

Use of High-Plex Data Reveals Novel Insights into the Tumour Microenvironment of Clear Cell Renal Cell Carcinoma
 , , , , ,
, , , , ,  and
and
Abstract
Simple Summary
Abstract
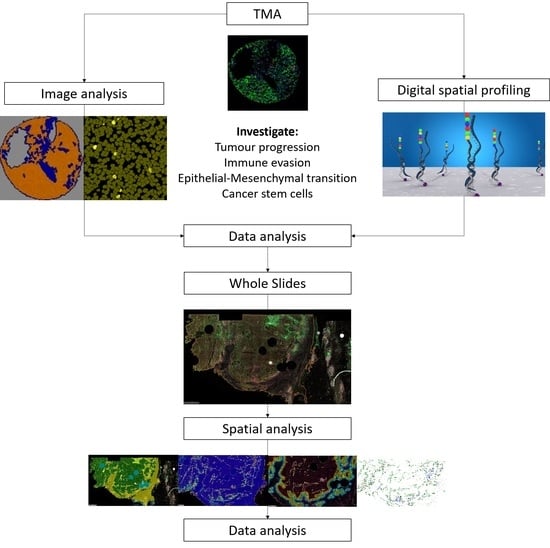
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Antibody Optimisation
2.3. Multiplex Immunofluorescence
2.4. GeoMx® Digital Spatial Profiling
2.4.1. Sample Preparation
2.4.2. DSP and nCounter® Readout
2.4.3. DSP Quality Control and Normalisation
2.5. Image Analysis
2.6. Statistical Analysis
3. Results (Tissue Microarray)
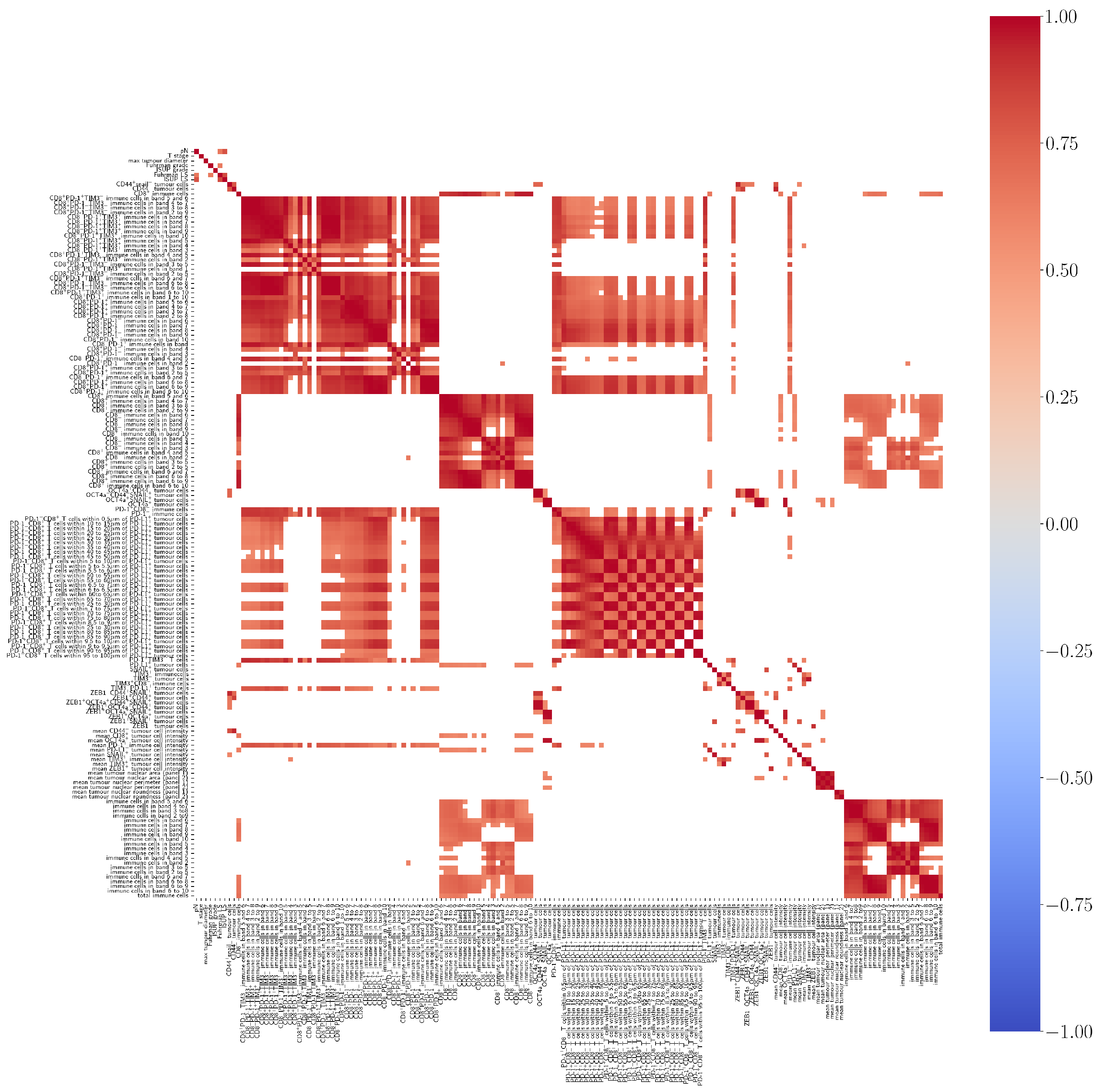
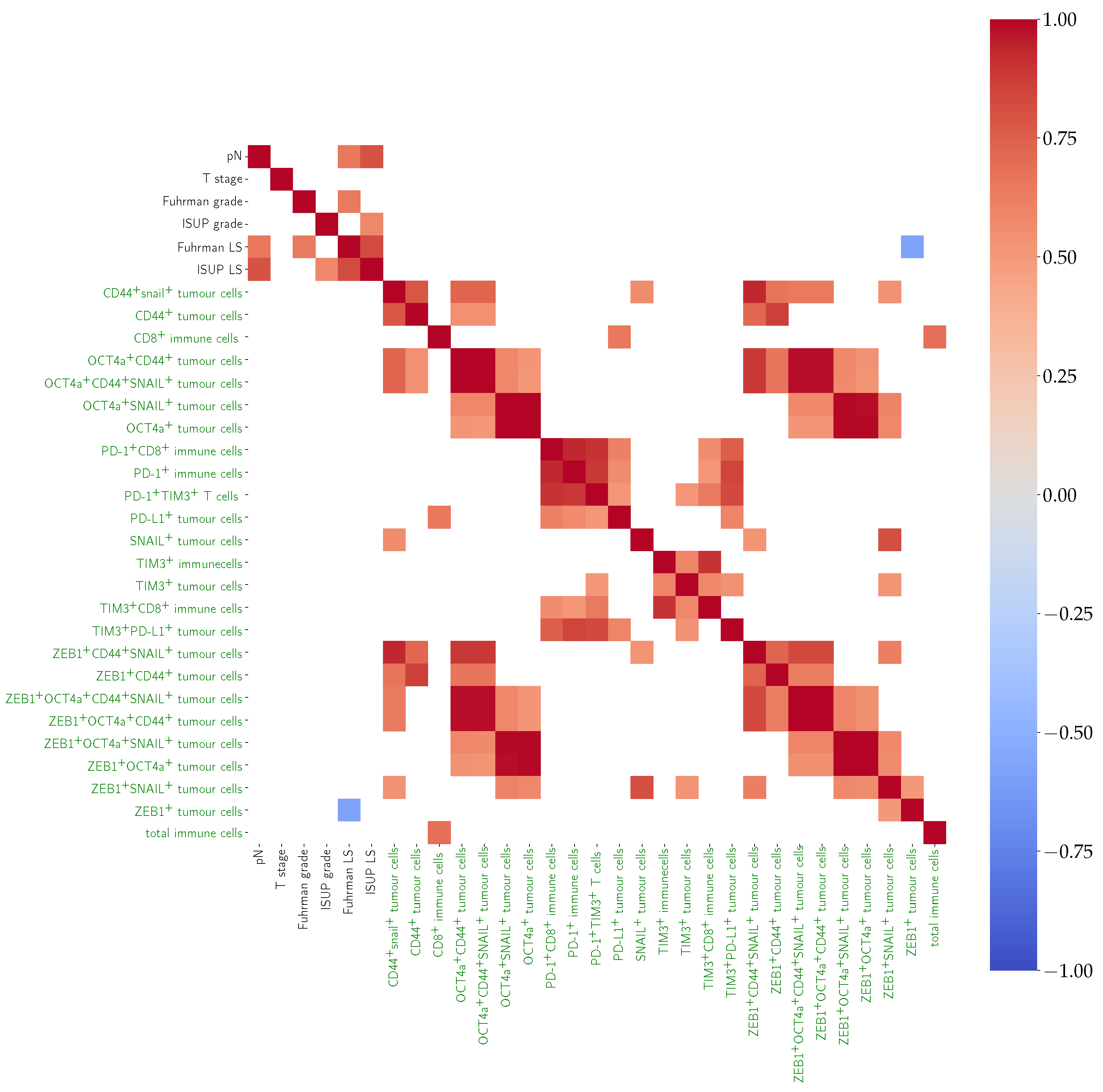
3.1. Pearson’s Correlation
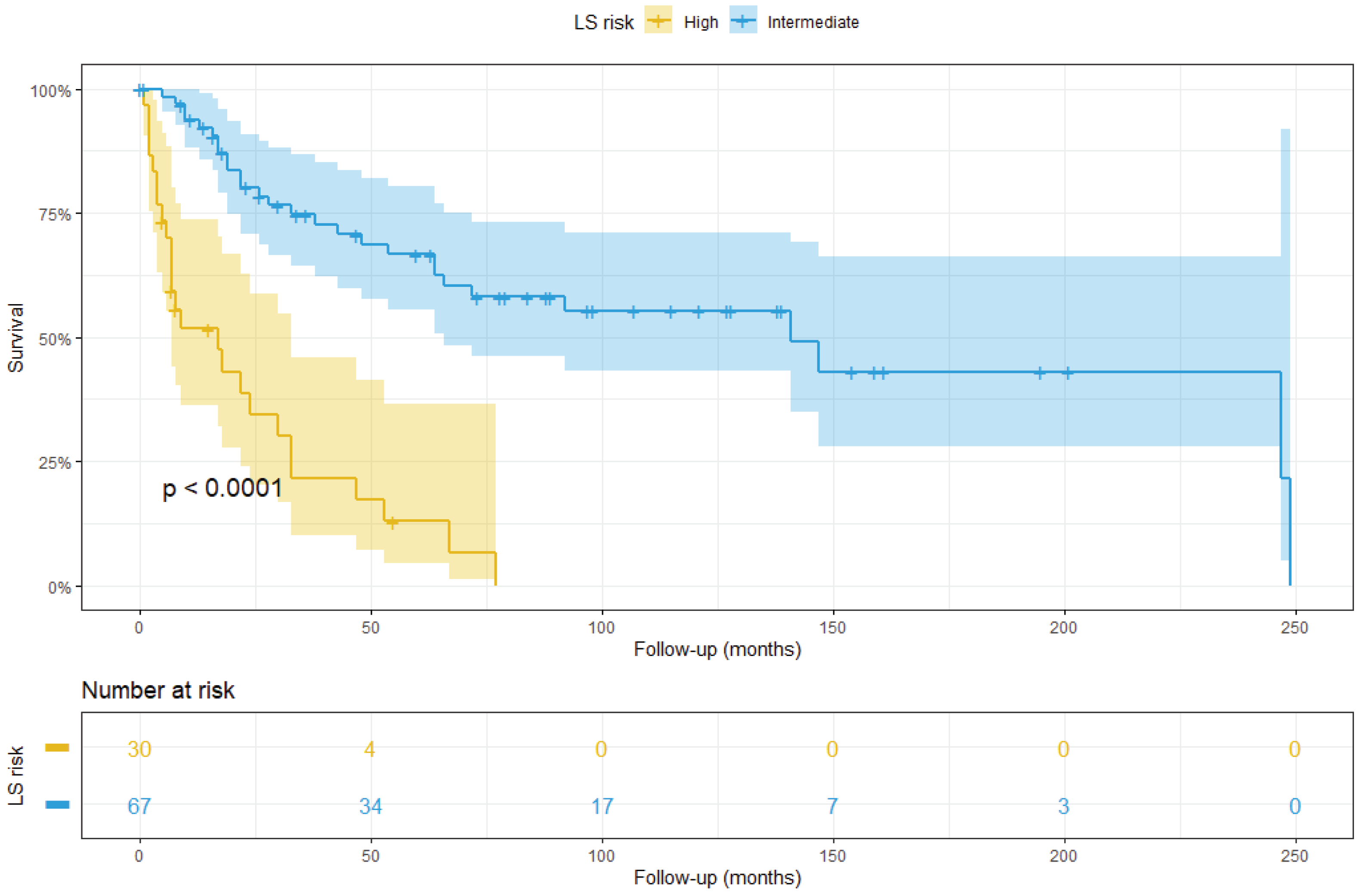
3.2. Survival Analysis
3.2.1. Univariate Cox Regression
3.2.2. Multivariate Cox Regression
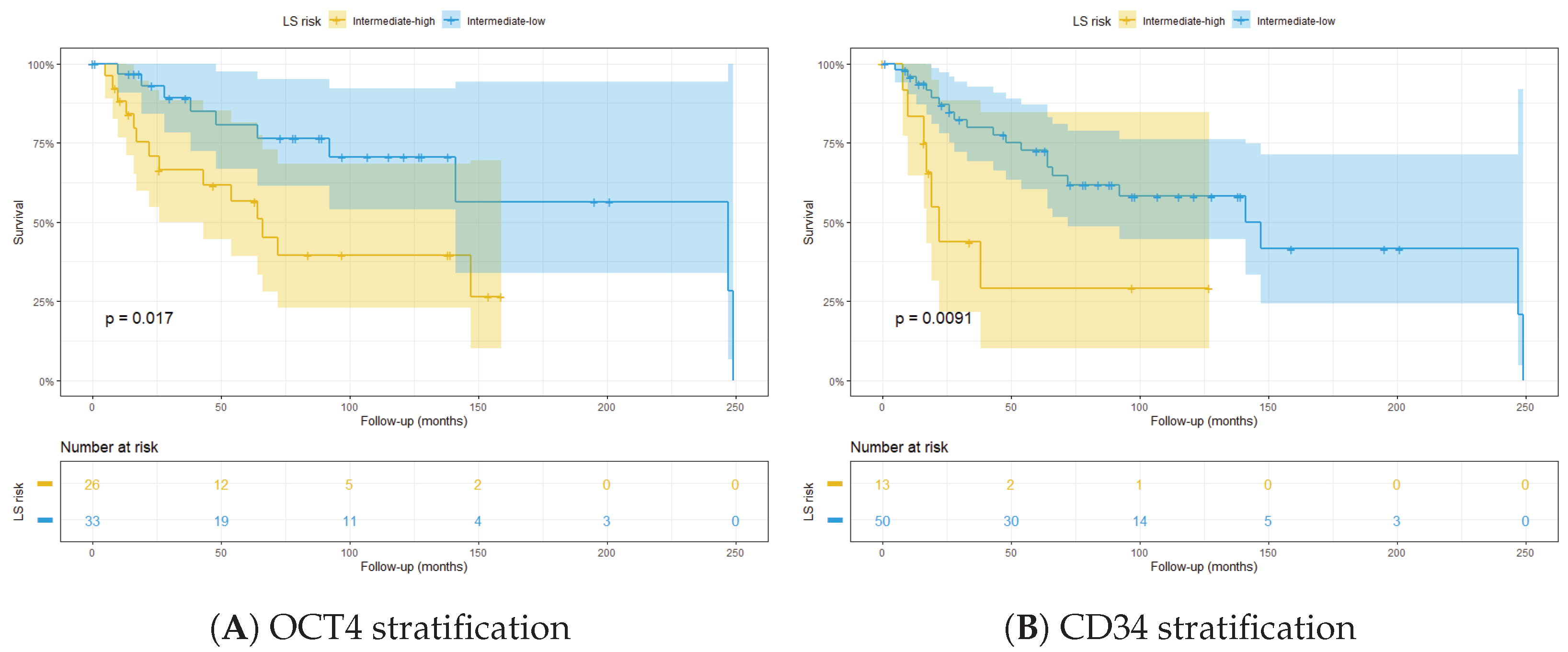
3.3. Integrated LS
4. Results (Whole Slides)
4.1. Pearson’s Correlation
4.2. Cox Regression
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Leibovich Score

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumour status | Score |
|---|---|
| pT1a | 0 |
| pT1b | 2 |
| pT2 | 3 |
| pT3a | 4 |
| pT3b | 4 |
| pT3c | 4 |
| pT4 | 4 |
| Lymph node involvement | Score |
| pNx | 0 |
| pN0 | 0 |
| pN1 | 2 |
| pN2 | 2 |
| Tumour size | Score |
| <10 cm | 0 |
| ≥10 cm | 1 |
| Nuclear grade | Score |
| 1 | 0 |
| 2 | 0 |
| 3 | 1 |
| 4 | 3 |
| Necrosis | Score |
| Absent | 0 |
| Present | 1 |
| Risk group | Score |
| Low | 0–2 |
| Intermediate | 3–5 |
| High | ≥6 |
Appendix B. Patient Information
| Gender | Male | Female | NA | ||
|---|---|---|---|---|---|
| 86 (57%) | 60 (40%) | 4 (3%) | |||
| Age (years) | Mean | Min | Max | ||
| 64 | 32 | 94 | |||
| Follow-up (months) | Mean | Min | Max | ||
| 60 | 4 | 252 | |||
| Status at end of FU | Dead | Alive | NA | ||
| 102 (68%) | 40 (26.6%) | 8 (5.3%) | |||
| Cancer-related death | Yes | No | NA | ||
| 75 (50%) | 75 (50%) | - | |||
| Metastasis at diagnosis | Yes | No | NA | ||
| 46 (30%) | 105 (70%) | - | |||
| Leibovich Score risk | High | Intermediate | Low | ||
| 17 (13.7%) | 99 (79.8%) | 8 (6.5%) | |||
| Lymph-node involvement | Yes | No | NA | ||
| 25 (16%) | 125 (84%) | - | |||
| pT Stage | 1 | 2 | 3 | 4 | NA |
| 8 (6.5%) | 11 (8.9%) | 100 (81.5%) | 4 (3.2%) | - | |
| ISUP Grade | 1 | 2 | 3 | 4 | NA |
| 19 (15.3%) | 49 (39.5%) | 30 (24.2%) | 3 (2.4%) | - |
Appendix C. Antibodies
| Antibody | Brand (Catalog No) | Species | Dilution | Target |
|---|---|---|---|---|
| CD163 | Cell Marque (MRQ-26) | Mouse | 1:3000 | M2 TAMs |
| HIF-2 | Abcam (ab8265) | Mouse | 1:400 | Hypoxia |
| CD105 | Abcam (Ab114052) | Mouse | 1:1500 | Blood vessels |
| CD8 | Agilent (M7 103) | Mouse | 1:200 | Cytotoxic T cells |
| CA9 | Novus Biological (NB100-417) | Rabbit | 1:100 | Tumour cells |
| Pan-Cadherin | Cell Signaling Technology (4068) | Rabbit | 1:100 | Tumour cells |
| -catenin | Cell Signaling Technology (8480S) | Rabbit | 1:100 | EMT |
| OCT4a | Cell Signaling Technology (C30A3) | Rabbit | 1:800 | CSLCs |
| SNAIL | Cell Signaling Technology (3879) | Rabbit | 1:500 | EMT |
| Vimentin | Cell Signaling Technology (5741) | Rabbit | 1:200 | EMT |
| ZEB1 | Cell Signaling Technology (70512) | Rabbit | 1:500 | EMT |
| PD-1 | Cell Signaling Technology (86163) | Rabbit | 1:500 | Immune escape |
| PD-L1 | Cell Signaling Technology (13684) | Rabbit | 1:500 | Immune escape |
| TIM-3 | Abcam (ab185703) | Rabbit | 1:500 | T cell exhaustion |
| LAG-3 | Novus Biological (NBP1-97657) | Mouse | 1:400 | T cell exhaustion |
| CD44 | Cell Signaling Technology (37259) | Rabbit | 1:500 | CSLCs |
| ZEB1 | Proteintech (21544-1-AP) | Rabbit | 1:500 | EMT |
| Antibody | Panel | Main Expression Site |
|---|---|---|
| b-2-microglobulin | Immune cell profiling | Tumour cells |
| CD11c | Immune cell profiling | Monocytes/macrophages |
| CD20 | Immune cell profiling | B cells |
| CD3 | Immune cell profiling | T cells |
| CD4 | Immune cell profiling | T-helper cells |
| CD45 | Immune cell profiling | Macrophages |
| CD56 | Immune cell profiling | NK cells |
| CD68 | Immune cell profiling | Macrophages |
| CD8 | Immune cell profiling | T cells |
| CTLA4 | Immune cell profiling | T cells |
| Fibronectin | Immune cell profiling | Fibroblasts |
| GZMB | Immune cell profiling | Cytotoxic T cells |
| HLA-DR | Immune cell profiling | APCs |
| PD-1 | Immune cell profiling | T cells |
| PD-L1 | Immune cell profiling | APCs, tumour cells |
| PD-L2 | Immune cell profiling | APCs, tumour cells |
| SMA | Immune cell profiling | Fibroblasts |
| CD127 | Immune activation status | Memory T cells |
| CD25 | Immune activation status | T cells |
| CD40 | Immune activation status | B cells/APCs |
| CD80 | Immune activation status | Myeloid cells |
| ICOS | Immune activation status | T cells |
| CD14 | Immune cell typing | Monocytes, myeloid cells |
| CD163 | Immune cell typing | M2 macrophages |
| CD34 | Immune cell typing | Hematopoietic cells |
| CD45RO | Immune cell typing | Memory T cells |
| FAP- | Immune cell typing | fibroblasts |
| ARG1 | Tumour marker | Tumour cells |
| Bcl-2 | Tumour marker | Tumour cells |
| EpCAM | Tumour marker | Tumour cells |
| Ki-67 | Tumour marker | Tumour cells |
| PanCK | Tumour marker | Tumour cells |
| TGFB1 | Tumour marker | Immune cells |
| 4-1BB | Drug target | Cytotoxic T cells |
| B7-H3 | Drug target | Tumour cells |
| GITR | Drug target | T cells |
| IDO1 | Drug target | Myeloid cells |
| LAG-3 | Drug target | T cells |
| OX40L | Drug target | Myeloid cells, T cells |
| STING | Drug target | Immune cells |
| TIM-3 | Drug target | T cells |
| VISTA | Drug target | Myeloid cells, macrophages |
| GADPH | Positive control | Housekeeper |
| Histone H3 | Positive control | Housekeeper |
| S6 | Positive control | Housekeeper |
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Eggener, S.E.; Yossepowitch, O.; Pettus, J.A.; Snyder, M.E.; Motzer, R.J.; Russo, P. Renal cell carcinoma recurrence after nephrectomy for localized disease: Predicting survival from time of recurrence. J. Clin. Oncol. 2006, 24, 3101–3106. [Google Scholar] [CrossRef] [PubMed]
- Padala, S.A.; Barsouk, A.; Thandra, K.C.; Saginala, K.; Mohammed, A.; Vakiti, A.; Rawla, P.; Barsouk, A. Epidemiology of renal cell carcinoma. World J. Oncol. 2020, 11, 79. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Lv, Y.; Gan, Z.; Zhang, Y.; Han, X.; Xu, Z. Identification of key genes involved in the metastasis of clear cell renal cell carcinoma. Oncol. Lett. 2019, 17, 4321–4328. [Google Scholar] [CrossRef] [PubMed]
- Moch, H.; Cubilla, A.L.; Humphrey, P.A.; Reuter, V.E.; Ulbright, T.M. The 2016 WHO classification of tumours of the urinary system and male genital organs—Part A: Renal, penile, and testicular tumours. Eur. Urol. 2016, 70, 93–105. [Google Scholar] [CrossRef]
- Warren, A.Y.; Harrison, D. WHO/ISUP classification, grading and pathological staging of renal cell carcinoma: Standards and controversies. World J. Urol. 2018, 36, 1913–1926. [Google Scholar] [CrossRef]
- Leibovich, B.C.; Blute, M.L.; Cheville, J.C.; Lohse, C.M.; Frank, I.; Kwon, E.D.; Weaver, A.L.; Parker, A.S.; Zincke, H. Prediction of progression after radical nephrectomy for patients with clear cell renal cell carcinoma: A stratification tool for prospective clinical trials. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2003, 97, 1663–1671. [Google Scholar] [CrossRef]
- Bektas, S.; Bahadir, B.; Kandemir, N.O.; Barut, F.; Gul, A.E.; Ozdamar, S.O. Intraobserver and interobserver variability of Fuhrman and modified Fuhrman grading systems for conventional renal cell carcinoma. Kaohsiung J. Med. Sci. 2009, 25, 596–600. [Google Scholar] [CrossRef]
- Ren, D.; Hua, Y.; Yu, B.; Ye, X.; He, Z.; Li, C.; Wang, J.; Mo, Y.; Wei, X.; Chen, Y.; et al. Predictive biomarkers and mechanisms underlying resistance to PD1/PD-L1 blockade cancer immunotherapy. Mol. Cancer 2020, 19, 19. [Google Scholar]
- Giraldo, N.A.; Becht, E.; Vano, Y.; Petitprez, F.; Lacroix, L.; Validire, P.; Sanchez-Salas, R.; Ingels, A.; Oudard, S.; Moatti, A.; et al. Tumor-infiltrating and peripheral blood T-cell immunophenotypes predict early relapse in localized clear cell renal cell carcinoma. Clin. Cancer Res. 2017, 23, 4416–4428. [Google Scholar] [CrossRef]
- Chen, X.; Wang, H.; Zhu, Y.J.; Wang, H.; Hu, X.Y.; Guo, J.M. Prognostic value of vimentin is associated with immunosuppression in metastatic renal cell carcinoma. Front. Oncol. 2020, 10, 1181. [Google Scholar]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Andreiana, B.C.; Stepan, A.E.; Mărgăritescu, C.; Tăisescu, O.; Osman, A.; Simionescu, C. Snail and E-cadherin immunoexpression in clear cell renal cell carcinoma. Curr. Health Sci. J. 2019, 45, 185. [Google Scholar]
- Wu, J.; Ru, N.; Zhang, Y.; Li, Y.; Wei, D.; Ren, Z.; Huang, X.; Chen, Z.; Bian, H. HAb18G/CD147 promotes epithelial–mesenchymal transition through TGF-β signaling and is transcriptionally regulated by Slug. Oncogene 2011, 30, 4410–4427. [Google Scholar] [CrossRef]
- Zhang, P.; Sun, Y.; Ma, L. ZEB1: At the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle 2015, 14, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Lin, H.H.; Tang, M.J.; Wang, Y.K. Vimentin contributes to epithelial-mesenchymal transition cancer cell mechanics by mediating cytoskeletal organization and focal adhesion maturation. Oncotarget 2015, 6, 15966. [Google Scholar] [CrossRef]
- O’Brien, S.J.; Fiechter, C.; Burton, J.; Hallion, J.; Paas, M.; Patel, A.; Patel, A.; Rochet, A.; Scheurlen, K.; Gardner, S.; et al. Long non-coding RNA ZFAS1 is a major regulator of epithelial-mesenchymal transition through miR-200/ZEB1/E-cadherin, vimentin signaling in colon adenocarcinoma. Cell Death Discov. 2021, 7, 61. [Google Scholar] [CrossRef]
- Kröger, C.; Afeyan, A.; Mraz, J.; Eaton, E.N.; Reinhardt, F.; Khodor, Y.L.; Thiru, P.; Bierie, B.; Ye, X.; Burge, C.B.; et al. Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc. Natl. Acad. Sci. USA 2019, 116, 7353–7362. [Google Scholar] [CrossRef]
- Myszczyszyn, A.; Czarnecka, A.M.; Matak, D.; Szymanski, L.; Lian, F.; Kornakiewicz, A.; Bartnik, E.; Kukwa, W.; Kieda, C.; Szczylik, C. The role of hypoxia and cancer stem cells in renal cell carcinoma pathogenesis. Stem Cell Rev. Rep. 2015, 11, 919–943. [Google Scholar] [CrossRef]
- Zöller, M. CD44: Can a cancer-initiating cell profit from an abundantly expressed molecule? Nat. Rev. Cancer 2011, 11, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ma, X.; Chen, L.; Gu, L.; Zhang, Y.; Zhang, F.; Ouyang, Y.; Gao, Y.; Huang, Q.; Zhang, X. Prognostic value of CD44 expression in renal cell carcinoma: A systematic review and meta-analysis. Sci. Rep. 2015, 5, 13157. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ma, C.; Komohara, Y.; Ohnishi, K.; Shimoji, T.; Kuwahara, N.; Sakumura, Y.; Matsuishi, K.; Fujiwara, Y.; Motoshima, T.; Takahashi, W.; et al. Infiltration of tumor-associated macrophages is involved in CD 44 expression in clear cell renal cell carcinoma. Cancer Sci. 2016, 107, 700–707. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, X.; Kang, B.; She, S.; Zhang, X.; Chen, C.; Li, W.; Chen, W.; Dan, S.; Pan, X.; et al. Endogenous authentic OCT4A proteins directly regulate FOS/AP-1 transcription in somatic cancer cells. Cell Death Dis. 2018, 9, 585. [Google Scholar] [CrossRef]
- Rasti, A.; Mehrazma, M.; Madjd, Z.; Abolhasani, M.; Zanjani, L.S.; Asgari, M. Co-expression of cancer stem cell markers OCT4 and NANOG predicts poor prognosis in renal cell carcinomas. Sci. Rep. 2018, 8, 11739. [Google Scholar] [CrossRef] [PubMed]
- Waggoner, S.N.; Cornberg, M.; Selin, L.K.; Welsh, R.M. Natural killer cells act as rheostats modulating antiviral T cells. Nature 2012, 481, 394–398. [Google Scholar] [CrossRef]
- Comber, J.D.; Philip, R. MHC class I antigen presentation and implications for developing a new generation of therapeutic vaccines. Ther. Adv. Vaccines 2014, 2, 77–89. [Google Scholar] [CrossRef]
- Caie, P.D.; Dimitriou, N.; Arandjelović, O. Precision medicine in digital pathology via image analysis and machine learning. In Artificial Intelligence and Deep Learning in Pathology; Elsevier: Amsterdam, The Netherlands, 2021; pp. 149–173. [Google Scholar]
- Geiss, G.K.; Bumgarner, R.E.; Birditt, B.; Dahl, T.; Dowidar, N.; Dunaway, D.L.; Fell, H.P.; Ferree, S.; George, R.D.; Grogan, T.; et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat. Biotechnol. 2008, 26, 317–325. [Google Scholar] [CrossRef]
- Merritt, C.R.; Ong, G.T.; Church, S.E.; Barker, K.; Danaher, P.; Geiss, G.; Hoang, M.; Jung, J.; Liang, Y.; McKay-Fleisch, J.; et al. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat. Biotechnol. 2020, 38, 586–599. [Google Scholar] [CrossRef]
- Chow, K.V.; Denning, K.C. A simple multiple variance ratio test. J. Econom. 1993, 58, 385–401. [Google Scholar] [CrossRef]
- Coelho, L.P. Jug: Software for parallel reproducible computation in python. J. Open Res. Softw. 2017, 5, 30. [Google Scholar] [CrossRef]
- Beechem, J.M. High-plex spatially resolved RNA and protein detection using digital spatial profiling: A technology designed for immuno-oncology biomarker discovery and translational research. In Biomarkers for Immunotherapy of Cancer; Springer: Berlin/Heidelberg, Germany, 2020; pp. 563–583. [Google Scholar]
- Raggi, C.; Mousa, H.; Correnti, M.; Sica, A.; Invernizzi, P. Cancer stem cells and tumor-associated macrophages: A roadmap for multitargeting strategies. Oncogene 2016, 35, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Harb, O.A.; Elfeky, M.A.; El Shafaay, B.S.; Taha, H.F.; Osman, G.; Harera, I.S.; Gertallah, L.M.; Abdelmonem, D.M.; Embaby, A. SPOP, ZEB-1 and E-cadherin expression in clear cell renal cell carcinoma (cc-RCC): Clinicopathological and prognostic significance. Pathophysiology 2018, 25, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Bi, T.; Ding, W.; Yu, C.; Jiang, C.; Yang, H.; Sun, X.; Yang, M. OCT4 potentiates radio-resistance and migration activity of rectal cancer cells by improving epithelial-mesenchymal transition in a ZEB1 dependent manner. BioMed Res. Int. 2018, 2018, 3424956. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Najafi, M.; Farhood, B.; Mortezaee, K.; Kharazinejad, E.; Majidpoor, J.; Ahadi, R. Hypoxia in solid tumors: A key promoter of cancer stem cell (CSC) resistance. J. Cancer Res. Clin. Oncol. 2020, 146, 19–31. [Google Scholar] [CrossRef]
- Xu, C.; Jackson, S.A. Machine learning and complex biological data. Genome Biol. 2019, 20, 76. [Google Scholar] [CrossRef]
- Bonferroni, C. Teoria statistica delle classi e calcolo delle probabilita. Pubbl. Ist. Super. Sci. Econ. Commericiali Firenze 1936, 8, 3–62. [Google Scholar]
- Grillo, F.; Bruzzone, M.; Pigozzi, S.; Prosapio, S.; Migliora, P.; Fiocca, R.; Mastracci, L. Immunohistochemistry on old archival paraffin blocks: Is there an expiry date? J. Clin. Pathol. 2017, 70, 988–993. [Google Scholar] [CrossRef]
- Bueno, R.; Hughes, E.; Wagner, S.; Gutin, A.S.; Lanchbury, J.S.; Zheng, Y.; Archer, M.A.; Gustafson, C.; Jones, J.T.; Rushton, K.; et al. Validation of a molecular and pathological model for five-year mortality risk in patients with early stage lung adenocarcinoma. J. Thorac. Oncol. 2015, 10, 67–73. [Google Scholar] [CrossRef][Green Version]
- Gupta, S.; Chen, T.; Destenaves, B. Quantitative RNA assessment and long-term stability in the FFPE tumor samples using Digital Spatial Profiler. Immuno-Oncol. Technol. 2022, 13, 100069. [Google Scholar] [CrossRef] [PubMed]












| Survival | Markers | p-Value | |
|---|---|---|---|
| 5yr | ↓ HLA-DR | ↓ CD8+LAG3+ T cells | |
| 5yr | ↓ ARG1 | ↑ OCT4+ tumour cells | |
| 5yr | ↓ TIM3 | ↑ OCT4+-catenin+ tumour cells | |
| 5yr | ↓ ARG1 | ↑ OCT4+vimentin+ tumour cells | |
| 5yr | ↑ OCT4+vimentin+ tumour cells | ↓ snail+vimentin+ tumour cells | |
| 5yr | ↓ HLA-DR | ↑ OCT4+vimentin+ tumour cells | |
| 5yr | ↓ CD8+TIM3+ T cells | ↑ TIM3+ cells | |
| overall | ↑ OCT4+vimentin+ tumour cells | ↓ PD-1+ cells | |
| overall | ↑ OCT4+ZEB1+-catenin+ tumour cells | ↓ snail+vimentin+ tumour cells | |
| overall | ↓ TIM3 | ↑ OCT4+-catenin+ tumour cells | |
| overall | ↑ OCT4+vimentin+ tumour cells | ↓ snail+vimentin+ tumour cells | |
| overall | ↑ OCT4+vimentin+ tumour cells | ↓ snail+ tumour cells | |
| overall | ↓ PD-1+ cells | ↓ snail+vimentin+ tumour cells |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Filippis, R.; Wölflein, G.; Um, I.H.; Caie, P.D.; Warren, S.; White, A.; Suen, E.; To, E.; Arandjelović, O.; Harrison, D.J. Use of High-Plex Data Reveals Novel Insights into the Tumour Microenvironment of Clear Cell Renal Cell Carcinoma. Cancers 2022, 14, 5387. https://doi.org/10.3390/cancers14215387
De Filippis R, Wölflein G, Um IH, Caie PD, Warren S, White A, Suen E, To E, Arandjelović O, Harrison DJ. Use of High-Plex Data Reveals Novel Insights into the Tumour Microenvironment of Clear Cell Renal Cell Carcinoma. Cancers. 2022; 14(21):5387. https://doi.org/10.3390/cancers14215387
Chicago/Turabian StyleDe Filippis, Raffaele, Georg Wölflein, In Hwa Um, Peter D. Caie, Sarah Warren, Andrew White, Elizabeth Suen, Emily To, Ognjen Arandjelović, and David J. Harrison. 2022. "Use of High-Plex Data Reveals Novel Insights into the Tumour Microenvironment of Clear Cell Renal Cell Carcinoma" Cancers 14, no. 21: 5387. https://doi.org/10.3390/cancers14215387
APA StyleDe Filippis, R., Wölflein, G., Um, I. H., Caie, P. D., Warren, S., White, A., Suen, E., To, E., Arandjelović, O., & Harrison, D. J. (2022). Use of High-Plex Data Reveals Novel Insights into the Tumour Microenvironment of Clear Cell Renal Cell Carcinoma. Cancers, 14(21), 5387. https://doi.org/10.3390/cancers14215387