

Genetic Characterization in High-Risk Individuals from a Low-Resource City of Peru

 , ,
, ,  , , , ,
, , , ,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Study Population
- Having an early onset of cancer (cancer diagnosis < 55 years of age) (n = 51)
- Having a late onset of cancer and familial history of cancer (n = 3)
- Being unaffected individuals with familial history of cancer (n = 47)
2.2. Next Generation Sequencing (NGS)
2.3. Variant Detection and Interpretation
2.4. Variant Validation by Cycling Temperature Capillary Electrophoresis (CTCE)
3. Results
3.1. Sociodemographic Characteristics of the Study Population
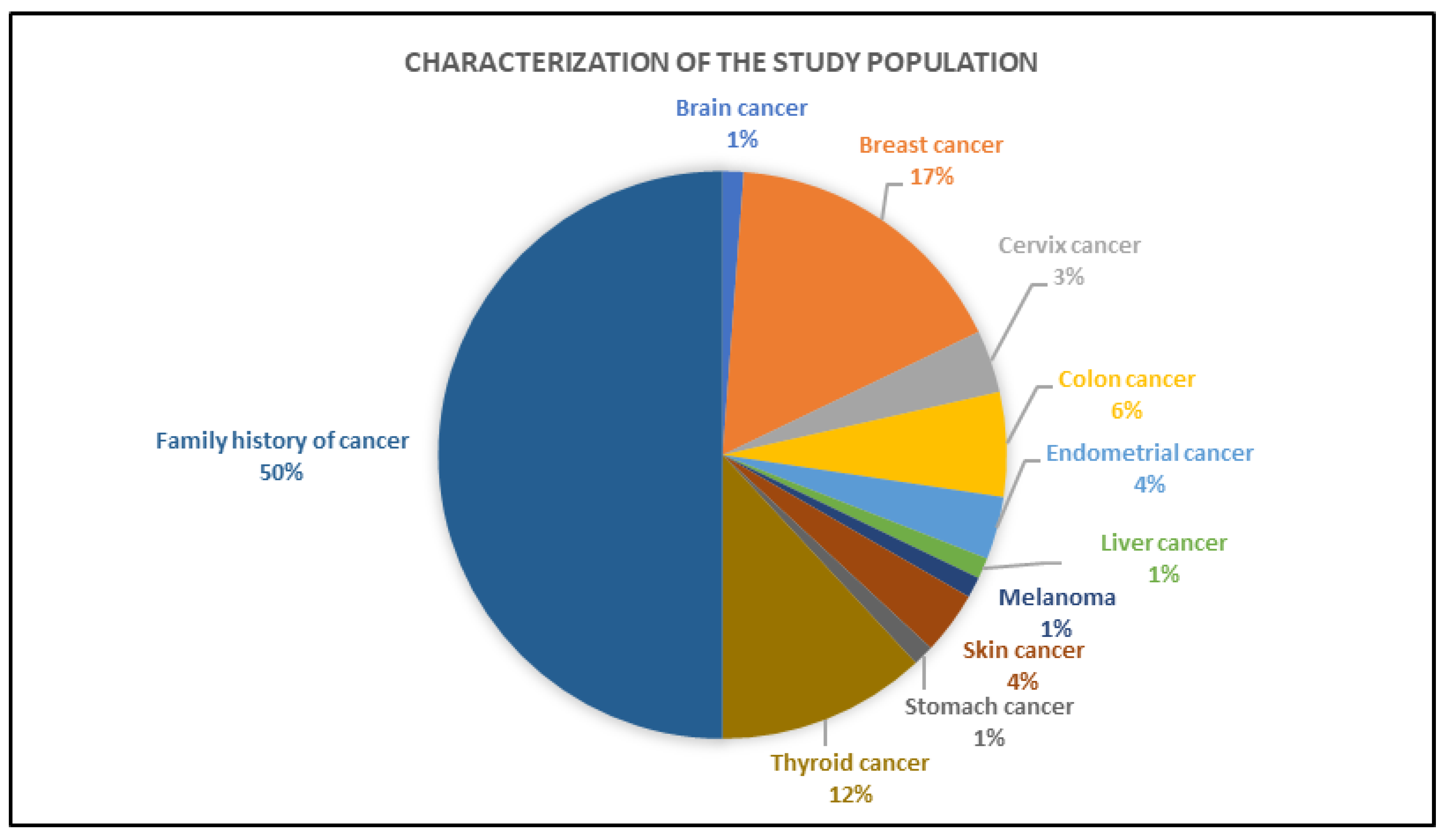
3.2. Clinical Characteristics of the Individuals Tested by Gene Panel
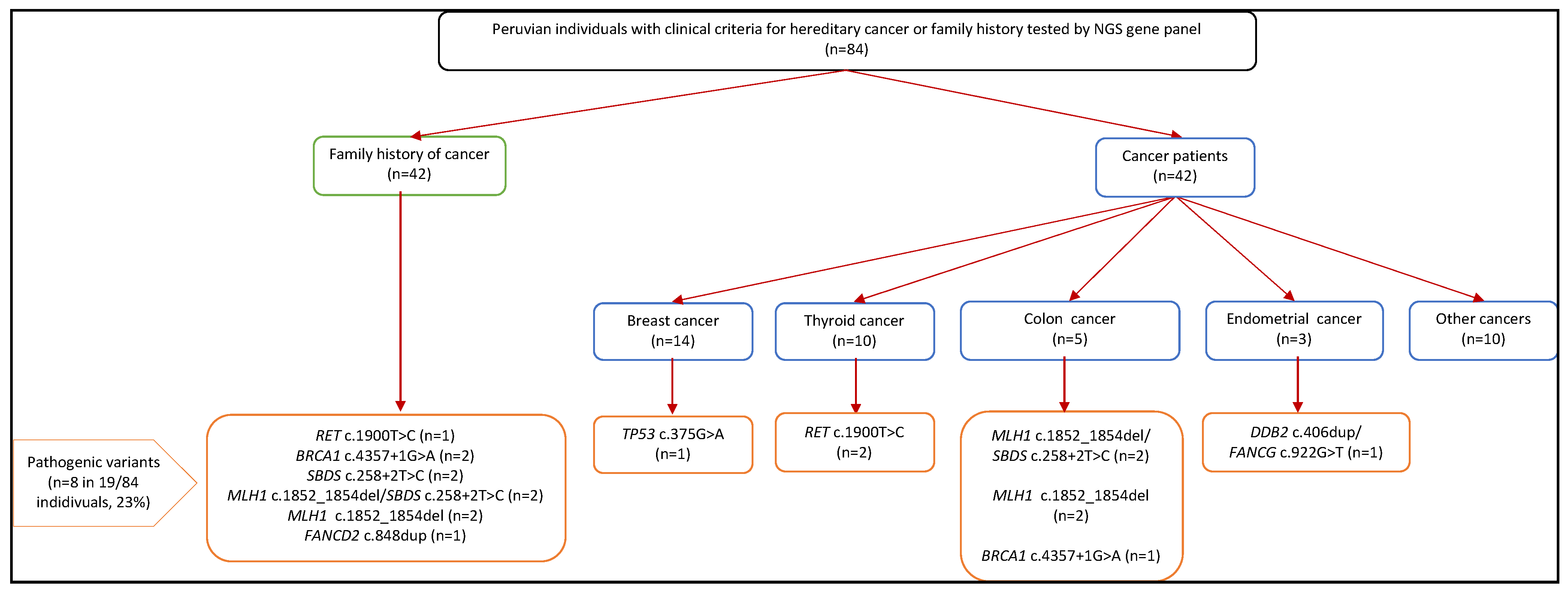
3.3. NGS Data Analysis
3.4. Pathogenic Germline Findings
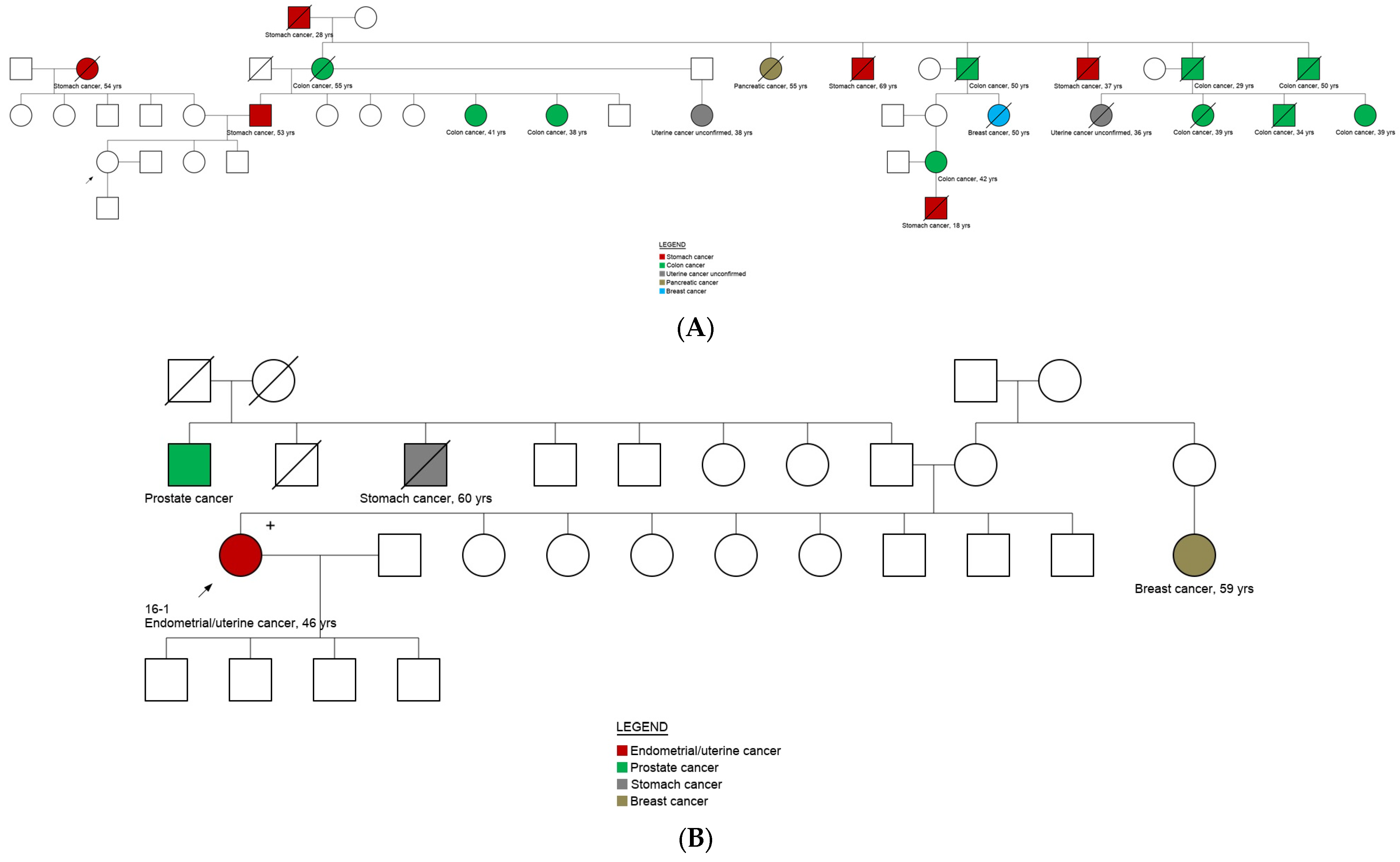
3.5. Genotype and Phenotype Correlation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dominguez-Valentin, M.; Nakken, S.; Tubeuf, H.; Vodak, D.; Ekstrøm, P.O.; Nissen, A.M.; Morak, M.; Holinski-Feder, E.; Martins, A.; Møller, P.; et al. Potentially pathogenic germline CHEK2 c.319+2T>A among multiple early-onset cancer families. Fam. Cancer 2018, 17, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Abeliovich, D.; Kaduri, L.; Lerer, I.; Weinberg, N.; Amir, G.; Sagi, M.; Zlotogora, J.; Heching, N.; Peretz, T. The founder mutations 185delAG and 5382insC in BRCA1 and 6174delT in BRCA2 appear in 60% of ovarian cancer and 30% of early-onset breast cancer patients among Ashkenazi women. Am. J. Hum. Genet. 1997, 60, 505–514. [Google Scholar]
- Newman, B.; Austin, M.A.; Lee, M.; King, M.C. Inheritance of human breast cancer: Evidence for autosomal dominant transmission in high-risk families. Proc. Natl. Acad. Sci. USA 1988, 85, 3044–3048. [Google Scholar] [CrossRef] [PubMed]
- Solis, N.; Zavaleta, E.; Wernhoff, P.; Dominguez-Barrera, C.; Dominguez-Valentin, M. Challenges to Bringing Personalized Medicine to a Low-Resource Setting in Peru. Int. J. Environ. Res. Public Health 2021, 18, 1470. [Google Scholar] [CrossRef]
- Aradhya, S.; Nussbaum, R.L. Genetics in mainstream medicine: Finally within grasp to influence healthcare globally. Mol. Genet. Genom. Med. 2018, 6, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Nakken, S.; Saveliev, V.; Hofmann, O.; Moller, P.; Myklebost, O.; Hovig, E. Cancer Predisposition Sequencing Reporter (CPSR): A flexible variant report engine for high-throughput germline screening in cancer. Int. J. Cancer 2021, 149, 1955–1960. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody MD, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Ekstrom, P.O.; Warren, D.J.; Thilly, W.G. Separation principles of cycling temperature capillary electrophoresis. Electrophoresis 2012, 33, 1162–1168. [Google Scholar] [CrossRef]
- Ekstrom, P.O.; Khrapko, K.; Li-Sucholeiki, X.C.; Hunter, I.W.; Thilly, W.G. Analysis of mutational spectra by denaturing capillary electrophoresis. Nat. Protoc. 2008, 3, 1153–1166. [Google Scholar] [CrossRef]
- Ekstrom, P.O.; Nakken, S.; Johansen, M.; Hovig, E. Automated amplicon design suitable for analysis of DNA variants by melting techniques. BMC Res. Notes. 2015, 8, 667. [Google Scholar] [CrossRef][Green Version]
- Hinselwood, D.C.; Abrahamsen, T.W.; Ekstrom, P.O. BRAF mutation detection and identification by cycling temperature capillary electrophoresis. Electrophoresis 2005, 26, 2553–2561. [Google Scholar] [CrossRef]
- Varley, J.M.; Chapman, P.; McGown, G.; Thorncroft, M.; White, G.R.; Greaves, M.J.; Scott, D.; Spreadborough, A.; Tricker, K.J.; Birch, J.M.; et al. Genetic and functional studies of a germline TP53 splicing mutation in a Li-Fraumeni-like family. Oncogene 1998, 16, 3291–3298. [Google Scholar] [CrossRef] [PubMed]
- Donis-Keller, H.; Dou, S.; Chi, D.; Carlson, K.M.; Toshima, K.; Lairmore, T.C.; Howe, J.R.; Moley, J.F.; Goodfellow, P.; Wells, S.A. Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Hum. Mol. Genet. 1993, 2, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Dror, Y.; Freedman, M.H. Shwachman-diamond syndrome. Br. J. Haematol. 2002, 118, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Shwachman, H.; Diamond, L.K.; Oski, F.A.; Khaw, K.T. The Syndrome of Pancreatic Insufficiency and Bone Marrow Dysfunction. J. Pediatr. 1964, 65, 645–663. [Google Scholar] [CrossRef]
- McGinnis, J.M.; Foege, W.H. Actual causes of death in the United States. JAMA 1993, 270, 2207–2212. [Google Scholar] [CrossRef]
- Davidson, K.W.; McGinn, T. Screening for Social Determinants of Health: The Known and Unknown. JAMA 2019, 322, 1037–1038. [Google Scholar] [CrossRef]
- Neoplásicas, I.R.d.E. Registro Hospitalario del Cáncer IREN Norte 2007–2021 [Internet]. Trujillo, La Libertad; 2022 [cited 4 June 2020]. Available online: http://www.irennorte.gob.pe/registro_hosp_ca.php (accessed on 20 December 2021).
- Perú: Población Estimada al 30 de Junio y Tasa de Crecimiento de Las Ciudades Capitales pd, 2011 y 2015. Perú: Estimaciones y Proyecciones de Población Total Por Sexo de Las Principales Ciudades, 2012-2015 (Report); Instituto Nacional de Estadística e Informática: Lima, Peru, 2015; March 2012. Retrieved 3 June 2015.
- Vidal, A.F.; Ferraz, R.S.; El-Husny, A.; Silva, C.S.; Vinasco-Sandoval, T.; Magalhaes, L.; Raiol-Moraes, M.; Barra, W.F.; Pereira, C.L.B.L.; de Assumpção, P.P.; et al. Comprehensive analysis of germline mutations in northern Brazil: A panel of 16 genes for hereditary cancer-predisposing syndrome investigation. BMC Cancer 2021, 21, 363. [Google Scholar] [CrossRef]
- Arslan Ates, E.; Turkyilmaz, A.; Alavanda, C.; Yildirim, O.; Guney, A.I. Multigene Panel Testing in Turkish Hereditary Cancer Syndrome Patients. Medeni. Med. J. 2022, 37, 150–158. [Google Scholar] [CrossRef]
- Ercoskun, P.; Yuce Kahraman, C.; Ozkan, G.; Tatar, A. Genetic Characterization of Hereditary Cancer Syndromes Based on Targeted Next-Generation Sequencing. Mol. Syndromol. 2022, 13, 123–131. [Google Scholar] [CrossRef]
- Krampitz, G.W.; Norton, J.A. RET gene mutations (genotype and phenotype) of multiple endocrine neoplasia type 2 and familial medullary thyroid carcinoma. Cancer Am. Cancer Soc. 2014, 120, 1920–1931. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Valentin, M.; Sampson, J.R.; Seppala, T.T.; Ten Broeke, S.W.; Plazzer, J.P.; Nakken, S.; Engel, C.; Aretz, S.; Jenkins, M.A.; Sunde, L.; et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: Findings from the Prospective Lynch Syndrome Database. Genet. Med. 2019, 22, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Network National Comprehensive Cancer Network. Genetic/Familial High-Risk Assessment: Colorectal. In NCNN Clinical Practice Guildeines in Oncology (NCCN Guidelines); National Comprehensive Cancer Network: Plymouth Meeting, PA, USA, 2021. [Google Scholar]
- Nelson, A.S.; Myers, K.C. Diagnosis, Treatment, and Molecular Pathology of Shwachman-Diamond Syndrome. Hematol. Oncol. Clin. N. Am. 2018, 32, 687–700. [Google Scholar] [CrossRef] [PubMed]
- Li, F.P.; Fraumeni, J.F., Jr.; Mulvihill, J.J.; Blattner, W.A.; Dreyfus, M.G.; Tucker, M.A.; Miller, R.W. A cancer family syndrome in twenty-four kindreds. Cancer Res. 1988, 48, 5358–5362. [Google Scholar] [PubMed]
- Peake, J.D.; Noguchi, E. Fanconi anemia: Current insights regarding epidemiology, cancer, and DNA repair. Hum. Genet. 2022. [Google Scholar] [CrossRef]
- Bogliolo, M.; Surralles, J. Fanconi anemia: A model disease for studies on human genetics and advanced therapeutics. Curr. Opin. Genet. Dev. 2015, 33, 32–40. [Google Scholar] [CrossRef]
- Gilson, P.; Drouot, G.; Witz, A.; Merlin, J.L.; Becuwe, P.; Harle, A. Emerging Roles of DDB2 in Cancer. Int. J. Mol. Sci. 2019, 20, 5168. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Characteristics | Overall (n = 101) | No Cancer Diagnosis (n = 47) | Cancer Diagnosis (n = 54) | ||||
|---|---|---|---|---|---|---|---|
| n | % | n | % | n | % | ||
| Sociodemographic characteristics | |||||||
| Age | |||||||
| <50 years | 79 | 78.2 | 44 | 55.7 | 35 | 44.3 | |
| >50 years | 22 | 21.8 | 3 | 13.6 | 19 | 86.4 | |
| Gender | |||||||
| Women | 84 | 83.2 | 35 | 41.7 | 49 | 58.3 | |
| Men | 17 | 16.8 | 12 | 70.6 | 5 | 29.4 | |
| Ethnicity * | |||||||
| White | 7 | 7.5 | 1 | 14.3 | 6 | 85.7 | |
| Non-white | 86 | 92.5 | 38 | 44.2 | 48 | 55.8 | |
| Educational level * | |||||||
| <7 years of education | 5 | 5.4 | 0 | 0.0 | 5 | 100.0 | |
| ≥7 years of education | 88 | 94.6 | 39 | 44.3 | 49 | 55.7 | |
| Professional level ** | |||||||
| College/University Student | 11 | 12.1 | 10 | 90.9 | 1 | 9.1 | |
| Undergraduate (finished) | 25 | 27.5 | 11 | 44.0 | 14 | 56.0 | |
| Technical (finished) | 12 | 13.2 | 5 | 41.7 | 7 | 58.3 | |
| Primary/secondary education | 43 | 47.3 | 11 | 25.6 | 32 | 74.4 | |
| Work condition * | |||||||
| Hired by a company | 19 | 20.4 | 9 | 47.4 | 10 | 52.6 | |
| Independent worker (freelance) | 8 | 8.6 | 2 | 25.0 | 6 | 75.0 | |
| Independent worker (entrepreneur) | 15 | 16.1 | 6 | 40.0 | 9 | 60.0 | |
| Domestic worker | 2 | 2.2 | 0 | 0.0 | 2 | 100.0 | |
| Housewife | 44 | 47.3 | 21 | 47.7 | 23 | 52.3 | |
| Not able to work | 5 | 5.4 | 1 | 20.0 | 4 | 80.0 | |
| Family income (monthly) * | |||||||
| High | 39 | 41.9 | 14 | 35.9 | 25 | 64.1 | |
| Low | 54 | 58.1 | 25 | 46.3 | 29 | 53.7 | |
| Family income source * | |||||||
| Head of home (1 person) | 52 | 55.9 | 22 | 42.3 | 30 | 57.7 | |
| Spouses (2 persons) | 28 | 30.1 | 12 | 42.9 | 16 | 57.1 | |
| Mixed (3 or more persons) | 13 | 14.0 | 5 | 38.5 | 8 | 61.5 | |
| House location * | |||||||
| Urban area | 85 | 91.4 | 35 | 41.2 | 50 | 58.8 | |
| Marginal-urban area | 1 | 1.1 | 0 | 0.0 | 1 | 100.0 | |
| Rural area | 7 | 7.5 | 4 | 57.1 | 3 | 42.9 | |
| Type of house * | |||||||
| For one family only | 42 | 45.2 | 15 | 35.7 | 27 | 64.3 | |
| For two or more families | 51 | 54.8 | 24 | 47.1 | 27 | 52.9 | |
| House tenancy * | |||||||
| Own house | 60 | 64.5 | 24 | 40.0 | 36 | 60.0 | |
| Rented house | 15 | 16.1 | 7 | 46.7 | 8 | 53.3 | |
| Occupied house (no payment) | 18 | 19.4 | 8 | 44.4 | 10 | 55.6 | |
| Type of family * | |||||||
| Nuclear (couple and children) | 46 | 49.5 | 17 | 37.0 | 29 | 63.0 | |
| Extended (more than one nuclear family) | 47 | 50.5 | 22 | 46.8 | 25 | 53.2 | |
| Number of persons per house * | |||||||
| Less than 3 persons | 5 | 5.4 | 2 | 40.0 | 3 | 60.0 | |
| 3 to 5 persons | 55 | 59.1 | 19 | 34.5 | 36 | 65.5 | |
| 6 or more persons | 33 | 35.5 | 18 | 54.5 | 15 | 45.5 | |
| Health services and insurance | |||||||
| Health services | |||||||
| ESSALUD III | 24 | 23.8 | 11 | 45.8 | 13 | 54.2 | |
| Hospital La Caleta | 73 | 72.3 | 35 | 47.9 | 38 | 52.1 | |
| Hospital Regional | 4 | 4.0 | 1 | 25.0 | 3 | 75.0 | |
| Health insurance * | |||||||
| Seguro Integral de Salud (SIS) | 57 | 61.3 | 22 | 38.6 | 35 | 61.4 | |
| Seguro Social de Salud (EsSalud) | 27 | 29.0 | 11 | 40.7 | 16 | 59.3 | |
| Private Insurance | 2 | 2.2 | 0 | 0.0 | 2 | 100.0 | |
| None | 7 | 7.5 | 6 | 85.7 | 1 | 14.3 | |
| Oncologic insurance * | |||||||
| Yes | 6 | 6.5 | 5 | 83.3 | 1 | 16.7 | |
| No | 87 | 93.5 | 34 | 39.1 | 53 | 60.9 | |
| Health and lifestyle | |||||||
| Chronic disease(s) * | |||||||
| Yes | 14 | 15.1 | 2 | 14.3 | 12 | 85.7 | |
| No | 79 | 84.9 | 37 | 46.8 | 42 | 53.2 | |
| Weight * | |||||||
| Normal | 59 | 63.4 | 30 | 50.8 | 29 | 49.2 | |
| Overweight or obesity | 34 | 36.6 | 9 | 26.5 | 25 | 73.5 | |
| Selection Criteria for the Study | Age at Cancer Diagnosis | Grade | Gene Variant | Protein Change | ACMG Classification | ACMG Evidence Code * | Pathogenicity Reported by | Reported Associated Phenotype | Inheritance Pattern |
|---|---|---|---|---|---|---|---|---|---|
| Breast | 34 | III | TP53 (ENST00000269305.4) c.375G>A | p.Thr125 = | Pathogenic | PM2_2, PP3 | Clinvar | Hereditary cancer-predisposing syndrome; Rhabdomyosarcoma (disease); Li-Fraumeni syndrome | Autosomal dominant |
| Endometrial | 45 | na | DDB2 (ENST00000256996.4) c.406dup | p.Ile136AsnfsTer30 | Pathogenic | PM2_2, PVS1_1 | CSPR | Xeroderma pigmentosum, group E, DDB-negative subtype | Autosomal recessive |
| FANCG (ENST00000378643.3) c.922G>T | p.Glu308Ter | Pathogenic | PM2_2, PVS1_1 | CSPR | Fanconi anemia, complementation group G | ||||
| Thyroid | 22 | na | RET (ENST00000355710.3) c.1900T>C | p.Cys634Arg | Pathogenic | PM2_1, PS1, PP3 | Clinvar | Multiple endocrine neoplasia type 2 | Autosomal dominant |
| Familial history of cancer | na | RET (ENST00000355710.3) c.1900T>C | p.Cys634Arg | Pathogenic | PM2_1, PS1, PP3 | Clinvar | Multiple endocrine neoplasia type 2 | Autosomal dominant | |
| Thyroid | 18 | na | RET (ENST00000355710.3) c.1900T>C | p.Cys634Arg | Pathogenic | PM2_1, PS1, PP3 | Clinvar | Multiple endocrine neoplasia type 2 | Autosomal dominant |
| Colon | 51 | III | BRCA1 (ENST00000471181.2) c.4357+1G>A | Pathogenic | PM2_1, PVS1_7, PP3 | Clinvar | Hereditary cancer-predisposing syndrome; Hereditary breast and ovarian cancer syndrome; Breast-ovarian cancer, familial 1 | Autosomal dominant | |
| Familial history of cancer | na | BRCA1 (ENST00000471181.2) c.4357+1G>A | Pathogenic | PM2_1, PVS1_7, PP3 | Clinvar | Hereditary cancer-predisposing syndrome; Hereditary breast and ovarian cancer syndrome; Breast-ovarian cancer, familial 1 | Autosomal dominant | ||
| Familial history of cancer | na | BRCA1 (ENST00000471181.2) c.4357+1G>A | Pathogenic | PM2_1, PVS1_7, PP3 | Clinvar | Hereditary cancer-predisposing syndrome; Hereditary breast and ovarian cancer syndrome; Breast-ovarian cancer, familial 1 | Autosomal dominant | ||
| Familial history of cancer | na | SBDS (ENST00000246868.2) c.258+2T>C | Pathogenic | BS1, PP3 | Clinvar | Shwachman-Diamond syndrome 1 | Autosomal recessive | ||
| Familial history of cancer | na | MLH1 (ENST00000231790.2) c.1852_1854del | p.Lys618del | Pathogenic | PM2_1, PS1, PM4 | Clinvar | Lynch syndrome | Autosomal dominant | |
| SBDS (ENST00000246868.2) c.258+2T>C | Pathogenic | BS1, PP3 | Clinvar | Shwachman-Diamond syndrome 1 | Autosomal recessive | ||||
| Colon | 41 | II | MLH1 (ENST00000231790.2) c.1852_1854del | p.Lys618del | Pathogenic | PM2_1, PS1, PM4 | Clinvar | Lynch syndrome | Autosomal dominant |
| SBDS (ENST00000246868.2) c.258+2T>C | Pathogenic | BS1, PP3 | Clinvar | Shwachman-Diamond syndrome 1 | Autosomal recessive | ||||
| Familial history of cancer | na | MLH1 (ENST00000231790.2) c.1852_1854del | p.Lys618del | Pathogenic | PM2_1, PS1, PM4 | Clinvar | Lynch syndrome | Autosomal dominant | |
| SBDS (ENST00000246868.2) c.258+2T>C | Pathogenic | BS1, PP3 | Clinvar | Shwachman-Diamond syndrome 1 | Autosomal recessive | ||||
| Familial history of cancer | na | SBDS (ENST00000246868.2) c.258+2T>C | Pathogenic | BS1, PP3 | Clinvar | Shwachman-Diamond syndrome 1 | Autosomal recessive | ||
| Colon | 38 | I | MLH1 (ENST00000231790.2) c.1852_1854del | p.Lys618del | Pathogenic | PM2_1, PS1, PM4 | Clinvar | Lynch syndrome | Autosomal dominant |
| SBDS (ENST00000246868.2) c.258+2T>C | Pathogenic | BS1, PP3 | Clinvar | Shwachman-Diamond syndrome 1 | Autosomal recessive | ||||
| Colon | 42 | I | MLH1 (ENST00000231790.2) c.1852_1854del | p.Lys618del | Pathogenic | PM2_1, PS1, PM4 | Clinvar | Lynch syndrome | Autosomal dominant |
| Colon | 39 | II | MLH1 (ENST00000231790.2) c.1852_1854del | p.Lys618del | Pathogenic | PM2_1, PS1, PM4 | Clinvar | Lynch syndrome | Autosomal dominant |
| Familial history of cancer | na | MLH1 (ENST00000231790.2) c.1852_1854del | p.Lys618del | Pathogenic | PM2_1, PS1, PM4 | Clinvar | Lynch syndrome | Autosomal dominant | |
| Familial history of cancer | na | MLH1 (ENST00000231790.2) c.1852_1854del | p.Lys618del | Pathogenic | PM2_1, PS1, PM4 | Clinvar | Lynch syndrome | Autosomal dominant | |
| Familial history of cancer | na | FANCD2 (ENST00000287647.3)c.848dup | p.Phe284ValfsTer13 | Pathogenic | PM2_2, PVS1_1 | CSPR | Fanconi anemia, complementation group D2 | Autosomal recessive |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zavaleta, E.; Solis, N.; Palacios, M.I.; Zevallos-Escobar, L.E.; Corales, E.V.; Bazo-Alvarez, J.C.; Dominguez-Barrera, C.; Campos, A.; Wernhoff, P.; Ekstrøm, P.O.; et al. Genetic Characterization in High-Risk Individuals from a Low-Resource City of Peru. Cancers 2022, 14, 5603. https://doi.org/10.3390/cancers14225603
Zavaleta E, Solis N, Palacios MI, Zevallos-Escobar LE, Corales EV, Bazo-Alvarez JC, Dominguez-Barrera C, Campos A, Wernhoff P, Ekstrøm PO, et al. Genetic Characterization in High-Risk Individuals from a Low-Resource City of Peru. Cancers. 2022; 14(22):5603. https://doi.org/10.3390/cancers14225603
Chicago/Turabian StyleZavaleta, Elizabeth, Nelly Solis, Maria Isabel Palacios, Liz Elva Zevallos-Escobar, Edison Vasquez Corales, Juan Carlos Bazo-Alvarez, Constantino Dominguez-Barrera, Anthony Campos, Patrik Wernhoff, Per Olaf Ekstrøm, and et al. 2022. "Genetic Characterization in High-Risk Individuals from a Low-Resource City of Peru" Cancers 14, no. 22: 5603. https://doi.org/10.3390/cancers14225603
APA StyleZavaleta, E., Solis, N., Palacios, M. I., Zevallos-Escobar, L. E., Corales, E. V., Bazo-Alvarez, J. C., Dominguez-Barrera, C., Campos, A., Wernhoff, P., Ekstrøm, P. O., Møller, P., Visnovska, T., Hovig, E., Balazar-Palacios, J., Alvarez-Valenzuela, K., Nakken, S., & Dominguez-Valentin, M. (2022). Genetic Characterization in High-Risk Individuals from a Low-Resource City of Peru. Cancers, 14(22), 5603. https://doi.org/10.3390/cancers14225603