
USP7 Inhibitors in Cancer Immunotherapy: Current Status and Perspective

 , , and
, , and 
Abstract
Simple Summary
Abstract
1. Introduction
2. DUBs: Cellular Functions and Their Role in the UPS System
3. USP7 as a Regulator of Anti-Tumor Immune Response
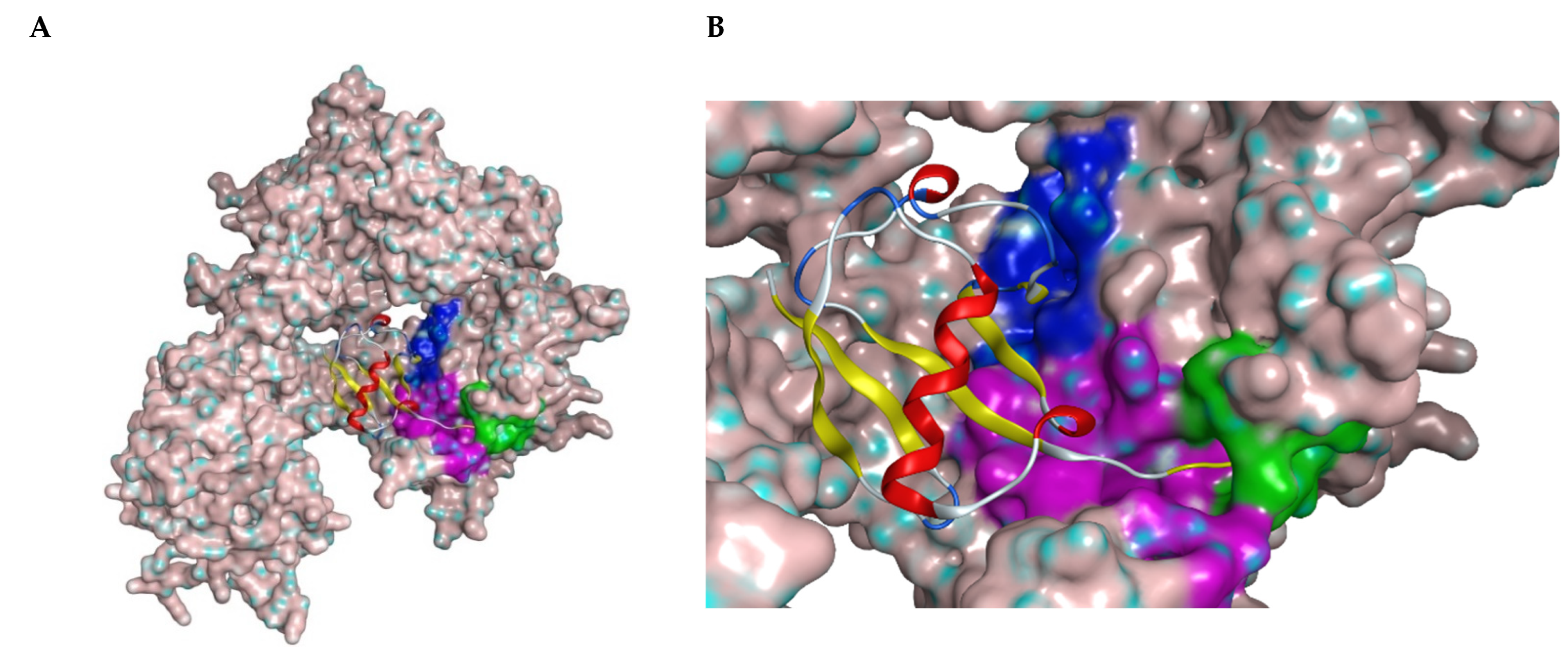
4. USP7 and the p53/MDM2 Axis
5. USP7 as a Therapeutic Target
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABCB1 | ATP binding cassette subfamily B member 1 |
| anti-CTLA4 | Anti-cytotoxic T lymphocytes A4 antibodies |
| Asp | Aspartic acid |
| CD8 | Cluster of differentiation 8 |
| CHK1 | Checkpoint kinase 1 |
| CLL | Chronic lymphocytic leukemia |
| CTLA | Cytotoxic T-lymphocyte-associated protein gene |
| Cys | Cysteine |
| DNA | Deoxyribonucleic acid |
| DNMT1 | DNA (cytosine-5)-methyltransferase 1 |
| DUBs | Deubiquitinases |
| Foxp3 | Forkhead box P3 |
| Gly | Glycine |
| HAUSP | Herpes virus-associated protease |
| HDAC | Histone deacetylase 1 |
| His | Histidine |
| IFN-γ | Interferon-gamma |
| IL | Interleukin |
| mAb | Monoclonal antibodies |
| MAPK | Mitogen-activated protein kinase |
| MDM2 | Mouse double minute 2 homolog |
| MGA | MAX dimerization protein |
| MINDY | Motif interacting with ubiquitin-containing novel deubiquitinase family proteases |
| MJD | Machado–Joseph (Josephin) domain containing proteases |
| MM | Multiple myeloma |
| NOXA | Phorbol-12-myristate-13-acetate-induced protein 1 |
| OTU | Ovarian tumor proteases |
| PD-1 | Programmed cell death protein |
| PD-L1 | Programmed death-ligand 1 |
| PDX | Patient-derived xenografts |
| PHIP1 | Pleckstrin homology domain interacting protein |
| PP2A | Protein phosphatase 2A |
| PROTAC | Proteolysis targeting chimera |
| PTM | Posttranslational modification |
| Raf-1 | RAF proto-oncogene serine/threonine-protein kinase |
| RNA | Ribonucleic acid |
| siRNA | Small interfering ribonucleic acid |
| TAMs | Tumor-associated macrophages |
| Teffs | Effector T cells |
| Tip60 | Tat-Interactive Protein |
| TME | Tumor microenvironment |
| TNBC | Triple-negative breast cancer |
| TNF-α | Tumor necrosis factor-alpha |
| TP53 | Tumor protein p53 |
| TRAF | Tumor necrosis factor Receptor-Associated Factor |
| Tregs | Regulatory T cells |
| Ub | Ubiquitin |
| UBL | Ubiquitin-like domain |
| UCH | Ubiquitin carboxyl-terminal hydrolases |
| UPS | Ubiquitin-proteasome system |
| USP | Ubiquitin-specific protease |
| USP7 | Ubiquitin-specific protease 7 |
| WT | Wild type |
| XIAP | X-linked inhibitor of apoptosis protein |
References
- Pickart, C.M. Mechanisms Underlying Ubiquitination. Annu. Rev. Biochem. 2001, 70, 503–533. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Ouyang, T.; Li, M.; Hong, T.; MHS, A.; Meng, W.; Zhang, N. Ubiquitin-Specific Peptidase 7: A Novel Deubiquitinase That Regulates Protein Homeostasis and Cancers. Front. Oncol. 2021, 11, 784672. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Liu, H.-M. Recent Advances in the Development of Ubiquitin-Specific-Processing Protease 7 (USP7) Inhibitors. Eur. J. Med. Chem. 2020, 191, 112107. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Young, M.A.; Donato, N.J. Emerging Potential of Therapeutic Targeting of Ubiquitin-Specific Proteases in the Treatment of Cancer. Cancer Res. 2014, 74, 4955–4966. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Kumar, S.; Wang, F.; Wang, H.; Chen, L.; Arsenault, P.; Mattern, M.; Weinstock, J. Chemical Approaches to Intervening in Ubiquitin Specific Protease 7 (USP7) Function for Oncology and Immune Oncology Therapies. J. Med. Chem. 2018, 61, 422–443. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, R.I.; Guedes, R.A.; Salvador, J.A.R. Highlights in USP7 Inhibitors for Cancer Treatment. Front. Chem. 2022, 10, 1005727. [Google Scholar] [CrossRef] [PubMed]
- Bailly, C.; Thuru, X.; Quesnel, B. Combined Cytotoxic Chemotherapy and Immunotherapy of Cancer: Modern Times. Nar. Cancer 2020, 2, zcaa002. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A Guide to Cancer Immunotherapy: From T Cell Basic Science to Clinical Practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 12. [Google Scholar] [CrossRef]
- Cogdill, A.P.; Andrews, M.C.; Wargo, J.A. Hallmarks of Response to Immune Checkpoint Blockade. Brit. J. Cancer 2017, 117, 136. [Google Scholar] [CrossRef]
- Osipov, A.; Murphy, A.; Zheng, L. Chapter Two From Immune Checkpoints to Vaccines: The Past, Present and Future of Cancer Immunotherapy. Adv. Cancer Res. 2019, 143, 63–144. [Google Scholar] [CrossRef] [PubMed]
- Borcoman, E.; Kanjanapan, Y.; Champiat, S.; Kato, S.; Servois, V.; Kurzrock, R.; Goel, S.; Bedard, P.; Tourneau, C.L. Novel Patterns of Response under Immunotherapy. Ann. Oncol. 2019, 30, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- van den Bulk, J.; Verdegaal, E.M.; Miranda, N.F. de Cancer Immunotherapy: Broadening the Scope of Targetable Tumours. Open Biol. 2018, 8, 180037. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Zhu, X.; Xu, P.; Li, Y. Pharmacological Inhibition of USP7 Promotes Antitumor Immunity and Contributes to Colon Cancer Therapy. Oncotargets 2019, 12, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Nixon, N.A.; Blais, N.; Ernst, S.; Kollmannsberger, C.; Bebb, G.; Butler, M.; Smylie, M.; Verma, S. Current Landscape of Immunotherapy in the Treatment of Solid Tumours, with Future Opportunities and Challenges. Curr. Oncol. 2018, 25, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Chen, N.; Li, L.; Du, N.; Bai, L.; Lv, Z.; Tian, H.; Cui, J. Mechanisms of Cancer Resistance to Immunotherapy. Front. Oncol. 2020, 10, 1290. [Google Scholar] [CrossRef] [PubMed]
- Hanna, J.; Guerra-Moreno, A.; Ang, J.; Micoogullari, Y. Protein Degradation and the Pathologic Basis of Disease. Am. J. Pathol. 2018, 189, 94–103. [Google Scholar] [CrossRef]
- Jang, H.H. Regulation of Protein Degradation by Proteasomes in Cancer. J. Cancer Prev. 2018, 23, 153–161. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, F. Post-Translational Modifications of Deubiquitinating Enzymes: Expanding the Ubiquitin Code. Front. Pharm. 2021, 12, 685011. [Google Scholar] [CrossRef]
- Varshavsky, A. The Ubiquitin System, an Immense Realm. Biochemistry 2012, 81, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Finley, D. Recognition and Processing of Ubiquitin-Protein Conjugates by the Proteasome. Biochemistry 2009, 78, 477–513. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Meng, T.; Chen, L.; Wei, W.; Wang, P. The Role of Ubiquitination in Tumorigenesis and Targeted Drug Discovery. Signal Transduct. Target. Ther. 2020, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A. The Ubiquitin System. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, K.D. Regulation of Ubiquitin-dependent Processes by Deubiquitinating Enzymes. FASEB J. 1997, 11, 1245–1256. [Google Scholar] [CrossRef]
- Park, C.-W.; Ryu, K.-Y. Cellular Ubiquitin Pool Dynamics and Homeostasis. BMB Rep. 2014, 47, 475–482. [Google Scholar] [CrossRef]
- Sun, S.-C. Deubiquitylation and Regulation of the Immune Response. Nat. Rev. Immunol. 2008, 8, 501–511. [Google Scholar] [CrossRef]
- Hanpude, P.; Bhattacharya, S.; Dey, A.K.; Maiti, T.K. Deubiquitinating Enzymes in Cellular Signaling and Disease Regulation. IUBMB Life 2015, 67, 544–555. [Google Scholar] [CrossRef]
- Harrigan, J.A.; Jacq, X.; Martin, N.M.; Jackson, S.P. Deubiquitylating Enzymes and Drug Discovery: Emerging Opportunities. Nat. Rev. Drug Discov. 2018, 17, 57–78. [Google Scholar] [CrossRef]
- Nijman, S.M.B.; Luna-Vargas, M.P.A.; Velds, A.; Brummelkamp, T.R.; Dirac, A.M.G.; Sixma, T.K.; Bernards, R. A Genomic and Functional Inventory of Deubiquitinating Enzymes. Cell 2005, 123, 773–786. [Google Scholar] [CrossRef]
- Pozhidaeva, A.; Bezsonova, I. USP7: Structure, Substrate Specificity, and Inhibition. DNA Repair 2019, 76, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Li, P.; Li, M.; Li, W.; Yao, T.; Wu, J.-W.; Gu, W.; Cohen, R.E.; Shi, Y. Crystal Structure of a UBP-Family Deubiquitinating Enzyme in Isolation and in Complex with Ubiquitin Aldehyde. Cell 2002, 111, 1041–1054. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Chakraborty, D.; Basu, M.; Ghosh, M.K. Emerging Insights into HAUSP (USP7) in Physiology, Cancer and Other Diseases. Signal Transduct. Target. Ther. 2018, 3, 17. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Meredith, M.; Orr, A.; Cross, A.; Kathoria, M.; Parkinson, J. A Novel Ubiquitin-specific Protease Is Dynamically Associated with the PML Nuclear Domain and Binds to a Herpesvirus Regulatory Protein. Embo J. 1997, 16, 1519–1530. [Google Scholar] [CrossRef]
- Holowaty, M.N.; Sheng, Y.; Nguyen, T.; Arrowsmith, C.; Frappier, L. Protein Interaction Domains of the Ubiquitin-Specific Protease, USP7/HAUSP*. J. Biol. Chem. 2003, 278, 47753–47761. [Google Scholar] [CrossRef]
- Kim, R.Q.; Sixma, T.K. Regulation of USP7: A High Incidence of E3 Complexes. J. Mol. Biol. 2017, 429, 3395–3408. [Google Scholar] [CrossRef]
- Hu, M.; Gu, L.; Li, M.; Jeffrey, P.D.; Gu, W.; Shi, Y. Structural Basis of Competitive Recognition of P53 and MDM2 by HAUSP/USP7: Implications for the Regulation of the P53–MDM2 Pathway. PLoS Biol. 2006, 4, e27. [Google Scholar] [CrossRef]
- Molland, K.; Zhou, Q.; Mesecar, A.D. A 2.2 Å Resolution Structure of the USP7 Catalytic Domain in a New Space Group Elaborates upon Structural Rearrangements Resulting from Ubiquitin Binding. Acta Cryst. Sect. F Struct. Biol. Commun. 2014, 70, 283–287. [Google Scholar] [CrossRef]
- Kim, R.Q.; van Dijk, W.J.; Sixma, T.K. Structure of USP7 Catalytic Domain and Three Ubl-Domains Reveals a Connector α-Helix with Regulatory Role. J. Struct. Biol. 2016, 195, 11–18. [Google Scholar] [CrossRef]
- Faesen, A.C.; Dirac, A.M.G.; Shanmugham, A.; Ovaa, H.; Perrakis, A.; Sixma, T.K. Mechanism of USP7/HAUSP Activation by Its C-Terminal Ubiquitin-like Domain and Allosteric Regulation by GMP-Synthetase. Mol. Cell 2011, 44, 147–159. [Google Scholar] [CrossRef]
- Rougé, L.; Bainbridge, T.W.; Kwok, M.; Tong, R.; Di Lello, P.; Wertz, I.E.; Maurer, T.; Ernst, J.A.; Murray, J. Molecular Understanding of USP7 Substrate Recognition and C-Terminal Activation. Structure 2016, 24, 1335–1345. [Google Scholar] [CrossRef] [PubMed]
- deLeeuw, R.J.; Kost, S.E.; Kakal, J.A.; Nelson, B.H. The Prognostic Value of FoxP3+ Tumor-Infiltrating Lymphocytes in Cancer: A Critical Review of the Literature. Clin. Cancer Res. 2012, 18, 3022–3029. [Google Scholar] [CrossRef] [PubMed]
- Shang, B.; Liu, Y.; Jiang, S.; Liu, Y. Prognostic Value of Tumor-Infiltrating FoxP3+ Regulatory T Cells in Cancers: A Systematic Review and Meta-Analysis. Sci. Rep. 2015, 5, 15179. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Mimura, Y.; Aoe, K.; Kobayashi, S.; Yamamoto, H.; Matsuda, E.; Okabe, K.; Matsumoto, T.; Sugi, K.; Ueoka, H. Prognostic Potential of FOXP3 Expression in Non-Small Cell Lung Cancer Cells Combined with Tumor-Infiltrating Regulatory T Cells. Lung Cancer 2012, 75, 95–101. [Google Scholar] [CrossRef]
- Salama, P.; Phillips, M.; Grieu, F.; Morris, M.; Zeps, N.; Joseph, D.; Platell, C.; Iacopetta, B. Tumor-Infiltrating FOXP3+ T Regulatory Cells Show Strong Prognostic Significance in Colorectal Cancer. J. Clin. Oncol. 2008, 27, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Shou, J.; Zhang, Z.; Lai, Y.; Chen, Z.; Huang, J. Worse Outcome in Breast Cancer with Higher Tumor-Infiltrating FOXP3+ Tregs: A Systematic Review and Meta-Analysis. Bmc Cancer 2016, 16, 687. [Google Scholar] [CrossRef]
- Finotello, F.; Trajanoski, Z. New Strategies for Cancer Immunotherapy: Targeting Regulatory T Cells. Genome Med. 2017, 9, 10. [Google Scholar] [CrossRef]
- Crunkhorn, S. Targeting Regulatory T Cells. Nat. Rev. Drug Discov. 2017, 16, 754. [Google Scholar] [CrossRef]
- Wang, L.; Kumar, S.; Dahiya, S.; Wang, F.; Wu, J.; Newick, K.; Han, R.; Samanta, A.; Beier, U.H.; Akimova, T.; et al. Ubiquitin-Specific Protease-7 Inhibition Impairs Tip60-Dependent Foxp3+ T-Regulatory Cell Function and Promotes Antitumor Immunity. Ebiomedicine 2016, 13, 99–112. [Google Scholar] [CrossRef]
- Chauhan, D.; Tian, Z.; Nicholson, B.; Kumar, K.G.S.; Zhou, B.; Carrasco, R.; McDermott, J.L.; Leach, C.A.; Fulcinniti, M.; Kodrasov, M.P.; et al. A Small Molecule Inhibitor of Ubiquitin-Specific Protease-7 Induces Apoptosis in Multiple Myeloma Cells and Overcomes Bortezomib Resistance. Cancer Cell 2012, 22, 345–358. [Google Scholar] [CrossRef]
- Dai, X.; Lu, L.; Deng, S.; Meng, J.; Wan, C.; Huang, J.; Sun, Y.; Hu, Y.; Wu, B.; Wu, G.; et al. USP7 Targeting Modulates Anti-Tumor Immune Response by Reprogramming Tumor-Associated Macrophages in Lung Cancer. Theranostics 2020, 10, 9332–9347. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, D.; Shiloh, A.; Luo, J.; Nikolaev, A.Y.; Qin, J.; Gu, W. Deubiquitination of P53 by HAUSP Is an Important Pathway for P53 Stabilization. Nature 2002, 416, 648–653. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.-M.; Cheng, G.; Cheng, X.-D.; Xu, Z.; Xu, B.; Zhang, W.-D.; Qin, J.-J. Targeting USP7-Mediated Deubiquitination of MDM2/MDMX-P53 Pathway for Cancer Therapy: Are We There Yet? Front. Cell Dev. Biol. 2020, 8, 233. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, J.; Chen, C.; Yuan, H.; Wen, X.; Sun, H. USP7: Target Validation and Drug Discovery for Cancer Therapy. Med. Chem. 2018, 14, 3–18. [Google Scholar] [CrossRef]
- Tavana, O.; Gu, W. Modulation of the P53/MDM2 Interplay by HAUSP Inhibitors. J. Mol. Cell Biol. 2017, 9, 45–52. [Google Scholar] [CrossRef]
- Hu, T.; Zhang, J.; Sha, B.; Li, M.; Wang, L.; Zhang, Y.; Liu, X.; Dong, Z.; Liu, Z.; Li, P.; et al. Targeting the Overexpressed USP7 Inhibits Esophageal Squamous Cell Carcinoma Cell Growth by Inducing NOXA-mediated Apoptosis. Mol. Carcinog. 2019, 58, 42–54. [Google Scholar] [CrossRef]
- Agathanggelou, A.; Smith, E.; Davies, N.J.; Kwok, M.; Zlatanou, A.; Oldreive, C.E.; Mao, J.; Costa, D.D.; Yadollahi, S.; Perry, T.; et al. USP7 Inhibition Alters Homologous Recombination Repair and Targets CLL Cells Independently of ATM/P53 Functional Status. Blood 2017, 130, 156–166. [Google Scholar] [CrossRef]
- Colland, F.; Formstecher, E.; Jacq, X.; Reverdy, C.; Planquette, C.; Conrath, S.; Trouplin, V.; Bianchi, J.; Aushev, V.N.; Camonis, J.; et al. Small-Molecule Inhibitor of USP7/HAUSP Ubiquitin Protease Stabilizes and Activates P53 in Cells. Mol. Cancer Ther. 2009, 8, 2286–2295. [Google Scholar] [CrossRef]
- Turnbull, A.P.; Ioannidis, S.; Krajewski, W.W.; Pinto-Fernandez, A.; Heride, C.; Martin, A.C.L.; Tonkin, L.M.; Townsend, E.C.; Buker, S.M.; Lancia, D.R.; et al. Molecular Basis of USP7 Inhibition by Selective Small-Molecule Inhibitors. Nature 2017, 550, 481–486. [Google Scholar] [CrossRef]
- Fang, D.D.; Tang, Q.; Kong, Y.; Wang, Q.; Gu, J.; Fang, X.; Zou, P.; Rong, T.; Wang, J.; Yang, D.; et al. MDM2 Inhibitor APG-115 Synergizes with PD-1 Blockade through Enhancing Antitumor Immunity in the Tumor Microenvironment. J. Immunother. Cancer 2019, 7, 327. [Google Scholar] [CrossRef]
- Al-Eidan, A.; Wang, Y.; Skipp, P.; Ewing, R.M. The USP7 Protein Interaction Network and Its Roles in Tumorigenesis. Genes Dis 2020, 9, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Khusbu, F.Y.; Chen, F.; Chen, H. Targeting Ubiquitin Specific Protease 7 in Cancer: A Deubiquitinase with Great Prospects. Cell Biochem. Funct. 2018, 36, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Do, H.-A.; Baek, K.-H. Protein Phosphatase 2A Regulated by USP7 Is Polyubiquitinated and Polyneddylated. Oncol. Rep. 2022, 48, 124. [Google Scholar] [CrossRef] [PubMed]
- Nie, L.; Wang, C.; Liu, X.; Teng, H.; Li, S.; Huang, M.; Feng, X.; Pei, G.; Hang, Q.; Zhao, Z.; et al. USP7 Substrates Identified by Proteomics Analysis Reveal the Specificity of USP. Gene Dev. 2022, 36, 1016–1030. [Google Scholar] [CrossRef]
- Saha, G.; Sarkar, S.; Mohanta, P.S.; Kumar, K.; Chakrabarti, S.; Basu, M.; Ghosh, M.K. USP7 Targets XIAP for Cancer Progression: Establishment of a P53-Independent Therapeutic Avenue for Glioma. Oncogene 2022, 1–15. [Google Scholar] [CrossRef]
- Lin, Y.-T.; Lin, J.; Liu, Y.-E.; Chen, Y.-C.; Liu, S.-T.; Hsu, K.-W.; Chen, D.-R.; Wu, H.-T. USP7 Induces Chemoresistance in Triple-Negative Breast Cancer via Deubiquitination and Stabilization of ABCB. Cells 2022, 11, 3294. [Google Scholar] [CrossRef]
- Park, H.-B.; Hwang, S.; Baek, K.-H. USP7 Regulates the ERK1/2 Signaling Pathway through Deubiquitinating Raf-1 in Lung Adenocarcinoma. Cell Death Dis. 2022, 13, 698. [Google Scholar] [CrossRef]
- Wang, Z.; Kang, W.; You, Y.; Pang, J.; Ren, H.; Suo, Z.; Liu, H.; Zheng, Y. USP7: Novel Drug Target in Cancer Therapy. Front. Pharm. 2019, 10, 427. [Google Scholar] [CrossRef]
- Sun, J.; Lin, L.; Zhang, J.; Hu, C.; Wang, J. The Prognostic Value of USP7 and P53 in Advanced Hypopharyngeal Carcinoma. Ann. Diagn. Pathol. 2021, 51, 151695. [Google Scholar] [CrossRef]
- Lu, J.; Zhao, H.; Yu, C.; Kang, Y.; Yang, X. Targeting Ubiquitin-Specific Protease 7 (USP7) in Cancer: A New Insight to Overcome Drug Resistance. Front. Pharm. 2021, 12, 648491. [Google Scholar] [CrossRef]
- Cartel, M.; Mouchel, P.-L.; Gotanègre, M.; David, L.; Bertoli, S.; Mas, V.M.-D.; Besson, A.; Sarry, J.-E.; Manenti, S.; Didier, C. Inhibition of Ubiquitin-Specific Protease 7 Sensitizes Acute Myeloid Leukemia to Chemotherapy. Leukemia 2021, 35, 417–432. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Qin, B.; Boughey, J.; Goetz, M.; Wang, L. Abstract 2980: The Role of USP7 and USP7 Inhibitor in HER2+ Breast Cancer Treatment. Cancer Res. 2019, 79, 2980. [Google Scholar] [CrossRef]
- Grunblatt, E.; Wu, N.; Zhang, H.; Liu, X.; Norton, J.P.; Ohol, Y.; Leger, P.; Hiatt, J.B.; Eastwood, E.C.; Thomas, R.; et al. MYCN Drives Chemoresistance in Small Cell Lung Cancer While USP7 Inhibition Can Restore Chemosensitivity. Gene Dev. 2020, 34, 1210–1226. [Google Scholar] [CrossRef]
- Bufalieri, F.; Severini, L.L.; Caimano, M.; Infante, P.; Marcotullio, L.D. DUBs Activating the Hedgehog Signaling Pathway: A Promising Therapeutic Target in Cancer. Cancers 2020, 12, 1518. [Google Scholar] [CrossRef]
- Harakandi, C.; Nininahazwe, L.; Xu, H.; Liu, B.; He, C.; Zheng, Y.-C.; Zhang, H. Recent Advances on the Intervention Sites Targeting USP7-MDM2-P53 in Cancer Therapy. Bioorg. Chem. 2021, 116, 105273. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Liu, Y.; Liu, H.-M. A Patent Review of Ubiquitin-Specific Protease 7 (USP7) Inhibitors (2014-Present). Expert Opin. Ther. Pat. 2022, 32, 753–767. [Google Scholar] [CrossRef]
- Nicholson, B.; Kumar, K.G.S. The Multifaceted Roles of USP7: New Therapeutic Opportunities. Cell Biochem. Biophys. 2011, 60, 61. [Google Scholar] [CrossRef]
- Cao, P.; Weinstock, J.; Kingsbury, W.D.; Leach, C.A.; Kizhakkethil-George, S.K.; Nicholson, B. Anti-Neoplastic Compounds, Compositions and Methods. U.S. Patent US-8680139-B2, 25 March 2014. [Google Scholar]
- Wang, F.; Wang, L.; Wu, J.; Sokirniy, I.; Nguyen, P.; Bregnard, T.; Weinstock, J.; Mattern, M.; Bezsonova, I.; Hancock, W.W.; et al. Active Site-Targeted Covalent Irreversible Inhibitors of USP7 Impair the Functions of Foxp3+ T-Regulatory Cells by Promoting Ubiquitination of Tip. PLoS ONE 2017, 12, e0189744. [Google Scholar] [CrossRef]
- Gavory, G.; O’Dowd, C.R.; Helm, M.D.; Flasz, J.; Arkoudis, E.; Dossang, A.; Hughes, C.; Cassidy, E.; McClelland, K.; Odrzywol, E.; et al. Discovery and Characterization of Highly Potent and Selective Allosteric USP7 Inhibitors. Nat. Chem. Biol. 2018, 14, 118–125. [Google Scholar] [CrossRef]
- Reverdy, C.; Conrath, S.; Lopez, R.; Planquette, C.; Atmanene, C.; Collura, V.; Harpon, J.; Battaglia, V.; Vivat, V.; Sippl, W.; et al. Discovery of Specific Inhibitors of Human USP7/HAUSP Deubiquitinating Enzyme. Chem. Biol. 2012, 19, 467–477. [Google Scholar] [CrossRef]
- Kategaya, L.; Lello, P.D.; Rougé, L.; Pastor, R.; Clark, K.R.; Drummond, J.; Kleinheinz, T.; Lin, E.; Upton, J.-P.; Prakash, S.; et al. USP7 Small-Molecule Inhibitors Interfere with Ubiquitin Binding. Nature 2017, 550, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Lello, P.D.; Pastor, R.; Murray, J.M.; Blake, R.A.; Cohen, F.; Crawford, T.D.; Drobnick, J.; Drummond, J.; Kategaya, L.; Kleinheinz, T.; et al. Discovery of Small-Molecule Inhibitors of Ubiquitin Specific Protease 7 (USP7) Using Integrated NMR and in Silico Techniques. J. Med. Chem. 2017, 60, 10056–10070. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage Activation and Polarization. Front. Biosci. 2008, 13, 453. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef]
- Weinstock, J.; Wu, J.; Cao, P.; Kingsbury, W.D.; McDermott, J.L.; Kodrasov, M.P.; McKelvey, D.M.; Kumar, K.G.S.; Goldenberg, S.J.; Mattern, M.R.; et al. Selective Dual Inhibitors of the Cancer-Related Deubiquitylating Proteases USP7 and USP. ACS Med. Chem. Lett. 2012, 3, 789–792. [Google Scholar] [CrossRef]
- Wang, Z.; Kang, W.; Li, O.; Qi, F.; Wang, J.; You, Y.; He, P.; Suo, Z.; Zheng, Y.; Liu, H.-M. Abrogation of USP7 Is an Alternative Strategy to Downregulate PD-L1 and Sensitize Gastric Cancer Cells to T Cells Killing. Acta Pharm. Sin. B 2021, 11, 694–707. [Google Scholar] [CrossRef]
- Das, D.S.; Ray, A.; Das, A.; Song, Y.; Tian, Z.; Oronsky, B.; Richardson, P.; Scicinski, J.; Chauhan, D.; Anderson, K.C. A Novel Hypoxia-Selective Epigenetic Agent RRx-001 Triggers Apoptosis and Overcomes Drug Resistance in Multiple Myeloma Cells. Leukemia 2016, 30, 2187–2197. [Google Scholar] [CrossRef]
- Du, Z.; Song, J.; Wang, Y.; Zhao, Y.; Guda, K.; Yang, S.; Kao, H.-Y.; Xu, Y.; Willis, J.; Markowitz, S.D.; et al. DNMT1 Stability Is Regulated by Proteins Coordinating Deubiquitination and Acetylation-Driven Ubiquitination. Sci. Signal 2010, 3, ra80. [Google Scholar] [CrossRef]
- Chaudhary, B.; Elkord, E. Regulatory T Cells in the Tumor Microenvironment and Cancer Progression: Role and Therapeutic Targeting. Nato Adv. Sci. Inst. Sci. 2016, 4, 28. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Marchesi, F.; Garlanda, C. Macrophages as Tools and Targets in Cancer Therapy. Nat. Rev. Drug Discov. 2022, 21, 799–820. [Google Scholar] [CrossRef]
- Leger, P.R.; Hu, D.X.; Biannic, B.; Bui, M.; Han, X.; Karbarz, E.; Maung, J.; Okano, A.; Osipov, M.; Shibuya, G.M.; et al. Discovery of Potent, Selective, and Orally Bioavailable Inhibitors of USP7 with In Vivo Antitumor Activity. J. Med. Chem. 2020, 63, 5398–5420. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.B.; Ketcham, J.M.; Reilly, M.K.; Biannic, B.; Bui, M.H.T.; Hu, D.X.; Wustrow, D.J.; Karbarz, E.; Han, X.; Shibuya, G.; et al. Ubiquitin-Specific-Processing Protease 7 (USP7) Modulators and Uses Thereof. U.S. Patent US-20210317134-A1, 14 October 2021. [Google Scholar]
- Biannic, B.; Han, X.; Hu, D.X.; Leger, P.R.; Maung, J.; Okano, A.; Schwarz, J.B.; Wustrow, D.J.; Young, K.; Cutler, G.; et al. Ubiquitin-Specific-Processing Protease 7 (USP7) Modulators and Uses Thereof. U.S. Patent US-11084829-B2, 10 August 2021. [Google Scholar]
- Varca, A.C.; Casalena, D.; Chan, W.C.; Hu, B.; Magin, R.S.; Roberts, R.M.; Liu, X.; Zhu, H.; Seo, H.-S.; Dhe-Paganon, S.; et al. Identification and Validation of Selective Deubiquitinase Inhibitors. Cell Chem. Biol. 2021, 28, 1758–1771.e13. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Fu, J.; Shi, Y.; Zhang, M.; Luo, G.; Luo, X.; Song, N.; Mi, T.; Yang, Y.; Li, J.; et al. Discovery of a Potent and Selective Degrader for USP. Angew. Chem. 2022, 134, 4395. [Google Scholar] [CrossRef]
- Murgai, A.; Sosič, I.; Gobec, M.; Lemnitzer, P.; Proj, M.; Wittenburg, S.; Voget, R.; Gütschow, M.; Krönke, J.; Steinebach, C. Targeting the Deubiquitinase USP7 for Degradation with PROTACs. Chem. Commun. 2022, 58, 8858–8861. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| USP7 Inhibitor | Mode of Action | Cancer Type In Vitro and/or In Vivo Studies |
|---|---|---|
 P5091 | Covalent Non-selective (dual USP7/47) IC50 (USP7) 4.2 μM IC50 (USP47) 4.3 μM | Multiple myeloma MM.1S, MM.1R, U266, KMS12PE, etc. Colorectal cancer HCT116 Prostate cancer PC3, LNCaP Ovarian cancer SKOV3, OVCAR-8, HeyA8 Urothelial bladder cancer J82, T24, KU-19-19 Esophageal squamous cell carcinoma HN5, HN30, UM-SCC-1, UM-SCC-25, etc. Chronic lympholytic leukemia CCRF-CEM Medulloblastoma D823, Daoy |
 P217564 | Covalent Non-selective (dual USP7/47) IC50 (USP7) 0.48 μM IC50 (USP47) 0.66 μM | Colorectal cancer HCT116 Prostate cancer PC3 |
 Almac4 | Non-covalent Selective IC50 (USP7) 0.0015 μM IC50 (USP47) > 50 μM | Colon cancer HCT116 Breast cancer LNCaP Osteosarcoma SJSA-1 Prostate cancer MCF7 |
 HBX19818 | Covalent Selective IC50 (USP7) 28.1 μM IC50 (USP47) NR 1 | Colon cancer HCT116 Osteosarcoma U2OS, SJSA-1 |
 GNE-6776 | Non-covalent Selective IC50 (USP7) 1.3 μM IC50 (USP47) > 200 μM | Colon cancer HCT116 Osteosarcoma SJSA-1 Acute myeloid leukemia EOL-1 Breast cancer MCF7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korenev, G.; Yakukhnov, S.; Druk, A.; Golovina, A.; Chasov, V.; Mirgayazova, R.; Ivanov, R.; Bulatov, E. USP7 Inhibitors in Cancer Immunotherapy: Current Status and Perspective. Cancers 2022, 14, 5539. https://doi.org/10.3390/cancers14225539
Korenev G, Yakukhnov S, Druk A, Golovina A, Chasov V, Mirgayazova R, Ivanov R, Bulatov E. USP7 Inhibitors in Cancer Immunotherapy: Current Status and Perspective. Cancers. 2022; 14(22):5539. https://doi.org/10.3390/cancers14225539
Chicago/Turabian StyleKorenev, Georgiy, Sergey Yakukhnov, Anastasia Druk, Anastasia Golovina, Vitaly Chasov, Regina Mirgayazova, Roman Ivanov, and Emil Bulatov. 2022. "USP7 Inhibitors in Cancer Immunotherapy: Current Status and Perspective" Cancers 14, no. 22: 5539. https://doi.org/10.3390/cancers14225539
APA StyleKorenev, G., Yakukhnov, S., Druk, A., Golovina, A., Chasov, V., Mirgayazova, R., Ivanov, R., & Bulatov, E. (2022). USP7 Inhibitors in Cancer Immunotherapy: Current Status and Perspective. Cancers, 14(22), 5539. https://doi.org/10.3390/cancers14225539