target in hematologic malignancies. Eur. J. Pharmacol. 2022, 920, 174831. [Google Scholar] [CrossRef] [PubMed]
- Passaro, D.; Irigoyen, M.; Catherinet, C.; Gachet, S.; Da Costa De Jesus, C.; Lasgi, C.; Tran Quang, C.; Ghysdael, J. CXCR4 Is Required for Leukemia-Initiating Cell Activity in T Cell Acute Lymphoblastic Leukemia. Cancer Cell 2015, 27, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Pitt, L.A.; Tikhonova, A.N.; Hu, H.; Trimarchi, T.; King, B.; Gong, Y.; Sanchez-Martin, M.; Tsirigos, A.; Littman, D.R.; Ferrando, A.A.; et al. CXCL12-Producing Vascular Endothelial Niches Control Acute T Cell Leukemia Maintenance. Cancer Cell 2015, 27, 755–768. [Google Scholar] [CrossRef]
- Liou, A.; Delgado-Martin, C.; Teachey, D.T.; Hermiston, M.L. The CXCR4/CXCL12 Axis Mediates Chemotaxis, Survival, and Chemoresistance in T-Cell Acute Lymphoblastic Leukemia. Blood 2014, 124, 3629. [Google Scholar] [CrossRef]
- Crazzolara, R.; Kreczy, A.; Mann, G.; Heitger, A.; Eibl, G.; Fink, F.M.; Möhle, R.; Meister, B. High expression of the chemokine receptor CXCR4 predicts extramedullary organ infiltration in childhood acute lymphoblastic leukaemia. Br. J. Haematol. 2001, 115, 545–553. [Google Scholar] [CrossRef]
- Jost, T.R.; Borga, C.; Radaelli, E.; Romagnani, A.; Perruzza, L.; Omodho, L.; Cazzaniga, G.; Biondi, A.; Indraccolo, S.; Thelen, M.; et al. Role of CXCR4-mediated bone marrow colonization in CNS infiltration by T cell acute lymphoblastic leukemia. J. Leukoc. Biol. 2016, 99, 1077–1087. [Google Scholar] [CrossRef]
- Peled, A.; Klein, S.; Beider, K.; Burger, J.A.; Abraham, M. Role of CXCL12 and CXCR4 in the pathogenesis of hematological malignancies. Cytokine 2018, 109, 11–16. [Google Scholar] [CrossRef]
- Lonetti, A.; Cappellini, A.; Bertaina, A.; Locatelli, F.; Pession, A.; Buontempo, F.; Evangelisti, C.; Evangelisti, C.; Orsini, E.; Zambonin, L.; et al. Improving nelarabine efficacy in T cell acute lymphoblastic leukemia by targeting aberrant PI3K/AKT/mTOR signaling pathway. J. Hematol. Oncol. 2016, 9, 114. [Google Scholar] [CrossRef] [PubMed]
- Sison, E.A.R.; Magoon, D.; Chevalier, E.; Dembowsky, K.; Brown, P. The Novel CXCR4 Antagonist POL5551 Decreases Surface CXCR4 (s-CXCR4) Expression, Inhibits Chemotaxis, and Enhances Chemosensitivity in Acute Lymphoblastic Leukemia (ALL). Blood 2012, 120, 780. [Google Scholar] [CrossRef]
- Xia, J.; Jotte, M.R.; Sun, S.; Uy, G.L.; Vainstein, A.; Sorani, E.; Bohana-Kashtan, O.; Shaw, S.; Link, D.C. The CXCR4 Antagonist, BL8040, Is Highly Active Against Human T-ALL in Preclinical Models. Blood 2018, 132, 2700. [Google Scholar] [CrossRef]
- Uy, G.L.; Kadia, T.M.; Stock, W.; Brammer, J.E.; Bohana-Kashtan, O.; Vainstein, A.; Sorani, E.; Chen, H.; DiPersio, J.F.; Link, D.C. CXCR4 Inhibition with BL-8040 in Combination with Nelarabine in Patients with Relapsed or Refractory T-Cell Acute Lymphoblastic Leukemia / Lymphoblastic Lymphoma. Blood 2019, 134, 2630. [Google Scholar] [CrossRef]
- van der Zwet, J.C.G.; Buijs-Gladdines, J.; Cordo, V.; Debets, D.O.; Smits, W.K.; Chen, Z.; Dylus, J.; Zaman, G.J.R.; Altelaar, M.; Oshima, K.; et al. MAPK-ERK is a central pathway in T-cell acute lymphoblastic leukemia that drives steroid resistance. Leukemia 2021, 35, 3394–3405. [Google Scholar] [CrossRef] [PubMed]
- Tabares-Seisdedos, R.; Rubenstein, J.L. Inverse cancer comorbidity: A serendipitous opportunity to gain insight into CNS disorders. Nat. Rev. Neurosci. 2013, 14, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Papayannidis, C.; DeAngelo, D.J.; Stock, W.; Huang, B.; Shaik, M.N.; Cesari, R.; Zheng, X.; Reynolds, J.M.; English, P.A.; Ozeck, M.; et al. A Phase 1 study of the novel gamma-secretase inhibitor PF-03084014 in patients with T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma. Blood Cancer J. 2015, 5, e350. [Google Scholar] [CrossRef] [PubMed]
- Borthakur, G.; Martinelli, G.; Raffoux, E.; Chevallier, P.; Chromik, J.; Lithio, A.; Smith, C.L.; Yuen, E.; Oakley, G.J., 3rd; Benhadji, K.A.; et al. Phase 1 study to evaluate Crenigacestat (LY3039478) in combination with dexamethasone in patients with T-cell acute lymphoblastic leukemia and lymphoma. Cancer 2021, 127, 372–380. [Google Scholar] [CrossRef]
- Pullarkat, V.A.; Lacayo, N.J.; Jabbour, E.; Rubnitz, J.E.; Bajel, A.; Laetsch, T.W.; Leonard, J.; Colace, S.I.; Khaw, S.L.; Fleming, S.A.; et al. Venetoclax and Navitoclax in Combination with Chemotherapy in Patients with Relapsed or Refractory Acute Lymphoblastic Leukemia and Lymphoblastic Lymphoma. Cancer Discov. 2021, 11, 1440–1453. [Google Scholar] [CrossRef] [PubMed]
- Place, A.E.; Goldsmith, K.; Bourquin, J.P.; Loh, M.L.; Gore, L.; Morgenstern, D.A.; Sanzgiri, Y.; Hoffman, D.; Zhou, Y.; Ross, J.A.; et al. Accelerating drug development in pediatric cancer: A novel Phase I study design of venetoclax in relapsed/refractory malignancies. Future Oncol. 2018, 14, 2115–2129. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Wunderle, L.; Badura, S.; Schleyer, E.; Bruggemann, M.; Serve, H.; Schnittger, S.; Gokbuget, N.; Pfeifer, H.; Wagner, S.; et al. A phase I study of a dual PI3-kinase/mTOR inhibitor BEZ235 in adult patients with relapsed or refractory acute leukemia. BMC Pharmacol. Toxicol. 2020, 21, 70. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Song, Z.; Zheng, G.; Nicolazzi, C.; Fromm, J.R.; Shehu, E.; Srinivasan, S.; Chen, X.; Zhu, C.; Blondel, M.C.; et al. Evaluation of Preclinical Activity of Isatuximab in Patients with Acute Lymphoblastic Leukemia. Mol. Cancer Ther. 2021, 20, 1916–1925. [Google Scholar] [CrossRef]
- Angiolillo, A.L.; Yu, A.L.; Reaman, G.; Ingle, A.M.; Secola, R.; Adamson, P.C. A phase II study of Campath-1H in children with relapsed or refractory acute lymphoblastic leukemia: A Children’s Oncology Group report. Pediatr. Blood Cancer 2009, 53, 978–983. [Google Scholar] [CrossRef]
- Ravandi, F.; Aribi, A.; O’Brien, S.; Faderl, S.; Jones, D.; Ferrajoli, A.; Huang, X.; York, S.; Pierce, S.; Wierda, W.; et al. Phase II study of alemtuzumab in combination with pentostatin in patients with T-cell neoplasms. J. Clin. Oncol. 2009, 27, 5425–5430. [Google Scholar] [CrossRef]
- Madrazo, E.; Gonzalez-Novo, R.; Ortiz-Placin, C.; Garcia de Lacoba, M.; Gonzalez-Murillo, A.; Ramirez, M.; Redondo-Munoz, J. Fast H3K9 methylation promoted by CXCL12 contributes to nuclear changes and invasiveness of T-acute lymphoblastic leukemia cells. Oncogene 2022, 41, 1324–1336. [Google Scholar] [CrossRef] [PubMed]
- Peirs, S.; Van der Meulen, J.; Van de Walle, I.; Taghon, T.; Speleman, F.; Poppe, B.; Van Vlierberghe, P. Epigenetics in T-cell acute lymphoblastic leukemia. Immunol. Rev. 2015, 263, 50–67. [Google Scholar] [CrossRef]
- Belver, L.; Ferrando, A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat. Rev. Cancer 2016, 16, 494–507. [Google Scholar] [CrossRef]
- Mackowska, N.; Drobna-Sledzinska, M.; Witt, M.; Dawidowska, M. DNA Methylation in T-Cell Acute Lymphoblastic Leukemia: In Search for Clinical and Biological Meaning. Int. J. Mol. Sci. 2021, 22, 1388. [Google Scholar] [CrossRef]
- Kurzer, J.H.; Weinberg, O.K. PHF6 Mutations in Hematologic Malignancies. Front. Oncol. 2021, 11, 704471. [Google Scholar] [CrossRef]
- Kitara, S.; Su, A.; Deluca, C.; Aversa, F.; Knoechel, B.; Jin, J.; Stegmaier, K.; Roti, G. Identification of EHMT2 As a New Target in T-Cell Acute Lymphoblastic Leukemia. Blood 2017, 130, 2548. [Google Scholar] [CrossRef]
- Zhou, M.; Zhou, K.; Cheng, L.; Chen, X.; Wang, J.; Wang, X.M.; Zhang, Y.; Yu, Q.; Zhang, S.; Wang, D.; et al. MBD2 Ablation Impairs Lymphopoiesis and Impedes Progression and Maintenance of T-ALL. Cancer Res. 2018, 78, 1632–1642. [Google Scholar] [CrossRef] [PubMed]
- Montanaro, A.; Kitara, S.; Cerretani, E.; Marchesini, M.; Rompietti, C.; Pagliaro, L.; Gherli, A.; Su, A.; Minchillo, M.L.; Caputi, M.; et al. Identification of an Epi-metabolic dependency on EHMT2/G9a in T-cell acute lymphoblastic leukemia. Cell Death Dis. 2022, 13, 551. [Google Scholar] [CrossRef] [PubMed]
- Triplett, T.A.; Cardenas, K.T.; Lancaster, J.N.; Hu, Z.; Selden, H.J.; Jasso, G.J.; Balasubramanyam, S.; Chan, K.; Li, L.; Chen, X.; et al. Endogenous dendritic cells from the tumor microenvironment support T-ALL growth via IGF1R activation. Proc. Natl. Acad. Sci. USA 2016, 113, E1016–E1025. [Google Scholar] [CrossRef] [PubMed]
- Lyu, A.; Triplett, T.A.; Nam, S.H.; Hu, Z.; Arasappan, D.; Godfrey, W.H.; Ames, R.Y.; Sarang, A.; Selden, H.J.; Lee, C.H.; et al. Tumor-associated myeloid cells provide critical support for T-ALL. Blood 2020, 136, 1837–1850. [Google Scholar] [CrossRef]
- Medyouf, H.; Gusscott, S.; Wang, H.; Tseng, J.C.; Wai, C.; Nemirovsky, O.; Trumpp, A.; Pflumio, F.; Carboni, J.; Gottardis, M.; et al. High-level IGF1R expression is required for leukemia-initiating cell activity in T-ALL and is supported by Notch signaling. J. Exp. Med. 2011, 208, 1809–1822. [Google Scholar] [CrossRef] [PubMed]
- Trimarchi, T.; Bilal, E.; Ntziachristos, P.; Fabbri, G.; Dalla-Favera, R.; Tsirigos, A.; Aifantis, I. Genome-wide mapping and characterization of Notch-regulated long noncoding RNAs in acute leukemia. Cell 2014, 158, 593–606. [Google Scholar] [CrossRef]
- Giambra, V.; Gusscott, S.; Gracias, D.; Song, R.; Lam, S.H.; Panelli, P.; Tyshchenko, K.; Jenkins, C.E.; Hoofd, C.; Lorzadeh, A.; et al. Epigenetic Restoration of Fetal-like IGF1 Signaling Inhibits Leukemia Stem Cell Activity. Cell Stem Cell 2018, 23, 714–726.e717. [Google Scholar] [CrossRef] [PubMed]
- Artico, L.L.; Laranjeira, A.B.A.; Campos, L.W.; Corrêa, J.R.; Zenatti, P.P.; Carvalheira, J.B.C.; Brambilla, S.R.; Nowill, A.E.; Brandalise, S.R.; Yunes, J.A. Physiologic IGFBP7 levels prolong IGF1R activation in acute lymphoblastic leukemia. Blood Adv. 2021, 5, 3633–3646. [Google Scholar] [CrossRef]
- Bartram, I.; Erben, U.; Ortiz-Tanchez, J.; Blunert, K.; Schlee, C.; Neumann, M.; Heesch, S.; Baldus, C.D. Inhibition of IGF1-R overcomes IGFBP7-induced chemotherapy resistance in T-ALL. BMC Cancer 2015, 15, 663. [Google Scholar] [CrossRef]
- Scupoli, M.T.; Vinante, F.; Krampera, M.; Vincenzi, C.; Nadali, G.; Zampieri, F.; Ritter, M.A.; Eren, E.; Santini, F.; Pizzolo, G. Thymic epithelial cells promote survival of human T-cell acute lymphoblastic leukemia blasts: The role of interleukin-7. Haematologica 2003, 88, 1229–1237. [Google Scholar] [PubMed]
- Silva, A.; Laranjeira, A.B.; Martins, L.R.; Cardoso, B.A.; Demengeot, J.; Yunes, J.A.; Seddon, B.; Barata, J.T. IL-7 contributes to the progression of human T-cell acute lymphoblastic leukemias. Cancer Res. 2011, 71, 4780–4789. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, D.; Melao, A.; van Boxtel, R.; Santos, C.I.; Silva, A.; Silva, M.C.; Cardoso, B.A.; Coffer, P.J.; Barata, J.T. STAT5 is essential for IL-7-mediated viability, growth, and proliferation of T-cell acute lymphoblastic leukemia cells. Blood Adv. 2018, 2, 2199–2213. [Google Scholar] [CrossRef]
- Zenatti, P.P.; Ribeiro, D.; Li, W.; Zuurbier, L.; Silva, M.C.; Paganin, M.; Tritapoe, J.; Hixon, J.A.; Silveira, A.B.; Cardoso, B.A.; et al. Oncogenic IL7R gain-of-function mutations in childhood T-cell acute lymphoblastic leukemia. Nat. Genet. 2011, 43, 932–939. [Google Scholar] [CrossRef]
- Shochat, C.; Tal, N.; Bandapalli, O.R.; Palmi, C.; Ganmore, I.; te Kronnie, G.; Cario, G.; Cazzaniga, G.; Kulozik, A.E.; Stanulla, M.; et al. Gain-of-function mutations in interleukin-7 receptor-alpha (IL7R) in childhood acute lymphoblastic leukemias. J. Exp. Med. 2011, 208, 901–908. [Google Scholar] [CrossRef]
- Zhang, J.; Ding, L.; Holmfeldt, L.; Wu, G.; Heatley, S.L.; Payne-Turner, D.; Easton, J.; Chen, X.; Wang, J.; Rusch, M.; et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012, 481, 157–163. [Google Scholar] [CrossRef]
- Treanor, L.M.; Zhou, S.; Janke, L.; Churchman, M.L.; Ma, Z.; Lu, T.; Chen, S.C.; Mullighan, C.G.; Sorrentino, B.P. Interleukin-7 receptor mutants initiate early T cell precursor leukemia in murine thymocyte progenitors with multipotent potential. J. Exp. Med. 2014, 211, 701–713. [Google Scholar] [CrossRef]
- Canté-Barrett, K.; Spijkers-Hagelstein, J.A.; Buijs-Gladdines, J.G.; Uitdehaag, J.C.; Smits, W.K.; van der Zwet, J.; Buijsman, R.C.; Zaman, G.J.; Pieters, R.; Meijerink, J.P. MEK and PI3K-AKT inhibitors synergistically block activated IL7 receptor signaling in T-cell acute lymphoblastic leukemia. Leukemia 2016, 30, 1832–1843. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Boissel, N.; Touzart, A.; Leguay, T.; Thonier, F.; Thomas, X.; Raffoux, E.; Huguet, F.; Villarese, P.; Fourrage, C.; et al. Adult T-cell acute lymphoblastic leukemias with IL7R pathway mutations are slow-responders who do not benefit from allogeneic stem-cell transplantation. Leukemia 2020, 34, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Buijs-Gladdines, J.G.; Cante-Barrett, K.; Stubbs, A.P.; Vroegindeweij, E.M.; Smits, W.K.; van Marion, R.; Dinjens, W.N.; Horstmann, M.; Kuiper, R.P.; et al. IL-7 Receptor Mutations and Steroid Resistance in Pediatric T cell Acute Lymphoblastic Leukemia: A Genome Sequencing Study. PLoS Med. 2016, 13, e1002200. [Google Scholar] [CrossRef]
- Silva, A.; Almeida, A.R.M.; Cachucho, A.; Neto, J.L.; Demeyer, S.; de Matos, M.; Hogan, T.; Li, Y.; Meijerink, J.; Cools, J.; et al. Overexpression of wild-type IL-7Rα promotes T-cell acute lymphoblastic leukemia/lymphoma. Blood 2021, 138, 1040–1052. [Google Scholar] [CrossRef]
- González-García, S.; Mosquera, M.; Fuentes, P.; Palumbo, T.; Escudero, A.; Pérez-Martínez, A.; Ramírez, M.; Corcoran, A.E.; Toribio, M.L. IL-7R is essential for leukemia-initiating cell activity of T-cell acute lymphoblastic leukemia. Blood 2019, 134, 2171–2182. [Google Scholar] [CrossRef]
- Tremblay, C.S.; Brown, F.C.; Collett, M.; Saw, J.; Chiu, S.K.; Sonderegger, S.E.; Lucas, S.E.; Alserihi, R.; Chau, N.; Toribio, M.L.; et al. Loss-of-function mutations of Dynamin 2 promote T-ALL by enhancing IL-7 signalling. Leukemia 2016, 30, 1993–2001. [Google Scholar] [CrossRef]
- Degryse, S.; de Bock, C.E.; Cox, L.; Demeyer, S.; Gielen, O.; Mentens, N.; Jacobs, K.; Geerdens, E.; Gianfelici, V.; Hulselmans, G.; et al. JAK3 mutants transform hematopoietic cells through JAK1 activation, causing T-cell acute lymphoblastic leukemia in a mouse model. Blood 2014, 124, 3092–3100. [Google Scholar] [CrossRef]
- de Bock, C.E.; Demeyer, S.; Degryse, S.; Verbeke, D.; Sweron, B.; Gielen, O.; Vandepoel, R.; Vicente, C.; Vanden Bempt, M.; Dagklis, A.; et al. HOXA9 Cooperates with Activated JAK/STAT Signaling to Drive Leukemia Development. Cancer Discov. 2018, 8, 616–631. [Google Scholar] [CrossRef] [PubMed]
- Pham, H.T.T.; Maurer, B.; Prchal-Murphy, M.; Grausenburger, R.; Grundschober, E.; Javaheri, T.; Nivarthi, H.; Boersma, A.; Kolbe, T.; Elabd, M.; et al. STAT5BN642H is a driver mutation for T cell neoplasia. J. Clin. Investig. 2018, 128, 387–401. [Google Scholar] [CrossRef] [PubMed]
- de Araujo, E.D.; Erdogan, F.; Neubauer, H.A.; Meneksedag-Erol, D.; Manaswiyoungkul, P.; Eram, M.S.; Seo, H.S.; Qadree, A.K.; Israelian, J.; Orlova, A.; et al. Structural and functional consequences of the STAT5B(N642H) driver mutation. Nat. Commun. 2019, 10, 2517. [Google Scholar] [CrossRef]
- Buonamici, S.; Trimarchi, T.; Ruocco, M.G.; Reavie, L.; Cathelin, S.; Mar, B.G.; Klinakis, A.; Lukyanov, Y.; Tseng, J.C.; Sen, F.; et al. CCR7 signalling as an essential regulator of CNS infiltration in T-cell leukaemia. Nature 2009, 459, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Alsadeq, A.; Fedders, H.; Vokuhl, C.; Belau, N.M.; Zimmermann, M.; Wirbelauer, T.; Spielberg, S.; Vossen-Gajcy, M.; Cario, G.; Schrappe, M.; et al. The role of ZAP70 kinase in acute lymphoblastic leukemia infiltration into the central nervous system. Haematologica 2017, 102, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Cuesta-Mateos, C.; Fuentes, P.; Schrader, A.; Juarez-Sanchez, R.; Loscertales, J.; Mateu-Albero, T.; Vega-Piris, L.; Espartero-Santos, M.; Marcos-Jimenez, A.; Sanchez-Lopez, B.A.; et al. CCR7 as a novel therapeutic target in t-cell PROLYMPHOCYTIC leukemia. Biomark. Res. 2020, 8, 54. [Google Scholar] [CrossRef]
- Erb, U.; Hikel, J.; Meyer, S.; Ishikawa, H.; Worst, T.S.; Nitschke, K.; Nuhn, P.; Porubsky, S.; Weiss, C.; Schroten, H.; et al. The Impact of Small Extracellular Vesicles on Lymphoblast Trafficking across the Blood-Cerebrospinal Fluid Barrier In Vitro. Int. J. Mol. Sci. 2020, 21, 5491. [Google Scholar] [CrossRef]
- Enting, R.H. Leptomeningeal neoplasia: Epidemiology, clinical presentation, CSF analysis and diagnostic imaging. Cancer Treat. Res. 2005, 125, 17–30. [Google Scholar] [CrossRef]
- Kato, I.; Nishinaka, Y.; Nakamura, M.; Akarca, A.U.; Niwa, A.; Ozawa, H.; Yoshida, K.; Mori, M.; Wang, D.; Morita, M.; et al. Hypoxic adaptation of leukemic cells infiltrating the CNS affords a therapeutic strategy targeting VEGFA. Blood 2017, 129, 3126–3129. [Google Scholar] [CrossRef]
- Savino, A.M.; Fernandes, S.I.; Olivares, O.; Zemlyansky, A.; Cousins, A.; Markert, E.K.; Barel, S.; Geron, I.; Frishman, L.; Birger, Y.; et al. Metabolic adaptation of acute lymphoblastic leukemia to the central nervous system microenvironment is dependent on Stearoyl CoA desaturase. Nat. Cancer 2020, 1, 998–1009. [Google Scholar] [CrossRef]
- Vanner, R.J.; Dobson, S.M.; Gan, O.I.; McLeod, J.; Schoof, E.M.; Grandal, I.; Wintersinger, J.A.; Garcia-Prat, L.; Hosseini, M.; Xie, S.Z.; et al. Multiomic Profiling of Central Nervous System Leukemia Identifies mRNA Translation as a Therapeutic Target. Blood Cancer Discov. 2022, 3, 16–31. [Google Scholar] [CrossRef]
- Krause, D.S.; Scadden, D.T. A hostel for the hostile: The bone marrow niche in hematologic neoplasms. Haematologica 2015, 100, 1376–1387. [Google Scholar] [CrossRef]
- Battula, V.L.; Le, P.M.; Sun, J.C.; Nguyen, K.; Yuan, B.; Zhou, X.; Sonnylal, S.; McQueen, T.; Ruvolo, V.; Michel, K.A.; et al. AML-induced osteogenic differentiation in mesenchymal stromal cells supports leukemia growth. JCI Insight 2017, 2, e90036. [Google Scholar] [CrossRef]
- Kumar, R.; Godavarthy, P.S.; Krause, D.S. The bone marrow microenvironment in health and disease at a glance. J. Cell Sci. 2018, 131, jcs201707. [Google Scholar] [CrossRef]
- Peaudecerf, L.; Lemos, S.; Galgano, A.; Krenn, G.; Vasseur, F.; Di Santo, J.P.; Ezine, S.; Rocha, B. Thymocytes may persist and differentiate without any input from bone marrow progenitors. J. Exp. Med. 2012, 209, 1401–1408. [Google Scholar] [CrossRef]
- Martins, V.C.; Ruggiero, E.; Schlenner, S.M.; Madan, V.; Schmidt, M.; Fink, P.J.; von Kalle, C.; Rodewald, H.R. Thymus-autonomous T cell development in the absence of progenitor import. J. Exp. Med. 2012, 209, 1409–1417. [Google Scholar] [CrossRef]
- Martins, V.C.; Busch, K.; Juraeva, D.; Blum, C.; Ludwig, C.; Rasche, V.; Lasitschka, F.; Mastitsky, S.E.; Brors, B.; Hielscher, T.; et al. Cell competition is a tumour suppressor mechanism in the thymus. Nature 2014, 509, 465–470. [Google Scholar] [CrossRef]
- Ballesteros-Arias, L.; Silva, J.G.; Paiva, R.A.; Carbonetto, B.; Faisca, P.; Martins, V.C. T Cell Acute Lymphoblastic Leukemia as a Consequence of Thymus Autonomy. J. Immunol. 2019, 202, 1137–1144. [Google Scholar] [CrossRef]
- Simone, J.V.; Verzosa, M.S.; Rudy, J.A. Initial features and prognosis in 363 children with acute lymphocytic leukemia. Cancer 1975, 36, 2099–2108. [Google Scholar] [CrossRef]
- Shuster, J.J.; Falletta, J.M.; Pullen, D.J.; Crist, W.M.; Humphrey, G.B.; Dowell, B.L.; Wharam, M.D.; Borowitz, M. Prognostic factors in childhood T-cell acute lymphoblastic leukemia: A Pediatric Oncology Group study. Blood 1990, 75, 166–173. [Google Scholar] [CrossRef]
- Majumdar, G.; Singh, A.K. Role of splenectomy in chronic lymphocytic leukaemia with massive splenomegaly and cytopenia. Leuk. Lymphoma 1992, 7, 131–134. [Google Scholar] [CrossRef]
- O’Dwyer, K.M. The challenge to further improvements in survival of patients with T-ALL: Current treatments and new insights from disease pathogenesis. Semin. Hematol. 2020, 57, 149–156. [Google Scholar] [CrossRef]
- Polgarova, K.; Otahal, P.; Salek, C.; Pytlik, R. Chimeric Antigen Receptor Based Cellular Therapy for Treatment Of T-Cell Malignancies. Front. Oncol. 2022, 12, 876758. [Google Scholar] [CrossRef]
- Samon, J.B.; Castillo-Martin, M.; Hadler, M.; Ambesi-Impiobato, A.; Paietta, E.; Racevskis, J.; Wiernik, P.H.; Rowe, J.M.; Jakubczak, J.; Randolph, S.; et al. Preclinical analysis of the gamma-secretase inhibitor PF-03084014 in combination with glucocorticoids in T-cell acute lymphoblastic leukemia. Mol. Cancer Ther. 2012, 11, 1565–1575. [Google Scholar] [CrossRef]
- Lopez-Nieva, P.; Gonzalez-Sanchez, L.; Cobos-Fernandez, M.A.; Cordoba, R.; Santos, J.; Fernandez-Piqueras, J. More Insights on the Use of gamma-Secretase Inhibitors in Cancer Treatment. Oncologist 2021, 26, e298–e305. [Google Scholar] [CrossRef]
- Wei, P.; Walls, M.; Qiu, M.; Ding, R.; Denlinger, R.H.; Wong, A.; Tsaparikos, K.; Jani, J.P.; Hosea, N.; Sands, M.; et al. Evaluation of selective gamma-secretase inhibitor PF-03084014 for its antitumor efficacy and gastrointestinal safety to guide optimal clinical trial design. Mol. Cancer Ther. 2010, 9, 1618–1628. [Google Scholar] [CrossRef]
- Baratta, M.G. Adjusting the focus on gamma-secretase inhibition. Nat. Rev. Cancer 2019, 19, 419. [Google Scholar] [CrossRef]
- Habets, R.A.; de Bock, C.E.; Serneels, L.; Lodewijckx, I.; Verbeke, D.; Nittner, D.; Narlawar, R.; Demeyer, S.; Dooley, J.; Liston, A.; et al. Safe targeting of T cell acute lymphoblastic leukemia by pathology-specific NOTCH inhibition. Sci. Transl. Med. 2019, 11, eaau6246. [Google Scholar] [CrossRef]
- Aste-Amezaga, M.; Zhang, N.; Lineberger, J.E.; Arnold, B.A.; Toner, T.J.; Gu, M.; Huang, L.; Vitelli, S.; Vo, K.T.; Haytko, P.; et al. Characterization of Notch1 antibodies that inhibit signaling of both normal and mutated Notch1 receptors. PLoS ONE 2010, 5, e9094. [Google Scholar] [CrossRef]
- Wu, Y.; Cain-Hom, C.; Choy, L.; Hagenbeek, T.J.; de Leon, G.P.; Chen, Y.; Finkle, D.; Venook, R.; Wu, X.; Ridgway, J.; et al. Therapeutic antibody targeting of individual Notch receptors. Nature 2010, 464, 1052–1057. [Google Scholar] [CrossRef]
- Agnusdei, V.; Minuzzo, S.; Frasson, C.; Grassi, A.; Axelrod, F.; Satyal, S.; Gurney, A.; Hoey, T.; Seganfreddo, E.; Basso, G.; et al. Therapeutic antibody targeting of Notch1 in T-acute lymphoblastic leukemia xenografts. Leukemia 2014, 28, 278–288. [Google Scholar] [CrossRef]
- Peirs, S.; Matthijssens, F.; Goossens, S.; Van de Walle, I.; Ruggero, K.; de Bock, C.E.; Degryse, S.; Cante-Barrett, K.; Briot, D.; Clappier, E.; et al. ABT-199 mediated inhibition of BCL-2 as a novel therapeutic strategy in T-cell acute lymphoblastic leukemia. Blood 2014, 124, 3738–3747. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- McEwan, A.; Pitiyarachchi, O.; Viiala, N. Relapsed/Refractory ETP-ALL Successfully Treated With Venetoclax and Nelarabine as a Bridge to Allogeneic Stem Cell Transplant. Hemasphere 2020, 4, e379. [Google Scholar] [CrossRef]
- Farhadfar, N.; Li, Y.; May, W.S.; Adams, C.B. Venetoclax and decitabine for treatment of relapsed T-cell acute lymphoblastic leukemia: A case report and review of literature. Hematol Oncol Stem Cell 2021, 14, 246–251. [Google Scholar] [CrossRef]
- Hantel, A.; Wynne, J.; Lacayo, N.; Khaw, S.L.; Rubnitz, J.; Mullighan, C.; Schmidt, M.; Zhou, Y.; Ross, J.A.; Rosenwinkel, L.; et al. Safety and Efficacy of the BCL Inhibitors Venetoclax and Navitoclax in Combination with Chemotherapy in Patients with Relapsed/Refractory Acute Lymphoblastic Leukemia and Lymphoblastic Lymphoma. Clin. Lymphoma Myeloma Leuk. 2018, 18, S184–S185. [Google Scholar] [CrossRef]
- Li, Z.; He, S.; Look, A.T. The MCL1-specific inhibitor S63845 acts synergistically with venetoclax/ABT-199 to induce apoptosis in T-cell acute lymphoblastic leukemia cells. Leukemia 2019, 33, 262–266. [Google Scholar] [CrossRef]
- Khan, S.; Zhang, X.; Lv, D.; Zhang, Q.; He, Y.; Zhang, P.; Liu, X.; Thummuri, D.; Yuan, Y.; Wiegand, J.S.; et al. A selective BCL-XL PROTAC degrader achieves safe and potent antitumor activity. Nat. Med. 2019, 25, 1938–1947. [Google Scholar] [CrossRef]
- Maude, S.L.; Dolai, S.; Delgado-Martin, C.; Vincent, T.; Robbins, A.; Selvanathan, A.; Ryan, T.; Hall, J.; Wood, A.C.; Tasian, S.K.; et al. Efficacy of JAK/STAT pathway inhibition in murine xenograft models of early T-cell precursor (ETP) acute lymphoblastic leukemia. Blood 2015, 125, 1759–1767. [Google Scholar] [CrossRef]
- Dubois, S.; Miljkovic, M.D.; Fleisher, T.; Hsu, J.; Waldmann, T.A.; Conlon, K.C. Transient Inhibition of the JAK1/3-STAT5 Pathway By Ruxolitinib Is Insufficient to Produce Clinical Benefit in Patients with Indolent Adult T-Cell Leukemia. Blood 2017, 130, 3840. [Google Scholar] [CrossRef]
- Verbeke, D.; Gielen, O.; Jacobs, K.; Boeckx, N.; De Keersmaecker, K.; Maertens, J.; Uyttebroeck, A.; Segers, H.; Cools, J. Ruxolitinib Synergizes With Dexamethasone for the Treatment of T-cell Acute Lymphoblastic Leukemia. Hemasphere 2019, 3, e310. [Google Scholar] [CrossRef]
- Delgado-Martin, C.; Meyer, L.K.; Huang, B.J.; Shimano, K.A.; Zinter, M.S.; Nguyen, J.V.; Smith, G.A.; Taunton, J.; Winter, S.S.; Roderick, J.R.; et al. JAK/STAT pathway inhibition overcomes IL7-induced glucocorticoid resistance in a subset of human T-cell acute lymphoblastic leukemias. Leukemia 2017, 31, 2568–2576. [Google Scholar] [CrossRef]
- Herbaux, C.; Kornauth, C.; Poulain, S.; Chong, S.J.F.; Collins, M.C.; Valentin, R.; Hackett, L.; Tournilhac, O.; Lemonnier, F.; Dupuis, J.; et al. BH3 profiling identifies ruxolitinib as a promising partner for venetoclax to treat T-cell prolymphocytic leukemia. Blood 2021, 137, 3495–3506. [Google Scholar] [CrossRef]
- Hales, E.C.; Orr, S.M.; Larson Gedman, A.; Taub, J.W.; Matherly, L.H. Notch1 receptor regulates AKT protein activation loop (Thr308) dephosphorylation through modulation of the PP2A phosphatase in phosphatase and tensin homolog (PTEN)-null T-cell acute lymphoblastic leukemia cells. J. Biol. Chem. 2013, 288, 22836–22848. [Google Scholar] [CrossRef]
- Shepherd, C.; Banerjee, L.; Cheung, C.W.; Mansour, M.R.; Jenkinson, S.; Gale, R.E.; Khwaja, A. PI3K/mTOR inhibition upregulates NOTCH-MYC signalling leading to an impaired cytotoxic response. Leukemia 2013, 27, 650–660. [Google Scholar] [CrossRef]
- Anand, P.; Guillaumet-Adkins, A.; Dimitrova, V.; Yun, H.; Drier, Y.; Sotudeh, N.; Rogers, A.; Ouseph, M.M.; Nair, M.; Potdar, S.; et al. Single-cell RNA-seq reveals developmental plasticity with coexisting oncogenic states and immune evasion programs in ETP-ALL. Blood 2021, 137, 2463–2480. [Google Scholar] [CrossRef]
- Joshi, I.; Minter, L.M.; Telfer, J.; Demarest, R.M.; Capobianco, A.J.; Aster, J.C.; Sicinski, P.; Fauq, A.; Golde, T.E.; Osborne, B.A. Notch signaling mediates G1/S cell-cycle progression in T cells via cyclin D3 and its dependent kinases. Blood 2009, 113, 1689–1698. [Google Scholar] [CrossRef]
- Sawai, C.M.; Freund, J.; Oh, P.; Ndiaye-Lobry, D.; Bretz, J.C.; Strikoudis, A.; Genesca, L.; Trimarchi, T.; Kelliher, M.A.; Clark, M.; et al. Therapeutic targeting of the cyclin D3:CDK4/6 complex in T cell leukemia. Cancer Cell 2012, 22, 452–465. [Google Scholar] [CrossRef]
- Bride, K.L.; Hu, H.; Tikhonova, A.; Fuller, T.J.; Vincent, T.L.; Shraim, R.; Li, M.M.; Carroll, W.L.; Raetz, E.A.; Aifantis, I.; et al. Rational drug combinations with CDK4/6 inhibitors in acute lymphoblastic Leukemia. Haematologica 2021, 107, 1746–1757. [Google Scholar] [CrossRef]
- Quintas-Cardama, A.; Tong, W.; Manshouri, T.; Vega, F.; Lennon, P.A.; Cools, J.; Gilliland, D.G.; Lee, F.; Cortes, J.; Kantarjian, H.; et al. Activity of tyrosine kinase inhibitors against human NUP214-ABL1-positive T cell malignancies. Leukemia 2008, 22, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Deenik, W.; Beverloo, H.B.; van der Poel-van de Luytgaarde, S.C.; Wattel, M.M.; van Esser, J.W.; Valk, P.J.; Cornelissen, J.J. Rapid complete cytogenetic remission after upfront dasatinib monotherapy in a patient with a NUP214-ABL1-positive T-cell acute lymphoblastic leukemia. Leukemia 2009, 23, 627–629. [Google Scholar] [CrossRef] [PubMed]
- Frismantas, V.; Dobay, M.P.; Rinaldi, A.; Tchinda, J.; Dunn, S.H.; Kunz, J.; Richter-Pechanska, P.; Marovca, B.; Pail, O.; Jenni, S.; et al. Ex vivo drug response profiling detects recurrent sensitivity patterns in drug-resistant acute lymphoblastic leukemia. Blood 2017, 129, e26–e37. [Google Scholar] [CrossRef]
- Shi, Y.; Beckett, M.C.; Blair, H.J.; Tirtakusuma, R.; Nakjang, S.; Enshaei, A.; Halsey, C.; Vormoor, J.; Heidenreich, O.; Krippner-Heidenreich, A.; et al. Phase II-like murine trial identifies synergy between dexamethasone and dasatinib in T-cell acute lymphoblastic leukemia. Haematologica 2021, 106, 1056–1066. [Google Scholar] [CrossRef]
- Cordo, V.; Meijer, M.T.; Hagelaar, R.; de Goeij-de Haas, R.R.; Poort, V.M.; Henneman, A.A.; Piersma, S.R.; Pham, T.V.; Oshima, K.; Ferrando, A.A.; et al. Phosphoproteomic profiling of T cell acute lymphoblastic leukemia reveals targetable kinases and combination treatment strategies. Nat. Commun. 2022, 13, 1048. [Google Scholar] [CrossRef]
- Hamilton, A.; Helgason, G.V.; Schemionek, M.; Zhang, B.; Myssina, S.; Allan, E.K.; Nicolini, F.E.; Muller-Tidow, C.; Bhatia, R.; Brunton, V.G.; et al. Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood 2012, 119, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Houshmand, M.; Simonetti, G.; Circosta, P.; Gaidano, V.; Cignetti, A.; Martinelli, G.; Saglio, G.; Gale, R.P. Chronic myeloid leukemia stem cells. Leukemia 2019, 33, 1543–1556. [Google Scholar] [CrossRef]
- Tembhare, P.R.; Sriram, H.; Khanka, T.; Chatterjee, G.; Panda, D.; Ghogale, S.; Badrinath, Y.; Deshpande, N.; Patkar, N.V.; Narula, G.; et al. Flow cytometric evaluation of CD38 expression levels in the newly diagnosed T-cell acute lymphoblastic leukemia and the effect of chemotherapy on its expression in measurable residual disease, refractory disease and relapsed disease: An implication for anti-CD38 immunotherapy. J. Immunother. Cancer 2020, 8, e000630. [Google Scholar] [CrossRef] [PubMed]
- Bride, K.L.; Vincent, T.L.; Im, S.Y.; Aplenc, R.; Barrett, D.M.; Carroll, W.L.; Carson, R.; Dai, Y.; Devidas, M.; Dunsmore, K.P.; et al. Preclinical efficacy of daratumumab in T-cell acute lymphoblastic leukemia. Blood 2018, 131, 995–999. [Google Scholar] [CrossRef] [PubMed]
- Vogiatzi, F.; Winterberg, D.; Lenk, L.; Buchmann, S.; Cario, G.; Schrappe, M.; Peipp, M.; Richter-Pechanska, P.; Kulozik, A.E.; Lentes, J.; et al. Daratumumab eradicates minimal residual disease in a preclinical model of pediatric T-cell acute lymphoblastic leukemia. Blood 2019, 134, 713–716. [Google Scholar] [CrossRef] [PubMed]
- Ofran, Y.; Ringelstein-Harlev, S.; Slouzkey, I.; Zuckerman, T.; Yehudai-Ofir, D.; Henig, I.; Beyar-Katz, O.; Hayun, M.; Frisch, A. Daratumumab for eradication of minimal residual disease in high-risk advanced relapse of T-cell/CD19/CD22-negative acute lymphoblastic leukemia. Leukemia 2020, 34, 293–295. [Google Scholar] [CrossRef]
- Vora, A.; Bhatla, T.; Teachey, D.; Bautista, F.; Moppett, J.; Velasco Puyó, P.; Micalizzi, C.; Rossig, C.; Shukla, N.; Gilad, G.; et al. Efficacy and Safety of Daratumumab in Pediatric and Young Adult Patients with Relapsed/Refractory T-Cell Acute Lymphoblastic Leukemia or Lymphoblastic Lymphoma: Results from Phase 2 DELPHINUS Study. In Proceedings of the 27th Annual Congress of the European Hematology Association, Vienna, Austria, 9–12 June 2022. [Google Scholar]
- Vakrmanova, B.; Novakova, M.; Riha, P.; Zaliova, M.; Fronkova, E.; Mejstrikova, E.; Stary, J.; Hrusak, O.; Sramkova, L. CD38: A target in relapsed/refractory acute lymphoblastic leukemia-Limitations in treatment and diagnostics. Pediatr. Blood Cancer 2022, 69, e29779. [Google Scholar] [CrossRef]
- Muller, K.; Vogiatzi, F.; Winterberg, D.; Rosner, T.; Lenk, L.; Bastian, L.; Gehlert, C.L.; Autenrieb, M.P.; Bruggemann, M.; Cario, G.; et al. Combining daratumumab with CD47 blockade prolongs survival in preclinical models of pediatric T-ALL. Blood 2022, 140, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Bayon-Calderon, F.; Toribio, M.L.; Gonzalez-Garcia, S. Facts and Challenges in Immunotherapy for T-Cell Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2020, 21, 7685. [Google Scholar] [CrossRef] [PubMed]
- Dearden, C.E.; Matutes, E.; Catovsky, D. Alemtuzumab in T-cell malignancies. Med. Oncol. 2002, 19, S27–S32. [Google Scholar] [CrossRef]
- Dearden, C. Alemtuzumab in peripheral T-cell malignancies. Cancer Biother. Radiopharm. 2004, 19, 391–398. [Google Scholar] [CrossRef]
- Akkapeddi, P.; Fragoso, R.; Hixon, J.A.; Ramalho, A.S.; Oliveira, M.L.; Carvalho, T.; Gloger, A.; Matasci, M.; Corzana, F.; Durum, S.K.; et al. A fully human anti-IL-7Ralpha antibody promotes antitumor activity against T-cell acute lymphoblastic leukemia. Leukemia 2019, 33, 2155–2168. [Google Scholar] [CrossRef]
- Hixon, J.A.; Andrews, C.; Kashi, L.; Kohnhorst, C.L.; Senkevitch, E.; Czarra, K.; Barata, J.T.; Li, W.; Schneider, J.P.; Walsh, S.T.R.; et al. New anti-IL-7Ralpha monoclonal antibodies show efficacy against T cell acute lymphoblastic leukemia in pre-clinical models. Leukemia 2020, 34, 35–49. [Google Scholar] [CrossRef]
- Gocho, Y.; Liu, J.; Hu, J.; Yang, W.; Dharia, N.V.; Zhang, J.; Shi, H.; Du, G.; John, A.; Lin, T.N.; et al. Network-based systems pharmacology reveals heterogeneity in LCK and BCL2 signaling and therapeutic sensitivity of T-cell acute lymphoblastic leukemia. Nat Cancer 2021, 2, 284–299. [Google Scholar] [CrossRef]
- Van Thillo, Q.; De Bie, J.; Seneviratne, J.A.; Demeyer, S.; Omari, S.; Balachandran, A.; Zhai, V.; Tam, W.L.; Sweron, B.; Geerdens, E.; et al. Oncogenic cooperation between TCF7-SPI1 and NRAS(G12D) requires beta-catenin activity to drive T-cell acute lymphoblastic leukemia. Nat. Commun. 2021, 12, 4164. [Google Scholar] [CrossRef]
- Fortini, M.E. Gamma-secretase-mediated proteolysis in cell-surface-receptor signalling. Nat. Reviews. Mol. Cell Biol. 2002, 3, 673–684. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Yang, K.; Zhang, H. Targeting Leukemia-Initiating Cells and Leukemic Niches: The Next Therapy Station for T-Cell Acute Lymphoblastic Leukemia? Cancers 2022, 14, 5655. https://doi.org/10.3390/cancers14225655
Zhang Z, Yang K, Zhang H. Targeting Leukemia-Initiating Cells and Leukemic Niches: The Next Therapy Station for T-Cell Acute Lymphoblastic Leukemia? Cancers. 2022; 14(22):5655. https://doi.org/10.3390/cancers14225655
Chicago/Turabian StyleZhang, Ziting, Kun Yang, and Han Zhang. 2022. "Targeting Leukemia-Initiating Cells and Leukemic Niches: The Next Therapy Station for T-Cell Acute Lymphoblastic Leukemia?" Cancers 14, no. 22: 5655. https://doi.org/10.3390/cancers14225655
APA StyleZhang, Z., Yang, K., & Zhang, H. (2022). Targeting Leukemia-Initiating Cells and Leukemic Niches: The Next Therapy Station for T-Cell Acute Lymphoblastic Leukemia? Cancers, 14(22), 5655. https://doi.org/10.3390/cancers14225655