
KRAS in NSCLC: State of the Art and Future Perspectives

 ,
, 
 and
and 
Abstract
Simple Summary
Abstract
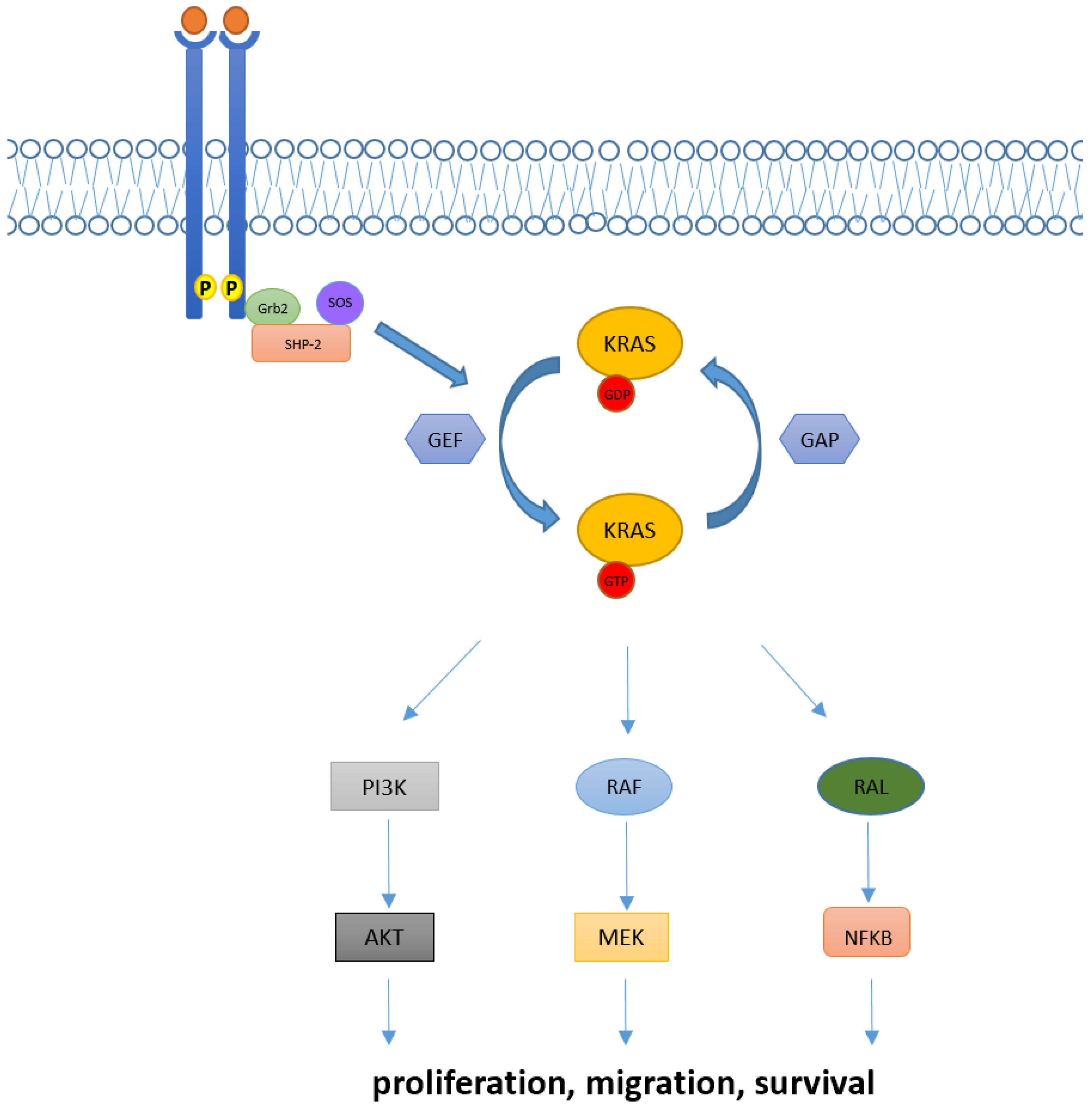
1. Introduction
1.1. Techniques for Detection of KRAS Mutations
1.2. KRAS in NSCLC: Different Alterations and Patients’ Characteristics
1.3. Predictive and Prognostic Role of KRAS Mutations in NSCLC Patients
1.4. Concurrent Molecular Alterations in KRAS Positive NSCLC Patients
2. Targeted Therapy in KRAS Mutant NSCLC
2.1. Sotorasib (AMG 510)
2.2. Adagrasib (MRTX849)

{kind=link}
{kind=link}
{kind=link}
| Drug/Dose | Phase | Population | N. of Enrolled Pts | N. of Previous Lines (Median) | Efficacy | Treatment-Related AEs |
|---|---|---|---|---|---|---|
| CodeBreak100 [61] Sotorasib 960 mg daily | Phase I/II |
| 126 | 2 | ORR: 37.1% DCR: 80.6% mPFS: 6.8 (5.1–8.2) mOS: 12.5 (10-NE) | G1-4: 69.8% G ≥ 3: 20.6% |
| KRISTAL-1 [66] Adagrasib 600 mg daily | Phase I/Ib |
| 116 | 2 | ORR: 42.9 DCR: 79.5 mPFS: 6.5 (4.7–8.4) mOS: 12.6 (9.2–19.2) | G1-4: 97.4% G ≥ 3: 44.8% |
2.3. Novel Molecules for Targeting KRAS Mutations
3. Resistance Mechanisms to KRAS Inhibitors
4. Immune Checkpoint Inhibitors (ICI) in KRAS Mutant NSCLC
5. Discussion
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ganti, A.K.; Klein, A.B.; Cotarla, I.; Seal, B.; Chou, E. Update of Incidence, Prevalence, Survival, and Initial Treatment in Patients With Non–Small Cell Lung Cancer in the US. JAMA Oncol. 2021, 7, 1824–1832. [Google Scholar] [CrossRef]
- Thai, A.A.; Solomon, B.J.; Sequist, L.V.; Gainor, J.F.; Heist, R.S. Lung Cancer. Lancet 2021, 398, 535–554. [Google Scholar] [CrossRef]
- KRAS—An Overview|ScienceDirect Topics. Available online: https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/kras (accessed on 31 July 2022).
- Plowman, S.J.; Berry, R.L.; Bader, S.A.; Luo, F.; Arends, M.J.; Harrison, D.J.; Hooper, M.L.; Patek, C.E. K-Ras 4A and 4B Are Co-Expressed Widely in Human Tissues, and Their Ratio Is Altered in Sporadic Colorectal Cancer. J. Exp. Clin. Cancer Res. 2006, 25, 259–267. [Google Scholar]
- Hancock, J.F. Ras Proteins: Different Signals from Different Locations. Nat. Rev. Mol. Cell Biol. 2003, 4, 373–385. [Google Scholar] [CrossRef]
- Westcott, P.M.K.; To, M.D. The Genetics and Biology of KRAS in Lung Cancer. Chin. J. Cancer 2013, 32, 63–70. [Google Scholar] [CrossRef]
- Pantsar, T. The Current Understanding of KRAS Protein Structure and Dynamics. Comput. Struct. Biotechnol. J. 2019, 18, 189–198. [Google Scholar] [CrossRef]
- Drugging an Undruggable Pocket on KRAS|PNAS. Available online: https://www.pnas.org/doi/10.1073/pnas.1904529116 (accessed on 22 September 2022).
- Dance, M.; Montagner, A.; Salles, J.-P.; Yart, A.; Raynal, P. The Molecular Functions of Shp2 in the Ras/Mitogen-Activated Protein Kinase (ERK1/2) Pathway. Cell. Signal. 2008, 20, 453–459. [Google Scholar] [CrossRef]
- Maertens, O.; Cichowski, K. An Expanding Role for RAS GTPase Activating Proteins (RAS GAPs) in Cancer. Adv. Biol. Regul. 2014, 55, 1–14. [Google Scholar] [CrossRef]
- Mosele, F.; Remon, J.; Mateo, J.; Westphalen, C.B.; Barlesi, F.; Lolkema, M.P.; Normanno, N.; Scarpa, A.; Robson, M.; Meric-Bernstam, F.; et al. Recommendations for the Use of Next-Generation Sequencing (NGS) for Patients with Metastatic Cancers: A Report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2020, 31, 1491–1505. [Google Scholar] [CrossRef]
- Gundry, C.N.; Vandersteen, J.G.; Reed, G.H.; Pryor, R.J.; Chen, J.; Wittwer, C.T. Amplicon Melting Analysis with Labeled Primers: A Closed-Tube Method for Differentiating Homozygotes and Heterozygotes. Clin. Chem. 2003, 49, 396–406. [Google Scholar] [CrossRef]
- Heid, C.A.; Stevens, J.; Livak, K.J.; Williams, P.M. Real Time Quantitative PCR. Genome Res. 1996, 6, 986–994. [Google Scholar] [CrossRef]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid Biopsies Come of Age: Towards Implementation of Circulating Tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef]
- Li, J.; Gan, S.; Blair, A.; Min, K.; Rehage, T.; Hoeppner, C.; Halait, H.; Brophy, V.H. A Highly Verified Assay for KRAS Mutation Detection in Tissue and Plasma of Lung, Colorectal, and Pancreatic Cancer. Arch. Pathol. Lab. Med. 2019, 143, 183–189. [Google Scholar] [CrossRef]
- Elazezy, M.; Joosse, S.A. Techniques of Using Circulating Tumor DNA as a Liquid Biopsy Component in Cancer Management. Comput. Struct. Biotechnol. J. 2018, 16, 370–378. [Google Scholar] [CrossRef]
- Gagan, J.; Van Allen, E.M. Next-Generation Sequencing to Guide Cancer Therapy. Genome Med. 2015, 7, 80. [Google Scholar] [CrossRef]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic Non-Small Cell Lung Cancer: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2018, 29, iv192–iv237. [Google Scholar] [CrossRef]
- Hagemann, I.S.; Devarakonda, S.; Lockwood, C.M.; Spencer, D.H.; Guebert, K.; Bredemeyer, A.J.; Al-Kateb, H.; Nguyen, T.T.; Duncavage, E.J.; Cottrell, C.E.; et al. Clinical Next-Generation Sequencing in Patients with Non-Small Cell Lung Cancer. Cancer 2015, 121, 631–639. [Google Scholar] [CrossRef]
- Tsoulos, N.; Papadopoulou, E.; Metaxa-Mariatou, V.; Tsaousis, G.; Efstathiadou, C.; Tounta, G.; Scapeti, A.; Bourkoula, E.; Zarogoulidis, P.; Pentheroudakis, G.; et al. Tumor Molecular Profiling of NSCLC Patients Using next Generation Sequencing. Oncol. Rep. 2017, 38, 3419–3429. [Google Scholar] [CrossRef][Green Version]
- Aggarwal, C.; Thompson, J.C.; Black, T.A.; Katz, S.I.; Fan, R.; Yee, S.S.; Chien, A.L.; Evans, T.L.; Bauml, J.M.; Alley, E.W.; et al. Clinical Implications of Plasma-Based Genotyping With the Delivery of Personalized Therapy in Metastatic Non-Small Cell Lung Cancer. JAMA Oncol. 2019, 5, 173–180. [Google Scholar] [CrossRef]
- Horn, L.; Whisenant, J.G.; Wakelee, H.; Reckamp, K.L.; Qiao, H.; Leal, T.A.; Du, L.; Hernandez, J.; Huang, V.; Blumenschein, G.R.; et al. Monitoring Therapeutic Response and Resistance: Analysis of Circulating Tumor DNA in Patients With ALK+ Lung Cancer. J. Thorac. Oncol. 2019, 14, 1901–1911. [Google Scholar] [CrossRef]
- Pascual, J.; Attard, G.; Bidard, F.-C.; Curigliano, G.; De Mattos-Arruda, L.; Diehn, M.; Italiano, A.; Lindberg, J.; Merker, J.D.; Montagut, C.; et al. ESMO Recommendations on the Use of Circulating Tumour DNA Assays for Patients with Cancer: A Report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2022, 33, 750–768. [Google Scholar] [CrossRef]
- Heitzer, E.; van den Broek, D.; Denis, M.G.; Hofman, P.; Hubank, M.; Mouliere, F.; Paz-Ares, L.; Schuuring, E.; Sültmann, H.; Vainer, G.; et al. Recommendations for a Practical Implementation of Circulating Tumor DNA Mutation Testing in Metastatic Non-Small-Cell Lung Cancer. ESMO Open 2022, 7, 100399. [Google Scholar] [CrossRef]
- IJzerman, M.J.; de Boer, J.; Azad, A.; Degeling, K.; Geoghegan, J.; Hewitt, C.; Hollande, F.; Lee, B.; To, Y.H.; Tothill, R.W.; et al. Towards Routine Implementation of Liquid Biopsies in Cancer Management: It Is Always Too Early, until Suddenly It Is Too Late. Diagnostics 2021, 11, 103. [Google Scholar] [CrossRef]
- Judd, J.; Abdel Karim, N.; Khan, H.; Naqash, A.R.; Baca, Y.; Xiu, J.; VanderWalde, A.M.; Mamdani, H.; Raez, L.E.; Nagasaka, M.; et al. Characterization of KRAS Mutation Subtypes in Non–Small Cell Lung Cancer. Mol. Cancer Ther. 2021, 20, 2577–2584. [Google Scholar] [CrossRef]
- Wood, K.; Hensing, T.; Malik, R.; Salgia, R. Prognostic and Predictive Value in KRAS in Non–Small-Cell Lung Cancer: A Review. JAMA Oncol. 2016, 2, 805–812. [Google Scholar] [CrossRef]
- Reck, M.; Carbone, D.P.; Garassino, M.; Barlesi, F. Targeting KRAS in Non-Small-Cell Lung Cancer: Recent Progress and New Approaches. Ann. Oncol. 2021, 32, 1101–1110. [Google Scholar] [CrossRef]
- Kwan, A.K.; Piazza, G.A.; Keeton, A.B.; Leite, C.A. The Path to the Clinic: A Comprehensive Review on Direct KRASG12C Inhibitors. J. Exp. Clin. Cancer Res. 2022, 41, 27. [Google Scholar] [CrossRef]
- Ihle, N.T.; Byers, L.A.; Kim, E.S.; Saintigny, P.; Lee, J.J.; Blumenschein, G.R.; Tsao, A.; Liu, S.; Larsen, J.E.; Wang, J.; et al. Effect of KRAS Oncogene Substitutions on Protein Behavior: Implications for Signaling and Clinical Outcome. J. Natl. Cancer Inst. 2012, 104, 228–239. [Google Scholar] [CrossRef]
- Désage, A.-L.; Léonce, C.; Swalduz, A.; Ortiz-Cuaran, S. Targeting KRAS Mutant in Non-Small Cell Lung Cancer: Novel Insights Into Therapeutic Strategies. Front. Oncol. 2022, 12, 796832. [Google Scholar] [CrossRef]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS Isoforms and Mutations in Cancer at a Glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef]
- Ruppert, A.-M.; Beau-Faller, M.; Debieuvre, D.; Ouafik, L.; Westeel, V.; Rouquette, I.; Mazières, J.; Bringuier, P.-P.; Monnet, I.; Escande, F.; et al. Outcomes of Patients With Advanced NSCLC From the Intergroupe Francophone de Cancérologie Thoracique Biomarkers France Study by KRAS Mutation Subtypes. JTO Clin. Res. Rep. 2020, 1, 100052. [Google Scholar] [CrossRef] [PubMed]
- Finn, S.P.; Addeo, A.; Dafni, U.; Thunnissen, E.; Bubendorf, L.; Madsen, L.B.; Biernat, W.; Verbeken, E.; Hernandez-Losa, J.; Marchetti, A.; et al. Prognostic Impact of KRAS G12C Mutation in Patients With NSCLC: Results From the European Thoracic Oncology Platform Lungscape Project. J. Thorac. Oncol. 2021, 16, 990–1002. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Zhang, E.W.; Strickland, M.R.; Mendoza, D.P.; Lipkin, L.; Lennerz, J.K.; Gainor, J.F.; Heist, R.S.; Digumarthy, S.R. Clinical and Imaging Features of Non-Small Cell Lung Cancer with G12C KRAS Mutation. Cancers 2021, 13, 3572. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Franchini, F.; Alexander, M.; Officer, A.; Wong, H.-L.; IJzerman, M.; Desai, J.; Solomon, B.J. Real World Outcomes in KRAS G12C Mutation Positive Non-Small Cell Lung Cancer. Lung Cancer 2020, 146, 310–317. [Google Scholar] [CrossRef]
- Yu, H.A.; Sima, C.S.; Shen, R.; Kass, S.; Gainor, J.; Shaw, A.; Hames, M.; Iams, W.; Aston, J.; Lovly, C.M.; et al. Prognostic Impact of KRAS Mutation Subtypes in 677 Patients with Metastatic Lung Adenocarcinomas. J. Thorac. Oncol. 2015, 10, 431–437. [Google Scholar] [CrossRef]
- Shepherd, F.A.; Lacas, B.; Le Teuff, G.; Hainaut, P.; Jänne, P.A.; Pignon, J.-P.; Le Chevalier, T.; Seymour, L.; Douillard, J.-Y.; Graziano, S.; et al. Pooled Analysis of the Prognostic and Predictive Effects of TP53 Comutation Status Combined With KRAS or EGFR Mutation in Early-Stage Resected Non-Small-Cell Lung Cancer in Four Trials of Adjuvant Chemotherapy. J. Clin. Oncol. 2017, 35, 2018–2027. [Google Scholar] [CrossRef]
- Goulding, R.E.; Chenoweth, M.; Carter, G.C.; Boye, M.E.; Sheffield, K.M.; John, W.J.; Leusch, M.S.; Muehlenbein, C.E.; Li, L.; Jen, M.-H.; et al. KRAS Mutation as a Prognostic Factor and Predictive Factor in Advanced/Metastatic Non-Small Cell Lung Cancer: A Systematic Literature Review and Meta-Analysis. Cancer Treat. Res. Commun. 2020, 24, 100200. [Google Scholar] [CrossRef]
- Rulli, E.; Marabese, M.; Torri, V.; Farina, G.; Veronese, S.; Bettini, A.; Longo, F.; Moscetti, L.; Ganzinelli, M.; Lauricella, C.; et al. Value of KRAS as Prognostic or Predictive Marker in NSCLC: Results from the TAILOR Trial. Ann. Oncol. 2015, 26, 2079–2084. [Google Scholar] [CrossRef]
- Svaton, M.; Fiala, O.; Pesek, M.; Bortlicek, Z.; Minarik, M.; Benesova, L.; Topolcan, O. The Prognostic Role of KRAS Mutation in Patients with Advanced NSCLC Treated with Second- or Third-Line Chemotherapy. Anticancer Res. 2016, 36, 1077–1082. [Google Scholar]
- Mellema, W.W.; Masen-Poos, L.; Smit, E.F.; Hendriks, L.E.L.; Aerts, J.G.; Termeer, A.; Goosens, M.J.; Smit, H.J.M.; van den Heuvel, M.M.; van der Wekken, A.J.; et al. Comparison of Clinical Outcome after First-Line Platinum-Based Chemotherapy in Different Types of KRAS Mutated Advanced Non-Small-Cell Lung Cancer. Lung Cancer 2015, 90, 249–254. [Google Scholar] [CrossRef]
- Arbour, K.C.; Jordan, E.; Kim, H.R.; Dienstag, J.; Yu, H.A.; Sanchez-Vega, F.; Lito, P.; Berger, M.; Solit, D.B.; Hellmann, M.; et al. Effects of Co-Occurring Genomic Alterations on Outcomes in Patients with KRAS-Mutant Non–Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 334–340. [Google Scholar] [CrossRef] [PubMed]
- La Fleur, L.; Falk-Sörqvist, E.; Smeds, P.; Berglund, A.; Sundström, M.; Mattsson, J.S.; Brandén, E.; Koyi, H.; Isaksson, J.; Brunnström, H.; et al. Mutation Patterns in a Population-Based Non-Small Cell Lung Cancer Cohort and Prognostic Impact of Concomitant Mutations in KRAS and TP53 or STK11. Lung Cancer 2019, 130, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Riely, G.J.; Jordan, E.; Kim, H.R.; Yu, H.A.; Berger, M.F.; Solit, D.B.; Kris, M.G.; Ni, A.; Arcila, M.E.; Ladanyi, M. Association of Outcomes and Co-Occuring Genomic Alterations in Patients with KRAS-Mutant Non-Small Cell Lung Cancer. J. Clin. Oncol. 2016, 34, 9019. [Google Scholar] [CrossRef]
- Passaro, A.; Attili, I.; Rappa, A.; Vacirca, D.; Ranghiero, A.; Fumagalli, C.; Guarize, J.; Spaggiari, L.; de Marinis, F.; Barberis, M.; et al. Genomic Characterization of Concurrent Alterations in Non-Small Cell Lung Cancer (NSCLC) Harboring Actionable Mutations. Cancers 2021, 13, 2172. [Google Scholar] [CrossRef]
- Skoulidis, F.; Byers, L.A.; Diao, L.; Papadimitrakopoulou, V.A.; Tong, P.; Izzo, J.; Behrens, C.; Kadara, H.; Parra, E.R.; Canales, J.R.; et al. Co-Occurring Genomic Alterations Define Major Subsets of KRAS-Mutant Lung Adenocarcinoma with Distinct Biology, Immune Profiles, and Therapeutic Vulnerabilities. Cancer Discov. 2015, 5, 860–877. [Google Scholar] [CrossRef]
- Mahoney, C.L.; Choudhury, B.; Davies, H.; Edkins, S.; Greenman, C.; van Haaften, G.; Mironenko, T.; Santarius, T.; Stevens, C.; Stratton, M.R.; et al. LKB1/KRAS Mutant Lung Cancers Constitute a Genetic Subset of NSCLC with Increased Sensitivity to MAPK and MTOR Signalling Inhibition. Br. J. Cancer 2009, 100, 370–375. [Google Scholar] [CrossRef]
- Schrock, A.B.; Frampton, G.M.; Suh, J.; Chalmers, Z.R.; Rosenzweig, M.; Erlich, R.L.; Halmos, B.; Goldman, J.; Forde, P.; Leuenberger, K.; et al. Characterization of 298 Patients with Lung Cancer Harboring MET Exon 14 Skipping Alterations. J. Thorac. Oncol. 2016, 11, 1493–1502. [Google Scholar] [CrossRef]
- Suzawa, K.; Offin, M.; Lu, D.; Kurzatkowski, C.; Vojnic, M.; Smith, R.S.; Sabari, J.K.; Tai, H.; Mattar, M.; Khodos, I.; et al. Activation of KRAS Mediates Resistance to Targeted Therapy in MET Exon 14-Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 1248–1260. [Google Scholar] [CrossRef]
- Zhu, Y.; Lin, X.; Li, X.; Wu, L.; Chen, H.; Wang, W.; Xu, C.; Shen, J.; Wei, J.; Du, K. Concurrent ROS1 Gene Rearrangement and KRAS Mutation in Lung Adenocarcinoma: A Case Report and Literature Review. Thorac. Cancer 2018, 9, 159–163. [Google Scholar] [CrossRef]
- Schmid, S.; Gautschi, O.; Rothschild, S.; Mark, M.; Froesch, P.; Klingbiel, D.; Reichegger, H.; Jochum, W.; Diebold, J.; Früh, M. Clinical Outcome of ALK-Positive Non–Small Cell Lung Cancer (NSCLC) Patients with De Novo EGFR or KRAS Co-Mutations Receiving Tyrosine Kinase Inhibitors (TKIs). J. Thorac. Oncol. 2017, 12, 681–688. [Google Scholar] [CrossRef]
- Chabon, J.J.; Simmons, A.D.; Lovejoy, A.F.; Esfahani, M.S.; Newman, A.M.; Haringsma, H.J.; Kurtz, D.M.; Stehr, H.; Scherer, F.; Karlovich, C.A.; et al. Circulating Tumour DNA Profiling Reveals Heterogeneity of EGFR Inhibitor Resistance Mechanisms in Lung Cancer Patients. Nat. Commun. 2016, 7, 11815. [Google Scholar] [CrossRef] [PubMed]
- Rachiglio, A.M.; Fenizia, F.; Piccirillo, M.C.; Galetta, D.; Crinò, L.; Vincenzi, B.; Barletta, E.; Pinto, C.; Ferraù, F.; Lambiase, M.; et al. The Presence of Concomitant Mutations Affects the Activity of EGFR Tyrosine Kinase Inhibitors in EGFR-Mutant Non-Small Cell Lung Cancer (NSCLC) Patients. Cancers 2019, 11, 341. [Google Scholar] [CrossRef] [PubMed]
- Xiu, W.; Zhang, Q.; Yu, M.; Huang, Y.; Huang, M. Case Report: Outcome of Osimertinib Treatment in Lung Adenocarcinoma Patients With Acquired KRAS Mutations. Front. Oncol. 2021, 11, 630256. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, T.; Ohmori, T.; Ohba, M.; Arata, S.; Murata, Y.; Kusumoto, S.; Ando, K.; Ishida, H.; Ohnishi, T.; Sasaki, Y. Distinct Afatinib Resistance Mechanisms Identified in Lung Adenocarcinoma Harboring an EGFR Mutation. Mol. Cancer Res. 2017, 15, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Ostrem, J.M.L.; Shokat, K.M. Direct Small-Molecule Inhibitors of KRAS: From Structural Insights to Mechanism-Based Design. Nat. Rev. Drug Discov. 2016, 15, 771–785. [Google Scholar] [CrossRef]
- Lito, P.; Solomon, M.; Li, L.-S.; Hansen, R.; Rosen, N. Allele-Specific Inhibitors Inactivate Mutant KRAS G12C by a Trapping Mechanism. Science 2016, 351, 604–608. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRAS G12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS G12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef]
- Ramalingam, S.; Skoulidis, F.; Govindan, R.; Velcheti, V.; Li, B.; Besse, B.; Dy, G.; Kim, D.; Schuler, M.; Vincent, M.; et al. P52.03 Efficacy of Sotorasib in KRAS p.G12C-Mutated NSCLC with Stable Brain Metastases: A Post-Hoc Analysis of CodeBreaK 100. J. Thorac. Oncol. 2021, 16, S1123. [Google Scholar] [CrossRef]
- Lumakras®/Lumykras® (Sotorasib) Demonstrates Superior Progression-Free Survival over Docetaxel in First Positive Phase 3 Trial of a Kras G12c Inhibitor in Non-Small Cell Lung Cancer. Available online: https://www.amgen.com/newsroom/press-releases/2022/09/lumakraslumykras-sotorasib-demonstrates-superior-progressionfree-survival-over-docetaxel-in-first-positive-phase-3-trial-of-a-kras-g12c-inhibitor-in-nonsmall-cell-lung-cancer (accessed on 20 September 2022).
- Nakajima, E.C.; Drezner, N.; Li, X.; Mishra-Kalyani, P.S.; Liu, Y.; Zhao, H.; Bi, Y.; Liu, J.; Rahman, A.; Wearne, E.; et al. FDA Approval Summary: Sotorasib for KRAS G12C-Mutated Metastatic NSCLC. Clin. Cancer Res. 2021, 28, 1482–1486. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.-H.I.; Jänne, P.A.; Leal, T.A.; Rybkin, I.I.; Sabari, J.K.; Barve, M.A.; Bazhenova, L.A.; Johnson, M.L.; Velastegui, K.L.; Cilliers, C.; et al. First-in-Human Phase I/IB Dose-Finding Study of Adagrasib (MRTX849) in Patients With Advanced KRASG12C Solid Tumors (KRYSTAL-1). J. Clin. Oncol. 2022, 40, 2530–2538. [Google Scholar] [CrossRef] [PubMed]
- Jänne, P.A.; Riely, G.J.; Gadgeel, S.M.; Heist, R.S.; Ou, S.-H.I.; Pacheco, J.M.; Johnson, M.L.; Sabari, J.K.; Leventakos, K.; Yau, E.; et al. Adagrasib in Non–Small-Cell Lung Cancer Harboring a KRASG12C Mutation. N. Engl. J. Med. 2022, 387, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Mirati Therapeutics Inc. A Randomized Phase 3 Study of MRTX849 Versus Docetaxel in Patients with Previously Treated Non-Small Cell Lung Cancer with KRAS G12C Mutation. 2022. Available online: https://clinicaltrials.gov/ (accessed on 3 November 2022).
- New Drug Application for Adagrasib Accepted by FDA for KRAS G12C+ NSCLC. Available online: https://www.cancernetwork.com/view/new-drug-application-for-adagrasib-accepted-by-fda-for-kras-g12c-nsclc (accessed on 21 September 2022).
- Evidence of Antitumor Effect with GDC-6036 Monotherapy in KRAS G12C+ NSCLC Revealed at 2022 WCLC. Available online: https://www.cancernetwork.com/view/evidence-of-antitumor-effect-with-gdc-6036-monotherapy-in-kras-g12c-nsclc-revealed-at-2022-wclc (accessed on 8 September 2022).
- A Study to Evaluate the Safety, Pharmacokinetics, and Activity of GDC-6036 Alone or in Combination in Participants with Advanced or Metastatic Solid Tumors with a KRAS G12C Mutation—No Study Results Posted—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/results/NCT04449874 (accessed on 23 September 2022).
- Cohen-Armon, M. Exclusive Modifications of NuMA in Malignant Epithelial Cells: A Potential Therapeutic Mechanism. Drug Discov. Today 2022, 27, 1205–1209. [Google Scholar] [CrossRef] [PubMed]
- Eli Lilly and Company. A Phase 1/2 Study of LY3499446 Administered to Patients with Advanced Solid Tumors with KRAS G12C Mutation. 2021. Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Novartis Pharmaceuticals. A Phase Ib/II Open-Label, Multi-Center Dose Escalation Study of JDQ443 in Patients with Advanced Solid Tumors Harboring the KRAS G12C Mutation. 2022. Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Jacobio Pharmaceuticals Co., Ltd. A Phase 1/2a Clinical Study to Evaluate the Safety, Tolerability, Pharmacokinetics and Antitumor Activity of JAB-21822 in Combination with JAB-3312 in Patients with Advanced Solid Tumors Harboring KRAS p.G12C Mutation. 2022. Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Boehringer Ingelheim. A Phase Ia/Ib, Open-Label, Multicentre Dose-Escalation and Expansion Study to Investigate the Safety, Pharmacokinetics and Preliminary Efficacy of BI 1823911 as a Monotherapy and in Combination with Other Anti-Cancer Therapies in Patients with Advanced or Metastatic Solid Tumours Expressing KRAS G12C Mutation. 2022. Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- InventisBio Co., Ltd. A Phase 1/2, Open Label Study to Evaluate the Safety, Tolerability, Pharmacokinetics and Efficacy of D-1553 in Combination with IN10018 in Subjects with Advanced or Metastatic Solid Tumors With KRasG12C Mutation. 2022. Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Merck Sharp & Dohme LLC. A Phase 1, Open-Label, Multicenter Study to Assess Safety, Tolerability, PK, and Efficacy of MK-1084 as Monotherapy and in Combination with Pembrolizumab in Subjects with KRASG12C Mutant Advanced Solid Tumors. 2022. Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Eli Lilly and Company. A Phase 1a/1b Study of LY3537982 in Patients with KRAS G12C-Mutant Advanced Solid Tumors. Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Nagasaka, M.; Potugari, B.; Nguyen, A.; Sukari, A.; Azmi, A.S.; Ou, S.-H.I. KRAS Inhibitors– Yes but What next? Direct Targeting of KRAS–Vaccines, Adoptive T Cell Therapy and Beyond. Cancer Treat. Rev. 2021, 101, 102309. [Google Scholar] [CrossRef]
- Koltun, E.; Cregg, J.; Rice, M.A.; Whalen, D.M.; Freilich, R.; Jiang, J.; Hansen, R.; Bermingham, A.; Knox, J.E.; Dinglasan, J.; et al. Abstract 1260: First-in-Class, Orally Bioavailable KRASG12V(ON) Tri-Complex Inhibitors, as Single Agents and in Combinations, Drive Profound Anti-Tumor Activity in Preclinical Models of KRASG12V Mutant Cancers. Cancer Res. 2021, 81, 1260. [Google Scholar] [CrossRef]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS Mutation: From Undruggable to Druggable in Cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef]
- Zhao, Y.; Murciano-Goroff, Y.R.; Xue, J.Y.; Ang, A.; Lucas, J.; Mai, T.T.; Da Cruz Paula, A.F.; Saiki, A.Y.; Mohn, D.; Achanta, P.; et al. Diverse Alterations Associated with Resistance to KRAS(G12C) Inhibition. Nature 2021, 599, 679–683. [Google Scholar] [CrossRef]
- Awad, M.M.; Liu, S.; Rybkin, I.I.; Arbour, K.C.; Dilly, J.; Zhu, V.W.; Johnson, M.L.; Heist, R.S.; Patil, T.; Riely, G.J.; et al. Acquired Resistance to KRAS G12C Inhibition in Cancer. N. Engl. J. Med. 2021, 384, 2382–2393. [Google Scholar] [CrossRef]
- Addario Lung Cancer Medical Institute. A Non-Interventional, Non-Treatment, Non-Randomized, Single Coordinating Center, Decentralized Bio-Specimen Collection Study in USA-Based Adult Subjects with Acquired Resistance to KRAS Inhibitors. Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Falchook, G.; Li, B.T.; Marrone, K.A.; Bestvina, C.M.; Langer, C.J.; Krauss, J.C.; Strickler, J.H.; Meloni, A.; Dai, T.; Varrieur, T.; et al. OA03.03 Sotorasib in Combination with RMC-4630, a SHP2 Inhibitor, in KRAS p.G12C-Mutated NSCLC and Other Solid Tumors. J. Thorac. Oncol. 2022, 17, S8. [Google Scholar] [CrossRef]
- Gandara, D.; Marrone, K.; Govindan, R.; Skoulidis, F.; Durm, G.; Clarke, J.; Frank, R.; Krauss, J.; Snyder, W.; Dai, T.; et al. Abstract P05-02: A Phase 1b Study Evaluating the Combination of Sotorasib, a KRASG12C Inhibitor, and Afatinib, a Pan-ErbB Tyrosine Kinase Inhibitor, in Advanced KRAS p.G12C Mutated Non-Small Cell Lung Cancer (NSCLC). Mol. Cancer Ther. 2021, 20, P05-02. [Google Scholar] [CrossRef]
- Chen, N.; Fang, W.; Lin, Z.; Peng, P.; Wang, J.; Zhan, J.; Hong, S.; Huang, J.; Liu, L.; Sheng, J.; et al. KRAS Mutation-Induced Upregulation of PD-L1 Mediates Immune Escape in Human Lung Adenocarcinoma. Cancer Immunol. Immunother. 2017, 66, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- D’Incecco, A.; Andreozzi, M.; Ludovini, V.; Rossi, E.; Capodanno, A.; Landi, L.; Tibaldi, C.; Minuti, G.; Salvini, J.; Coppi, E.; et al. PD-1 and PD-L1 Expression in Molecularly Selected Non-Small-Cell Lung Cancer Patients. Br. J. Cancer 2015, 112, 95–102. [Google Scholar] [CrossRef]
- Liu, C.; Zheng, S.; Jin, R.; Wang, X.; Wang, F.; Zang, R.; Xu, H.; Lu, Z.; Huang, J.; Lei, Y.; et al. The Superior Efficacy of Anti-PD-1/PD-L1 Immunotherapy in KRAS-Mutant Non-Small Cell Lung Cancer That Correlates with an Inflammatory Phenotype and Increased Immunogenicity. Cancer Lett. 2020, 470, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.E.; El-Refai, S.M.; Sha, W.; Puccini, A.; Grothey, A.; George, T.J.; Hwang, J.J.; O’Neil, B.; Barrett, A.S.; Kadakia, K.C.; et al. Landscape of KRASG12C, Associated Genomic Alterations, and Interrelation With Immuno-Oncology Biomarkers in KRAS-Mutated Cancers. JCO Precis. Oncol. 2022, 6, e2100245. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Li, G.; Wang, Y.; Wang, Y.; Zhao, S.; Haihong, P.; Zhao, H.; Wang, Y. PD-L1 Expression in Lung Cancer and Its Correlation with Driver Mutations: A Meta-Analysis. Sci. Rep. 2017, 7, 10255. [Google Scholar] [CrossRef]
- Petrelli, F.; Maltese, M.; Tomasello, G.; Conti, B.; Borgonovo, K.; Cabiddu, M.; Ghilardi, M.; Ghidini, M.; Passalacqua, R.; Barni, S.; et al. Clinical and Molecular Predictors of PD-L1 Expression in Non–Small-Cell Lung Cancer: Systematic Review and Meta-Analysis. Clin. Lung Cancer 2018, 19, 315–322. [Google Scholar] [CrossRef]
- Herbst, R.S.; Lopes, G.; Kowalski, D.M.; Kasahara, K.; Wu, Y.-L.; De Castro, G.; Cho, B.C.; Turna, H.Z.; Cristescu, R.; Aurora-Garg, D.; et al. LBA4 Association of KRAS Mutational Status with Response to Pembrolizumab Monotherapy given as First-Line Therapy for PD-L1-Positive Advanced Non-Squamous NSCLC in Keynote-042. Ann. Oncol. 2019, 30, xi63–xi64. [Google Scholar] [CrossRef]
- Sun, L.; Hsu, M.; Cohen, R.B.; Langer, C.J.; Mamtani, R.; Aggarwal, C. Association Between KRAS Variant Status and Outcomes With First-Line Immune Checkpoint Inhibitor–Based Therapy in Patients With Advanced Non–Small-Cell Lung Cancer. JAMA Oncol. 2021, 7, 937–939. [Google Scholar] [CrossRef]
- Jeanson, A.; Tomasini, P.; Souquet-Bressand, M.; Brandone, N.; Boucekine, M.; Grangeon, M.; Chaleat, S.; Khobta, N.; Milia, J.; Mhanna, L.; et al. Efficacy of Immune Checkpoint Inhibitors in KRAS-Mutant Non-Small Cell Lung Cancer (NSCLC). J. Thorac. Oncol. 2019, 14, 1095–1101. [Google Scholar] [CrossRef]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus Docetaxel in Patients with Previously Treated Non-Small-Cell Lung Cancer (OAK): A Phase 3, Open-Label, Multicentre Randomised Controlled Trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef]
- Lee, C.K.; Man, J.; Lord, S.; Cooper, W.; Links, M.; Gebski, V.; Herbst, R.S.; Gralla, R.J.; Mok, T.; Yang, J.C.-H. Clinical and Molecular Characteristics Associated With Survival Among Patients Treated With Checkpoint Inhibitors for Advanced Non–Small Cell Lung Carcinoma: A Systematic Review and Meta-Analysis. JAMA Oncol. 2018, 4, 210. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, H.S.; Kim, B.J. Prognostic Value of KRAS Mutation in Advanced Non-Small-Cell Lung Cancer Treated with Immune Checkpoint Inhibitors: A Meta-Analysis and Review. Oncotarget 2017, 8, 48248–48252. [Google Scholar] [CrossRef] [PubMed]
- Landre, T.; Justeau, G.; Assié, J.-B.; Chouahnia, K.; Davoine, C.; Taleb, C.; Chouaïd, C.; Duchemann, B. Anti-PD-(L)1 for KRAS-Mutant Advanced Non-Small-Cell Lung Cancers: A Meta-Analysis of Randomized-Controlled Trials. Cancer Immunol. Immunother. 2022, 71, 719–726. [Google Scholar] [CrossRef]
- Guaitoli, G.; Tiseo, M.; Di Maio, M.; Friboulet, L.; Facchinetti, F. Immune Checkpoint Inhibitors in Oncogene-Addicted Non-Small Cell Lung Cancer: A Systematic Review and Meta-Analysis. Transl. Lung Cancer Res. 2021, 10, 2890–2916. [Google Scholar] [CrossRef]
- Mazieres, J.; Drilon, A.; Lusque, A.; Mhanna, L.; Cortot, A.B.; Mezquita, L.; Thai, A.A.; Mascaux, C.; Couraud, S.; Veillon, R.; et al. Immune Checkpoint Inhibitors for Patients with Advanced Lung Cancer and Oncogenic Driver Alterations: Results from the IMMUNOTARGET Registry. Ann. Oncol. 2019, 30, 1321–1328. [Google Scholar] [CrossRef]
- Mirati Therapeutics Inc. A Phase 1/2 Multiple Expansion Cohort Trial of MRTX849 in Patients with Advanced Solid Tumors with KRAS G12C Mutation KRYSTAL-1. Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Mirati Therapeutics Inc. A Phase 2 Trial of MRTX849 Monotherapy and in Combination with Pembrolizumab in Patients with Advanced Non-Small Cell Lung Cancer with KRAS G12C Mutation. Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Li, B.T.; Falchook, G.S.; Durm, G.A.; Burns, T.F.; Skoulidis, F.; Ramalingam, S.S.; Spira, A.; Bestvina, C.M.; Goldberg, S.B.; Veluswamy, R.; et al. OA03.06 CodeBreaK 100/101: First Report of Safety/Efficacy of Sotorasib in Combination with Pembrolizumab or Atezolizumab in Advanced KRAS p.G12C NSCLC. J. Thorac. Oncol. 2022, 17, S10–S11. [Google Scholar] [CrossRef]
- Liu, C.; Zheng, S.; Wang, Z.; Wang, S.; Wang, X.; Yang, L.; Xu, H.; Cao, Z.; Feng, X.; Xue, Q.; et al. KRAS-G12D Mutation Drives Immune Suppression and the Primary Resistance of Anti-PD-1/PD-L1 Immunotherapy in Non-Small Cell Lung Cancer. Cancer Commun. 2022, 42, 828–847. [Google Scholar] [CrossRef]
- Ricciuti, B.; Alessi, J.V.; Elkrief, A.; Wang, X.; Cortellini, A.; Li, Y.Y.; Vaz, V.R.; Gupta, H.; Pecci, F.; Barrichello, A.; et al. Dissecting the Clinicopathologic, Genomic, and Immunophenotypic Correlates of KRASG12D-Mutated Non-Small-Cell Lung Cancer. Ann. Oncol. 2022, 33, 1029–1040. [Google Scholar] [CrossRef]
- Arbour, K.C.; Rizvi, H.; Plodkowski, A.J.; Hellmann, M.D.; Knezevic, A.; Heller, G.; Yu, H.A.; Ladanyi, M.; Kris, M.G.; Arcila, M.E.; et al. Treatment Outcomes and Clinical Characteristics of Patients with KRAS-G12C-Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. 2021, 27, 2209–2215. [Google Scholar] [CrossRef]
- Dong, Z.-Y.; Zhong, W.-Z.; Zhang, X.-C.; Su, J.; Xie, Z.; Liu, S.-Y.; Tu, H.-Y.; Chen, H.-J.; Sun, Y.-L.; Zhou, Q.; et al. Potential Predictive Value of TP53 and KRAS Mutation Status for Response to PD-1 Blockade Immunotherapy in Lung Adenocarcinoma. Clin. Cancer Res. 2017, 23, 3012–3024. [Google Scholar] [CrossRef] [PubMed]
- Kadara, H.; Choi, M.; Zhang, J.; Parra, E.R.; Rodriguez-Canales, J.; Gaffney, S.G.; Zhao, Z.; Behrens, C.; Fujimoto, J.; Chow, C.; et al. Whole-Exome Sequencing and Immune Profiling of Early-Stage Lung Adenocarcinoma with Fully Annotated Clinical Follow-Up. Ann. Oncol. 2017, 28, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Xu, B.; Zhang, H.; Fang, S. Lung Adenocarcinoma Patients with KEAP1 Mutation Harboring Low Immune Cell Infiltration and Low Activity of Immune Environment. Thorac. Cancer 2021, 12, 2458–2467. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [PubMed]
- Ricciuti, B.; Arbour, K.C.; Lin, J.J.; Vajdi, A.; Vokes, N.; Hong, L.; Zhang, J.; Tolstorukov, M.Y.; Li, Y.Y.; Spurr, L.F.; et al. Diminished Efficacy of Programmed Death-(Ligand)1 Inhibition in STK11- and KEAP1-Mutant Lung Adenocarcinoma Is Affected by KRAS Mutation Status. J. Thorac. Oncol. 2022, 17, 399–410. [Google Scholar] [CrossRef]
- Biton, J.; Mansuet-Lupo, A.; Pécuchet, N.; Alifano, M.; Ouakrim, H.; Arrondeau, J.; Boudou-Rouquette, P.; Goldwasser, F.; Leroy, K.; Goc, J.; et al. TP53, STK11, and EGFR Mutations Predict Tumor Immune Profile and the Response to Anti–PD-1 in Lung Adenocarcinoma. Clin. Cancer Res. 2018, 24, 5710–5723. [Google Scholar] [CrossRef]
- Assoun, S.; Theou-Anton, N.; Nguenang, M.; Cazes, A.; Danel, C.; Abbar, B.; Pluvy, J.; Gounant, V.; Khalil, A.; Namour, C.; et al. Association of TP53 Mutations with Response and Longer Survival under Immune Checkpoint Inhibitors in Advanced Non-Small-Cell Lung Cancer. Lung Cancer 2019, 132, 65–71. [Google Scholar] [CrossRef]
- Sotorasib Improves PFS in KRAS G12C-Mutated NSCLC. Available online: https://dailyreporter.esmo.org/esmo-congress-2022/top/sotorasib-improves-pfs-versus-docetaxel-in-patients-with-pre-treated-kras-g12c-mutated-nsclc (accessed on 16 September 2022).
- Amgen. A Phase 1b/2, Protocol Evaluating the Safety, Tolerability, Pharmacokinetics, and Efficacy of Sotorasib Monotherapy and in Combination with Other Anti-Cancer Therapies in Subjects with Advanced Solid Tumors with KRAS p.G12C Mutation (CodeBreak 101). Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Mirati Therapeutics Inc. A Phase 1/2 Trial of MRTX849 in Combination with TNO155 in Patients with Advanced Solid Tumors with KRAS G12C Mutation KRYSTAL 2. Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Verastem, Inc. A Phase 1/2 Study of VS-6766 in Combination with Sotorasib in Patients with KRAS G12C Mutant Non-Small Cell Lung Cancer (NSCLC). Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Mirati Therapeutics Inc. A Phase 1/1b Trial of MRTX849 in Combination with BI 1701963 in Patients with Advanced Solid Tumors with KRAS G12C Mutation. Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Ruess, D.A.; Heynen, G.J.; Ciecielski, K.J.; Ai, J.; Berninger, A.; Kabacaoglu, D.; Görgülü, K.; Dantes, Z.; Wörmann, S.M.; Diakopoulos, K.N.; et al. Mutant KRAS-Driven Cancers Depend on PTPN11/SHP2 Phosphatase. Nat. Med. 2018, 24, 954–960. [Google Scholar] [CrossRef]
- Mainardi, S.; Mulero-Sánchez, A.; Prahallad, A.; Germano, G.; Bosma, A.; Krimpenfort, P.; Lieftink, C.; Steinberg, J.D.; de Wit, N.; Gonçalves-Ribeiro, S.; et al. SHP2 Is Required for Growth of KRAS-Mutant Non-Small-Cell Lung Cancer in Vivo. Nat. Med. 2018, 24, 961–967. [Google Scholar] [CrossRef]
- Revolution Medicines, Inc. A Phase 2, Open-Label, Multicenter Study of the Combination of RMC-4630 and Sotorasib for Non-Small Cell Lung Cancer Subjects with KRASG12C Mutation after Failure of Prior Standard Therapies. Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Moll, H.P.; Pranz, K.; Musteanu, M.; Grabner, B.; Hruschka, N.; Mohrherr, J.; Aigner, P.; Stiedl, P.; Brcic, L.; Laszlo, V.; et al. Afatinib Restrains K-RAS Driven Lung Tumorigenesis. Sci. Transl. Med. 2018, 10, eaao2301. [Google Scholar] [CrossRef]
- Li, S.; Liu, S.; Deng, J.; Akbay, E.A.; Hai, J.; Ambrogio, C.; Zhang, L.; Zhou, F.; Jenkins, R.W.; Adeegbe, D.O.; et al. Assessing Therapeutic Efficacy of MEK Inhibition in a KRASG12C-Driven Mouse Model of Lung Cancer. Clin. Cancer Res. 2018, 24, 4854–4864. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Shcherba, M.; Pendurti, G.; Liang, Y.; Piperdi, B.; Perez-Soler, R. Targeting the PI3K/AKT/MTOR Pathway: Potential for Lung Cancer Treatment. Lung Cancer Manag. 2014, 3, 67–75. [Google Scholar] [CrossRef]
- Coma, S.; Chowdhury, S.; Pachter, J.A. Abstract 1263: Dual RAF/MEK Inhibitor VS-6766 Enhances Antitumor Efficacy of KRAS-G12C Inhibitors through a Vertical Pathway Inhibition Strategy. Cancer Res. 2021, 81, 1263. [Google Scholar] [CrossRef]
- Cáceres-Gutiérrez, R.E.; Alfaro-Mora, Y.; Andonegui, M.A.; Díaz-Chávez, J.; Herrera, L.A. The Influence of Oncogenic RAS on Chemotherapy and Radiotherapy Resistance Through DNA Repair Pathways. Front. Cell. Dev. Biol. 2022, 10, 751367. [Google Scholar] [CrossRef] [PubMed]
- Southwest Oncology Group. A Phase II Study of AMG 510 in Participants with Previously Treated Stage IV or Recurrent KRAS G12C Mutated Non-Squamous Non-Small Cell Lung Cancer (ECOG-ACRIN LUNG-MAP SUB-STUDY). Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Fundación GECP. Phase II Clinical Trial of AMG510 (Sotorasib) in Stage III Unresectable NSCLC KRAS p.G12C Patients and Medically Ineligible for Concurrent Chemo-Radiotherapy. Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Fox Chase Cancer Center. Neoadjuvant Sotorasib in KRAS G12C Mutated, Resectable, Stage Ib-IIIA Non-Small Cell Lung Cancer (NSCLC). Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- M.D. Anderson Cancer Center. A Phase II Study of Neoadjuvant Sotorasib in Combination with Cisplatin or Carboplatin and Pemetrexed for Surgically Resectable Stage IIA-IIIB Non-Squamous Non-Small Cell Lung Cancer with a KRAS p.G12C Mutation. Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins. Phase 2 Trial of Neoadjuvant KRAS G12C Directed Therapy with Adagrasib (MRTX849) with or without Nivolumab in Resectable Non-Small Cell Lung Cancer (Neo-KAN). Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Amgen. A Phase 2, Multicenter, Open-Label Study of Sotorasib (AMG 510) in Subjects with Stage IV NSCLC Whose Tumors Harbor a KRAS G12C Mutation in Need of First-Line Treatment (CodeBreaK 201). Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Ceddia, S.; Landi, L.; Cappuzzo, F. KRAS-Mutant Non-Small-Cell Lung Cancer: From Past Efforts to Future Challenges. Int. J. Mol. Sci. 2022, 23, 9391. [Google Scholar] [CrossRef] [PubMed]
- Adderley, H.; Blackhall, F.H.; Lindsay, C.R. KRAS-Mutant Non-Small Cell Lung Cancer: Converging Small Molecules and Immune Checkpoint Inhibition. EBioMedicine 2019, 41, 711–716. [Google Scholar] [CrossRef]
- Addeo, A.; Banna, G.L.; Friedlaender, A. KRAS G12C Mutations in NSCLC: From Target to Resistance. Cancers 2021, 13, 2541. [Google Scholar] [CrossRef]
- Xie, M.; Xu, X.; Fan, Y. KRAS-Mutant Non-Small Cell Lung Cancer: An Emerging Promisingly Treatable Subgroup. Front. Oncol. 2021, 11, 672612. [Google Scholar] [CrossRef]



| Trial | Phase | Setting | Stage | Pts | Treatment | Primary Endpoints |
|---|---|---|---|---|---|---|
| NCT04449874 [70] | Phase I | Naive or pretreated | Stage IV | 498 | GDC-6036 (KRAS G12C inhibitor) +/− other molecules:
| Safety |
| NCT04699188 [73] | Phase Ib/II | Pretreated | Unresectable/Metastatic | 425 | JDQ-443 (KRAS G12C inhibitor) + other molecules:
| Dose esclataion: DLT, safety and tolerability, dose intensity by treatement Dose expansion: ORR |
| NCT04956640 [78] | Phase Ia/Ib | Pretreated (also after G12C inhibitors) | Locally advanced, unresectable, metastatic | 360 | LY3537982 (KRAS G12C inhibitor) + other molecules:
| Phase 1a: RP2D, DLT Phase 1b: safety and tolerability |
| NCT04973163 [75] | Phase Ia/Ib | Pretreated | Locally advanced, metastatic | 72 | BI 1823911 (KRAS G12C inhibitor)+/− BI 1701963 (SOS1 inhibitor) | Dose escalation: DLT Dose confirmation and expansion: OR (BOR of confirmed CR or PR) |
| NCT05288205 [74] | Phase I/IIa | Preatreated | Locally advanced, metastatic | 124 | JAB-21822 (KRAS G12C inhibitor) + JAB-3312 (SHP2 Inhibitor) | RP2D, MTD, safety |
| NCT05379946 [76] | Phase I/II | Preatreated | Locally advanced, unresectable, metastatic | 92 | D-1553 (KRAS G12C inhibitor) + IN10018 (FAK inhibitor) | Safety, ORR |
| NCT05067283 [77] | Phase I | Preatreated | Unresectable, metastatic | 264 | MK-1084 (KRAS G12C inhibitor) +/− pembrolizumab | DLT, safety |
| Trial | Phase | Setting | Stage | Pts | Treatment | Primary Endpoints |
|---|---|---|---|---|---|---|
| NCT04185883 [116] (CodeBreak 101) | Phase Ib/II | Pretreated, naïve from KRAS G12C ihibitors | Stage IV | 1054 | Sotorasib +/− other molecules
| Phase Ib: Cmax, Tmax, AUC, ORR, DCR, DoR, PFS, Duration of Stable Disease, TTR, OS Phase II: safety, Cmax, Tmax, AUC, DCR, DoR, TTR, OS |
| NCT03785249 [102] (KRYSTAL-1) | Phase I/II | Naive or pretreated | Stage IV | 740 | Adagrasib +/− other molecules:
| Safety, blood plasma concetration, ORR |
| NCT04613596 [103] (KRYSTAL-7) | Phase II | Treatment naïve | Unresectable/metastatic | 250 | Adagrasib +/− pembrolizumab | ORR |
| NCT04330664 [117] (KRYSTAL-2) | Phase I/II | Naïve or pretreated | Unresectable/metastatic | 86 | Adagrasib + TNO155 (SHP2 inhibitor) | Safety, blood plasma concentration |
| NCT05074810 [118] | Phase I/II | Pretreated | Stage IIIB-IV | 53 | Sotorasib + VS-6766 (RAF/MEK inhibitor) | PartA: RP2D, DLTs Part B: ORR |
| NCT04975256 [119] | Phase I/Ib | Naïve or pretreated | Unresectable/metastatic | 100 | Adagrasib + BI 1701963 (SOS1 inhibitor) | Safety, blood plasma concentration, DLT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cascetta, P.; Marinello, A.; Lazzari, C.; Gregorc, V.; Planchard, D.; Bianco, R.; Normanno, N.; Morabito, A. KRAS in NSCLC: State of the Art and Future Perspectives. Cancers 2022, 14, 5430. https://doi.org/10.3390/cancers14215430
Cascetta P, Marinello A, Lazzari C, Gregorc V, Planchard D, Bianco R, Normanno N, Morabito A. KRAS in NSCLC: State of the Art and Future Perspectives. Cancers. 2022; 14(21):5430. https://doi.org/10.3390/cancers14215430
Chicago/Turabian StyleCascetta, Priscilla, Arianna Marinello, Chiara Lazzari, Vanesa Gregorc, David Planchard, Roberto Bianco, Nicola Normanno, and Alessandro Morabito. 2022. "KRAS in NSCLC: State of the Art and Future Perspectives" Cancers 14, no. 21: 5430. https://doi.org/10.3390/cancers14215430
APA StyleCascetta, P., Marinello, A., Lazzari, C., Gregorc, V., Planchard, D., Bianco, R., Normanno, N., & Morabito, A. (2022). KRAS in NSCLC: State of the Art and Future Perspectives. Cancers, 14(21), 5430. https://doi.org/10.3390/cancers14215430