
Trabectedin Is Active against Two Novel, Patient-Derived Solitary Fibrous Pleural Tumor Cell Lines and Synergizes with Ponatinib

 ,
,  ,
,  , , ,
, , , 
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
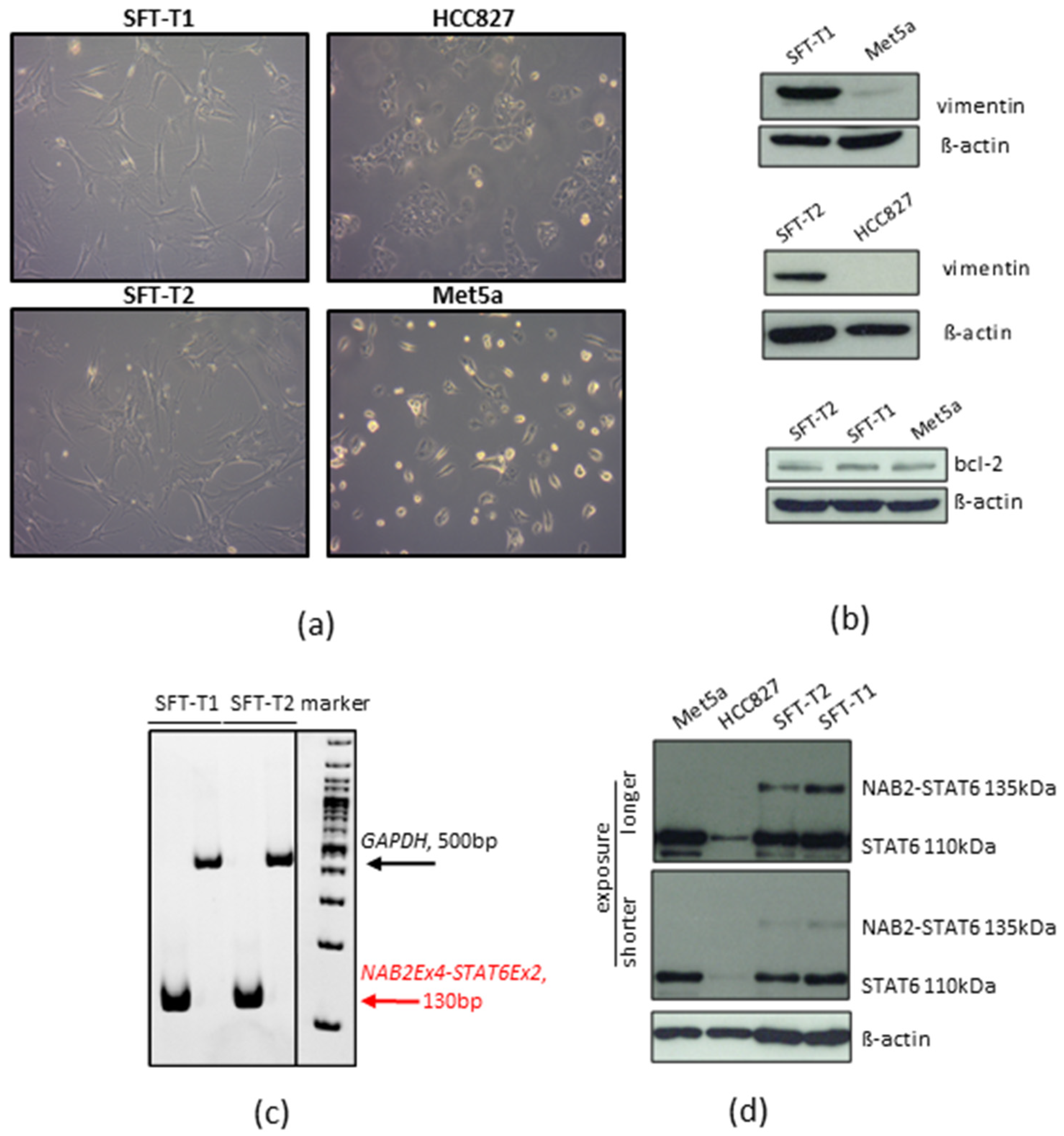
2.1. Establishment of Patient-Derived Cell Lines
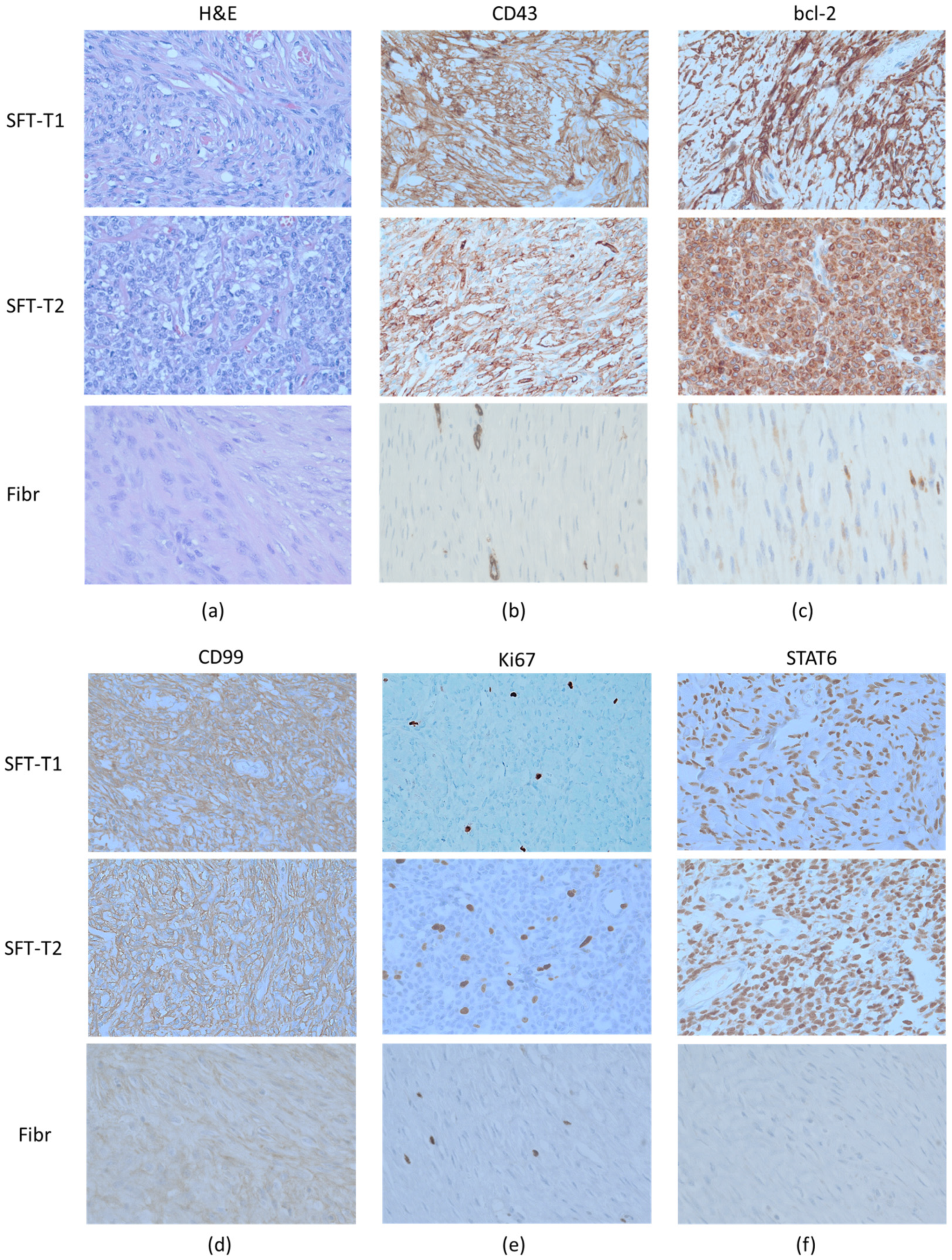
2.2. Immunohistochemistry
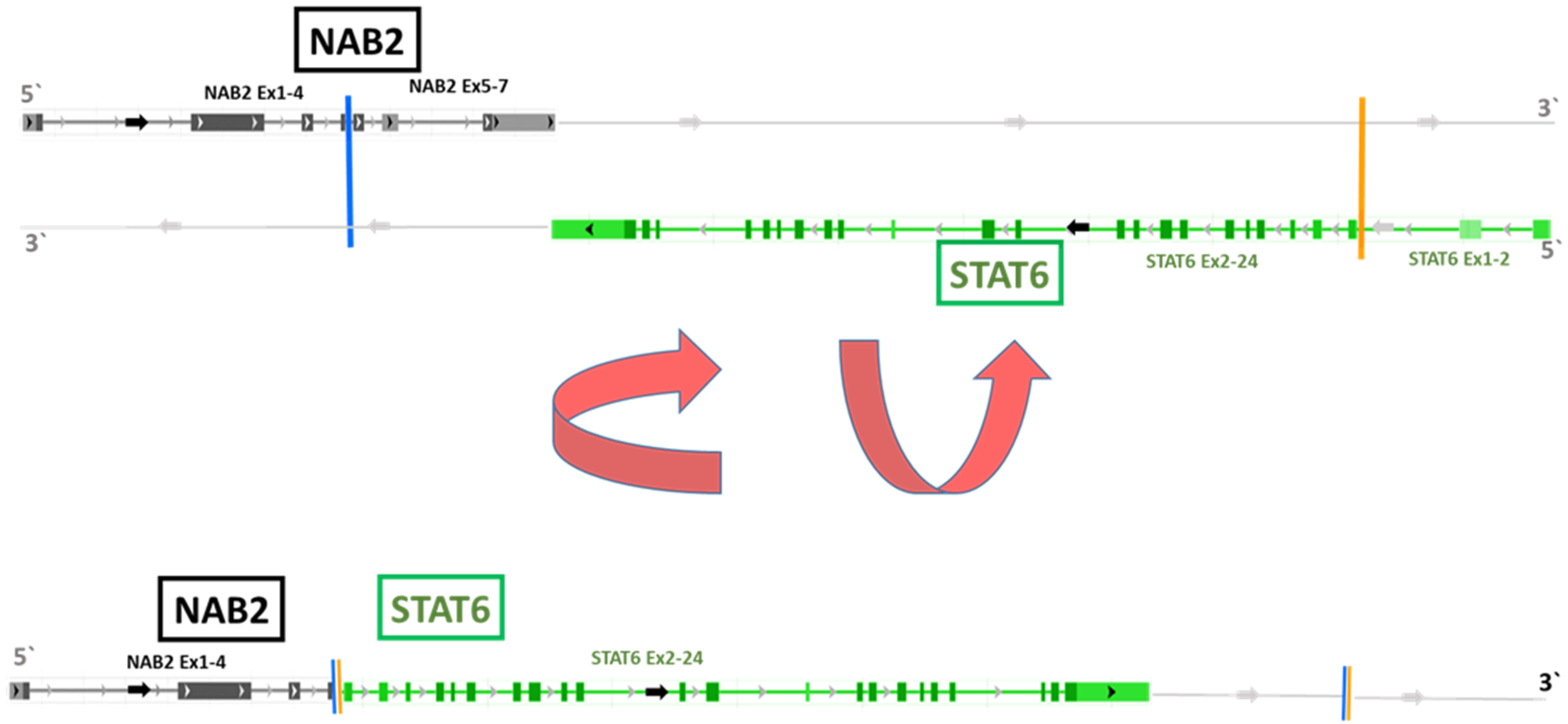
2.3. Detection of the NAB2-STAT6 Fusion
2.4. Drugs
2.5. Determination of Cell Proliferation
2.6. Cell Viability Assay
2.7. Clonogenicity (Clone Formation) Assay
2.8. Western Blot Analyses
2.9. RNA Isolation, Reverse Transcription into cDNA, and RT-PCR
2.10. Statistical Analyses
3. Results
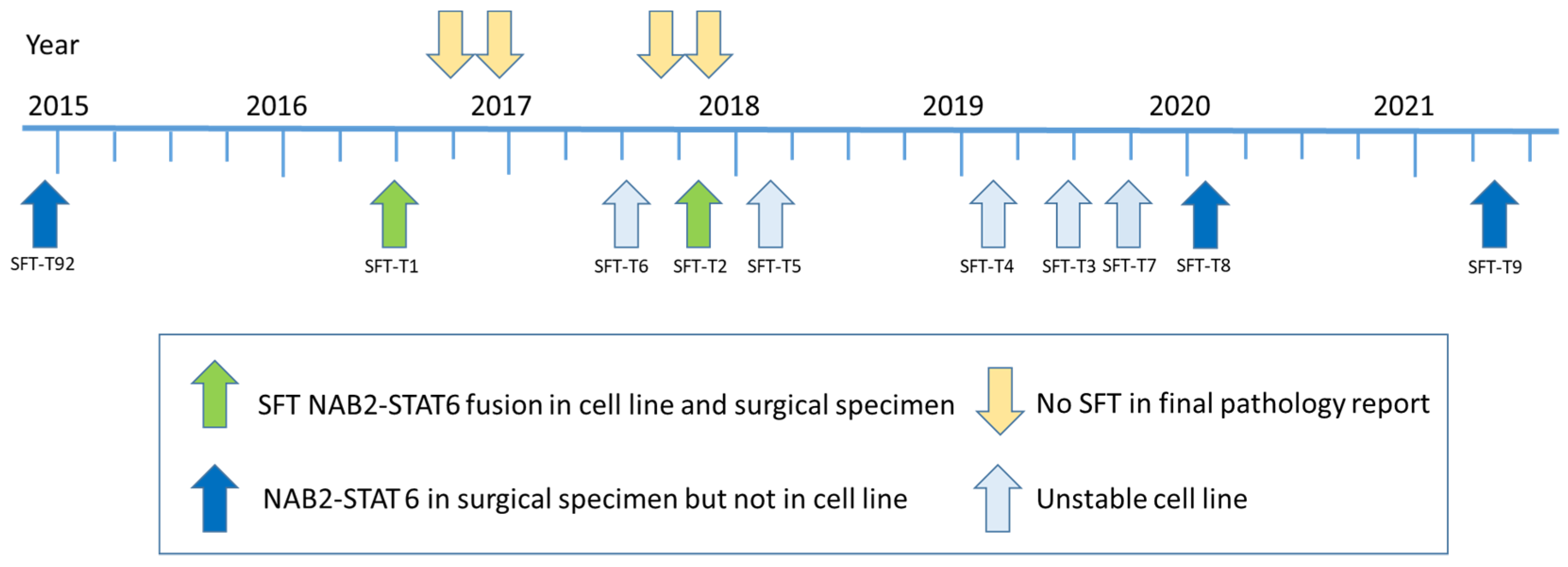
3.1. Establishment of Novel SFT Models from Surgical Specimens
3.2. Screening for Treatment Responses in Novel SFT Cell Models
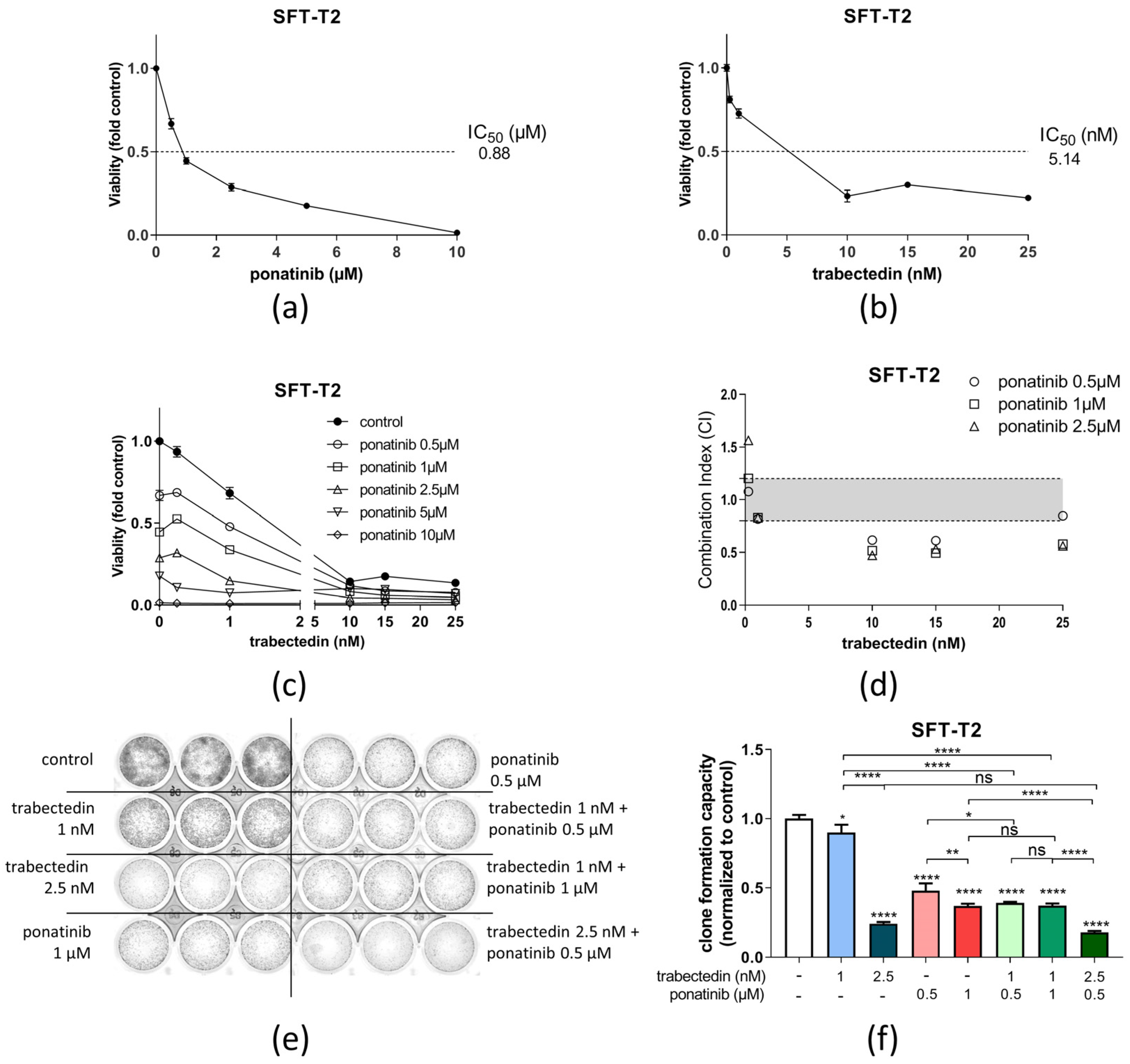
3.3. Ponatinib and Trabectedin Are Active against SFT Cell Growth and Synergize In Vitro
3.4. Ponatinib Treatment Targets Fibroblast Growth Factor Receptor Downstream Signaling in SFT Cells
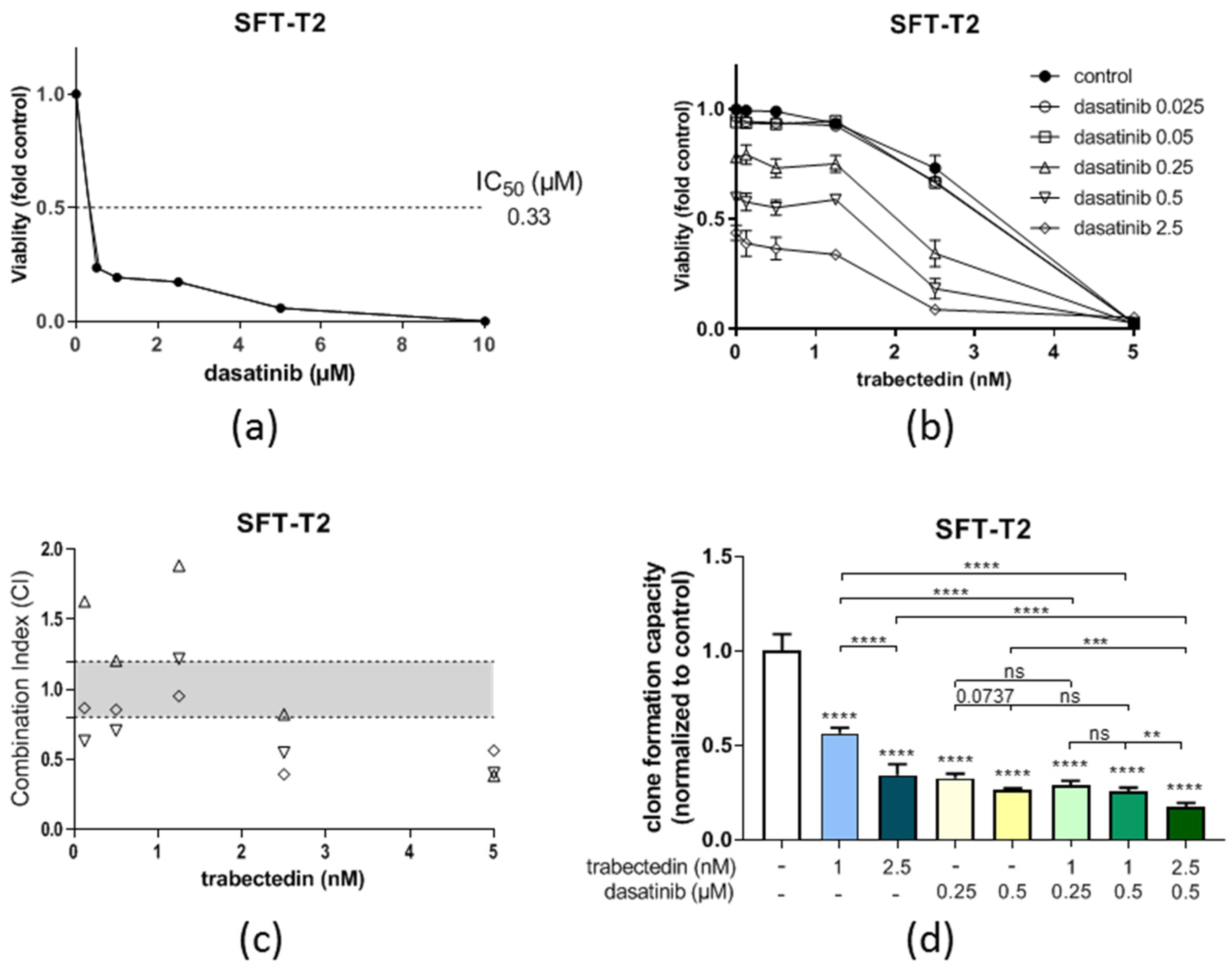
3.5. Dasatinib Is Active against SFT Growth In Vitro and Enhances Trabectedin Activity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stiller, C.A.; Trama, A.; Serraino, D.; Rossi, S.; Navarro, C.; Chirlaque, M.D.; Casali, P.G.; Group, R.W. Descriptive epidemiology of sarcomas in Europe: Report from the RARECARE project. Eur. J. Cancer. 2013, 49, 684–695. [Google Scholar] [CrossRef] [PubMed]
- England, D.M.; Hochholzer, L.; McCarthy, M.J. Localized benign and malignant fibrous tumors of the pleura. A clinicopathologic review of 223 cases. Am. J. Surg. Pathol. 1989, 13, 640–658. [Google Scholar] [CrossRef] [PubMed]
- de Perrot, M.; Fischer, S.; Brundler, M.A.; Sekine, Y.; Keshavjee, S. Solitary fibrous tumors of the pleura. Ann. Thorac. Surg. 2002, 74, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Abu Arab, W. Solitary fibrous tumours of the pleura. Eur. J. Cardiothorac. Surg. 2012, 41, 587–597. [Google Scholar] [CrossRef]
- Kallen, M.E.; Hornick, J.L. The 2020 WHO Classification: What’s New in Soft Tissue Tumor Pathology? Am. J. Surg. Pathol. 2021, 45, e1–e23. [Google Scholar] [CrossRef]
- Chmielecki, J.; Crago, A.M.; Rosenberg, M.; O’Connor, R.; Walker, S.R.; Ambrogio, L.; Auclair, D.; McKenna, A.; Heinrich, M.C.; Frank, D.A.; et al. Whole-exome sequencing identifies a recurrent NAB2-STAT6 fusion in solitary fibrous tumors. Nat. Genet. 2013, 45, 131–132. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.M.; Kalyana-Sundaram, S.; Cao, X.; Lonigro, R.J.; Sung, Y.S.; Chen, C.L.; Zhang, L.; Wang, R.; Su, F.; et al. Identification of recurrent NAB2-STAT6 gene fusions in solitary fibrous tumor by integrative sequencing. Nat. Genet. 2013, 45, 180–185. [Google Scholar] [CrossRef]
- Yoshida, A.; Tsuta, K.; Ohno, M.; Yoshida, M.; Narita, Y.; Kawai, A.; Asamura, H.; Kushima, R. STAT6 immunohistochemistry is helpful in the diagnosis of solitary fibrous tumors. Am. J. Surg. Pathol. 2014, 38, 552–559. [Google Scholar] [CrossRef]
- Bertucci, F.; Bouvier-Labit, C.; Finetti, P.; Metellus, P.; Adelaide, J.; Mokhtari, K.; Figarella-Branger, D.; Decouvelaere, A.V.; Miquel, C.; Coindre, J.M.; et al. Gene expression profiling of solitary fibrous tumors. PLoS ONE 2013, 8, e64497. [Google Scholar] [CrossRef]
- Hajdu, M.; Singer, S.; Maki, R.G.; Schwartz, G.K.; Keohan, M.L.; Antonescu, C.R. IGF2 over-expression in solitary fibrous tumours is independent of anatomical location and is related to loss of imprinting. J. Pathol. 2010, 221, 300–307. [Google Scholar] [CrossRef]
- Sun, Y.; Naito, Z.; Ishiwata, T.; Maeda, S.; Sugisaki, Y.; Asano, G. Basic FGF and Ki-67 proteins useful for immunohistological diagnostic evaluations in malignant solitary fibrous tumor. Pathol. Int. 2003, 53, 284–290. [Google Scholar] [CrossRef]
- Ghanim, B.; Hess, S.; Bertoglio, P.; Celik, A.; Bas, A.; Oberndorfer, F.; Melfi, F.; Mussi, A.; Klepetko, W.; Pirker, C.; et al. Intrathoracic solitary fibrous tumor—An international multicenter study on clinical outcome and novel circulating biomarkers. Sci. Rep. 2017, 7, 12557. [Google Scholar] [CrossRef] [PubMed]
- DeVito, N.; Henderson, E.; Han, G.; Reed, D.; Bui, M.M.; Lavey, R.; Robinson, L.; Zager, J.S.; Gonzalez, R.J.; Sondak, V.K.; et al. Clinical Characteristics and Outcomes for Solitary Fibrous Tumor (SFT): A Single Center Experience. PLoS ONE 2015, 10, e0140362. [Google Scholar] [CrossRef] [PubMed]
- Levard, A.; Derbel, O.; Meeus, P.; Ranchere, D.; Ray-Coquard, I.; Blay, J.Y.; Cassier, P.A. Outcome of patients with advanced solitary fibrous tumors: The Centre Leon Berard experience. BMC Cancer 2013, 13, 109. [Google Scholar] [CrossRef]
- Schoffski, P.; Timmermans, I.; Hompes, D.; Stas, M.; Sinnaeve, F.; De Leyn, P.; Coosemans, W.; Van Raemdonck, D.; Hauben, E.; Sciot, R.; et al. Clinical Presentation, Natural History, and Therapeutic Approach in Patients with Solitary Fibrous Tumor: A Retrospective Analysis. Sarcoma 2020, 2020, 1385978. [Google Scholar] [CrossRef] [PubMed]
- Martin-Broto, J.; Mondaza-Hernandez, J.L.; Moura, D.S.; Hindi, N. A Comprehensive Review on Solitary Fibrous Tumor: New Insights for New Horizons. Cancers 2021, 13, 2913. [Google Scholar] [CrossRef]
- Park, M.S.; Ravi, V.; Conley, A.; Patel, S.R.; Trent, J.C.; Lev, D.C.; Lazar, A.J.; Wang, W.L.; Benjamin, R.S.; Araujo, D.M. The role of chemotherapy in advanced solitary fibrous tumors: A retrospective analysis. Clin. Sarcoma Res. 2013, 3, 1–7. [Google Scholar] [CrossRef]
- Valentin, T.; Fournier, C.; Penel, N.; Bompas, E.; Chaigneau, L.; Isambert, N.; Chevreau, C. Sorafenib in patients with progressive malignant solitary fibrous tumors: A subgroup analysis from a phase II study of the French Sarcoma Group (GSF/GETO). Investig. New Drugs 2013, 31, 1626–1627. [Google Scholar] [CrossRef]
- Khalifa, J.; Ouali, M.; Chaltiel, L.; Le Guellec, S.; Le Cesne, A.; Blay, J.Y.; Cousin, P.; Chaigneau, L.; Bompas, E.; Piperno-Neumann, S.; et al. Efficacy of trabectedin in malignant solitary fibrous tumors: A retrospective analysis from the French Sarcoma Group. BMC Cancer 2015, 15, 700. [Google Scholar] [CrossRef]
- Hoda, M.A.; Mohamed, A.; Ghanim, B.; Filipits, M.; Hegedus, B.; Tamura, M.; Berta, J.; Kubista, B.; Dome, B.; Grusch, M.; et al. Temsirolimus inhibits malignant pleural mesothelioma growth in vitro and in vivo: Synergism with chemotherapy. J. Thorac. Oncol. 2011, 6, 852–863. [Google Scholar] [CrossRef]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Berger, W.; Elbling, L.; Micksche, M. Expression of the major vault protein LRP in human non-small-cell lung cancer cells: Activation by short-term exposure to antineoplastic drugs. Int. J. Cancer 2000, 88, 293–300. [Google Scholar] [CrossRef]
- Berger, W.; Hauptmann, E.; Elbling, L.; Vetterlein, M.; Kokoschka, E.M.; Micksche, M. Possible role of the multidrug resistance-associated protein (MRP) in chemoresistance of human melanoma cells. Int. J. Cancer 1997, 71, 108–115. [Google Scholar] [CrossRef]
- Guseva, N.V.; Tanas, M.R.; Stence, A.A.; Sompallae, R.; Schade, J.C.; Bossler, A.D.; Bellizzi, A.M.; Ma, D. The NAB2-STAT6 gene fusion in solitary fibrous tumor can be reliably detected by anchored multiplexed PCR for targeted next-generation sequencing. Cancer Genet. 2016, 209, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Demicco, E.G.; Wagner, M.J.; Maki, R.G.; Gupta, V.; Iofin, I.; Lazar, A.J.; Wang, W.L. Risk assessment in solitary fibrous tumors: Validation and refinement of a risk stratification model. Mod. Pathol. 2017, 30, 1433–1442. [Google Scholar] [CrossRef] [PubMed]
- Park, H.K.; Yu, D.B.; Sung, M.; Oh, E.; Kim, M.; Song, J.Y.; Lee, M.S.; Jung, K.; Noh, K.W.; An, S.; et al. Molecular changes in solitary fibrous tumor progression. J. Mol. Med. 2019, 97, 1413–1425. [Google Scholar] [CrossRef]
- Or, C.R.; Chang, Y.; Lin, W.C.; Lee, W.C.; Su, H.L.; Cheung, M.W.; Huang, C.P.; Ho, C.; Chang, C.C. Obatoclax, a Pan-BCL-2 Inhibitor, Targets Cyclin D1 for Degradation to Induce Antiproliferation in Human Colorectal Carcinoma Cells. Int. J. Mol. Sci. 2016, 18, 44. [Google Scholar] [CrossRef]
- Steele, T.M.; Talbott, G.C.; Sam, A.; Tepper, C.G.; Ghosh, P.M.; Vinall, R.L. Obatoclax, a BH3 Mimetic, Enhances Cisplatin-Induced Apoptosis and Decreases the Clonogenicity of Muscle Invasive Bladder Cancer Cells via Mechanisms That Involve the Inhibition of Pro-Survival Molecules as Well as Cell Cycle Regulators. Int. J. Mol. Sci. 2019, 20, 1285. [Google Scholar] [CrossRef]
- D’Incalci, M.; Badri, N.; Galmarini, C.M.; Allavena, P. Trabectedin, a drug acting on both cancer cells and the tumour microenvironment. Br. J. Cancer 2014, 111, 646–650. [Google Scholar] [CrossRef]
- Musumeci, F.; Greco, C.; Grossi, G.; Molinari, A.; Schenone, S. Recent Studies on Ponatinib in Cancers Other Than Chronic Myeloid Leukemia. Cancers 2018, 10, 430. [Google Scholar] [CrossRef]
- Schelch, K.; Hoda, M.A.; Klikovits, T.; Munzker, J.; Ghanim, B.; Wagner, C.; Garay, T.; Laszlo, V.; Setinek, U.; Dome, B.; et al. Fibroblast growth factor receptor inhibition is active against mesothelioma and synergizes with radio- and chemotherapy. Am. J. Respir. Crit. Care Med. 2014, 190, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Zeng, P.; Schmaier, A. Ponatinib and other CML Tyrosine Kinase Inhibitors in Thrombosis. Int. J. Mol. Sci. 2020, 21, 6556. [Google Scholar] [CrossRef] [PubMed]
- Schuetze, S.M.; Bolejack, V.; Choy, E.; Ganjoo, K.N.; Staddon, A.P.; Chow, W.A.; Tawbi, H.A.; Samuels, B.L.; Patel, S.R.; von Mehren, M.; et al. Phase 2 study of dasatinib in patients with alveolar soft part sarcoma, chondrosarcoma, chordoma, epithelioid sarcoma, or solitary fibrous tumor. Cancer 2017, 123, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.W.; Kwon, S.S.; Kim, Y.D. Malignant solitary fibrous tumor of the pleura slowly growing over 17 years: Case report. J. Cardiothorac. Surg. 2014, 9, 113. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Lana, D.; Gambarotti, M.; Ferrari, C.; Sbaraglia, M.; Pedrini, E.; Pazzaglia, L.; Sangiorgi, L.; Bartolotti, I.; Dei Tos, A.P.; et al. Clinical, Histological, and Molecular Features of Solitary Fibrous Tumor of Bone: A Single Institution Retrospective Review. Cancers 2021, 13, 2470. [Google Scholar] [CrossRef]
- Apra, C.; El Arbi, A.; Montero, A.S.; Parker, F.; Knafo, S. Spinal Solitary Fibrous Tumors: An Original Multicenter Series and Systematic Review of Presentation, Management, and Prognosis. Cancers 2022, 14, 2839. [Google Scholar] [CrossRef]
- Stacchiotti, S.; Saponara, M.; Frapolli, R.; Tortoreto, M.; Cominetti, D.; Provenzano, S.; Negri, T.; Dagrada, G.P.; Gronchi, A.; Colombo, C.; et al. Patient-derived solitary fibrous tumour xenografts predict high sensitivity to doxorubicin/dacarbazine combination confirmed in the clinic and highlight the potential effectiveness of trabectedin or eribulin against this tumour. Eur. J. Cancer 2017, 76, 84–92. [Google Scholar] [CrossRef]
- Dagrada, G.P.; Spagnuolo, R.D.; Mauro, V.; Tamborini, E.; Cesana, L.; Gronchi, A.; Stacchiotti, S.; Pierotti, M.A.; Negri, T.; Pilotti, S. Solitary fibrous tumors: Loss of chimeric protein expression and genomic instability mark dedifferentiation. Mod. Pathol. 2015, 28, 1074–1083. [Google Scholar] [CrossRef]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell. 2013, 23, 249–262. [Google Scholar] [CrossRef]
- Evangelisti, G.; Barra, F.; D’Alessandro, G.; Tantari, M.; Stigliani, S.; Della Corte, L.; Bifulco, G.; Ferrero, S. Trabectedin for the therapy of ovarian cancer. Drugs Today 2020, 56, 669–688. [Google Scholar] [CrossRef]
- Nakano, K.; Takahashi, S. Translocation-Related Sarcomas. Int. J. Mol. Sci. 2018, 19, 3784. [Google Scholar] [CrossRef] [PubMed]
- Chaigneau, L.; Kalbacher, E.; Thiery-Vuillemin, A.; Fagnoni-Legat, C.; Isambert, N.; Aherfi, L.; Pauchot, J.; Delroeux, D.; Servagi-Vernat, S.; Mansi, L.; et al. Efficacy of trabectedin in metastatic solitary fibrous tumor. Rare Tumors 2011, 3, e29. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Iwata, S.; Wakamatsu, T.; Hayakawa, K.; Yonemoto, T.; Wasa, J.; Oka, H.; Ueda, T.; Tanaka, S. Efficacy and safety of trabectedin for patients with unresectable and relapsed soft-tissue sarcoma in Japan: A Japanese Musculoskeletal Oncology Group study. Cancer 2020, 126, 1253–1263. [Google Scholar] [CrossRef] [PubMed]
- Vogl, M.; Rosenmayr, A.; Bohanes, T.; Scheed, A.; Brndiar, M.; Stubenberger, E.; Ghanim, B. Biomarkers for Malignant Pleural Mesothelioma-A Novel View on Inflammation. Cancers 2021, 13, 658. [Google Scholar] [CrossRef]
- Stacchiotti, S.; Tortoreto, M.; Baldi, G.G.; Grignani, G.; Toss, A.; Badalamenti, G.; Cominetti, D.; Morosi, C.; Dei Tos, A.P.; Festinese, F.; et al. Preclinical and clinical evidence of activity of pazopanib in solitary fibrous tumour. Eur. J. Cancer 2014, 50, 3021–3028. [Google Scholar] [CrossRef]
- Maruzzo, M.; Martin-Liberal, J.; Messiou, C.; Miah, A.; Thway, K.; Alvarado, R.; Judson, I.; Benson, C. Pazopanib as first line treatment for solitary fibrous tumours: The Royal Marsden Hospital experience. Clin. Sarcoma Res. 2015, 5, 1–7. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SFT# | Age | Sex | Risk Group | CD34 | bcl-2 | CD99 | STAT6 | Ki67 | NAB2-STAT6 | NAB2-STAT6 |
|---|---|---|---|---|---|---|---|---|---|---|
| Tumor | Cell Line | |||||||||
| T92 | 72 | male | intermediate | + | + | + | + | 15 | + | − |
| T1 | 50 | male | low | + | + | + | + | 1 | + | + |
| T2 | 45 | female | intermediate | + | + | + | + | 5 | + | + |
| T8 | 34 | female | intermediate | + | + | + | + | 20 | + | − |
| T9 | 76 | female | high | + | + | + | + | 25 | + | − |
| IC50 ± SD | |||
|---|---|---|---|
| Drug | SFT-T1 | SFT-T2 | Main Target(s) |
| Cisplatin (µM) | 16.38 ± 1.91 | 14.64 ± 4.91 | DNA synthesis |
| Paclitaxel (µM) | 0.032 ± 0.04 | 0.451 ± 0.57 | cytoskeletal components |
| Vincristine (nM) | 37.50 ± 9.09 | 36.54 ± 10.08 | microtubule formation |
| Venetoclax (µM) | 27.91 ± 5.34 | 18.03 ± 4.64 | bcl-2 |
| Obatoclax (nM) | 258.55 ± 84.24 | 237.69 ± 33.68 | bcl-2 |
| Doxorubicin (nM) | 555.36 ± 327.33 | 410.51 ± 193.32 | DNA topoisomerase II |
| Imatinib (µM) | 27.28 ± 6.92 | 18.18 ± 2.70 | v-Abl, c-Kit, and PDGFR |
| Stattic (µM) | 3.04 ± 1.09 | 5.30 ± 1.52 | STAT3 |
| Trabectedin (nM) | 4.36 ± 0.73 | 3.54 ± 0.98 | DNA damaging, blocks DNA binding of FUS-CHOP |
| Ponatinib (µM) | 0.47 ± 0.07 | 1.89 ± 0.98 | FGFR, Abl, PDGFRα, VEGFR2, and Src |
| Nintedanib (µM) | 3.59 ± 0.89 | 3.63 ± 1.84 | VEGFR1-3, FGFR1-3, PDGFR α, and β |
| Dasatinib (µM) | 0.39 ± 0.02 | 0.40 ± 0.19 | Abl, Src, and c-Kit |
| PD173074 (µM) | 2.32 ± 0.73 | 2.46 ± 0.46 | FGFR1 and VEGFR2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghanim, B.; Baier, D.; Pirker, C.; Müllauer, L.; Sinn, K.; Lang, G.; Hoetzenecker, K.; Berger, W. Trabectedin Is Active against Two Novel, Patient-Derived Solitary Fibrous Pleural Tumor Cell Lines and Synergizes with Ponatinib. Cancers 2022, 14, 5602. https://doi.org/10.3390/cancers14225602
Ghanim B, Baier D, Pirker C, Müllauer L, Sinn K, Lang G, Hoetzenecker K, Berger W. Trabectedin Is Active against Two Novel, Patient-Derived Solitary Fibrous Pleural Tumor Cell Lines and Synergizes with Ponatinib. Cancers. 2022; 14(22):5602. https://doi.org/10.3390/cancers14225602
Chicago/Turabian StyleGhanim, Bahil, Dina Baier, Christine Pirker, Leonhard Müllauer, Katharina Sinn, Gyoergy Lang, Konrad Hoetzenecker, and Walter Berger. 2022. "Trabectedin Is Active against Two Novel, Patient-Derived Solitary Fibrous Pleural Tumor Cell Lines and Synergizes with Ponatinib" Cancers 14, no. 22: 5602. https://doi.org/10.3390/cancers14225602
APA StyleGhanim, B., Baier, D., Pirker, C., Müllauer, L., Sinn, K., Lang, G., Hoetzenecker, K., & Berger, W. (2022). Trabectedin Is Active against Two Novel, Patient-Derived Solitary Fibrous Pleural Tumor Cell Lines and Synergizes with Ponatinib. Cancers, 14(22), 5602. https://doi.org/10.3390/cancers14225602