Simple Summary
Rat sarcoma virus (RAS) GTP-ase proteins represent a key element in cellular proliferation, growth, and differentiation. Three different isoforms of RAS proteins have been identified to date (KRAS, NRAS, HRAS) and mutations in KRAS are frequently found in human cancers, among which the Non-Small Cell Lung Cancer (NSCLC). In this review we assess molecular, prognostic, and clinico-pathological characteristics of KRAS mutations in NSCLC patients. Next, we present current therapeutic strategies for KRAS mutant NSCLC patients and mechanisms of acquired resistance identified to date. We then focus on the role of immune-checkpoint inhibitors in KRAS mutant NSCLC patients. Finally, we will overview ongoing trials and future needs for this subpopulation cohort.
Abstract
In NSCLC, KRAS mutations occur in up to 30% of all cases, most frequently at codon 12 and 13. KRAS mutations have been linked to adenocarcinoma histology, positive smoking history, and Caucasian ethnicity, although differences have been described across KRAS mutational variants subtypes. KRAS mutations often concur with other molecular alterations, notably TP53, STK11, and KEAP1, which could play an important role in treatment efficacy and patient outcomes. For many years, KRAS mutations have been considered undruggable mainly due to a high toxicity profile and low specificity of compounds. Sotorasib and adagrasib are novel KRAS inhibitors that recently gained FDA approval for pre-treated KRAS mutant NSCLC patients, and other molecules such as GDC-6036 are currently being investigated with promising results. Despite their approval, the efficacy of these drugs is lower than expected and progression among responders has been reported. Mechanisms of acquired resistance to anti-KRAS molecules typically involves either on target secondary mutations (e.g., G12, G13, Q61H, R68S, H95, Y96C, V8L) or off-target alterations. Ongoing trials are currently evaluating strategies for implementing efficacy and overcoming acquired resistance to these compounds. Finally, the efficacy of immune-checkpoint inhibitors still needs to be completely assessed and responses to anti-PD-1/PD-L1 agents may strongly depend on concomitant mutations.
Keywords:
KRAS; NSCLC; lung cancer; adagrasib; MRTX849; sotorasib; AMG 510; immune-checkpoint inhibitors; acquired resistance 1. Introduction
With almost 1.8 million deaths per year, lung cancer remains the leading cause of cancer-related death worldwide. Non-small cell lung cancer (NSCLC) accounts for 85% of all diagnosed cases, with adenocarcinoma being the most common histological subtype [1,2]. Progress in molecular characterization and development of new targeted therapies have changed the treatment scenario of NSCLC in recent years. So far, molecular alterations thought to be undruggable for many years, such as KRAS mutations, may now benefit from targeted agents.
KRAS (Kirsten Rat sarcoma virus) proteins are small GTP-ases belonging to the wider family of RAS proteins. The KRAS gene is located in the short arm of chromosome 12 (12 p) and its transcripts may further undergo an alternative splicing process, resulting in two distinct proteins of approximately 21 KDa each: KRAS 4A and KRAS 4B [3]. Even though differences in physiological expression have been reported in some preclinical data [4], little is known about the real impact of these alternatively spliced variants in NSCLC.
Structurally, all RAS isoforms have a highly conserved catalytic region, implied in GTP bounding and hydrolysis. Differences across RAS isoforms are mainly founded at the C-terminal hyper variable region (HVR) domain, responsible for post-transcriptional modifications and interaction with the phospholipidic membrane bilayer [5,6].
In physiological conditions, KRAS proteins are key elements in transducing signaling from activated receptor tyrosine kinase (RTK) to downstream intracellular pathways. Indeed, intracellular KRAS proteins may present in two distinct functional forms: KRAS-GTP and KRAS-GDP, which correspond to “active” and “inactive” states, respectively. Transition from the inactive state (KRAS-GDP) to active state (KRAS-GTP) is usually mediated by the guanine nucleotide exchange factors (GEFs) via the releasing of guanosine diphosphate (GDP) and the subsequent binding of guanosine triphosphate (GTP). Many GEFs proteins have been identified to date, with the GEF Son of Sevenless (SOS1) playing a crucial role in KRAS activation. Of note, this process determines key structural modification in KRAS proteins mainly involving the switch pockets I and II, which serve as the binding interface for effector proteins [7,8]. However, the transition from the inactive to active form is complex and usually requires other proteins (e.g., the adaptor protein Grb2 and the SOS1/Grb2 recruiter SHP-2) which directly interact with upstream phosphorylated RTKs [9]. Contrarily, transition from the KRAS-GTP to KRAS-GDP binding state is facilitated by GTPase activating proteins (GAPs) such as NF1, which accelerate the hydrolysis of GTP to GDP [10].
Downstream pathways of KRAS normally include the RAF-MEK-ERK mitogen-activated protein kinase (MAPK) pathway, the phosphatidylinositol 3-kinase (PI3K)-AKT-mTOR, and the RAS-like (RAL) pathways [5], whose deregulations have been linked with abnormal cellular proliferation, migration, and death (Figure 1).
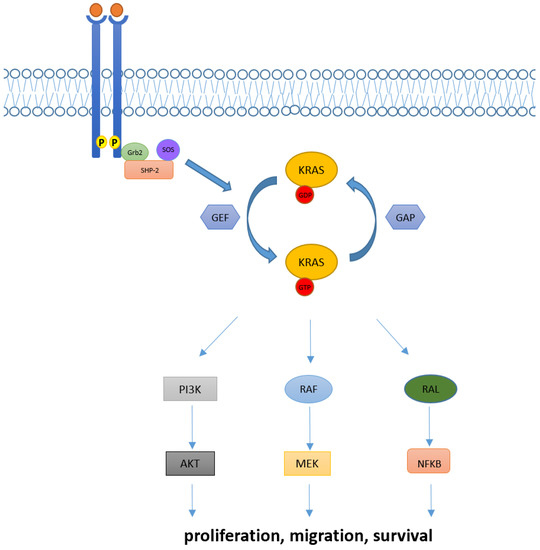
Figure 1.
KRAS activation and its downstream pathway. KRAS proteins are key elements in transducing extracellular signaling to downstream pathways. Intracellular KRAS proteins may present in two functional forms, KRAS-GTP and KRAS-GDP, which correspond to their active and inactive state, respectively. Transition from the KRAS-GDP to KRAS-GTP binding state is usually mediated by GEFs proteins and occurs via other adaptor and recruiter proteins, such as Grb2 and SHP-2. Conversely, transition from KRAS-GDP to KRAS-GTP usually requires GAPs proteins. Downstream signaling of KRAS normally includes RAF-MEK-ERK, PI3K/AKT and RAL pathways, implied in the regulation of cellular proliferation, migration, and death.
The current review provides an overview of the biology and clinic-pathological characteristics of KRAS mutant NSCLC patients. Moreover, we discuss therapeutic strategies that have been explored in these subgroups of patients and the clinical data leading to the approval of sotorasib and adagrasib, the first compounds that efficiently inhibit KRAS G12C mutation. We then focus on acquired resistance mechanisms evidenced to date and we further assess the role of immune checkpoint inhibitors in KRAS mutant patients. Finally, we look into ongoing trials and new KRAS inhibitors, which are currently being tested.
1.1. Techniques for Detection of KRAS Mutations
The assessment of the KRAS status in patients affected by colorectal cancer and NSCLC is mandatory for a correct therapeutical management [11]. The main techniques allowing KRAS point mutations are polymerase chain reaction (PCR) based methods, such as high-resolution melting and real time RCP (RT.-PCR), and next generation sequencing (NGS) [12,13,14]. RT-PCR allows the highly sensitive qualitative detection and identification of a fixed number of mutations in specific areas of the gene [13]. Highly verified assays, such as Cobas Mutation Test V2, reaches a 100% level of accuracy in tissue samples, analyzing exons 2, 3, and 4 of the KRAS gene [15]. NGS technologies, being DNA-based or RNA-based, enable the simultaneous analysis of a high variety of genes and loci, at the price of reducing sensitivity, when compared to PCR [16]. With the advent of new molecular targets, an exhaustive characterization of lung cancer patients has become essential, making NGS an extremely valuable tool [17,18,19,20].
The sensitivity of KRAS mutation detection has been evaluated and compared in various types of specimens, such as tissue, plasma, and other body fluids, finding a high rate of concordance between tissue and liquid biopsy both for PCR and NGS [21,22]. The use of liquid biopsy is of great relevance for the management of advanced NSCLC, enabling the identification of KRAS mutations at the baseline, but also to picture disease heterogeneity, to evaluate response to treatment in time and to identify molecular mechanisms of resistance to targeted molecules [22,23,24]. Overall, tissue-based testing remains the preferred test for most cancer patients, although liquid biopsy may be chosen when faster results will be clinically important, or when obtaining tissue specimens is not possible or inappropriate [23] However, it is important to point out that reimbursement for broad coverage is still limited [25].
1.2. KRAS in NSCLC: Different Alterations and Patients’ Characteristics
In NSCLC, KRAS mutations are founded in up to 30% of all cases and most frequently involve the codon 12 (90% of cases), while less common mutations are observed in codon 13 (2–6%) and 61 (1%) [26,27]. Within codon 12 mutations, the one resulting in the substitution of glycine with cysteine (KRAS G12C) represents the most prevalent KRAS alteration in NSCLC (40% of all KRAS mutant cases), followed by substitution of glycine with valine (KRAS G12V, 21%), and substitution of glycine with aspartic acid (KRAS G12D, 17%). Indeed, other point mutation in codon 12 such as G12A/R/S are rare [26,27,28]. Functionally, mutations at codon 12 and 13 result in reduced GAP-mediated hydrolysis via allosteric bloc that hinders the GTP-ase site of KRAS, whereas alterations in codon 61 abolish both intrinsic and GAP-mediated GTP hydrolysis of KRAS [29]. Of note, KRAS mutant variants can differently activate their downstream pathway. Indeed, the hydrophobic G12C and G12V preferentially activate the RAL pathway, while the hydrophilic G12D mainly acts via PI3K-AKT signaling. KRAS G12C weakens the interaction with PI3K, and the bulky aspartic acid in the G12D prevents the formation of the binding with RelA/RelB [30]. Besides NSCLC, KRAS mutations are also frequent in other tumor types, such as pancreatic and colorectal adenocarcinoma (88% and 50% of all cases, respectively) [28,31], with KRAS G12D mutations being the most common alteration among these histotypes (32% and 46% of all cases, respectively) [32].
In lung cancer patients, KRAS mutations have been associated with Caucasian ethnicity (Caucasian vs. Asian population: 26% vs. 11%), female sex (female vs. males: 31.35 vs. 23.7%, p < 0.0001), adenocarcinoma histology (adenocarcinoma vs. squamous cell carcinoma: 37.2% vs. 4.4%), and a positive smoking history (smokers vs. non-smokers: 30% vs. 11%) [26,28]. However, these characteristics may strongly depend on single mutant KRAS variants. As such, KRAS G12C/G12A/G12V/G13C mutations are more frequently found in current or former smokers, while prevalence of KRAS G12D/G12S/G13D mutations is higher among non-smokers [26,33]. Furthermore, the median age for KRAS G12C patients seems to be significantly lower than that observed for other KRAS mutants (KRAS G12C vs. other KRAS-MUT: 63.1 vs. 65.9 years, p = 0.0092) [34], although this evidence has not been further confirmed in other reports [26]. Finally, the metastatic pattern could also slightly differ across single KRAS mutant variants, since KRAS G12C mutants are more likely associated with lung metastasis (KRAS G12C vs. non–G12C: 38% vs. 21%; p = 0.043) and less associated with either pleural metastasis or lymphangitic carcinomatosis (KRAS G12C vs. non–G12C: 4% vs. 39%, p = 0.0001) [35]. Of note, KRAS mutant patients do not have a higher brain tropism than those without KRAS alterations, and the same incidence of brain metastasis has been observed across different KRAS mutant variants (KRAS mut vs. KRAS wild-type: 33% vs. 40%, p = 0.17; KRAS G12C vs. KRAS other: 40% vs. 41%, p = 0.74) [36].
1.3. Predictive and Prognostic Role of KRAS Mutations in NSCLC Patients
Multiple studies have evaluated the predictive and prognostic role of KRAS mutations in NSCLC.
In an early setting, different reports have demonstrated similar overall survival (OS) and disease-free survival (DFS) in KRAS mutants versus wild type patients (OS HR = 1.17, DFS HR = 1.04) [37,38]. However, these results were not further confirmed and other reports eventually demonstrated a worse OS and DFS for KRAS mutant patients (DFS p = 0.038, OS HR = 1.30; p = 0.002) [37].
It has been asked if the prognostic and predictive significance of KRAS mutation could depend on specific variants. Indeed, Finn et al. recently demonstrated a negative prognostic impact for KRAS G12C completely resected patients compared to both the KRAS wild type and other mutant variants (OS KRAS G12C versus other KRAS HR = 1.39, p = 0.031; KRAS G12C versus no KRAS HR = 1.32, p = 0.028) [34]. Contrarily, no prognostic nor predictive value for codon 12 mutations has been identified in a pooled meta-analysis conducted in early stage disease, while codon 13 mutations have been associated with a possible deleterious effect to adjuvant chemotherapy [37].
In an advanced setting, a meta-analysis of 43 selected studies demonstrated an impaired OS and progression-free survival (PFS) for KRAS mutants (OS HR = 1.71; 95% CI [1.07, 2.84]; PFS HR = 1.18; 95% CI [1.02, 1.36]). However, these results were only observed when considering observational studies and not further confirmed in randomized clinical trials (OS HR = 1.10; 95% CI [0.88–1.38]; PFS HR = 1.03; 95% CI [0.80–1.33]) [39]. Likewise, discordant results about the prognostic and predictive role of KRAS mutations have been reported in the advanced setting [27,40,41]. Furthermore, no differences in OS have been observed across different KRAS mutations, although a better response to taxanes has been reported for KRAS G12V mutants [33,42].
In conclusion, the prognostic and predictive role of KRAS mutations in NSCLC is still a matter of debate and discordant results provided so far need further assessment.
1.4. Concurrent Molecular Alterations in KRAS Positive NSCLC Patients
Concurrent molecular alterations are often found in KRAS mutant NSCLC patients. In the largest cohort available to date, Riely et al. described a median of seven concomitant mutations in KRAS positive NSCLC patients, the most frequent being TP53 (39%), STK11 (30%), KEAP1 (24%), RBM10 (15%), and PTPRD (15%). Curiously, those harboring concomitant mutations in either STK11 or KEAP1 had a shorter OS (STK11: HR = 2, p = 0.006; KEAP1: HR = 2.8, p < 0.001) and the negative prognostic impact of STK11 and KEAP1 co-alterations has been further confirmed in multiple studies [43,44]. Of note, neither the TP53 status nor number of concurrent mutations affected OS [45]. In another retrospective study, Passaro et al. demonstrated that 77% of patients with KRAS mutations had a concurrent molecular alteration. Interestingly, the percentage of concomitant alterations did not differ between G12C and non G-12C KRAS mutants. Again, the most frequently reported co-alteration was TP53 (23%) followed by STK11 (15%). Importantly, alterations in CDK2/4 and receptor tyrosine-kinase (RTK) have also been described in KRAS mutant cases [46].
Importantly, concurrent alterations might affect the tumor microenvironment with consequent alterations in the immunogenic phenotype. As such, Skoulidis et al. demonstrated a lower immunogenic infiltrate in STK11 positive tumors, while the opposite was seen for concomitant TP53-mutated tumors [47].
It has also been demonstrated that co-mutations could differentially activate downstream pathways. Indeed, engineered mice harboring KRAS G12D alone or with concomitant TP53 or STK11 mutations, respectively, hyper-activated MEK/ERK or AKT/SRC pathways [48].
Concurrent alterations with other key oncogenic drivers have also been described. Of note, KRAS mutations are found in approximately 3% of MET ex14 alterations [49,50], and a few cases of concomitant KRAS/ALK and KRAS/ROS1 alterations have been reported [51,52]. Interestingly, KRAS mutations have been found as mechanisms of resistance to the first, second, and third generation of EGFR TKI [53,54,55,56].
2. Targeted Therapy in KRAS Mutant NSCLC
As mentioned above, KRAS mutations have been considered undruggable for many years. Difficulties in targeting this protein were mainly linked to its smooth surface coupled with its high affinity to GTP, which made the targeting of the GDP/GTP binding site very challenging [29]. The subsequent discovery of the switch pocket II of KRAS protein represented a breakthrough in the treatment of KRAS mutations and molecules that specifically bind cysteine residues of KRAS mutant proteins were further developed. Because this binding preferentially occurs in the RAS-GDP form, KRAS G12C inhibitors mainly act by trapping KRAS-GDP proteins and consequently by impairing their transition to the KRAS-GTP active state [29,57,58,59] (Figure 2). Even though different molecules have been developed to date, sotorasib and adagrasib are the two having received regulatory approval to date (Figure 3).
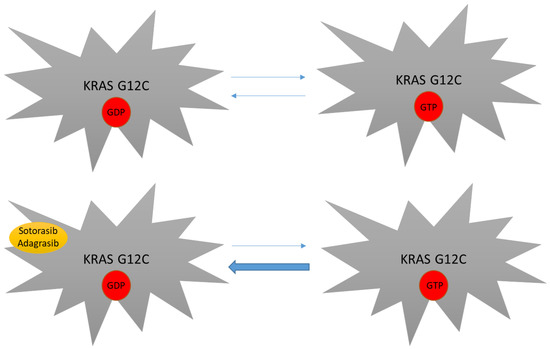
Figure 2.
Mechanism of action of novel KRAS inhibitors. KRAS G12C mutants still depend on their GTP binding state in order to be fully active. By covalently binding the switch pocket II, sotorasib and adagrasib reduce KRAS G12C-GDP availability, thus impairing transition to the KRAS-GTP active form.
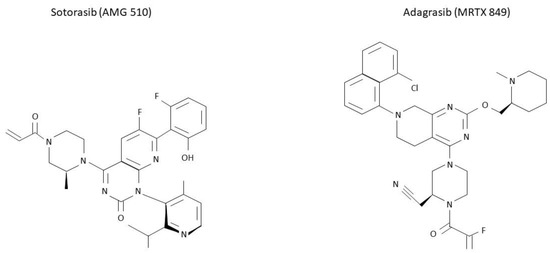
Figure 3.
Molecular structure of sotorasib and adagrasib. KRAS G12C inhibitors sotorasib and adagrasib have been recently approved for a similar setting. However, differences in their molecular structure are here presented.
2.1. Sotorasib (AMG 510)
Sotorasib (AMG 510) was one of the first molecules entered in clinical trials. In the single-arm, phase I-II CodeBreaK100 trial, the efficacy and safety of sotorasib were evaluated in different types of KRAS G12C mutant cancers [60,61]. The phase I trial enrolled 129 patients, 59 of whom affected by advanced NSCLC [60]. The primary endpoint was safety. KRAS G12C-mutated NSCLC patients were heavily pre-treated, with a median of three (range 0–11) previous lines of anticancer therapy for metastatic disease. Treatment-related adverse events (AEs) of any grade were observed in 56.6% of patients. Grade 3 treatment-related AEs included an increase in the alanine aminotransferase (ALT) level (4.7%), diarrhea (3.9%), anemia (3.1%), and an increase in the aspartate aminotransferase (AST) level (2.3%). AEs for any cause were reported in 96.9% of cases, mainly diarrhea (29.5%), fatigue (23.3%), and nausea (20.9%). Grade 3 or higher of all related AEs were reported in 68 patients (52.7%), consisting of anemia, liver enzymes alteration, and diarrhea. No dose-limiting toxic effects nor toxic deaths were observed. The dose of 960 mg administered daily was identified as the dose for the expansion cohort. As for efficacy in NSCLC, an overall response rate (ORR) of 32.2% and a disease control rate (DCR) of 88.1% were observed, with 71.2% of patients showing tumor-shrinkage at the first assessment. After a median follow-up of 11.7 months (range 4.8 to 21.2), median time to response (mTTR) was 1.4 months (range 1.1 to 9.5), median duration of response (mDoR) was 10.9 months (range 1.1+ to 13.6), median PFS (mPFS) was 6.3 months (range 0.0+ to 14.9).
Given the aforementioned results, the phase II of this trial started [61] (Table 1). This second part of the trial enrolled 126 patients with NSCLC harboring KRAS G12C mutation, who had progressed after anti-PD-1/PD-L1 molecules and platinum-based chemotherapy alone or combined. Patients with stable brain metastases were allowed. The primary endpoint was ORR, key secondary endpoints were DoR, DCR, TTR, PFS and OS. Sotorasib was administered at the identified dose of 960 mg orally once daily. Median number of systemic therapy was 2, patients receiving more than 3 previous lines were excluded. After a median follow up of 15.3 months (range 1.1–18.4+), 37.1% of patients obtained complete response (CR) or partial response (PR) (4 CR, 3.2%, 42 PR, 33.9%), and DCR was 80.6%. Responders had rapid and durable benefit (mTTR 1.4 months, range 1.2–10.1; mDoR 11.1 months, range 6.9-NE). mPFS was 6.8 months (95% CI, range 5.1 to 8.2), mOS was 12.5 months (95% CI, range 10.0 to NE). As for safety, treatment-related AEs were consistent with previous data and observed in overall 69.8% of patients. Again, most common AEs were diarrhea (31.7%), nausea (19%), fatigue (11%), and liver enzyme alteration. (15.1%). Grade 3 AEs were observed in 19.8% of patients and grade 4 in 1 patient (0.8%, consisting in pneumonitis and dyspnea); no toxic deaths were reported. Treatment modification (interruption, dose reduction, or both) occurred in 22.2% of patients and discontinuation of experimental therapy was reported in 7.1% of cases.
In terms of concurrent alterations, responses to sotorasib were evaluated in those harboring co-mutation in either STK11 or KEAP1 (STK11+/KEAP 1− or STK11−/KEAP1+) as well as in those with concomitant STK11 and KEAP1 co-alterations (STK11+/KEAP1+). Responses were higher in the STK11+/KEAP1− patients (50%), while worst outcomes were seen in STK11−/KEAP1+ mutants (14%). Responses for STK11+/KEAP1+ patients were placed in the middle (23%). Of note, RR was not significantly different in TP53-mutated versus wild-type patients (RR in TP53-mutated vs. wild-type: 39% vs. 40%), while KEAP1− mutated patients had detrimental results compared to KEAP1 wild-type after sotorasib exposure (RR in KEAP1-mutated vs. wild-type: 20% vs. 44%).
Interestingly, a post-hoc analysis demonstrated intracranial efficacy of sotorasib. Of note, among 40 patients with evaluable brain metastasis at baseline, ORR was 25% and DCR was 87.5% [62].
Preliminary results of the CodeBreak 200 trial were presented at ESMO 2022. This was a global, open-label, randomized phase III trial evaluating the efficacy of sotorasib versus docetaxel in KRAS mutant NSCLC patients who progressed after prior platinum-based chemotherapy and a checkpoint inhibitor. The primary endpoint was PFS, key secondary endpoints included OS, DCR, ORR. After study initiation and per regulatory guidance, initial planned enrolment was reduced and the crossover to sotorasib was allowed in docetaxel progressing patients. At a median follow-up of 17.7 months, sotorasib demonstrated a longer PFS compared to docetaxel (HR= 0.66 [95% CI: 0.51, 0.86], p = 0.002), with a 1-y reported PFS of 24.8% versus 10.1% in sotorasib and docetaxel arm, respectively. Among secondary endpoints, ORR and DCR were in favor of sotorasib (ORR sotorasib vs. docetaxel: 28.1% vs. 13.2%; p < 0.001; DCR sotorasib vs. docetaxel: 82.5% vs. 60.3%), while no differences in terms of OS were evidenced. In terms of safety, sotorasib was better tolerated than docetaxel and, consistently with the aforementioned results, most frequently reported Grade ≥3 secondary effects of sotorasib arm were diarrhea (33.7%), nausea (14.2%), fatigue (6.5%), hepatic enzyme alterations (ALT/AST increase in 10.1% of cases each) [63].
Based on these results, sotorasib received accelerated FDA approval in May 2021 for the treatment of patients who had received at least one prior line of systemic therapy [64].
2.2. Adagrasib (MRTX849)
Adagrasib (MRTX849) was the second KRAS G12C inhibitor that was entered in clinic trials [59]. The phase I/Ib dose-finding component of the first-in-human KRYSTAL-1 trial evaluated the safety, pharmacokinetics (PK), and clinical activity of adagrasib in KRAS G12C mutant advanced solid tumors [65]. In this trial, a total of 25 mutant patients were enrolled and 18 (72%) of them were affected by NSCLC. After PK and safety assessment, 600 mg twice a day was chosen as the recommended phase II dose (RP2D). Overall, 23 patients (92.0%) experienced treatment-related AEs and most frequently nausea (76.0%), diarrhea (72.0.0%), vomiting (48.0%), and fatigue (40.0%). Severe AEs (G ≥ 3) were reported in 36% of patients and the most common consisted in fatigue (15.0%). Additionally, one grade 5 pneumonitis occurred in one patient with underlying radiation and underlying pneumonitis. Furthermore, nausea (25.0%), diarrhea (20%), vomiting (20%) and fatigue (20%) were the main AEs responsible for treatment interruption or dose reduction at 600 mg twice daily. Among 15 evaluable patients with NSCLC treated with adagrasib at RP2D, ORR was 53.3%. After a median follow-up time of 19.6 months, mDOR was 16.4 months (95% CI, 3.1 to NE; range 2.8–16.9), while mPFS was 11.1 months (95% CI, range 2.6 to NE).
Given these results, the expansion phase of KRISTAL-1 trial was conducted [66] (Table 1). This second part of the trial enrolled a total of 116 KRAS mutant NSCLC patients. Patients enrolled were pre-treated with two median lines of systemic therapies and 14 (12.1%) of them received at least four previous systemic regimens. Globally, 98.3% of included patients received chemotherapy and immunotherapy as previous lines of therapy. Among 112 patients with measurable disease at the baseline, 48 (42.9%) showed an objective response and DCR was observed in 79.5% of cases. In particular, one patient obtained a CR (0.9%), 47 (42%) had PR, and 41 had (36.6%) stable disease (SD) as their best response. mDoR was 8.5 months (95% CI, 6.2–13.8) and mPFS was 6.5 months (95% CI, range 4.7–8.4). At the last update, after a median follow-up of 15.6 months, mOS was 12.6 months (95% CI, range 9.2–19.2). Considering patients with stable, previously treated CNS metastases, an intracranial confirmed response rate of 33.3% (95% CI, range 8.0–51.8) was reported. Further analysis of patients carrying co-occurring alterations in STK11, KEAP1, or both, confirmed a detrimental effect in those harboring KEAP1 co-mutations (ORR 14.3% in STK11-/KEAP1+). As for safety, treatment-related AEs occurred in 97.4% of patients, mainly grade 1 or 2, while 44.8% of them were G3 or higher. Most common treatment-related AEs were diarrhea (62.9%), nausea (62.1%), vomiting (47.4%), fatigue (40.5%), an increased alanine aminotransferase (ALT) level (27.6%), an increased blood creatinine level (25.9%), and an increased aspartate aminotransferase (AST) level (25.0%). Furthermore, two fatal events were reported (one cardiac failure and one pulmonary hemorrhage) and drug discontinuation related to AEs was observed in 8 (6.9%) patients, while AEs led to dose reduction and dose interruption in 51.7% and 61.2% of all cases, respectively.
In terms of intracranial responses, ORR of adagrasib was 33.3% and among patients with brain metastasis at baseline the mDoR was 11.2 months. The efficacy of adagrasib is currently being investigated in a phase 3 trial versus docetaxel in pretreated patients (KRISTAL-12, NCT04685135) [67].
Based on these results, adagrasib received FDA approval for the treatment of previously treated KRAS NSCLC patients [68].

Table 1.
Efficacy and safety of treatment of sotorasib and adagrasib in the CodeBreak100 and the KRISTAL-1 trials.
Table 1.
Efficacy and safety of treatment of sotorasib and adagrasib in the CodeBreak100 and the KRISTAL-1 trials.
| Drug/Dose | Phase | Population | N. of Enrolled Pts | N. of Previous Lines (Median) | Efficacy | Treatment-Related AEs |
|---|---|---|---|---|---|---|
| CodeBreak100 [61] Sotorasib 960 mg daily | Phase I/II |
| 126 | 2 | ORR: 37.1% DCR: 80.6% mPFS: 6.8 (5.1–8.2) mOS: 12.5 (10-NE) | G1-4: 69.8% G ≥ 3: 20.6% |
| KRISTAL-1 [66] Adagrasib 600 mg daily | Phase I/Ib |
| 116 | 2 | ORR: 42.9 DCR: 79.5 mPFS: 6.5 (4.7–8.4) mOS: 12.6 (9.2–19.2) | G1-4: 97.4% G ≥ 3: 44.8% |
ORR: objective response rate; PFS: progression-free survival (months, 95% CI); OS: overall survival (months, 95%CI).
2.3. Novel Molecules for Targeting KRAS Mutations
Novel KRAS G12C inhibitors are currently being evaluated in multiple phase I–II clinical trials.
A new anti-KRAS inhibitor (JNJ-74699157) has been recently tested in a phase I trial. Ten patients, including 5 NSCLC cases, have been enrolled. Due to the high toxicity profile (grade 3–4 increased blood creatinine phosphokinase) and the lack of clinical efficacy, this trial has been prematurely interrupted. Similarly, the phase I evaluating LY3499446 compound has been stopped because of unexpected toxicity [29].
The efficacy and safety of GDC-6036 has been recently presented at 2022 WCLC. In the phase I trial evaluating this novel molecule, GDC-6036 was given to 135 patients with advanced solid tumors, including 59 patients affected by KRAS-mutated NSCLC. In the NSCLC cohort, the median age was 67 years, and the majority received prior checkpoint inhibitor therapy (86%) and platinum-based chemotherapy (90%). The median number of prior therapies was 1 (range, 0–5). As for safety, grade treatment-related AEs were observed in 88.1% of all patients, the most common being nausea (76.3%), diarrhea (61%), vomiting (54.2%), fatigue (23.7%), decreased appetite (15.3%), increased alanine aminotransferase (13.6%), increased aspartate aminotransferase (10.2%), and dyspepsia (6.8%). Severe treatment-related AEs were observed in 16.9% of all patients and the most frequent were increased alanine aminotransferase (6.8%), increased aspartate aminotransferase (5.1%), diarrhea (3.4%), and fatigue (1.7%). No dose-limiting toxicities were reported. As for efficacy, a confirmed ORR was reported in 46% of cases [69,70].
Other anti-KRAS G12C inhibitors are currently being evaluated, including PJ34, a molecule that modifies the nuclear mitotic apparatus (NuMA). In NSCLC cell lines, this molecule efficiently determined cancer cell death via an exclusive structural faults inserted in their mitotic spindle. Due to a more selective mechanism of action, PJ34 eventually spared healthy NSCLC cells, with a potentially better tolerance in clinic [71]. However, no ongoing clinical trial is currently evaluating the efficacy and safety of PJ34. Other KRAS inhibitors in ongoing trials are listed in Table 2, with no results posted to date [72,73,74,75,76,77,78]. Of note, these novel compounds will also be tested in patients who progressed after previous KRAS G12C inhibitors, and combinational strategies with other molecular targets implied in resistance will also be evaluated (see moreover).
Table 2.
Ongoing trials evaluating novel inhibitors for KRAS-G12 mutant advanced NSCLC.
Molecular targeting of KRAS mutations other than G12C still represents a pioneer domain. KRAS G12D inhbitors (e.g., MRTX1133 and JAB-22000) as well as KRAS G12V molecules (e.g., JAB23000) are currently being investigated with promising preclinical results [79,80]. However, no clinical data are available to date.
3. Resistance Mechanisms to KRAS Inhibitors
Despite these encouraging results, patients exposed to KRAS inhibitors may further progress. Two different mechanisms of acquired resistance to KRAS inhibitors have been identified to date: on-target and off-target mechanisms. On-target mechanism of resistance typically involves the switch pocket II, thus impairing drug binding. In KRAS-mutated NSCLC cell lines, patterns of on-target acquired mutation varied after exposure of either sotorasib or adagrasib. Indeed, KRAS G13D was the most frequently observed on-target mutation in sotorasib-resistant clones (23%), while KRAS Q99L represented the most common mutation after adagrasib exposure (52.8%). Of note, sotorasib-and adagrasib-resistant clones were both associated with Y96D and A59S point mutations [31]. Importantly, a new KRAS inhibitor RM-018 proved preclinical evidence of efficacy in overcoming secondary acquired KRAS G12C/Y96D mutations [31,81], although these data need to be validated in prospective clinical trials. Other KRAS point mutations identified to date in pre-clinical setting include R68S/M, H95D/Q/R and Y96C. Importantly, cross-resistance to sotorasib and adagrasib have been demonstrated for Y96C and Y96D mutants. At the opposite, G13D and A59S secondary mutations remained partially sensitive to adagrasib, while Q99L and H95D/Q/R gathered responsiveness to sotorasib [31]. In clinic, reported cases of acquired G12D/R/V/W, G13D, Q61H, R68S, H95D/Q/R, and Y96C point mutations have been described after progression to adagrasib, while patients progressing after sotorasib further develop G12D/V/F, V8L point mutations [82,83].
Multiple off-target mechanisms of resistance have been identified to date and include: bypass signaling pathways, epithelial-to-mesenchymal transformation (EMT) and transition to senescence.
Activation of bypass signaling pathways typically involves MET amplifications, activating mutation in NRAS, BRAF, MAP2K1, and RET, fusions of ALK, RET, BRAF, RAF1, and FGFR3 as well as loss-of-function of NF1 and PTEN. Of note, these alterations were equally identified after progression to both sotorasib and adagrasib, and were also detected in cell-free DNA [81,82,83].
Epithelial-to-mesenchymal transformation (EMT) has also been reported as mechanisms of resistance both in preclinical and in clinical settings. In sotorasib resistant clones, EMT was associated with E-cadherin downregulation and vimentin upregulation, while patients progressing after adagrasib experienced transition from adenocarcinoma to squamous cell carcinoma histology [83]. Of note, RTK activation (and particularly IGFR1 and FGFR) has been reported in sotorasib resistant cells which displayed EMT [81].
Finally, activation of the aurora kinase signaling could represent a key element explaining transition to senescence as acquired mechanism of resistance. As such, ongoing trials are currently evaluating the efficacy of anti-aurora kinases combined to novel KRAS inhibitors after progression to either sotorasib or adagrasib (see above) [81].
Although significant progresses in this field have been made, mechanisms of resistance to KRAS inhibitors are to date only barely understood. Importantly, a putative mechanism of resistance to adagrasib has been found in only about 45% of cases, while 41% of them displaying more than one molecular alteration [83]. With the aim of improving the knowledge in this field, the ongoing SPARK trial is currently assessing molecular alterations detected via ctDNA after progression to KRAS inhibitors [84].
Another key issue is how to treat patients after progression, since subsequent lines of therapy could strongly depend on the mechanism of resistance identified at progression. A possible scenario could be adding KRAS inhibitors to targeted compounds depending on the acquired mechanism or resistance. Of note, preliminary results obtained from the phase Ib-II CodeBreak 101 trial demonstrated the efficacy of sotorasib combined with either afatinib or the SHP-2 inhibitor RMC-4630 in patients progressing after KRAS inhibitors, thus highlighting a subset of patients who could benefit from this combined approach at progression to KRAS inhibitors [85,86].
4. Immune Checkpoint Inhibitors (ICI) in KRAS Mutant NSCLC
Efficacy of ICIs in KRAS mutant patients still needs to be fully assessed.
Biologically, KRAS mutations have been associated with a pro-inflammatory tumor microenvironment mainly via higher PD-1/PD-L1 expression, CD8+ tumor-infiltrating lymphocytes (TILs) and TMB [87,88,89], and other data also demonstrated that PD-L1 expression is higher in KRAS mutant versus wild-type cases (PD-L1 in KRAS mutant vs. wild-type: 65.3% vs. 58.5%, p = 0.0002) [90]. However, discrepancies have been reported, with some meta-analysis rather disconfirming a significant association between PD-L1 status and KRAS mutations [91,92].
Consistently with these contrasting results, the clinical outcomes of immunotherapy-treated KRAS mutant patients are equivocal.
In an exploratory analysis of the Keynote-042 trial, KRAS mutant patients benefited from first-line pembrolizumab [93], and a further cohort study also confirmed a longer OS in KRAS mutant patients compared with wild-type after ICI exposure (mOS KRAS mutants vs. wild-type: 21.1 vs. 13.6 months, p = 0.03) [94]. In strike contrast, a multicentric retrospective study found no significant differences in terms of ORR, PFS, and OS between KRAS mutants versus wild-type patients (ORR KRAS mutants vs. wild-type: 18.7 vs. 14.4%, p = 0.348; mPFS KRAS mutants vs. wild-type; 3.09 vs. 2.66 months, p = 0.584; OS KRAS mutants vs. wild-type: 14.29 vs. 11.14 months, p = 0.682) [95], and both KRAS mutant and wild-type equally benefited from atezolizumab in the subgroup analysis of the OAK trial [96]. Likewise, discordant results were also observed in three meta-analyses evaluating the efficacy of ICI in KRAS mutant NSCLC patients [97,98,99,100].
Compared to other oncogenic drivers, KRAS mutants have been associated with higher responses to ICIs. In the IMMUNOTARGET registry, KRAS positive patients experienced the highest ORR (26%) and long term responses were frequently observed in KRAS mutant cases compared to those with other molecular drivers (12 months PFS KRAS: 25.6%, MET 23.4%, BRAF 18.0%, EGFR 6.4%, ALK 5.9%, HER2 13.6%, and RET 7.0%). Of note, KRAS mutants had the lower rate of rapid progression to ICI (<2 months) compared to those harboring other oncogenic alterations (KRAS 36%, EGFR 44.8%, ALK 45.5%, ROS1 42.9%, RET 43.8%) [101].
Given these contradictory results, it has been wondered if the efficacy of both immune checkpoint and KRAS inhibitors could be boosted up by adding these two molecular classes. So far, multiple ongoing trials are currently evaluating this treatment strategy without any reported data [102,103]. Encouraging results have been recently presented for the CodeBreak 100/101 phase Ib trial. In this trial, adding either pembrolizumab or atezolizumab to sotorasib resulted in a confirmed response of 39% (17/58 patients; range 0–67), and responders had a mDOR of 17.9 months (range 1.5+–23.4). For all patients, mOS was 15.7 months. Importantly, treatment-related AEs were observed in 79–100% of patients and hepatotoxicity was the most frequently observed grade 3–4 AE (30–50%) [104]. Other ongoing trials are evaluating this combinational strategy, without any reported results to date.
As already mentioned, differences in clinical and pathological characteristics have been identified across single KRAS mutant variants. Thus, it has been wondered if the immune infiltrate could also vary depending on KRAS point mutations.
In preclinical data, KRAS G12D mutants have been linked to a “cold” immune phenotype mainly via PD-L1 downregulation, reduced secretion of pro-inflammatory chemokines (CXCL10/CXCL11), and decreased CD8+ TILs [105]. Furthermore, a lower level of PD-L1 expression and tumor mutation burden (TMB) in KRAS G12D has also emerged in a more recent study (PD-L1 tumor proportion score KRAS G12D vs. non G-12D: 1% vs. 5%, p < 0.01; TMB G12D vs. non-G12D: median 8.4 versus 9.9 mt/Mb, p < 0.0001) [106]. Again, these data are not univocal and other studies denied any difference in PD-L1 expression across KRAS mutational subgroups [90]. In the aforementioned IMMUNOTARGET registry, no differences in terms of PFS were observed when comparing KRAS G12C to other KRAS mutations or KRAS G12D to other KRAS mutations (p = 0.47) or G12D (p = 0.40) and similar outcomes were observed when comparing KRAS G12D, G13D, and G12S versus KRAS G12C, G12A, G12V, and G13C mutations [101]. Similarly, other retrospective data excluded any influence on treatment outcomes across single mutant KRAS variants [107]. Conversely, other data highlighted worse treatment outcomes for KRAS G12D compared to non-G12D in those receiving single agent ICI but not after chemo-immunotherapy combinations [106].
It has been supposed that the co-mutational pattern could play a role in pro-immunogenic infiltrate. Indeed, lower levels of PD-L1 expression, TILs infiltration, and higher rates of T-cell exhaustion have been reported for concomitant STK11/KEAP1 co-mutations, thus underlying a subset of KRAS patients that could not benefice from immunotherapy [76,77,78,79]. On the contrary, concomitant KRAS/TP53 mutants have been associated with an increased expression of PD-L1 and a higher mutational load, as well as with a decreased expression of non–PD-L1 immune inhibitory checkpoints, such as Lymphocyte Activating 3 (LAG3) and V-Set Domain Containing T Cell Activation Inhibitor 1 (VTCN1) [108,109].
Clinical evidence also supported these data. As such, patients with concurrent alterations in either STK11 or KEAP1 had a significantly worse response to ICI treatment compared to those without these concomitant alterations [110,111,112]. Similarly, patients with TP53 mutations experienced better outcomes from ICI treatment than those harboring TP53 wild-type in multiple series [108,113,114].
5. Discussion
Sotorasib and adagrasib represents the firsts anti KRAS molecules entered in clinical trials and receiving FDA approval for the treatment of KRAS-positive NSCLC patients in subsequent lines.
Although these two drugs have been evaluated in similar subsets of patients, noteworthy differences in terms of toxicities emerged so far. As such, all grade treatment-related AEs experienced with sotorasib were significantly lower than adagrasib (all grade treatment-related AEs sotorasib: 69.8%; all grade treatment-related AEs adagrasib: 97.4%), and two cases of grade 5 treatment-related AEs have been reported in the KRYSTAL-1 trial, while no fatal event was described among sotorasib treated patients. The reasons underlying this supposed discrepancy in the toxicity profile need to be fully understood and further validated. Importantly, efficacy was similar among both these molecules, and sotorasib recently confirmed its superiority over docetaxel in the phase III CodeBreak 200 trial [63].
Despite these encouraging results, however, responses observed are at around 40% of cases, which is considerably lower than that observed with other oncogenic drivers. As such, two questions may arise: Which would be the reasons behind this suspected lower efficacy? How can it be improved?
Regarding the first question, we need to remember that KRAS is only one of the three RAS isoforms that normally co-exist in human tissues and that its downstream pathway could be activated via multiple bypass cascades. Therefore, the blockage of KRAS mutants could be easily bypassed via other wild-type RAS isoforms or further escape pathways, such as ERBB family members (see moreover). Furthermore, Passaro A. pointed out that the mechanism of action of KRAS inhibitors significantly differs from that observed for other TKI inhibitors, since it blocks KRAS mutants in their GDP inactive form [115]. Whether and how this difference in the mechanism of action could impact drug effectiveness still needs to be fully understood. Moreover, besides KEAP1 and STK11, little is known about the predictive role of other concomitant mutants and the real impact of concomitant mutations other than STK11/KEAP1/TP53 in determining KRAS inhibitor’s efficacy still needs to be assessed. Additionally, it is still an open question to what extent patients with other oncogenic drivers (e.g., EGFR, METex14, ALK, ROS1) who develop KRAS mutations as the mechanism of escape could effectively benefit from KRAS inhibitors.
Regarding the second question, multiple ongoing trials are currently evaluating the efficacy of combining sotorasib or adagrasib with other agents (Table 3). One potential combination strategy includes molecules that target either the upstream or downstream signaling pathway.
Table 3.
Ongoing trials evaluating sotorasib/adagrasib combined with other agents in KRAS-G12 mutant advanced NSCLC.
As mentioned above, KRAS activation may involve other proteins besides Grb2 and SOS-1, such as the ubiquitously expressed phosphatase SHP-2. The role of SHP-2 in activating the KRAS pathway is still controversial but evidence suggests that it could serve as a scaffold protein to Grb2/SOS1 after RTK phosphorylation, thus aiding in KRAS upstream transduction signaling [9]. Furthermore, SHP-2 inhibition resulted in a higher cell senescence and lower grade of tumor development in preclinical models of KRAS mutant NSCLC [120,121], with a synergistic effect observed when combining adagrasib with the anti-SHP-2 agent RMC-4550 [59].
Primary results of the ongoing phase Ib-II multiple cohorts CodeBreaK 101 trial were recently presented. In this trial, patients with advanced solid tumors harboring KRAS G12C received sotorasib either alone or combined with other molecules, including the SHP-2 inhibitor RMC-4630 [116]. The primary endpoint was safety and tolerability, key secondary endpoints included ORR and pharmacokinetics. Results from the cohort of patients treated with sotorasib and the SHP-2 inhibitor RMC-4630 have been recently presented. Among the 11 NSCLC patients included, 3 (27%) had PR and 7 (64%) has disease control and this percentage was higher among KRAS inhibitor naïve patients (75% PR and 100% disease control). The safety profile was manageable, and main AEs consisted of diarrhea and transaminase increase [85]. Other ongoing trials are evaluating the efficacy of anti-SHP2 or anti SOS-1 molecules, with no data reported yet [117,119,122].
Another potential strategy in targeting the upstream pathway could rely on the inhibition of EGFR. In contrast with the previous thought of KRAS mutant independency from upstream RTK, recent data demonstrate that the genetic abrogation of EGFR resulted in impaired tumor growth of KRAS mutant NSCLC models. Of note, EGFR inhibition was rapidly overcome via alternative pathways that include the activation and overexpression of other ERBB family members [123]. On thisasis, it has been supposed that co-targeting KRAS with a pan-ERRB inhibitor such as afatinib could potentiate KRAS-inhibitors’ efficacy. Indeed, preclinical evidence pointed out a significant reduction of tumor growth when afatinib was combined with anti-KRAS molecules [59].
The combination of sotorasib and afatinib is currently being investigated in the aforementioned CodeBreak 101 trial. Firsts results were presented at AACR in 2021. Afatinib was given in two different scheduled (either 20 mg or 30 mg daily, cohort 1 and cohort 2 respectively) and combined to sotorasib 960 mg daily. A total of 33 NSCLC patients were enrolled, of whom 10 patients were in cohort 1 (afatinib 20 mg + sotorasib 960 mg) and 23 patients in cohort 2 (afatinib 30 mg + sotorasib 960 mg). In both cohorts, patients received a median number of two prior therapies (range 0–7; 66.7% ≥ 2 prior lines) and five patients (15.2%) received prior sotorasib. The combination of sotorasib and afatinib resulted in an ORR of 20% (cohort 1) and 34.8% (cohort 2), DCR was also higher in cohort 2 than cohort 1 (DCR 70.0% in cohort 1, DCR 73.9% in cohort 2). In terms of toxicity, all grade treatment-related AEs were mainly gastrointestinal (diarrhea 69.7%, nausea 21.2%, vomiting 18.2%). Grade ≥3 treatment-related AEs were reported in 30% of patients in each cohort, with diarrhea being the most frequent (21.2%). Eight patients (24.2%) discontinued sotorasib and/or afatinib due to an AE [86]. Other trials are still ongoing and will further assess the efficacy and safety of KRAS inhibitors combined with EGFR/panERBB inhibitors [102].
As what concerns downstream pathways, preclinical experiments demonstrated that KRAS mutants are associated with higher ERK1/2 phosphorylation and that sensitiveness to MEK inhibitor is higher for G12C than for G12D mutants [124]. Furthermore, PI3K can directly be activated by RAS proteins, thus representing a direct RAS effector [125]. As such, the combination with anti-KRAS and inhibitors that insist on RAF/MEK or PI3K/AKT pathway have been tested in preclinical settings, with promising data [59,126]. Multiple ongoing trials are currently assessing the efficacy and safety of combination treatment with sotorasib/adagrasib and molecules which either inhibit PI3K/ATK/mTOR or RAF/MEK pathways, with no reported data available [116,118].
Preclinical data demonstrated that KRAS mutations might induce chemo and radio-resistance via multiple mechanisms, such as the induction of senescence, imbalance in ROS production, and activation of mechanisms of DNA repair [127]. Importantly, resistance to radiation has also been demonstrated in some clinical reports [81]. As such, blocking KRAS mutations could eventually reverse radiation resistance, with obvious positive consequences. Clinical trials evaluating radiation combined with sotorasib are ongoing and would further elucidate this potential approach [128,129].
Finally, other open questions remain, such as which would be the best timing for treatment with KRAS inhibitors and which would be the best therapeutic option in those who progressed to KRAS inhibitors. In this context, ongoing trials will further elucidate the effectiveness of KRAS inhibitors in neoadjuvant, locally advanced, and first line settings [103,129,130,131,132,133].
In conclusion, KRAS mutations in NSCLC represent a topic of growing interest and, not surprisingly, literature reviews on this subject have exponentially increased over time. Nevertheless, we think that our work could be of added value for multiple reasons. First, it represents a very up-to-date article about the clinical efficacy of KRAS inhibitors, including the most recent trials results. This is particularly remarkable if we consider that the last review on this topic dates back to August 2022 and that the treatment scenario rapidly evolved since then [134]. Secondarily, many previous reports alternatively focused on specific aspects within this topic, such as mechanisms of resistance rather than therapeutic agents’ efficacy [135,136,137], leaving behind some other important factors that should have been conversely considered. Nevertheless, two recent reviews may display some similarities with our paper. In their paper, Huang et al. also described the heterogeneity of clinical characteristics in KRAS mutants, co-mutant alterations, immature clinical data from KRAS inhibitors, as well as combinational strategies. Nonetheless, their paper has a rather preclinical approach and barely mentioned novel KRAS inhibitors such as GCD-6036, while completely missing clinical data on the efficacy of immune checkpoint inhibitor’s in KRAS mutant patients [81]. In contrast, Kwan et al. published a clinically focused article on KRAS G12C inhibitors and clinical data from novel KRAS inhibitors as well as combinational strategies and these were brilliantly reported [29]. Strikingly, no reference to either co-mutations or clinical heterogeneity across KRAS mutant patients has been described. Of note, none of these two aforementioned papers referred to some aspects that were conversely described in our review, such as the prognostic and predictive significance of KRAS mutations and diagnostic tools for detecting KRAS mutations.
With the aim of presenting an updated clinical overview on the state of the art of KRAS mutations in NSCLC, we think that our review extensively summarized the main hot topics connected with this subject and could thus represent a resource of added value.
6. Conclusions
Sotorasib and adagrasib are the first KRAS inhibitors that received regulatory approval for the treatment of KRAS G12C mutant NSCLC patients in subsequent settings. Evidence supporting these approvals mainly derive from phase I/II trials, while sotorasib recently demonstrated its superiority over docetaxel in the phase III CodeBreak 200 trial. Despite these data, the efficacy of these new compounds is lower than that observed for other molecular drivers. Furthermore, the efficacy of ICIs in this subpopulation is not yet completely established and could strongly be influenced by concomitant alterations as well as single KRAS mutational variants. Open issues concerning the increase of efficacy, the definition of mechanisms of resistance, and the best timing for treatment with anti-KRAS molecules are currently being assessed in ongoing trials.
Author Contributions
Conceptualization, A.M. (Arianna Marinello) and P.C.; writing, P.C., A.M. (Arianna Marinello), C.L., V.G., R.B., D.P., A.M. (Alessandro Morabito) and N.N., review and editing, P.C., A.M. (Arianna Marinello) and A.M. (Alessandro Morabito); visualization, P.C. and A.M. (Arianna Marinello); supervision, A.M. (Alessandro Morabito), N.N., D.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
Alessandro Morabito declares the following conflicts of interest: Speaker’s fee: MSD, BMS, Boehringer, Pfizer, Roche, AstraZeneca, Novartis; Advisory Board: Takeda, Eli-Lilly. Nicola Normanno declares the following personal financial interests (speaker’s fee and/or advisory boards): MSD, Qiagen, Bayer, Biocartis, Incyte, Roche, BMS, MERCK, Thermofisher, Boehringer Ingelheim, Astrazeneca, Sanofi, Eli Lilly; Institutional financial interests (financial support to research projets): MERCK, Sysmex, Thermofisher, QIAGEN, Roche, Astrazeneca, Biocartis. Non-financial interests: President, International Quality Network for Pathology (IQN Path); President, Italian Cancer Society (SIC). David Planchard declares the following conflicts of interest: Consulting, advisory role and/or lectures: Abbvie, AstraZeneca, Bristol-Myers Squibb, Boehringer Ingelheim, Celgene, Daiichi Sankyo, Eli Lilly, Janssen, Merck, Novartis, Pfizer, prIME Oncology, Peer CME, Roche; Honoraria: Abbvie, AstraZeneca, Bristol-Myers Squibb, Boehringer Ingelheim, Celgene, Eli Lilly, Janssen, Merck, Novartis, Peer CME, Pfizer, prIME Oncology, Roche; Clinical trials research as principal or co-investigator, institutional financial interests: Abbvie, AstraZeneca, Bristol-Myers Squibb, Boehringer Ingelheim, Daiichi Sankyo, Eli Lilly, Janssen, MedImmune, Merck, Novartis, Novocure, Pfizer, Roche, Sanofi-Aventis, Taiho Pharma; Travel, accommodation and/or expenses: AstraZeneca, Novartis, Pfizer, Roche. All the other Authors declare no conflicts of interest.
References
- Ganti, A.K.; Klein, A.B.; Cotarla, I.; Seal, B.; Chou, E. Update of Incidence, Prevalence, Survival, and Initial Treatment in Patients With Non–Small Cell Lung Cancer in the US. JAMA Oncol. 2021, 7, 1824–1832. [Google Scholar] [CrossRef]
- Thai, A.A.; Solomon, B.J.; Sequist, L.V.; Gainor, J.F.; Heist, R.S. Lung Cancer. Lancet 2021, 398, 535–554. [Google Scholar] [CrossRef]
- KRAS—An Overview|ScienceDirect Topics. Available online: https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/kras (accessed on 31 July 2022).
- Plowman, S.J.; Berry, R.L.; Bader, S.A.; Luo, F.; Arends, M.J.; Harrison, D.J.; Hooper, M.L.; Patek, C.E. K-Ras 4A and 4B Are Co-Expressed Widely in Human Tissues, and Their Ratio Is Altered in Sporadic Colorectal Cancer. J. Exp. Clin. Cancer Res. 2006, 25, 259–267. [Google Scholar]
- Hancock, J.F. Ras Proteins: Different Signals from Different Locations. Nat. Rev. Mol. Cell Biol. 2003, 4, 373–385. [Google Scholar] [CrossRef]
- Westcott, P.M.K.; To, M.D. The Genetics and Biology of KRAS in Lung Cancer. Chin. J. Cancer 2013, 32, 63–70. [Google Scholar] [CrossRef]
- Pantsar, T. The Current Understanding of KRAS Protein Structure and Dynamics. Comput. Struct. Biotechnol. J. 2019, 18, 189–198. [Google Scholar] [CrossRef]
- Drugging an Undruggable Pocket on KRAS|PNAS. Available online: https://www.pnas.org/doi/10.1073/pnas.1904529116 (accessed on 22 September 2022).
- Dance, M.; Montagner, A.; Salles, J.-P.; Yart, A.; Raynal, P. The Molecular Functions of Shp2 in the Ras/Mitogen-Activated Protein Kinase (ERK1/2) Pathway. Cell. Signal. 2008, 20, 453–459. [Google Scholar] [CrossRef]
- Maertens, O.; Cichowski, K. An Expanding Role for RAS GTPase Activating Proteins (RAS GAPs) in Cancer. Adv. Biol. Regul. 2014, 55, 1–14. [Google Scholar] [CrossRef]
- Mosele, F.; Remon, J.; Mateo, J.; Westphalen, C.B.; Barlesi, F.; Lolkema, M.P.; Normanno, N.; Scarpa, A.; Robson, M.; Meric-Bernstam, F.; et al. Recommendations for the Use of Next-Generation Sequencing (NGS) for Patients with Metastatic Cancers: A Report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2020, 31, 1491–1505. [Google Scholar] [CrossRef]
- Gundry, C.N.; Vandersteen, J.G.; Reed, G.H.; Pryor, R.J.; Chen, J.; Wittwer, C.T. Amplicon Melting Analysis with Labeled Primers: A Closed-Tube Method for Differentiating Homozygotes and Heterozygotes. Clin. Chem. 2003, 49, 396–406. [Google Scholar] [CrossRef]
- Heid, C.A.; Stevens, J.; Livak, K.J.; Williams, P.M. Real Time Quantitative PCR. Genome Res. 1996, 6, 986–994. [Google Scholar] [CrossRef]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid Biopsies Come of Age: Towards Implementation of Circulating Tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef]
- Li, J.; Gan, S.; Blair, A.; Min, K.; Rehage, T.; Hoeppner, C.; Halait, H.; Brophy, V.H. A Highly Verified Assay for KRAS Mutation Detection in Tissue and Plasma of Lung, Colorectal, and Pancreatic Cancer. Arch. Pathol. Lab. Med. 2019, 143, 183–189. [Google Scholar] [CrossRef]
- Elazezy, M.; Joosse, S.A. Techniques of Using Circulating Tumor DNA as a Liquid Biopsy Component in Cancer Management. Comput. Struct. Biotechnol. J. 2018, 16, 370–378. [Google Scholar] [CrossRef]
- Gagan, J.; Van Allen, E.M. Next-Generation Sequencing to Guide Cancer Therapy. Genome Med. 2015, 7, 80. [Google Scholar] [CrossRef]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic Non-Small Cell Lung Cancer: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2018, 29, iv192–iv237. [Google Scholar] [CrossRef]
- Hagemann, I.S.; Devarakonda, S.; Lockwood, C.M.; Spencer, D.H.; Guebert, K.; Bredemeyer, A.J.; Al-Kateb, H.; Nguyen, T.T.; Duncavage, E.J.; Cottrell, C.E.; et al. Clinical Next-Generation Sequencing in Patients with Non-Small Cell Lung Cancer. Cancer 2015, 121, 631–639. [Google Scholar] [CrossRef]
- Tsoulos, N.; Papadopoulou, E.; Metaxa-Mariatou, V.; Tsaousis, G.; Efstathiadou, C.; Tounta, G.; Scapeti, A.; Bourkoula, E.; Zarogoulidis, P.; Pentheroudakis, G.; et al. Tumor Molecular Profiling of NSCLC Patients Using next Generation Sequencing. Oncol. Rep. 2017, 38, 3419–3429. [Google Scholar] [CrossRef][Green Version]
- Aggarwal, C.; Thompson, J.C.; Black, T.A.; Katz, S.I.; Fan, R.; Yee, S.S.; Chien, A.L.; Evans, T.L.; Bauml, J.M.; Alley, E.W.; et al. Clinical Implications of Plasma-Based Genotyping With the Delivery of Personalized Therapy in Metastatic Non-Small Cell Lung Cancer. JAMA Oncol. 2019, 5, 173–180. [Google Scholar] [CrossRef]
- Horn, L.; Whisenant, J.G.; Wakelee, H.; Reckamp, K.L.; Qiao, H.; Leal, T.A.; Du, L.; Hernandez, J.; Huang, V.; Blumenschein, G.R.; et al. Monitoring Therapeutic Response and Resistance: Analysis of Circulating Tumor DNA in Patients With ALK+ Lung Cancer. J. Thorac. Oncol. 2019, 14, 1901–1911. [Google Scholar] [CrossRef]
- Pascual, J.; Attard, G.; Bidard, F.-C.; Curigliano, G.; De Mattos-Arruda, L.; Diehn, M.; Italiano, A.; Lindberg, J.; Merker, J.D.; Montagut, C.; et al. ESMO Recommendations on the Use of Circulating Tumour DNA Assays for Patients with Cancer: A Report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2022, 33, 750–768. [Google Scholar] [CrossRef]
- Heitzer, E.; van den Broek, D.; Denis, M.G.; Hofman, P.; Hubank, M.; Mouliere, F.; Paz-Ares, L.; Schuuring, E.; Sültmann, H.; Vainer, G.; et al. Recommendations for a Practical Implementation of Circulating Tumor DNA Mutation Testing in Metastatic Non-Small-Cell Lung Cancer. ESMO Open 2022, 7, 100399. [Google Scholar] [CrossRef]
- IJzerman, M.J.; de Boer, J.; Azad, A.; Degeling, K.; Geoghegan, J.; Hewitt, C.; Hollande, F.; Lee, B.; To, Y.H.; Tothill, R.W.; et al. Towards Routine Implementation of Liquid Biopsies in Cancer Management: It Is Always Too Early, until Suddenly It Is Too Late. Diagnostics 2021, 11, 103. [Google Scholar] [CrossRef]
- Judd, J.; Abdel Karim, N.; Khan, H.; Naqash, A.R.; Baca, Y.; Xiu, J.; VanderWalde, A.M.; Mamdani, H.; Raez, L.E.; Nagasaka, M.; et al. Characterization of KRAS Mutation Subtypes in Non–Small Cell Lung Cancer. Mol. Cancer Ther. 2021, 20, 2577–2584. [Google Scholar] [CrossRef]
- Wood, K.; Hensing, T.; Malik, R.; Salgia, R. Prognostic and Predictive Value in KRAS in Non–Small-Cell Lung Cancer: A Review. JAMA Oncol. 2016, 2, 805–812. [Google Scholar] [CrossRef]
- Reck, M.; Carbone, D.P.; Garassino, M.; Barlesi, F. Targeting KRAS in Non-Small-Cell Lung Cancer: Recent Progress and New Approaches. Ann. Oncol. 2021, 32, 1101–1110. [Google Scholar] [CrossRef]
- Kwan, A.K.; Piazza, G.A.; Keeton, A.B.; Leite, C.A. The Path to the Clinic: A Comprehensive Review on Direct KRASG12C Inhibitors. J. Exp. Clin. Cancer Res. 2022, 41, 27. [Google Scholar] [CrossRef]
- Ihle, N.T.; Byers, L.A.; Kim, E.S.; Saintigny, P.; Lee, J.J.; Blumenschein, G.R.; Tsao, A.; Liu, S.; Larsen, J.E.; Wang, J.; et al. Effect of KRAS Oncogene Substitutions on Protein Behavior: Implications for Signaling and Clinical Outcome. J. Natl. Cancer Inst. 2012, 104, 228–239. [Google Scholar] [CrossRef]
- Désage, A.-L.; Léonce, C.; Swalduz, A.; Ortiz-Cuaran, S. Targeting KRAS Mutant in Non-Small Cell Lung Cancer: Novel Insights Into Therapeutic Strategies. Front. Oncol. 2022, 12, 796832. [Google Scholar] [CrossRef]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS Isoforms and Mutations in Cancer at a Glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef]
- Ruppert, A.-M.; Beau-Faller, M.; Debieuvre, D.; Ouafik, L.; Westeel, V.; Rouquette, I.; Mazières, J.; Bringuier, P.-P.; Monnet, I.; Escande, F.; et al. Outcomes of Patients With Advanced NSCLC From the Intergroupe Francophone de Cancérologie Thoracique Biomarkers France Study by KRAS Mutation Subtypes. JTO Clin. Res. Rep. 2020, 1, 100052. [Google Scholar] [CrossRef] [PubMed]
- Finn, S.P.; Addeo, A.; Dafni, U.; Thunnissen, E.; Bubendorf, L.; Madsen, L.B.; Biernat, W.; Verbeken, E.; Hernandez-Losa, J.; Marchetti, A.; et al. Prognostic Impact of KRAS G12C Mutation in Patients With NSCLC: Results From the European Thoracic Oncology Platform Lungscape Project. J. Thorac. Oncol. 2021, 16, 990–1002. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Zhang, E.W.; Strickland, M.R.; Mendoza, D.P.; Lipkin, L.; Lennerz, J.K.; Gainor, J.F.; Heist, R.S.; Digumarthy, S.R. Clinical and Imaging Features of Non-Small Cell Lung Cancer with G12C KRAS Mutation. Cancers 2021, 13, 3572. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Franchini, F.; Alexander, M.; Officer, A.; Wong, H.-L.; IJzerman, M.; Desai, J.; Solomon, B.J. Real World Outcomes in KRAS G12C Mutation Positive Non-Small Cell Lung Cancer. Lung Cancer 2020, 146, 310–317. [Google Scholar] [CrossRef]
- Yu, H.A.; Sima, C.S.; Shen, R.; Kass, S.; Gainor, J.; Shaw, A.; Hames, M.; Iams, W.; Aston, J.; Lovly, C.M.; et al. Prognostic Impact of KRAS Mutation Subtypes in 677 Patients with Metastatic Lung Adenocarcinomas. J. Thorac. Oncol. 2015, 10, 431–437. [Google Scholar] [CrossRef]
- Shepherd, F.A.; Lacas, B.; Le Teuff, G.; Hainaut, P.; Jänne, P.A.; Pignon, J.-P.; Le Chevalier, T.; Seymour, L.; Douillard, J.-Y.; Graziano, S.; et al. Pooled Analysis of the Prognostic and Predictive Effects of TP53 Comutation Status Combined With KRAS or EGFR Mutation in Early-Stage Resected Non-Small-Cell Lung Cancer in Four Trials of Adjuvant Chemotherapy. J. Clin. Oncol. 2017, 35, 2018–2027. [Google Scholar] [CrossRef]
- Goulding, R.E.; Chenoweth, M.; Carter, G.C.; Boye, M.E.; Sheffield, K.M.; John, W.J.; Leusch, M.S.; Muehlenbein, C.E.; Li, L.; Jen, M.-H.; et al. KRAS Mutation as a Prognostic Factor and Predictive Factor in Advanced/Metastatic Non-Small Cell Lung Cancer: A Systematic Literature Review and Meta-Analysis. Cancer Treat. Res. Commun. 2020, 24, 100200. [Google Scholar] [CrossRef]
- Rulli, E.; Marabese, M.; Torri, V.; Farina, G.; Veronese, S.; Bettini, A.; Longo, F.; Moscetti, L.; Ganzinelli, M.; Lauricella, C.; et al. Value of KRAS as Prognostic or Predictive Marker in NSCLC: Results from the TAILOR Trial. Ann. Oncol. 2015, 26, 2079–2084. [Google Scholar] [CrossRef]
- Svaton, M.; Fiala, O.; Pesek, M.; Bortlicek, Z.; Minarik, M.; Benesova, L.; Topolcan, O. The Prognostic Role of KRAS Mutation in Patients with Advanced NSCLC Treated with Second- or Third-Line Chemotherapy. Anticancer Res. 2016, 36, 1077–1082. [Google Scholar]
- Mellema, W.W.; Masen-Poos, L.; Smit, E.F.; Hendriks, L.E.L.; Aerts, J.G.; Termeer, A.; Goosens, M.J.; Smit, H.J.M.; van den Heuvel, M.M.; van der Wekken, A.J.; et al. Comparison of Clinical Outcome after First-Line Platinum-Based Chemotherapy in Different Types of KRAS Mutated Advanced Non-Small-Cell Lung Cancer. Lung Cancer 2015, 90, 249–254. [Google Scholar] [CrossRef]
- Arbour, K.C.; Jordan, E.; Kim, H.R.; Dienstag, J.; Yu, H.A.; Sanchez-Vega, F.; Lito, P.; Berger, M.; Solit, D.B.; Hellmann, M.; et al. Effects of Co-Occurring Genomic Alterations on Outcomes in Patients with KRAS-Mutant Non–Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 334–340. [Google Scholar] [CrossRef] [PubMed]
- La Fleur, L.; Falk-Sörqvist, E.; Smeds, P.; Berglund, A.; Sundström, M.; Mattsson, J.S.; Brandén, E.; Koyi, H.; Isaksson, J.; Brunnström, H.; et al. Mutation Patterns in a Population-Based Non-Small Cell Lung Cancer Cohort and Prognostic Impact of Concomitant Mutations in KRAS and TP53 or STK11. Lung Cancer 2019, 130, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Riely, G.J.; Jordan, E.; Kim, H.R.; Yu, H.A.; Berger, M.F.; Solit, D.B.; Kris, M.G.; Ni, A.; Arcila, M.E.; Ladanyi, M. Association of Outcomes and Co-Occuring Genomic Alterations in Patients with KRAS-Mutant Non-Small Cell Lung Cancer. J. Clin. Oncol. 2016, 34, 9019. [Google Scholar] [CrossRef]
- Passaro, A.; Attili, I.; Rappa, A.; Vacirca, D.; Ranghiero, A.; Fumagalli, C.; Guarize, J.; Spaggiari, L.; de Marinis, F.; Barberis, M.; et al. Genomic Characterization of Concurrent Alterations in Non-Small Cell Lung Cancer (NSCLC) Harboring Actionable Mutations. Cancers 2021, 13, 2172. [Google Scholar] [CrossRef]
- Skoulidis, F.; Byers, L.A.; Diao, L.; Papadimitrakopoulou, V.A.; Tong, P.; Izzo, J.; Behrens, C.; Kadara, H.; Parra, E.R.; Canales, J.R.; et al. Co-Occurring Genomic Alterations Define Major Subsets of KRAS-Mutant Lung Adenocarcinoma with Distinct Biology, Immune Profiles, and Therapeutic Vulnerabilities. Cancer Discov. 2015, 5, 860–877. [Google Scholar] [CrossRef]
- Mahoney, C.L.; Choudhury, B.; Davies, H.; Edkins, S.; Greenman, C.; van Haaften, G.; Mironenko, T.; Santarius, T.; Stevens, C.; Stratton, M.R.; et al. LKB1/KRAS Mutant Lung Cancers Constitute a Genetic Subset of NSCLC with Increased Sensitivity to MAPK and MTOR Signalling Inhibition. Br. J. Cancer 2009, 100, 370–375. [Google Scholar] [CrossRef]
- Schrock, A.B.; Frampton, G.M.; Suh, J.; Chalmers, Z.R.; Rosenzweig, M.; Erlich, R.L.; Halmos, B.; Goldman, J.; Forde, P.; Leuenberger, K.; et al. Characterization of 298 Patients with Lung Cancer Harboring MET Exon 14 Skipping Alterations. J. Thorac. Oncol. 2016, 11, 1493–1502. [Google Scholar] [CrossRef]
- Suzawa, K.; Offin, M.; Lu, D.; Kurzatkowski, C.; Vojnic, M.; Smith, R.S.; Sabari, J.K.; Tai, H.; Mattar, M.; Khodos, I.; et al. Activation of KRAS Mediates Resistance to Targeted Therapy in MET Exon 14-Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 1248–1260. [Google Scholar] [CrossRef]
- Zhu, Y.; Lin, X.; Li, X.; Wu, L.; Chen, H.; Wang, W.; Xu, C.; Shen, J.; Wei, J.; Du, K. Concurrent ROS1 Gene Rearrangement and KRAS Mutation in Lung Adenocarcinoma: A Case Report and Literature Review. Thorac. Cancer 2018, 9, 159–163. [Google Scholar] [CrossRef]
- Schmid, S.; Gautschi, O.; Rothschild, S.; Mark, M.; Froesch, P.; Klingbiel, D.; Reichegger, H.; Jochum, W.; Diebold, J.; Früh, M. Clinical Outcome of ALK-Positive Non–Small Cell Lung Cancer (NSCLC) Patients with De Novo EGFR or KRAS Co-Mutations Receiving Tyrosine Kinase Inhibitors (TKIs). J. Thorac. Oncol. 2017, 12, 681–688. [Google Scholar] [CrossRef]
- Chabon, J.J.; Simmons, A.D.; Lovejoy, A.F.; Esfahani, M.S.; Newman, A.M.; Haringsma, H.J.; Kurtz, D.M.; Stehr, H.; Scherer, F.; Karlovich, C.A.; et al. Circulating Tumour DNA Profiling Reveals Heterogeneity of EGFR Inhibitor Resistance Mechanisms in Lung Cancer Patients. Nat. Commun. 2016, 7, 11815. [Google Scholar] [CrossRef] [PubMed]
- Rachiglio, A.M.; Fenizia, F.; Piccirillo, M.C.; Galetta, D.; Crinò, L.; Vincenzi, B.; Barletta, E.; Pinto, C.; Ferraù, F.; Lambiase, M.; et al. The Presence of Concomitant Mutations Affects the Activity of EGFR Tyrosine Kinase Inhibitors in EGFR-Mutant Non-Small Cell Lung Cancer (NSCLC) Patients. Cancers 2019, 11, 341. [Google Scholar] [CrossRef] [PubMed]
- Xiu, W.; Zhang, Q.; Yu, M.; Huang, Y.; Huang, M. Case Report: Outcome of Osimertinib Treatment in Lung Adenocarcinoma Patients With Acquired KRAS Mutations. Front. Oncol. 2021, 11, 630256. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, T.; Ohmori, T.; Ohba, M.; Arata, S.; Murata, Y.; Kusumoto, S.; Ando, K.; Ishida, H.; Ohnishi, T.; Sasaki, Y. Distinct Afatinib Resistance Mechanisms Identified in Lung Adenocarcinoma Harboring an EGFR Mutation. Mol. Cancer Res. 2017, 15, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Ostrem, J.M.L.; Shokat, K.M. Direct Small-Molecule Inhibitors of KRAS: From Structural Insights to Mechanism-Based Design. Nat. Rev. Drug Discov. 2016, 15, 771–785. [Google Scholar] [CrossRef]
- Lito, P.; Solomon, M.; Li, L.-S.; Hansen, R.; Rosen, N. Allele-Specific Inhibitors Inactivate Mutant KRAS G12C by a Trapping Mechanism. Science 2016, 351, 604–608. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRAS G12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS G12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef]
- Ramalingam, S.; Skoulidis, F.; Govindan, R.; Velcheti, V.; Li, B.; Besse, B.; Dy, G.; Kim, D.; Schuler, M.; Vincent, M.; et al. P52.03 Efficacy of Sotorasib in KRAS p.G12C-Mutated NSCLC with Stable Brain Metastases: A Post-Hoc Analysis of CodeBreaK 100. J. Thorac. Oncol. 2021, 16, S1123. [Google Scholar] [CrossRef]
- Lumakras®/Lumykras® (Sotorasib) Demonstrates Superior Progression-Free Survival over Docetaxel in First Positive Phase 3 Trial of a Kras G12c Inhibitor in Non-Small Cell Lung Cancer. Available online: https://www.amgen.com/newsroom/press-releases/2022/09/lumakraslumykras-sotorasib-demonstrates-superior-progressionfree-survival-over-docetaxel-in-first-positive-phase-3-trial-of-a-kras-g12c-inhibitor-in-nonsmall-cell-lung-cancer (accessed on 20 September 2022).
- Nakajima, E.C.; Drezner, N.; Li, X.; Mishra-Kalyani, P.S.; Liu, Y.; Zhao, H.; Bi, Y.; Liu, J.; Rahman, A.; Wearne, E.; et al. FDA Approval Summary: Sotorasib for KRAS G12C-Mutated Metastatic NSCLC. Clin. Cancer Res. 2021, 28, 1482–1486. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.-H.I.; Jänne, P.A.; Leal, T.A.; Rybkin, I.I.; Sabari, J.K.; Barve, M.A.; Bazhenova, L.A.; Johnson, M.L.; Velastegui, K.L.; Cilliers, C.; et al. First-in-Human Phase I/IB Dose-Finding Study of Adagrasib (MRTX849) in Patients With Advanced KRASG12C Solid Tumors (KRYSTAL-1). J. Clin. Oncol. 2022, 40, 2530–2538. [Google Scholar] [CrossRef] [PubMed]
- Jänne, P.A.; Riely, G.J.; Gadgeel, S.M.; Heist, R.S.; Ou, S.-H.I.; Pacheco, J.M.; Johnson, M.L.; Sabari, J.K.; Leventakos, K.; Yau, E.; et al. Adagrasib in Non–Small-Cell Lung Cancer Harboring a KRASG12C Mutation. N. Engl. J. Med. 2022, 387, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Mirati Therapeutics Inc. A Randomized Phase 3 Study of MRTX849 Versus Docetaxel in Patients with Previously Treated Non-Small Cell Lung Cancer with KRAS G12C Mutation. 2022. Available online: https://clinicaltrials.gov/ (accessed on 3 November 2022).
- New Drug Application for Adagrasib Accepted by FDA for KRAS G12C+ NSCLC. Available online: https://www.cancernetwork.com/view/new-drug-application-for-adagrasib-accepted-by-fda-for-kras-g12c-nsclc (accessed on 21 September 2022).
- Evidence of Antitumor Effect with GDC-6036 Monotherapy in KRAS G12C+ NSCLC Revealed at 2022 WCLC. Available online: https://www.cancernetwork.com/view/evidence-of-antitumor-effect-with-gdc-6036-monotherapy-in-kras-g12c-nsclc-revealed-at-2022-wclc (accessed on 8 September 2022).
- A Study to Evaluate the Safety, Pharmacokinetics, and Activity of GDC-6036 Alone or in Combination in Participants with Advanced or Metastatic Solid Tumors with a KRAS G12C Mutation—No Study Results Posted—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/results/NCT04449874 (accessed on 23 September 2022).
- Cohen-Armon, M. Exclusive Modifications of NuMA in Malignant Epithelial Cells: A Potential Therapeutic Mechanism. Drug Discov. Today 2022, 27, 1205–1209. [Google Scholar] [CrossRef] [PubMed]
- Eli Lilly and Company. A Phase 1/2 Study of LY3499446 Administered to Patients with Advanced Solid Tumors with KRAS G12C Mutation. 2021. Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Novartis Pharmaceuticals. A Phase Ib/II Open-Label, Multi-Center Dose Escalation Study of JDQ443 in Patients with Advanced Solid Tumors Harboring the KRAS G12C Mutation. 2022. Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Jacobio Pharmaceuticals Co., Ltd. A Phase 1/2a Clinical Study to Evaluate the Safety, Tolerability, Pharmacokinetics and Antitumor Activity of JAB-21822 in Combination with JAB-3312 in Patients with Advanced Solid Tumors Harboring KRAS p.G12C Mutation. 2022. Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Boehringer Ingelheim. A Phase Ia/Ib, Open-Label, Multicentre Dose-Escalation and Expansion Study to Investigate the Safety, Pharmacokinetics and Preliminary Efficacy of BI 1823911 as a Monotherapy and in Combination with Other Anti-Cancer Therapies in Patients with Advanced or Metastatic Solid Tumours Expressing KRAS G12C Mutation. 2022. Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- InventisBio Co., Ltd. A Phase 1/2, Open Label Study to Evaluate the Safety, Tolerability, Pharmacokinetics and Efficacy of D-1553 in Combination with IN10018 in Subjects with Advanced or Metastatic Solid Tumors With KRasG12C Mutation. 2022. Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Merck Sharp & Dohme LLC. A Phase 1, Open-Label, Multicenter Study to Assess Safety, Tolerability, PK, and Efficacy of MK-1084 as Monotherapy and in Combination with Pembrolizumab in Subjects with KRASG12C Mutant Advanced Solid Tumors. 2022. Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Eli Lilly and Company. A Phase 1a/1b Study of LY3537982 in Patients with KRAS G12C-Mutant Advanced Solid Tumors. Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Nagasaka, M.; Potugari, B.; Nguyen, A.; Sukari, A.; Azmi, A.S.; Ou, S.-H.I. KRAS Inhibitors– Yes but What next? Direct Targeting of KRAS–Vaccines, Adoptive T Cell Therapy and Beyond. Cancer Treat. Rev. 2021, 101, 102309. [Google Scholar] [CrossRef]
- Koltun, E.; Cregg, J.; Rice, M.A.; Whalen, D.M.; Freilich, R.; Jiang, J.; Hansen, R.; Bermingham, A.; Knox, J.E.; Dinglasan, J.; et al. Abstract 1260: First-in-Class, Orally Bioavailable KRASG12V(ON) Tri-Complex Inhibitors, as Single Agents and in Combinations, Drive Profound Anti-Tumor Activity in Preclinical Models of KRASG12V Mutant Cancers. Cancer Res. 2021, 81, 1260. [Google Scholar] [CrossRef]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS Mutation: From Undruggable to Druggable in Cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef]
- Zhao, Y.; Murciano-Goroff, Y.R.; Xue, J.Y.; Ang, A.; Lucas, J.; Mai, T.T.; Da Cruz Paula, A.F.; Saiki, A.Y.; Mohn, D.; Achanta, P.; et al. Diverse Alterations Associated with Resistance to KRAS(G12C) Inhibition. Nature 2021, 599, 679–683. [Google Scholar] [CrossRef]
- Awad, M.M.; Liu, S.; Rybkin, I.I.; Arbour, K.C.; Dilly, J.; Zhu, V.W.; Johnson, M.L.; Heist, R.S.; Patil, T.; Riely, G.J.; et al. Acquired Resistance to KRAS G12C Inhibition in Cancer. N. Engl. J. Med. 2021, 384, 2382–2393. [Google Scholar] [CrossRef]
- Addario Lung Cancer Medical Institute. A Non-Interventional, Non-Treatment, Non-Randomized, Single Coordinating Center, Decentralized Bio-Specimen Collection Study in USA-Based Adult Subjects with Acquired Resistance to KRAS Inhibitors. Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Falchook, G.; Li, B.T.; Marrone, K.A.; Bestvina, C.M.; Langer, C.J.; Krauss, J.C.; Strickler, J.H.; Meloni, A.; Dai, T.; Varrieur, T.; et al. OA03.03 Sotorasib in Combination with RMC-4630, a SHP2 Inhibitor, in KRAS p.G12C-Mutated NSCLC and Other Solid Tumors. J. Thorac. Oncol. 2022, 17, S8. [Google Scholar] [CrossRef]
- Gandara, D.; Marrone, K.; Govindan, R.; Skoulidis, F.; Durm, G.; Clarke, J.; Frank, R.; Krauss, J.; Snyder, W.; Dai, T.; et al. Abstract P05-02: A Phase 1b Study Evaluating the Combination of Sotorasib, a KRASG12C Inhibitor, and Afatinib, a Pan-ErbB Tyrosine Kinase Inhibitor, in Advanced KRAS p.G12C Mutated Non-Small Cell Lung Cancer (NSCLC). Mol. Cancer Ther. 2021, 20, P05-02. [Google Scholar] [CrossRef]
- Chen, N.; Fang, W.; Lin, Z.; Peng, P.; Wang, J.; Zhan, J.; Hong, S.; Huang, J.; Liu, L.; Sheng, J.; et al. KRAS Mutation-Induced Upregulation of PD-L1 Mediates Immune Escape in Human Lung Adenocarcinoma. Cancer Immunol. Immunother. 2017, 66, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- D’Incecco, A.; Andreozzi, M.; Ludovini, V.; Rossi, E.; Capodanno, A.; Landi, L.; Tibaldi, C.; Minuti, G.; Salvini, J.; Coppi, E.; et al. PD-1 and PD-L1 Expression in Molecularly Selected Non-Small-Cell Lung Cancer Patients. Br. J. Cancer 2015, 112, 95–102. [Google Scholar] [CrossRef]
- Liu, C.; Zheng, S.; Jin, R.; Wang, X.; Wang, F.; Zang, R.; Xu, H.; Lu, Z.; Huang, J.; Lei, Y.; et al. The Superior Efficacy of Anti-PD-1/PD-L1 Immunotherapy in KRAS-Mutant Non-Small Cell Lung Cancer That Correlates with an Inflammatory Phenotype and Increased Immunogenicity. Cancer Lett. 2020, 470, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.E.; El-Refai, S.M.; Sha, W.; Puccini, A.; Grothey, A.; George, T.J.; Hwang, J.J.; O’Neil, B.; Barrett, A.S.; Kadakia, K.C.; et al. Landscape of KRASG12C, Associated Genomic Alterations, and Interrelation With Immuno-Oncology Biomarkers in KRAS-Mutated Cancers. JCO Precis. Oncol. 2022, 6, e2100245. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Li, G.; Wang, Y.; Wang, Y.; Zhao, S.; Haihong, P.; Zhao, H.; Wang, Y. PD-L1 Expression in Lung Cancer and Its Correlation with Driver Mutations: A Meta-Analysis. Sci. Rep. 2017, 7, 10255. [Google Scholar] [CrossRef]
- Petrelli, F.; Maltese, M.; Tomasello, G.; Conti, B.; Borgonovo, K.; Cabiddu, M.; Ghilardi, M.; Ghidini, M.; Passalacqua, R.; Barni, S.; et al. Clinical and Molecular Predictors of PD-L1 Expression in Non–Small-Cell Lung Cancer: Systematic Review and Meta-Analysis. Clin. Lung Cancer 2018, 19, 315–322. [Google Scholar] [CrossRef]
- Herbst, R.S.; Lopes, G.; Kowalski, D.M.; Kasahara, K.; Wu, Y.-L.; De Castro, G.; Cho, B.C.; Turna, H.Z.; Cristescu, R.; Aurora-Garg, D.; et al. LBA4 Association of KRAS Mutational Status with Response to Pembrolizumab Monotherapy given as First-Line Therapy for PD-L1-Positive Advanced Non-Squamous NSCLC in Keynote-042. Ann. Oncol. 2019, 30, xi63–xi64. [Google Scholar] [CrossRef]
- Sun, L.; Hsu, M.; Cohen, R.B.; Langer, C.J.; Mamtani, R.; Aggarwal, C. Association Between KRAS Variant Status and Outcomes With First-Line Immune Checkpoint Inhibitor–Based Therapy in Patients With Advanced Non–Small-Cell Lung Cancer. JAMA Oncol. 2021, 7, 937–939. [Google Scholar] [CrossRef]
- Jeanson, A.; Tomasini, P.; Souquet-Bressand, M.; Brandone, N.; Boucekine, M.; Grangeon, M.; Chaleat, S.; Khobta, N.; Milia, J.; Mhanna, L.; et al. Efficacy of Immune Checkpoint Inhibitors in KRAS-Mutant Non-Small Cell Lung Cancer (NSCLC). J. Thorac. Oncol. 2019, 14, 1095–1101. [Google Scholar] [CrossRef]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus Docetaxel in Patients with Previously Treated Non-Small-Cell Lung Cancer (OAK): A Phase 3, Open-Label, Multicentre Randomised Controlled Trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef]
- Lee, C.K.; Man, J.; Lord, S.; Cooper, W.; Links, M.; Gebski, V.; Herbst, R.S.; Gralla, R.J.; Mok, T.; Yang, J.C.-H. Clinical and Molecular Characteristics Associated With Survival Among Patients Treated With Checkpoint Inhibitors for Advanced Non–Small Cell Lung Carcinoma: A Systematic Review and Meta-Analysis. JAMA Oncol. 2018, 4, 210. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, H.S.; Kim, B.J. Prognostic Value of KRAS Mutation in Advanced Non-Small-Cell Lung Cancer Treated with Immune Checkpoint Inhibitors: A Meta-Analysis and Review. Oncotarget 2017, 8, 48248–48252. [Google Scholar] [CrossRef] [PubMed]
- Landre, T.; Justeau, G.; Assié, J.-B.; Chouahnia, K.; Davoine, C.; Taleb, C.; Chouaïd, C.; Duchemann, B. Anti-PD-(L)1 for KRAS-Mutant Advanced Non-Small-Cell Lung Cancers: A Meta-Analysis of Randomized-Controlled Trials. Cancer Immunol. Immunother. 2022, 71, 719–726. [Google Scholar] [CrossRef]
- Guaitoli, G.; Tiseo, M.; Di Maio, M.; Friboulet, L.; Facchinetti, F. Immune Checkpoint Inhibitors in Oncogene-Addicted Non-Small Cell Lung Cancer: A Systematic Review and Meta-Analysis. Transl. Lung Cancer Res. 2021, 10, 2890–2916. [Google Scholar] [CrossRef]
- Mazieres, J.; Drilon, A.; Lusque, A.; Mhanna, L.; Cortot, A.B.; Mezquita, L.; Thai, A.A.; Mascaux, C.; Couraud, S.; Veillon, R.; et al. Immune Checkpoint Inhibitors for Patients with Advanced Lung Cancer and Oncogenic Driver Alterations: Results from the IMMUNOTARGET Registry. Ann. Oncol. 2019, 30, 1321–1328. [Google Scholar] [CrossRef]
- Mirati Therapeutics Inc. A Phase 1/2 Multiple Expansion Cohort Trial of MRTX849 in Patients with Advanced Solid Tumors with KRAS G12C Mutation KRYSTAL-1. Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Mirati Therapeutics Inc. A Phase 2 Trial of MRTX849 Monotherapy and in Combination with Pembrolizumab in Patients with Advanced Non-Small Cell Lung Cancer with KRAS G12C Mutation. Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Li, B.T.; Falchook, G.S.; Durm, G.A.; Burns, T.F.; Skoulidis, F.; Ramalingam, S.S.; Spira, A.; Bestvina, C.M.; Goldberg, S.B.; Veluswamy, R.; et al. OA03.06 CodeBreaK 100/101: First Report of Safety/Efficacy of Sotorasib in Combination with Pembrolizumab or Atezolizumab in Advanced KRAS p.G12C NSCLC. J. Thorac. Oncol. 2022, 17, S10–S11. [Google Scholar] [CrossRef]
- Liu, C.; Zheng, S.; Wang, Z.; Wang, S.; Wang, X.; Yang, L.; Xu, H.; Cao, Z.; Feng, X.; Xue, Q.; et al. KRAS-G12D Mutation Drives Immune Suppression and the Primary Resistance of Anti-PD-1/PD-L1 Immunotherapy in Non-Small Cell Lung Cancer. Cancer Commun. 2022, 42, 828–847. [Google Scholar] [CrossRef]
- Ricciuti, B.; Alessi, J.V.; Elkrief, A.; Wang, X.; Cortellini, A.; Li, Y.Y.; Vaz, V.R.; Gupta, H.; Pecci, F.; Barrichello, A.; et al. Dissecting the Clinicopathologic, Genomic, and Immunophenotypic Correlates of KRASG12D-Mutated Non-Small-Cell Lung Cancer. Ann. Oncol. 2022, 33, 1029–1040. [Google Scholar] [CrossRef]
- Arbour, K.C.; Rizvi, H.; Plodkowski, A.J.; Hellmann, M.D.; Knezevic, A.; Heller, G.; Yu, H.A.; Ladanyi, M.; Kris, M.G.; Arcila, M.E.; et al. Treatment Outcomes and Clinical Characteristics of Patients with KRAS-G12C-Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. 2021, 27, 2209–2215. [Google Scholar] [CrossRef]
- Dong, Z.-Y.; Zhong, W.-Z.; Zhang, X.-C.; Su, J.; Xie, Z.; Liu, S.-Y.; Tu, H.-Y.; Chen, H.-J.; Sun, Y.-L.; Zhou, Q.; et al. Potential Predictive Value of TP53 and KRAS Mutation Status for Response to PD-1 Blockade Immunotherapy in Lung Adenocarcinoma. Clin. Cancer Res. 2017, 23, 3012–3024. [Google Scholar] [CrossRef] [PubMed]
- Kadara, H.; Choi, M.; Zhang, J.; Parra, E.R.; Rodriguez-Canales, J.; Gaffney, S.G.; Zhao, Z.; Behrens, C.; Fujimoto, J.; Chow, C.; et al. Whole-Exome Sequencing and Immune Profiling of Early-Stage Lung Adenocarcinoma with Fully Annotated Clinical Follow-Up. Ann. Oncol. 2017, 28, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Xu, B.; Zhang, H.; Fang, S. Lung Adenocarcinoma Patients with KEAP1 Mutation Harboring Low Immune Cell Infiltration and Low Activity of Immune Environment. Thorac. Cancer 2021, 12, 2458–2467. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [PubMed]
- Ricciuti, B.; Arbour, K.C.; Lin, J.J.; Vajdi, A.; Vokes, N.; Hong, L.; Zhang, J.; Tolstorukov, M.Y.; Li, Y.Y.; Spurr, L.F.; et al. Diminished Efficacy of Programmed Death-(Ligand)1 Inhibition in STK11- and KEAP1-Mutant Lung Adenocarcinoma Is Affected by KRAS Mutation Status. J. Thorac. Oncol. 2022, 17, 399–410. [Google Scholar] [CrossRef]
- Biton, J.; Mansuet-Lupo, A.; Pécuchet, N.; Alifano, M.; Ouakrim, H.; Arrondeau, J.; Boudou-Rouquette, P.; Goldwasser, F.; Leroy, K.; Goc, J.; et al. TP53, STK11, and EGFR Mutations Predict Tumor Immune Profile and the Response to Anti–PD-1 in Lung Adenocarcinoma. Clin. Cancer Res. 2018, 24, 5710–5723. [Google Scholar] [CrossRef]
- Assoun, S.; Theou-Anton, N.; Nguenang, M.; Cazes, A.; Danel, C.; Abbar, B.; Pluvy, J.; Gounant, V.; Khalil, A.; Namour, C.; et al. Association of TP53 Mutations with Response and Longer Survival under Immune Checkpoint Inhibitors in Advanced Non-Small-Cell Lung Cancer. Lung Cancer 2019, 132, 65–71. [Google Scholar] [CrossRef]
- Sotorasib Improves PFS in KRAS G12C-Mutated NSCLC. Available online: https://dailyreporter.esmo.org/esmo-congress-2022/top/sotorasib-improves-pfs-versus-docetaxel-in-patients-with-pre-treated-kras-g12c-mutated-nsclc (accessed on 16 September 2022).
- Amgen. A Phase 1b/2, Protocol Evaluating the Safety, Tolerability, Pharmacokinetics, and Efficacy of Sotorasib Monotherapy and in Combination with Other Anti-Cancer Therapies in Subjects with Advanced Solid Tumors with KRAS p.G12C Mutation (CodeBreak 101). Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Mirati Therapeutics Inc. A Phase 1/2 Trial of MRTX849 in Combination with TNO155 in Patients with Advanced Solid Tumors with KRAS G12C Mutation KRYSTAL 2. Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Verastem, Inc. A Phase 1/2 Study of VS-6766 in Combination with Sotorasib in Patients with KRAS G12C Mutant Non-Small Cell Lung Cancer (NSCLC). Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Mirati Therapeutics Inc. A Phase 1/1b Trial of MRTX849 in Combination with BI 1701963 in Patients with Advanced Solid Tumors with KRAS G12C Mutation. Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Ruess, D.A.; Heynen, G.J.; Ciecielski, K.J.; Ai, J.; Berninger, A.; Kabacaoglu, D.; Görgülü, K.; Dantes, Z.; Wörmann, S.M.; Diakopoulos, K.N.; et al. Mutant KRAS-Driven Cancers Depend on PTPN11/SHP2 Phosphatase. Nat. Med. 2018, 24, 954–960. [Google Scholar] [CrossRef]
- Mainardi, S.; Mulero-Sánchez, A.; Prahallad, A.; Germano, G.; Bosma, A.; Krimpenfort, P.; Lieftink, C.; Steinberg, J.D.; de Wit, N.; Gonçalves-Ribeiro, S.; et al. SHP2 Is Required for Growth of KRAS-Mutant Non-Small-Cell Lung Cancer in Vivo. Nat. Med. 2018, 24, 961–967. [Google Scholar] [CrossRef]
- Revolution Medicines, Inc. A Phase 2, Open-Label, Multicenter Study of the Combination of RMC-4630 and Sotorasib for Non-Small Cell Lung Cancer Subjects with KRASG12C Mutation after Failure of Prior Standard Therapies. Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Moll, H.P.; Pranz, K.; Musteanu, M.; Grabner, B.; Hruschka, N.; Mohrherr, J.; Aigner, P.; Stiedl, P.; Brcic, L.; Laszlo, V.; et al. Afatinib Restrains K-RAS Driven Lung Tumorigenesis. Sci. Transl. Med. 2018, 10, eaao2301. [Google Scholar] [CrossRef]
- Li, S.; Liu, S.; Deng, J.; Akbay, E.A.; Hai, J.; Ambrogio, C.; Zhang, L.; Zhou, F.; Jenkins, R.W.; Adeegbe, D.O.; et al. Assessing Therapeutic Efficacy of MEK Inhibition in a KRASG12C-Driven Mouse Model of Lung Cancer. Clin. Cancer Res. 2018, 24, 4854–4864. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Shcherba, M.; Pendurti, G.; Liang, Y.; Piperdi, B.; Perez-Soler, R. Targeting the PI3K/AKT/MTOR Pathway: Potential for Lung Cancer Treatment. Lung Cancer Manag. 2014, 3, 67–75. [Google Scholar] [CrossRef]
- Coma, S.; Chowdhury, S.; Pachter, J.A. Abstract 1263: Dual RAF/MEK Inhibitor VS-6766 Enhances Antitumor Efficacy of KRAS-G12C Inhibitors through a Vertical Pathway Inhibition Strategy. Cancer Res. 2021, 81, 1263. [Google Scholar] [CrossRef]
- Cáceres-Gutiérrez, R.E.; Alfaro-Mora, Y.; Andonegui, M.A.; Díaz-Chávez, J.; Herrera, L.A. The Influence of Oncogenic RAS on Chemotherapy and Radiotherapy Resistance Through DNA Repair Pathways. Front. Cell. Dev. Biol. 2022, 10, 751367. [Google Scholar] [CrossRef] [PubMed]
- Southwest Oncology Group. A Phase II Study of AMG 510 in Participants with Previously Treated Stage IV or Recurrent KRAS G12C Mutated Non-Squamous Non-Small Cell Lung Cancer (ECOG-ACRIN LUNG-MAP SUB-STUDY). Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Fundación GECP. Phase II Clinical Trial of AMG510 (Sotorasib) in Stage III Unresectable NSCLC KRAS p.G12C Patients and Medically Ineligible for Concurrent Chemo-Radiotherapy. Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Fox Chase Cancer Center. Neoadjuvant Sotorasib in KRAS G12C Mutated, Resectable, Stage Ib-IIIA Non-Small Cell Lung Cancer (NSCLC). Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- M.D. Anderson Cancer Center. A Phase II Study of Neoadjuvant Sotorasib in Combination with Cisplatin or Carboplatin and Pemetrexed for Surgically Resectable Stage IIA-IIIB Non-Squamous Non-Small Cell Lung Cancer with a KRAS p.G12C Mutation. Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins. Phase 2 Trial of Neoadjuvant KRAS G12C Directed Therapy with Adagrasib (MRTX849) with or without Nivolumab in Resectable Non-Small Cell Lung Cancer (Neo-KAN). Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Amgen. A Phase 2, Multicenter, Open-Label Study of Sotorasib (AMG 510) in Subjects with Stage IV NSCLC Whose Tumors Harbor a KRAS G12C Mutation in Need of First-Line Treatment (CodeBreaK 201). Available online: https://clinicaltrials.gov/ (accessed on 24 September 2022).
- Ceddia, S.; Landi, L.; Cappuzzo, F. KRAS-Mutant Non-Small-Cell Lung Cancer: From Past Efforts to Future Challenges. Int. J. Mol. Sci. 2022, 23, 9391. [Google Scholar] [CrossRef] [PubMed]
- Adderley, H.; Blackhall, F.H.; Lindsay, C.R. KRAS-Mutant Non-Small Cell Lung Cancer: Converging Small Molecules and Immune Checkpoint Inhibition. EBioMedicine 2019, 41, 711–716. [Google Scholar] [CrossRef]
- Addeo, A.; Banna, G.L.; Friedlaender, A. KRAS G12C Mutations in NSCLC: From Target to Resistance. Cancers 2021, 13, 2541. [Google Scholar] [CrossRef]
- Xie, M.; Xu, X.; Fan, Y. KRAS-Mutant Non-Small Cell Lung Cancer: An Emerging Promisingly Treatable Subgroup. Front. Oncol. 2021, 11, 672612. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).