


Identification of Factors Driving Doxorubicin-Resistant Ewing Tumor Cells to Survival

 , ,
, ,  ,
,
Abstract
Simple Summary
Abstract
1. Background
2. Materials and Methods
2.1. Materials
2.2. Primary Tissue Samples
2.3. Cell Cultures
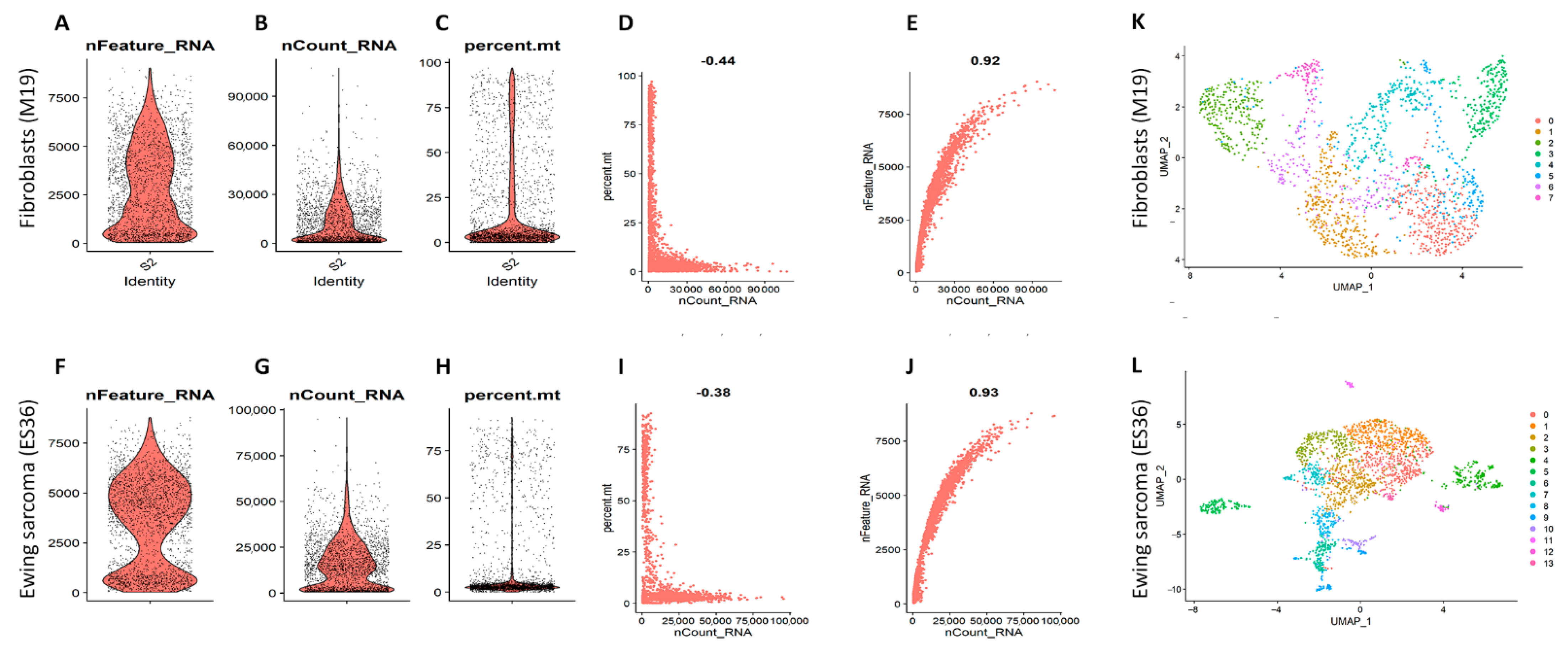
2.4. cDNA Library Preparation and Single-Cell RNA-Seq
2.5. Proteomic Data Generation and Analysis
2.6. Data Source, Data Processing, and Data Distribution
2.7. Functional GO Enrichment Analysis
2.8. Experimental Validation of Fold-Change Values Using Semiquantitative RT-PCR Analysis
2.9. Statistical Analyses
2.10. Availability of Data and Materials
3. Results
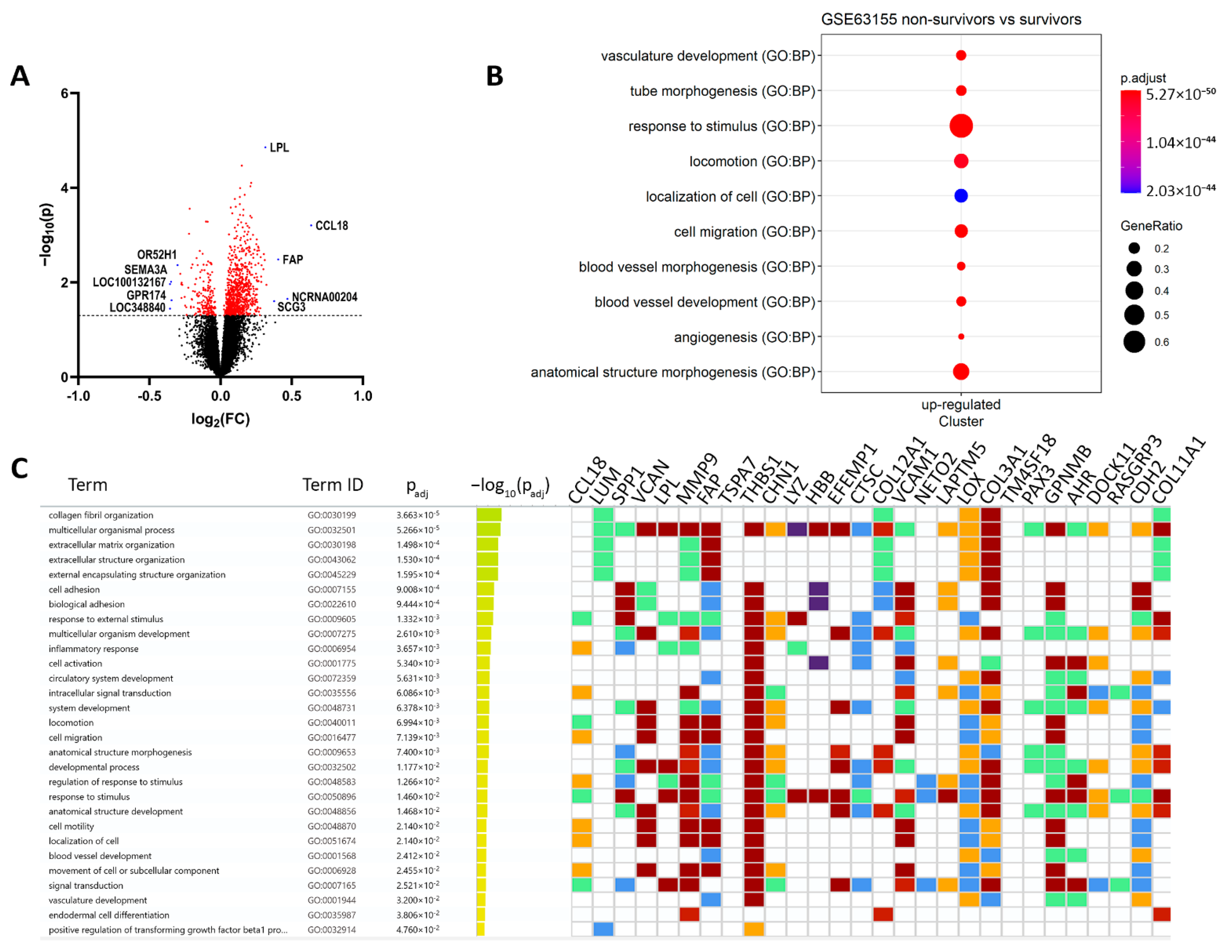
3.1. Hypoxia and Metastatic Traits Distinguish Tumor Cells from Non-Neoplastic Cells
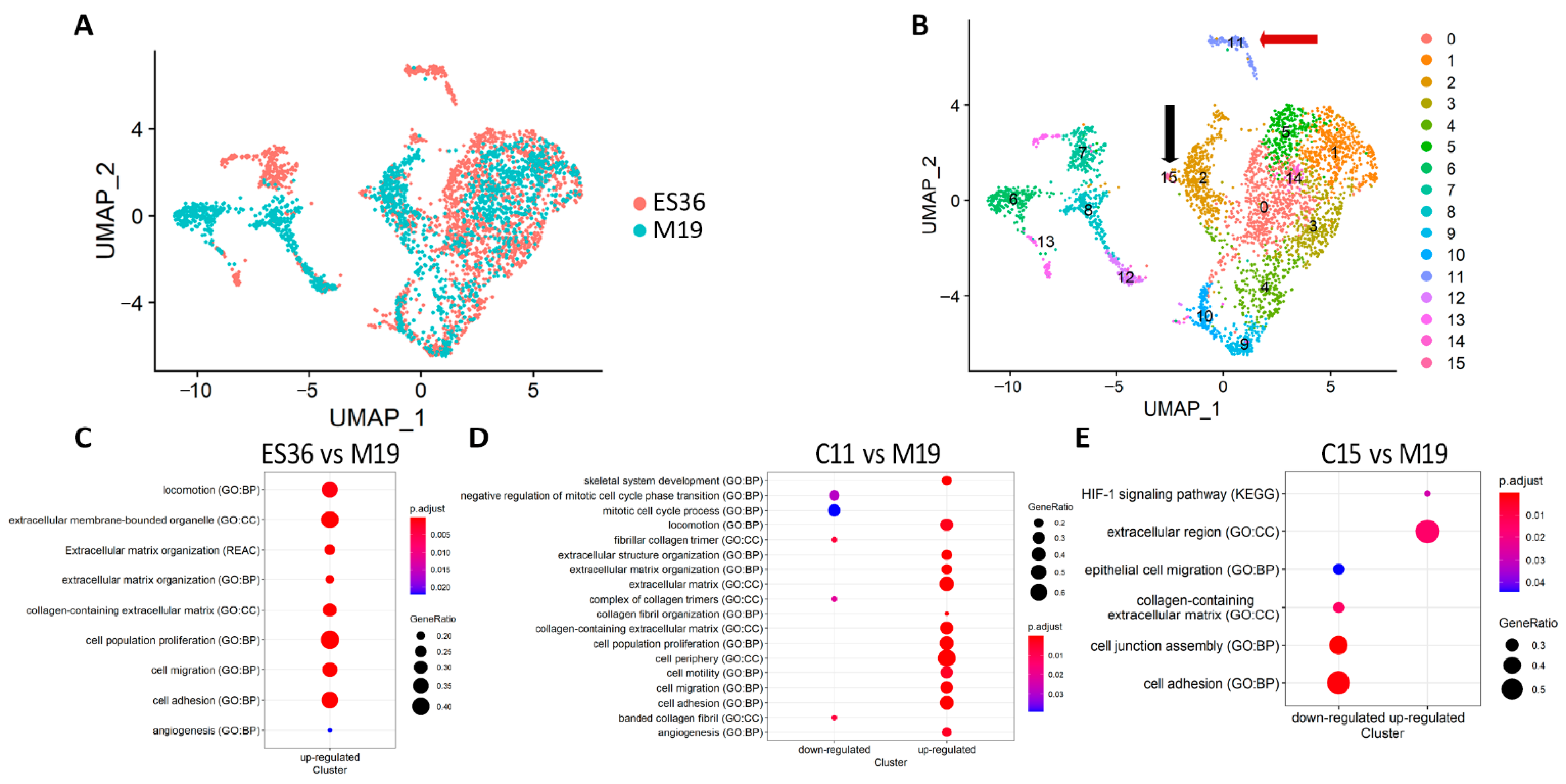
3.2. Embryonic Fibroblasts (M19) and ES (ES36) Cells Are Genetically Close
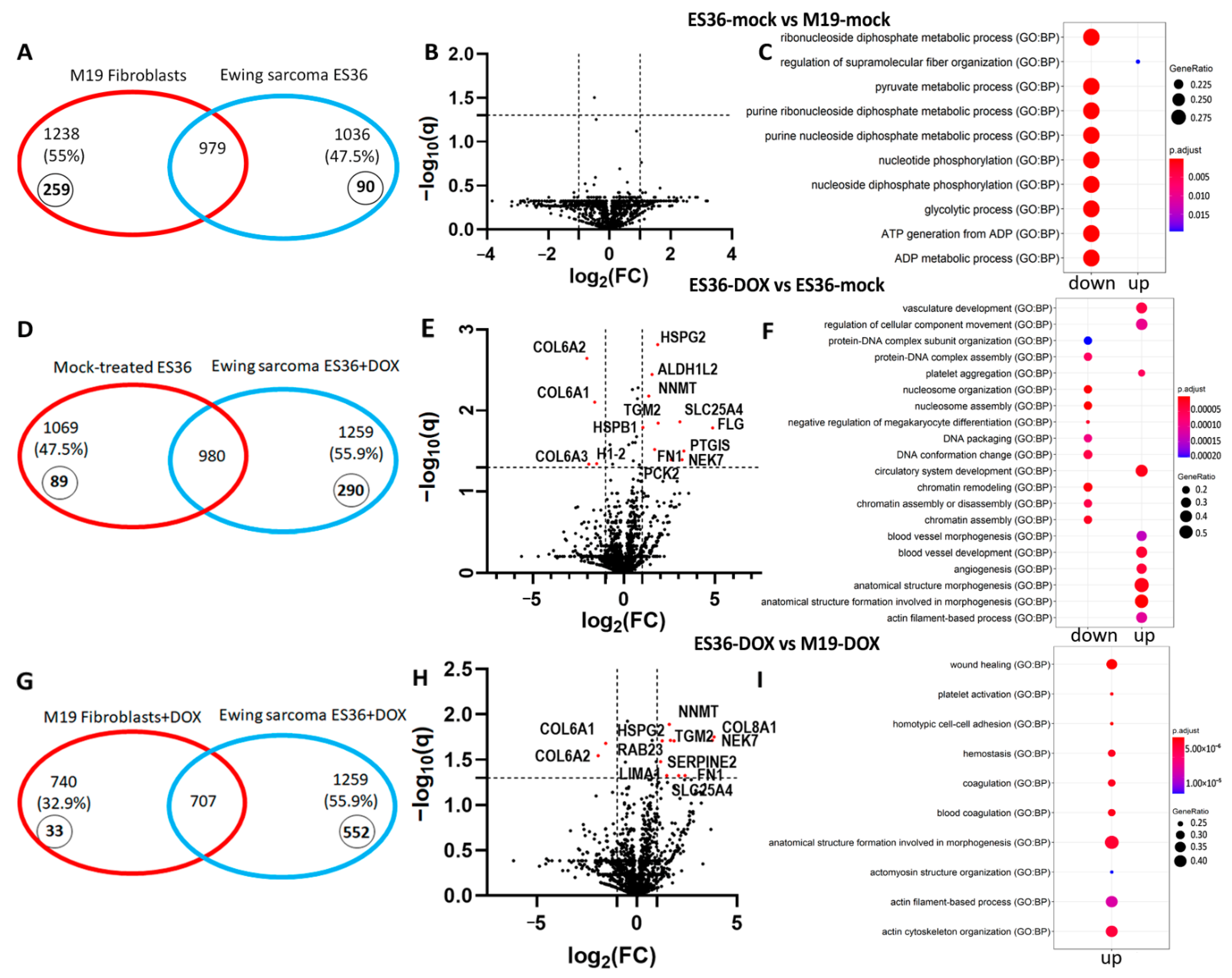
3.3. Proteomic Profiling of DOX-Treated ES36 Cells Provides a Unique Signature
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koelsche, C.; Kriegsmann, M.; Kommoss, F.K.F.; Stichel, D.; Kriegsmann, K.; Vokuhl, C.; Grunewald, T.G.P.; Romero-Perez, L.; Kirchner, T.; de Alava, E.; et al. DNA methylation profiling distinguishes Ewing-like sarcoma with EWSR1-NFATc2 fusion from Ewing sarcoma. J. Cancer Res. Clin. Oncol. 2019, 145, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Sankar, S.; Lessnick, S.L. Promiscuous partnerships in Ewing’s sarcoma. Cancer Genet. 2011, 204, 351–365. [Google Scholar] [CrossRef]
- Shulman, D.S.; Whittle, S.B.; Surdez, D.; Bailey, K.M.; de Alava, E.; Yustein, J.T.; Shlien, A.; Hayashi, M.; Bishop, A.J.R.; Crompton, B.D.; et al. An international working group consensus report for the prioritization of molecular biomarkers for Ewing sarcoma. NPJ Precis. Oncol. 2022, 6, 65. [Google Scholar] [CrossRef] [PubMed]
- Miser, J.S.; Goldsby, R.E.; Chen, Z.; Krailo, M.D.; Tarbell, N.J.; Link, M.P.; Fryer, C.J.; Pritchard, D.J.; Gebhardt, M.C.; Dickman, P.S.; et al. Treatment of metastatic Ewing sarcoma/primitive neuroectodermal tumor of bone: Evaluation of increasing the dose intensity of chemotherapy—A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2007, 49, 894–900. [Google Scholar] [CrossRef]
- Leavey, P.J.; Mascarenhas, L.; Marina, N.; Chen, Z.; Krailo, M.; Miser, J.; Brown, K.; Tarbell, N.; Bernstein, M.L.; Granowetter, L.; et al. Prognostic factors for patients with Ewing sarcoma (EWS) at first recurrence following multi-modality therapy: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2008, 51, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Hong, T.; Liu, W.; Dong, S.; Wang, H.; Tang, Z.R.; Li, W.; Wang, B.; Hu, Z.; Liu, Q.; et al. Development of a Machine Learning-Based Predictive Model for Lung Metastasis in Patients With Ewing Sarcoma. Front. Med. 2022, 9, 807382. [Google Scholar] [CrossRef]
- Margulies, B.S.; DeBoyace, S.D.; Damron, T.A.; Allen, M.J. Ewing’s sarcoma of bone tumor cells produces MCSF that stimulates monocyte proliferation in a novel mouse model of Ewing’s sarcoma of bone. Bone 2015, 79, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.; Knowles, H.J.; Athanasou, N.A. Ewing sarcoma cells express RANKL and support osteoclastogenesis. J. Pathol. 2011, 225, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Neve, A.; Cantatore, F.P.; Maruotti, N.; Corrado, A.; Ribatti, D. Extracellular matrix modulates angiogenesis in physiological and pathological conditions. BioMed Res. Int. 2014, 2014, 756078. [Google Scholar] [CrossRef] [PubMed]
- Perut, F.; Carta, F.; Bonuccelli, G.; Grisendi, G.; Di Pompo, G.; Avnet, S.; Sbrana, F.V.; Hosogi, S.; Dominici, M.; Kusuzaki, K.; et al. Carbonic anhydrase IX inhibition is an effective strategy for osteosarcoma treatment. Expert Opin. Ther. Targets 2015, 19, 1593–1605. [Google Scholar] [CrossRef] [PubMed]
- Hatano, M.; Matsumoto, Y.; Fukushi, J.; Matsunobu, T.; Endo, M.; Okada, S.; Iura, K.; Kamura, S.; Fujiwara, T.; Iida, K.; et al. Cadherin-11 regulates the metastasis of Ewing sarcoma cells to bone. Clin. Exp. Metastasis 2015, 32, 579–591. [Google Scholar] [CrossRef]
- Supuran, C.T.; Alterio, V.; Di Fiore, A.; D’Ambrosio, K.; Carta, F.; Monti, S.M.; De Simone, G. Inhibition of carbonic anhydrase IX targets primary tumors, metastases, and cancer stem cells: Three for the price of one. Med. Res. Rev. 2018, 38, 1799–1836. [Google Scholar] [CrossRef] [PubMed]
- Vieira, G.M.; Roberto, G.M.; Lira, R.C.; Engel, E.E.; Tone, L.G.; Brassesco, M.S. Prognostic value and functional role of ROCK2 in pediatric Ewing sarcoma. Oncol. Lett. 2018, 15, 2296–2304. [Google Scholar] [CrossRef]
- Hetland, T.E.; Nymoen, D.A.; Emilsen, E.; Kaern, J.; Trope, C.G.; Florenes, V.A.; Davidson, B. MGST1 expression in serous ovarian carcinoma differs at various anatomic sites, but is unrelated to chemoresistance or survival. Gynecol. Oncol. 2012, 126, 460–465. [Google Scholar] [CrossRef] [PubMed]
- van Kuijk, S.J.; Yaromina, A.; Houben, R.; Niemans, R.; Lambin, P.; Dubois, L.J. Prognostic Significance of Carbonic Anhydrase IX Expression in Cancer Patients: A Meta-Analysis. Front. Oncol. 2016, 6, 69. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.K.; Liu, J.; Chen, C.; Yang, K.Y.; Deng, Y.T.; Jiang, Y. Locally advanced malignant solitary fibrous tumour successfully treated with conversion chemotherapy, operation and postoperative radiotherapy: A case report. J. Int. Med. Res. 2021, 49, 300060521996940. [Google Scholar] [CrossRef]
- Ulanet, D.B.; Ludwig, D.L.; Kahn, C.R.; Hanahan, D. Insulin receptor functionally enhances multistage tumor progression and conveys intrinsic resistance to IGF-1R targeted therapy. Proc. Natl. Acad. Sci. USA 2010, 107, 10791–10798. [Google Scholar] [CrossRef] [PubMed]
- Scotlandi, K.; Remondini, D.; Castellani, G.; Manara, M.C.; Nardi, F.; Cantiani, L.; Francesconi, M.; Mercuri, M.; Caccuri, A.M.; Serra, M.; et al. Overcoming resistance to conventional drugs in Ewing sarcoma and identification of molecular predictors of outcome. J. Clin. Oncol. 2009, 27, 2209–2216. [Google Scholar] [CrossRef]
- Yarovinsky, T.O.; Gorlina, N.K.; Cheredeev, A.N.; Kozlov, I.G.; Zorin, N.A.; Zorina, R.M. Alpha2-Macroglobulin Modulates Interactions between Lymphocytes and Fibroblasts. Russ. J. Immunol. 2001, 6, 1–8. [Google Scholar]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed]
- Volchenboum, S.L.; Andrade, J.; Huang, L.; Barkauskas, D.A.; Krailo, M.; Womer, R.B.; Ranft, A.; Potratz, J.; Dirksen, U.; Triche, T.J.; et al. Gene Expression Profiling of Ewing Sarcoma Tumors Reveals the Prognostic Importance of Tumor-Stromal Interactions: A Report from the Children’s Oncology Group. J. Pathol. Clin. Res. 2015, 1, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.P.; Ding, S.Y.; Sun, Y.Y.; Li, Y.H.; Fu, W.N. MYCT1 Inhibits the Adhesion and Migration of Laryngeal Cancer Cells Potentially Through Repressing Collagen VI. Front. Oncol. 2020, 10, 564733. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Chen, Z.; Zou, M.; Wan, R.; Wu, T.; Luo, Y.; Wu, G.; Wang, W.; Liu, T. Prognosis and Immune Infiltration of Chromobox Family Genes in Sarcoma. Front. Oncol. 2021, 11, 657595. [Google Scholar] [CrossRef]
- Nakatani, F.; Ferracin, M.; Manara, M.C.; Ventura, S.; Del Monaco, V.; Ferrari, S.; Alberghini, M.; Grilli, A.; Knuutila, S.; Schaefer, K.L.; et al. miR-34a predicts survival of Ewing’s sarcoma patients and directly influences cell chemo-sensitivity and malignancy. J. Pathol. 2012, 226, 796–805. [Google Scholar] [CrossRef]
- Krueger, S.; Kellner, U.; Buehling, F.; Roessner, A. Cathepsin L antisense oligonucleotides in a human osteosarcoma cell line: Effects on the invasive phenotype. Cancer Gene Ther. 2001, 8, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Yeh, L.T.; Lin, C.W.; Lu, K.H.; Hsieh, Y.H.; Yeh, C.B.; Yang, S.F.; Yang, J.S. Niclosamide Suppresses Migration and Invasion of Human Osteosarcoma Cells by Repressing TGFBI Expression via the ERK Signaling Pathway. Int. J. Mol. Sci. 2022, 23, 484. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Mu, T. LncRNA LINC00958 promotes tumor progression through miR-4306/CEMIP axis in osteosarcoma. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 3182–3199. [Google Scholar] [CrossRef]
- Park, I.C.; Lee, S.Y.; Jeon, D.G.; Lee, J.S.; Hwang, C.S.; Hwang, B.G.; Lee, S.H.; Hong, W.S.; Hong, S.I. Enhanced expression of cathepsin L in metastatic bone tumors. J. Korean Med. Sci. 1996, 11, 144–148. [Google Scholar] [CrossRef]
- Wiggers, F.; Wohl, S.; Dubovetskyi, A.; Rosenblum, G.; Zheng, W.; Hofmann, H. Diffusion of a disordered protein on its folded ligand. Proc. Natl. Acad. Sci. USA 2021, 118, e2106690118. [Google Scholar] [CrossRef]
- Tang, M.; Liu, P.; Wu, X.; Gong, J.; Weng, J.; Gao, G.; Liu, Y.; Gan, L. COL3A1 and Its Related Molecules as Potential Biomarkers in the Development of Human Ewing’s Sarcoma. BioMed Res. Int. 2021, 2021, 7453500. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, J.; Liu, W.; Ding, L.; Cheng, D.; Xiao, H. Identification of Common Oncogenic Genes and Pathways Both in Osteosarcoma and Ewing’s Sarcoma Using Bioinformatics Analysis. J. Immunol. Res. 2022, 2022, 3655908. [Google Scholar] [CrossRef] [PubMed]
- Fellenberg, J.; Bernd, L.; Delling, G.; Witte, D.; Zahlten-Hinguranage, A. Prognostic significance of drug-regulated genes in high-grade osteosarcoma. Mod. Pathol. 2007, 20, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.H.; Fu, L.; Chen, J.; Wei, F.; Shi, W.X. Decreased expression of ferritin light chain in osteosarcoma and its correlation with epithelial-mesenchymal transition. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 2580–2587. [Google Scholar] [CrossRef]
- Mao, M.; Wang, W. SerpinE2 promotes multiple cell proliferation and drug resistance in osteosarcoma. Mol. Med. Rep. 2016, 14, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Brooks, M.; Mo, Q.; Krasnow, R.; Ho, P.L.; Lee, Y.C.; Xiao, J.; Kurtova, A.; Lerner, S.; Godoy, G.; Jian, W.; et al. Positive association of collagen type I with non-muscle invasive bladder cancer progression. Oncotarget 2016, 7, 82609–82619. [Google Scholar] [CrossRef]
- Yang, L.; Jing, J.; Sun, L.; Yue, Y. Exploring prognostic genes in ovarian cancer stage-related coexpression network modules. Medicine 2018, 97, e11895. [Google Scholar] [CrossRef]
- Lovitt, C.J.; Shelper, T.B.; Avery, V.M. Doxorubicin resistance in breast cancer cells is mediated by extracellular matrix proteins. BMC Cancer 2018, 18, 41. [Google Scholar] [CrossRef]
- Guzel Tanoglu, E.; Ozturk, S. miR-145 suppresses epithelial-mesenchymal transition by targeting stem cells in Ewing sarcoma cells. Bratisl. Lek. Listy 2021, 122, 71–77. [Google Scholar] [CrossRef]
- Wiles, E.T.; Bell, R.; Thomas, D.; Beckerle, M.; Lessnick, S.L. ZEB2 Represses the Epithelial Phenotype and Facilitates Metastasis in Ewing Sarcoma. Genes Cancer 2013, 4, 486–500. [Google Scholar] [CrossRef]
- Garofalo, C.; Manara, M.C.; Nicoletti, G.; Marino, M.T.; Lollini, P.L.; Astolfi, A.; Pandini, G.; Lopez-Guerrero, J.A.; Schaefer, K.L.; Belfiore, A.; et al. Efficacy of and resistance to anti-IGF-1R therapies in Ewing’s sarcoma is dependent on insulin receptor signaling. Oncogene 2011, 30, 2730–2740. [Google Scholar] [CrossRef]
- Hong, S.H.; Tilan, J.U.; Galli, S.; Izycka-Swieszewska, E.; Polk, T.; Horton, M.; Mahajan, A.; Christian, D.; Jenkins, S.; Acree, R.; et al. High neuropeptide Y release associates with Ewing sarcoma bone dissemination—In vivo model of site-specific metastases. Oncotarget 2015, 6, 7151–7165. [Google Scholar] [CrossRef] [PubMed]
- Sainz-Jaspeado, M.; Lagares-Tena, L.; Lasheras, J.; Navid, F.; Rodriguez-Galindo, C.; Mateo-Lozano, S.; Notario, V.; Sanjuan, X.; Garcia Del Muro, X.; Fabra, A.; et al. Caveolin-1 modulates the ability of Ewing’s sarcoma to metastasize. Mol. Cancer Res. 2010, 8, 1489–1500. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Dominguez, D.J.; Hajji, N.; Lopez-Alemany, R.; Sanchez-Molina, S.; Figuerola-Bou, E.; Moron Civanto, F.J.; Rello-Varona, S.; Andres-Leon, E.; Benito, A.; Keun, H.C.; et al. Selective histone methyltransferase G9a inhibition reduces metastatic development of Ewing sarcoma through the epigenetic regulation of NEU1. Oncogene 2022, 41, 2638–2650. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ma, Y.; Gao, H.; Peng, T.; Shi, H.; Tang, Y.; Li, H.; Chen, L.; Hu, K.; Han, A. A novel HDGF-ALCAM axis promotes the metastasis of Ewing sarcoma via regulating the GTPases signaling pathway. Oncogene 2021, 40, 731–745. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Alemany, R.; Tirado, O.M. Metastasis Assessment in Ewing Sarcoma Using Orthotopic Xenografts. Methods Mol. Biol. 2021, 2226, 201–213. [Google Scholar] [CrossRef]
- Choo, S.; Wang, P.; Newbury, R.; Roberts, W.; Yang, J. Reactivation of TWIST1 contributes to Ewing sarcoma metastasis. Pediatr. Blood Cancer 2018, 65, e26721. [Google Scholar] [CrossRef]
- Cerra, C.; Harris, M.A.; Hawkins, C.J. Establishment and Characterisation of Metastatic Extraskeletal Ewing Sarcoma Mouse Models. In Vivo 2021, 35, 3097–3106. [Google Scholar] [CrossRef]
- Grossman, T.R.; Gamliel, A.; Wessells, R.J.; Taghli-Lamallem, O.; Jepsen, K.; Ocorr, K.; Korenberg, J.R.; Peterson, K.L.; Rosenfeld, M.G.; Bodmer, R.; et al. Over-expression of DSCAM and COL6A2 cooperatively generates congenital heart defects. PLoS Genet. 2011, 7, e1002344. [Google Scholar] [CrossRef]
- McIlwain, C.C.; Townsend, D.M.; Tew, K.D. Glutathione S-transferase polymorphisms: Cancer incidence and therapy. Oncogene 2006, 25, 1639–1648. [Google Scholar] [CrossRef]
- Drozd, E.; Krzyszton-Russjan, J.; Marczewska, J.; Drozd, J.; Bubko, I.; Bielak, M.; Lubelska, K.; Wiktorska, K.; Chilmonczyk, Z.; Anuszewska, E.; et al. Up-regulation of glutathione-related genes, enzyme activities and transport proteins in human cervical cancer cells treated with doxorubicin. Biomed. Pharmacother. 2016, 83, 397–406. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
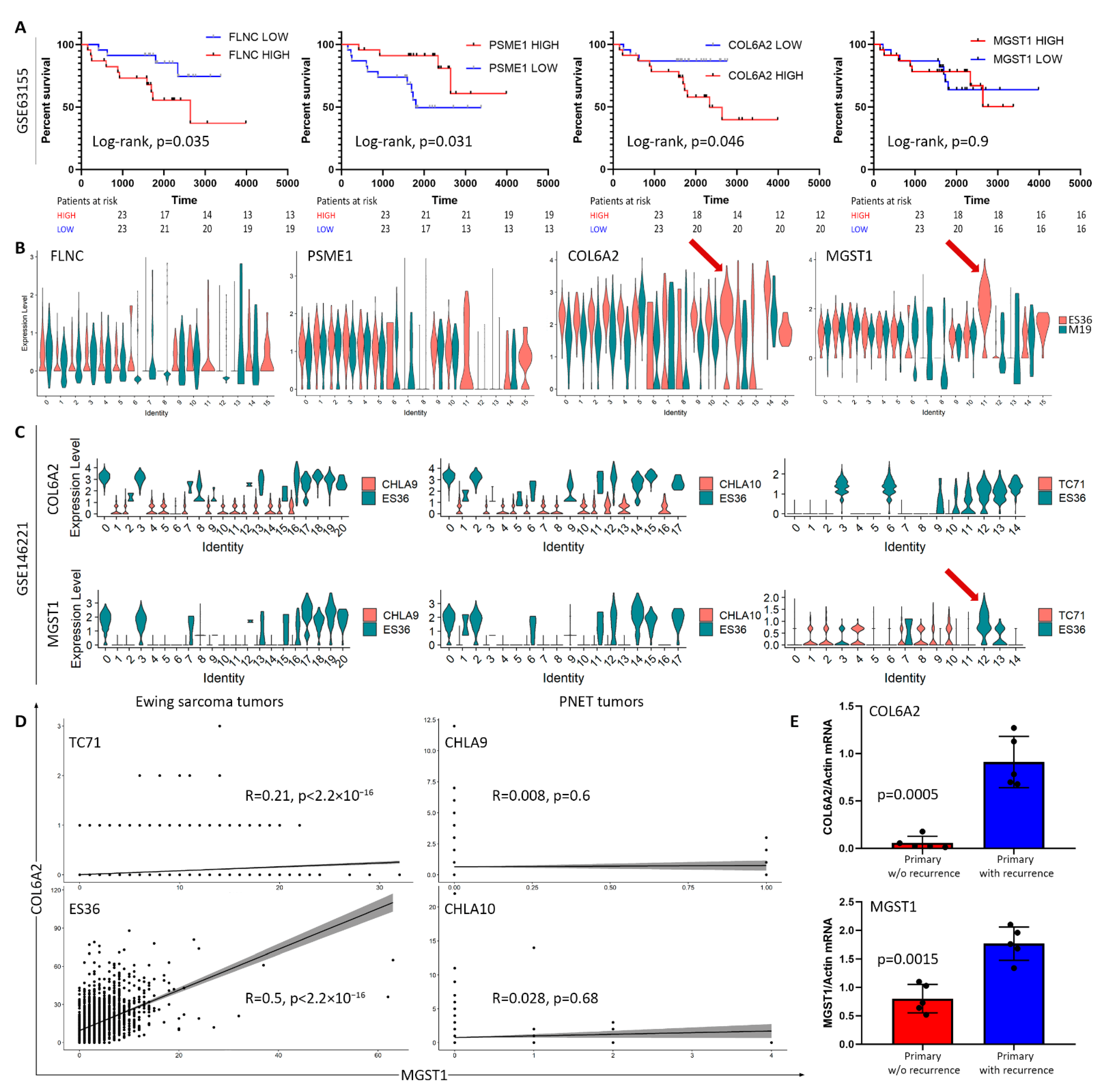{kind=link}
| Univariate | Multivariate | |||||
|---|---|---|---|---|---|---|
| Beta | HR (95% CI) | p.Value | Beta | HR (95% CI) | p.Value | |
| Sex | −0.47 | 0.62 (0.21–1.9) | 0.4 | −2.358 | 0.095 (0.005–1.84) | 0.120 |
| Age_at_enrollment_days | 0.00033 | 1 (1–1) | 0.07 | −0.004 | 0.996 (0.98–1.0) | 0.465 |
| Age | 0.11 | 1.1 (0.99–1.3) | 0.075 | 1.637 | 5.14 (0.085–309.385) | 0.433 |
| Primary_tumor_site | −0.28 | 0.76 (0.35–1.6) | 0.47 | 1.502 | 4.492 (0.377–53.53) | 0.235 |
| Efs_event | 2 | 7.7 (3.4–17) | 9.5e-07 | 3.941 | 51.476 (3.736–709.32) | 0.003 |
| Tumor_content | 0.77 | 2.2 (0.57–8.2) | 0.26 | −0.562 | 0.57 (0.088–3.676) | 0.554 |
| Description | ID | p Value | Count/Gene ID | |
|---|---|---|---|---|
| Downregulated | locomotion | GO:0040011 | 1.14 × 10−26 | CCL18/VCAN/MMP9/FAP/THBS1/CHN1/VCAM1/LOX/COL3A1/GPNMB/CDH2/SULF1/GJA1/ACTA2/TGFBR2/SMOC2/CCL21/MMP2/DCLK1/AKT3/CD200/MAP1B/RND3/KDR/MCC/LEF1/SGK1/SDC2/ENPP2/PECAM1/KITLG/CCR1/ADAMTS1/ITGA6/EPS8/ADAMTS9/EFNB2/CD34/THY1/SEMA6A/LYN/ALX1/S100A8/ITGA9/MTUS1/NRP1/PLVAP/PRKD1/ETS1/LYVE1/CDH5/PLXNC1/PODXL/ITGA2/FCER1G/TLR4/TEK/NRP2/PDGFRB/LAMB1/ELMO1/EMP2/MCTP1/FLT1/DOCK2/PTPRC/XBP1/CHL1/SATB2/PLXNA2/DLC1 |
| angiogenesis | GO:0001525 | 4.39 × 10−26 | FAP/THBS1/GPNMB/SULF1/THBS2/PTPRB/TGFBR2/SMOC2/MMP2/CALCRL/CYBB/ADAM12/AKT3/ANPEP/KDR/LEF1/ENPP2/COL15A1/CEMIP2/TGFBI/APLNR/ADAMTS1/ADAMTS9/EFNB2/CD34/THY1/SEMA6A/SAT1/NRP1/PRKD1/ETS1/COL4A1/CDH5/EMCN/EPAS1/TEK/COL4A2/NRP2/PDGFRB/EMP2/FLT1/XBP1 | |
| positive regulation of cell migration | GO:0030335 | 2.52 × 10−19 | MMP9/THBS1/GPNMB/ACTA2/TGFBR2/SMOC2/CCL21/MMP2/AKT3/KDR/LEF1/ENPP2/PECAM1/KITLG/CCR1/ADAMTS1/ITGA6/THY1/SEMA6A/LYN/NRP1/PLVAP/PRKD1/ETS1/LYVE1/CDH5/PODXL/ITGA2/TLR4/TEK/NRP2/PDGFRB/LAMB1/FLT1/PTPRC/XBP1 | |
| cell differentiation | GO:0030154 | 6.24 × 10−16 | SPP1/VCAN/LPL/MMP9/CHN1/EFEMP1/COL12A1/VCAM1/LOX/COL3A1/DOCK11/CDH2/COL11A1/KRT10/SULF1/STEAP4/GJA1/A2M/ACTA2/TGFBR2/CCL21/MMP2/CD53/GPM6B/DCLK1/ADAM12/PLEK/MAP1B/ANPEP/KDR/ESRP1/LEF1/SGK1/SDC2/PECAM1/COL15A1/KITLG/CCR1/TGFBI/APLNR/ITGA6/MAP2/ADAMTS9/MTSS1/EFNB2/CD34/CD36/THY1/SEMA6A/LYN/HEY2/RAI14/ALX1/S100A8/RPS6KA2/SPRY4/NRP1/PRKD1/TMEM119/ETS1/PRICKLE1/COL4A1/CDH5/PLXNC1/PODXL/ITGA2/SLC6A6/EPAS1/FCER1G/TLR4/TEK/COL4A2/FRMD6/NRP2/PDGFRB/LAMB1/EMP2/FLT1/FARP1/DOCK2/PTPRC/XBP1/CHL1/RPS6KA3/SATB2/PLXNA2/TAGLN/BHLHE40 | |
| extracellular matrix organization | GO:0030198 | 8.07 × 10−14 | LUM/MMP9/FAP/COL12A1/LOX/COL3A1/COL11A1/SULF1/SMOC2/MMP2/GPM6B/COL14A1/MMP16/COL15A1/TGFBI/ADAMTS1/ITGA6/ADAMTS9/ITGA9/FBLN5/COL4A1/ITGA2/COL4A2/LAMB1/NID2 | |
| cell-substrate adhesion | GO:0031589 | 7.69 × 10−11 | THBS1/VCAM1/COL3A1/EDIL3/CCL21/GPM6B/VWF/KDR/ITGA6/ADAMTS9/CD34/CD36/THY1/SPRY4/NRP1/FBLN5/LYVE1/ITGA2/TEK/LAMB1/EMP2/NID2/DLC1 | |
| chemotaxis | GO:0006935 | 7.74 × 10−11 | CCL18/THBS1/CHN1/VCAM1/LOX/GPNMB/SMOC2/CCL21/KDR/LEF1/ENPP2/CCR1/EFNB2/SEMA6A/LYN/S100A8/ITGA9/MTUS1/NRP1/PRKD1/PLXNC1/ITGA2/FCER1G/NRP2/PDGFRB/FLT1/DOCK2/CHL1/PLXNA2 | |
| cell-matrix adhesion | GO:0007160 | 1.55 × 10−9 | THBS1/VCAM1/COL3A1/CCL21/GPM6B/KDR/ADAMTS9/CD34/CD36/THY1/NRP1/FBLN5/LYVE1/ITGA2/TEK/EMP2/NID2/DLC1 | |
| cell-substrate junction assembly | GO:0007044 | 0.0002 | THBS1/GPM6B/KDR/ITGA6/THY1/NRP1/ITGA2/TEK/DLC1 | |
| collagen-containing extracellular matrix | GO:0062023 | 3.43 × 10−21 | LUM/VCAN/MMP9/THBS1/EFEMP1/COL12A1/CTSC/COL3A1/CDH2/COL11A1/EDIL3/SULF1/A2M/THBS2/SMOC2/MMP2/VWF/COL14A1/SDC2/SPON1/PCOLCE/COL15A1/TGFBI/ADAMTS1/SPARCL/ADAMTS9/S100A8/BGN/HAPLN1/FBLN5/COL4A1/COL4A2/LAMB1/NID2 | |
| extracellular exosome | GO:0070062 | 6.20 × 10−10 | LUM/SPP1/MMP9/THBS1/LYZ/HBB/EFEMP1/COL12A1/CTSC/VCAM1/PPIC/EDIL3/KRT10/STEAP4/A2M/ACTA2/CD53/VWF/ANPEP/MAN1A1/PECAM1/PCOLCE/COL15A1/CEMIP2/TGFBI/EPS8/PRSS23/THY1/LYN/S100A8/CD14/BGN/PLVAP/MYO1B/ENTPD1/FBLN5/LYVE1/RFTN1/PODXL/NT5E/FCGR3A/COL4A2/LAMB1/DOCK2/RAB27B/PTPRC/MARCKS/CHL1/NID2/TNFSF10/PYGL | |
| collagen trimer | GO:0005581 | 1.90 × 10−6 | LUM/COL12A1/LOX/COL3A1/COL11A1/COL14A1/COL15A1/CD36/COL4A1/COL4A2 | |
| collagen binding | GO:0005518 | 1.53 × 10−8 | LUM/MMP9/THBS1/LOX/VWF/COL14A1/PCOLCE/TGFBI/SPARCL1/ITGA2/NID2 | |
| glycosaminoglycan binding | GO:0005539 | 3.92 × 10−8 | VCAN/LPL/THBS1/GPNMB/COL11A1/SULF1/THBS2/TGFBR2/SMOC2/PCOLCE/ADAMTS1/NRP1/BGN/HAPLN1/LYVE1/NRP2/PTPRC | |
| Degradation of the extracellular matrix | REAC:R-HSA-1474228 | 6.00 × 10−8 | SPP1/MMP9/COL12A1/COL3A1/COL11A1/A2M/MMP2/COL14A1/MMP16/COL15A1/ADAMTS1/ADAMTS9/COL4A1/COL4A2/LAMB1 | |
| Collagen formation | REAC:R-HSA-1474290 | 4.10 × 10−6 | MMP9/COL12A1/LOX/COL3A1/COL11A1/COL14A1/PCOLCE/COL15A1/ITGA6/COL4A1/COL4A2 | |
| Collagen degradation | REAC:R-HSA-1442490 | 2.99 × 10−5 | MMP9/COL12A1/COL3A1/COL11A1/MMP2/COL14A1/COL15A1/COL4A1/COL4A2 | |
| Collagen chain trimerization | REAC:R-HSA-8948216 | 0.0004 | COL12A1/COL3A1/COL11A1/COL14A1/COL15A1/COL4A1/COL4A2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yakushov, S.; Menyailo, M.; Denisov, E.; Karlina, I.; Zainullina, V.; Kirgizov, K.; Romantsova, O.; Timashev, P.; Ulasov, I. Identification of Factors Driving Doxorubicin-Resistant Ewing Tumor Cells to Survival. Cancers 2022, 14, 5498. https://doi.org/10.3390/cancers14225498
Yakushov S, Menyailo M, Denisov E, Karlina I, Zainullina V, Kirgizov K, Romantsova O, Timashev P, Ulasov I. Identification of Factors Driving Doxorubicin-Resistant Ewing Tumor Cells to Survival. Cancers. 2022; 14(22):5498. https://doi.org/10.3390/cancers14225498
Chicago/Turabian StyleYakushov, Semyon, Maxim Menyailo, Evgeny Denisov, Irina Karlina, Viktoria Zainullina, Kirill Kirgizov, Olga Romantsova, Peter Timashev, and Ilya Ulasov. 2022. "Identification of Factors Driving Doxorubicin-Resistant Ewing Tumor Cells to Survival" Cancers 14, no. 22: 5498. https://doi.org/10.3390/cancers14225498
APA StyleYakushov, S., Menyailo, M., Denisov, E., Karlina, I., Zainullina, V., Kirgizov, K., Romantsova, O., Timashev, P., & Ulasov, I. (2022). Identification of Factors Driving Doxorubicin-Resistant Ewing Tumor Cells to Survival. Cancers, 14(22), 5498. https://doi.org/10.3390/cancers14225498