

Artificial Intelligence Predicted Overall Survival and Classified Mature B-Cell Neoplasms Based on Immuno-Oncology and Immune Checkpoint Panels




Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
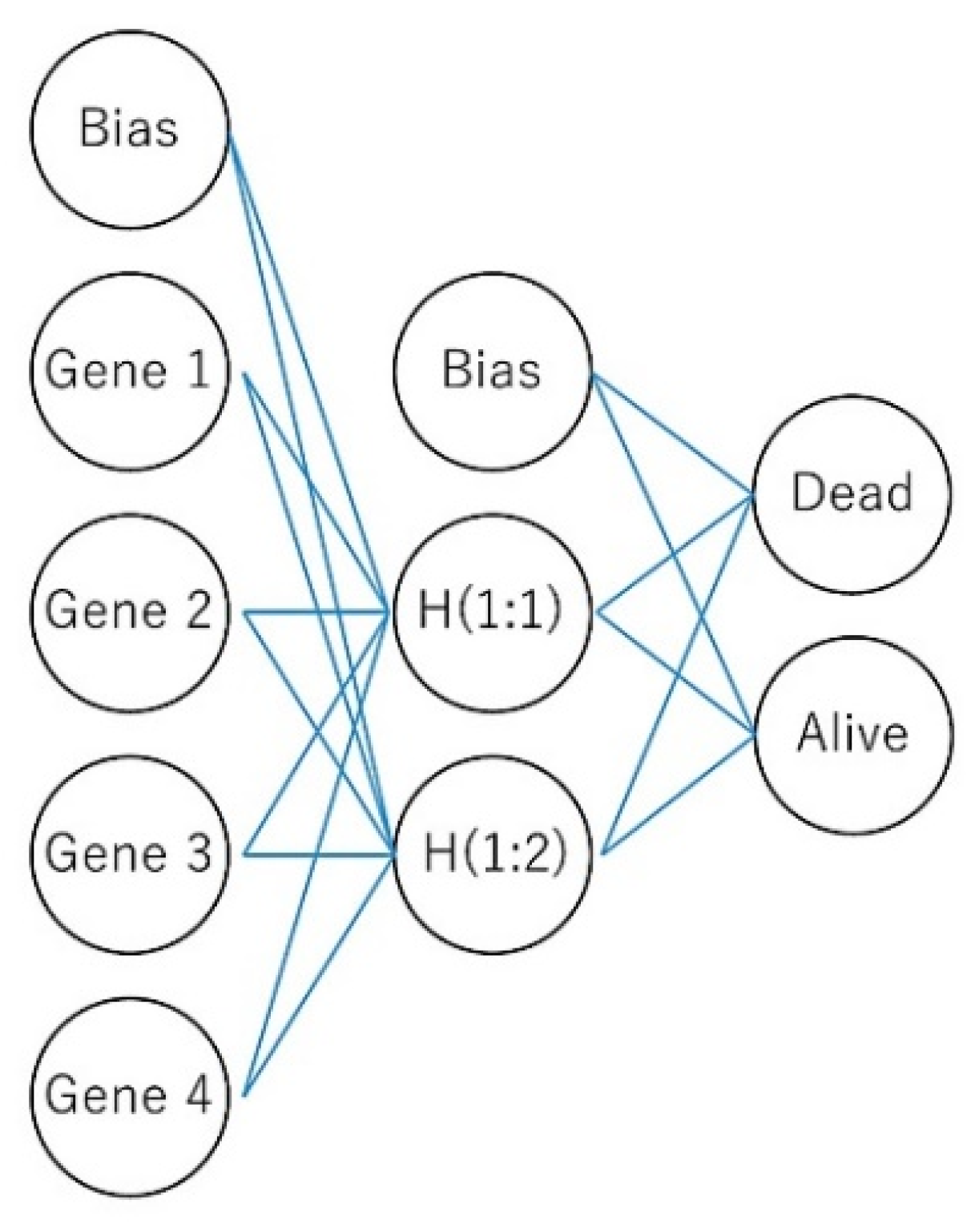
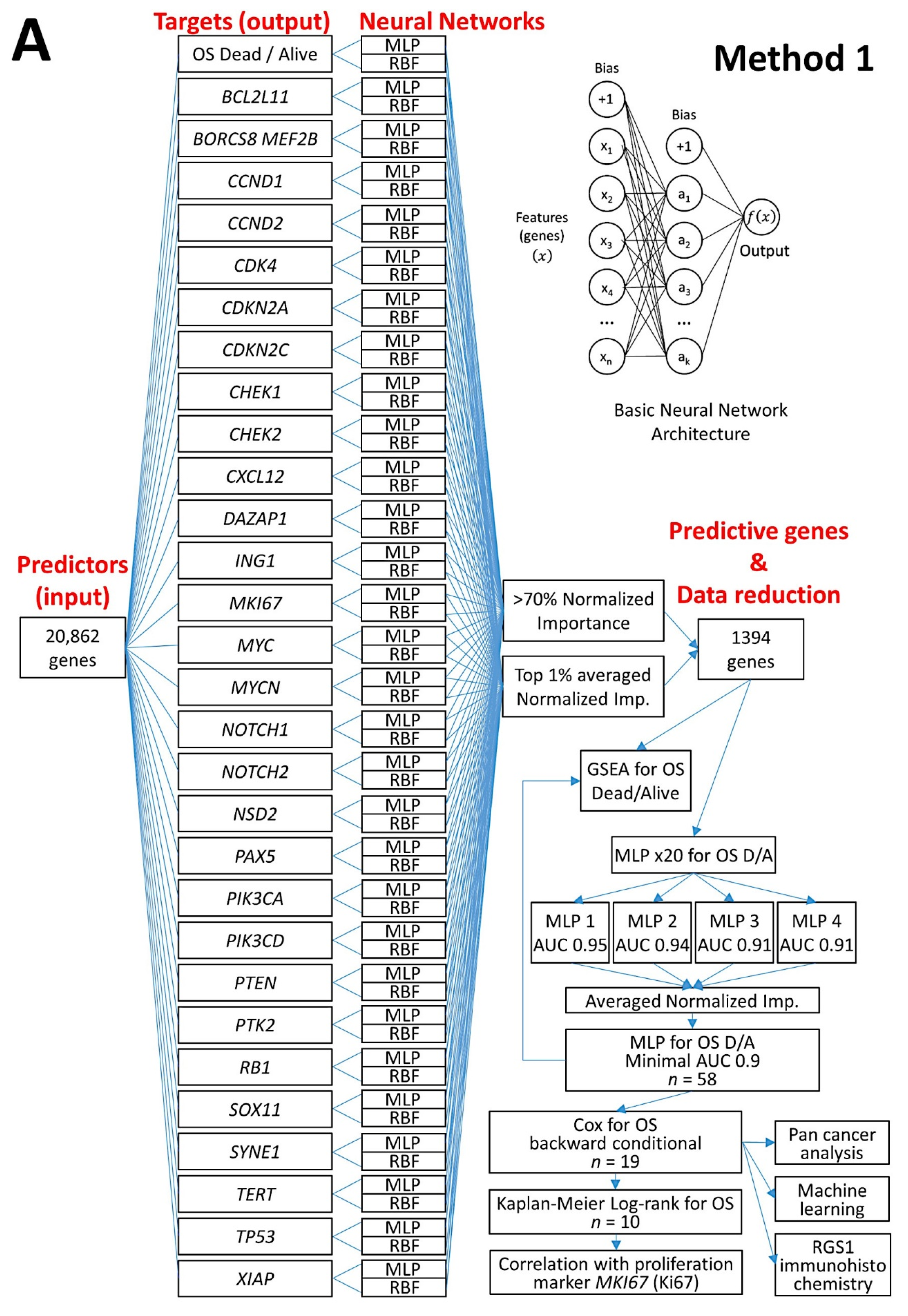
2.1. Machine Learning and Neural Networks
2.2. Multilayer Perceptron Artificial Neural Network
- I
- Number of layers, discounting the input layer.
- Ji
- Number of units in layer i. J0 = P,Ji = R, discounting the bias unit.
- wi:j,k
- Weight leading from layer i–1, unit j to layer i, unit k. No weights connect and the bias ; that is, there is no wi:j,0 for any j.
- γi(c)
- Activation function for layer i.
- w
- Weight vector containing all weights (w1:0,1, w1:0,2, …, wI:JI−1,JI).
2.3. Differential Gene Expression Using the GEOR2 Software
2.4. Gene Set Enrichment Analysis
2.5. Conventional Statistical Analyses
2.6. Risk Groups
2.7. Hardware
2.8. Datasets and Immunohistochemical Procedures
3. Results
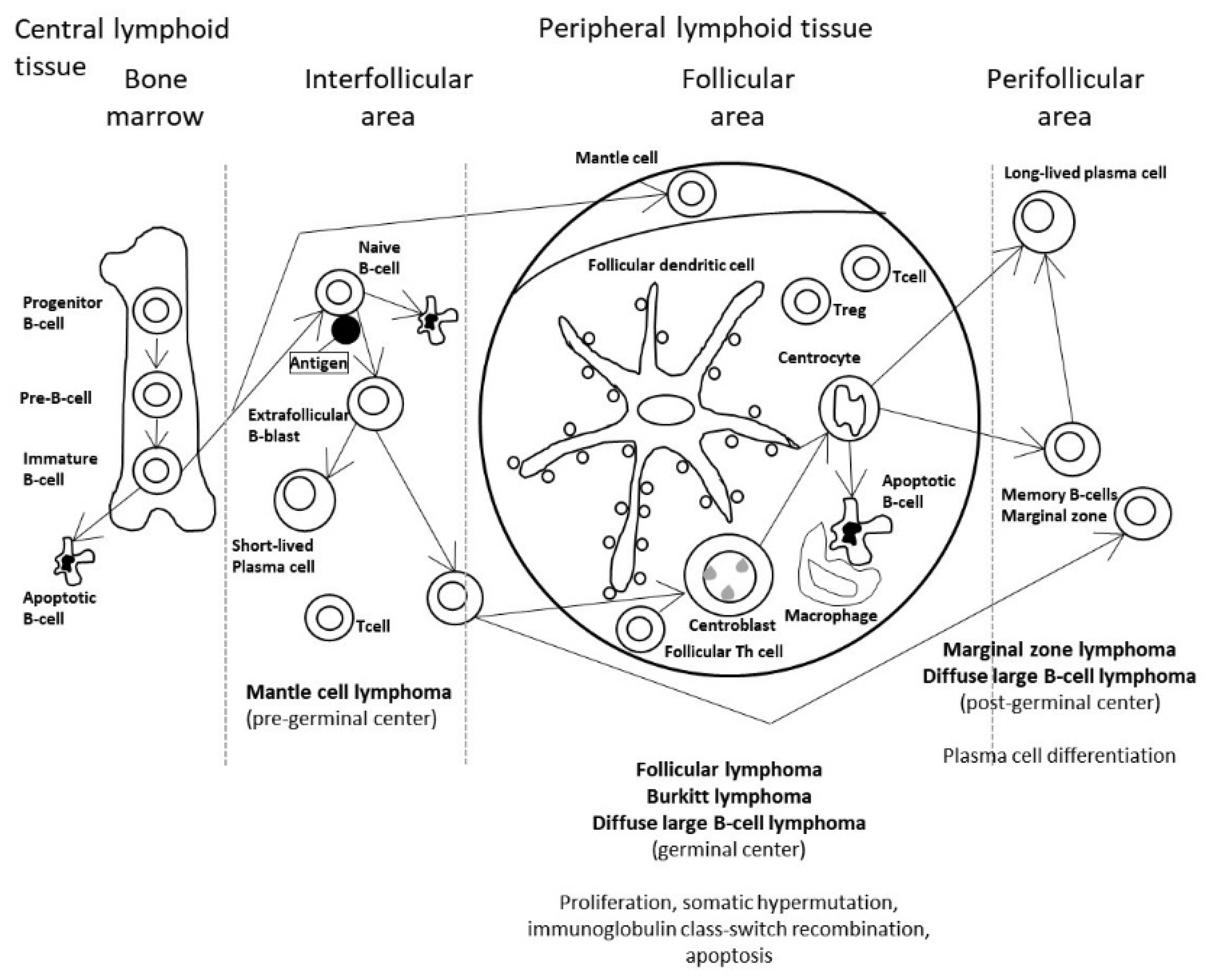
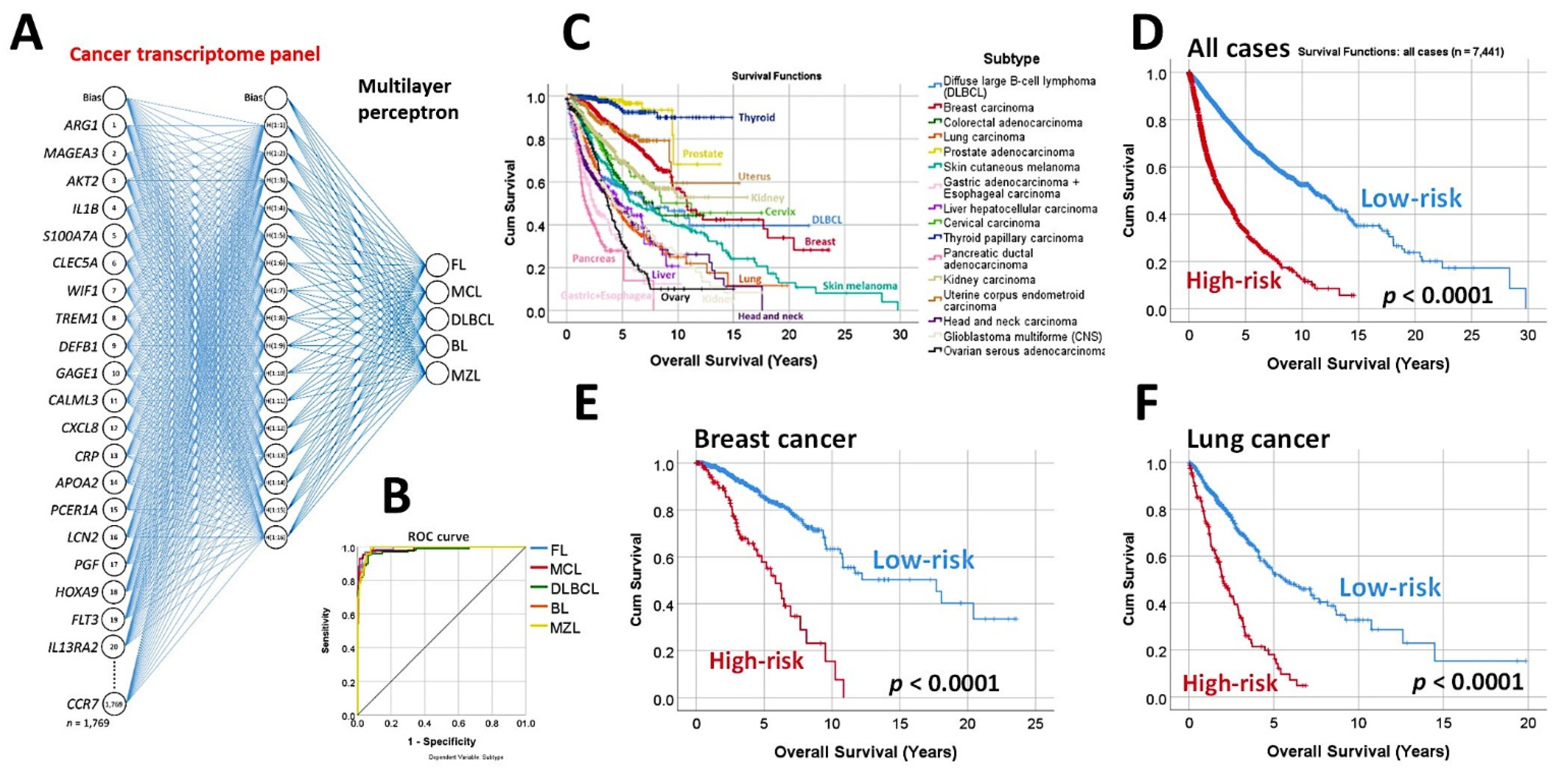
3.1. Predictive Classification of Non-Hodgkin Lymphomas
- Using the whole array of 20,863 and a cancer transcriptome panel, the lymphoma subtypes were predicted by a neural network with high accuracy [19].
3.2. Follicular Lymphoma, Immune Response, and Microenvironment
- An algorithm combined two types of neural networks (multilayer perceptron and radial basis function) to predict the overall survival, in combination with other clinically relevant variables [29].
- These variables were more than 60 years, the number of extranodal sites > 1, LDH-level ratio > 1, stage > 2, IPI score 2−3, with translocation (14;18) positive, immune response ratio 2:1 high (≥0.97), and overall survival up to 5 years vs alive from 10 years [29].
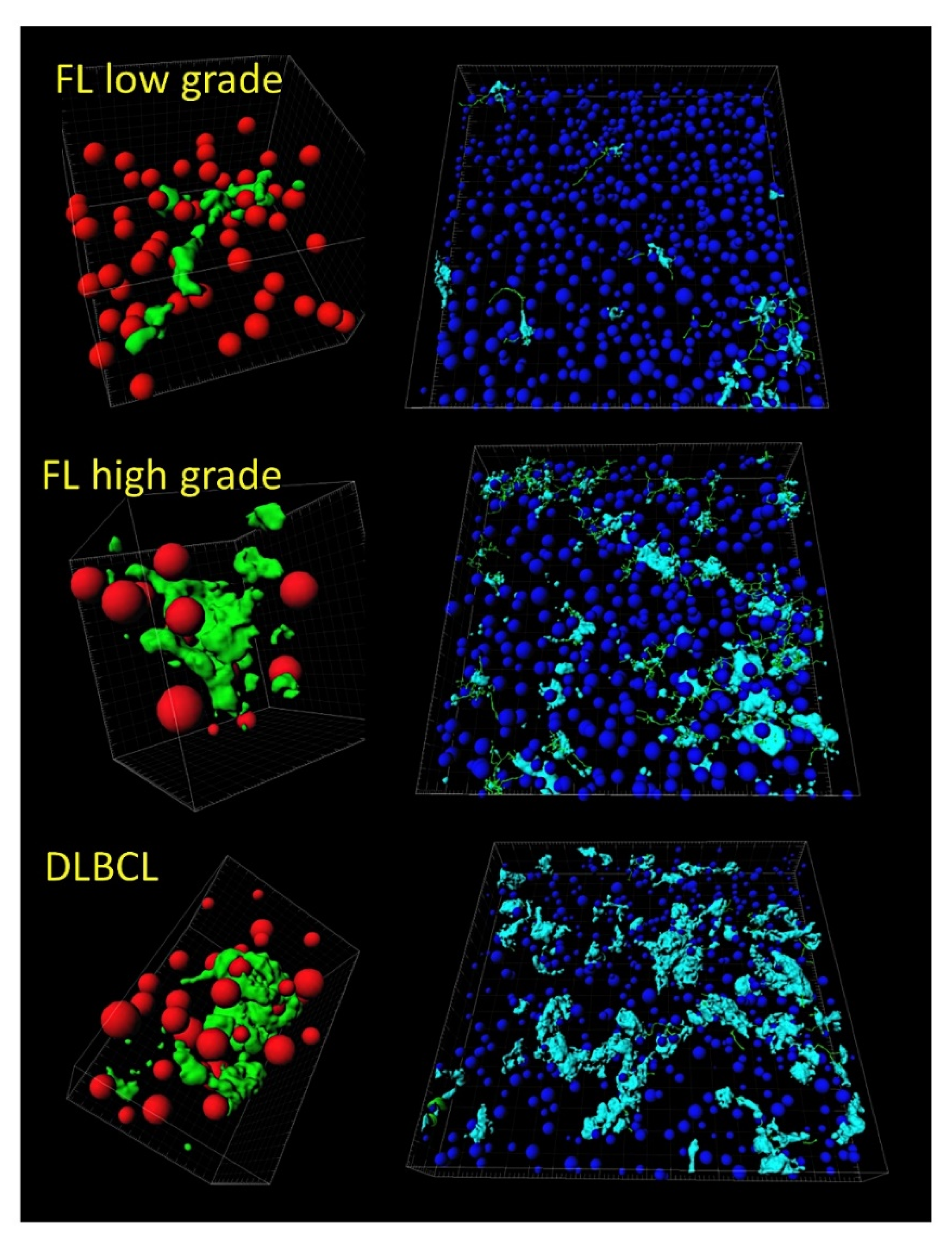
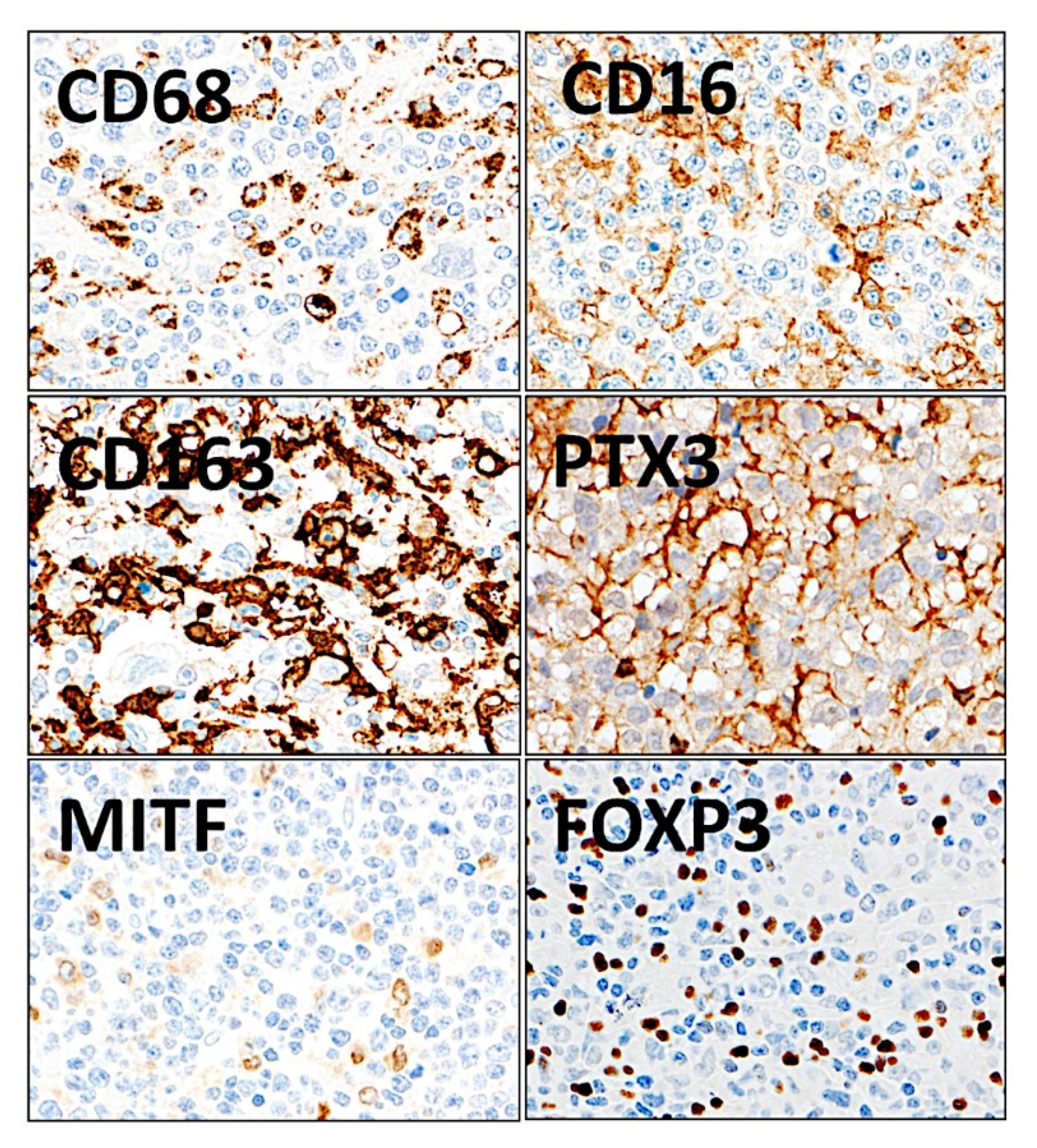
- Tridimensional (3D) analysis of tumor-associated macrophages (TAMs) of follicular lymphoma and transformation to diffuse large B-cell lymphoma was associated with increased numbers of TAMs, which created a network-like structure (Figure 7).
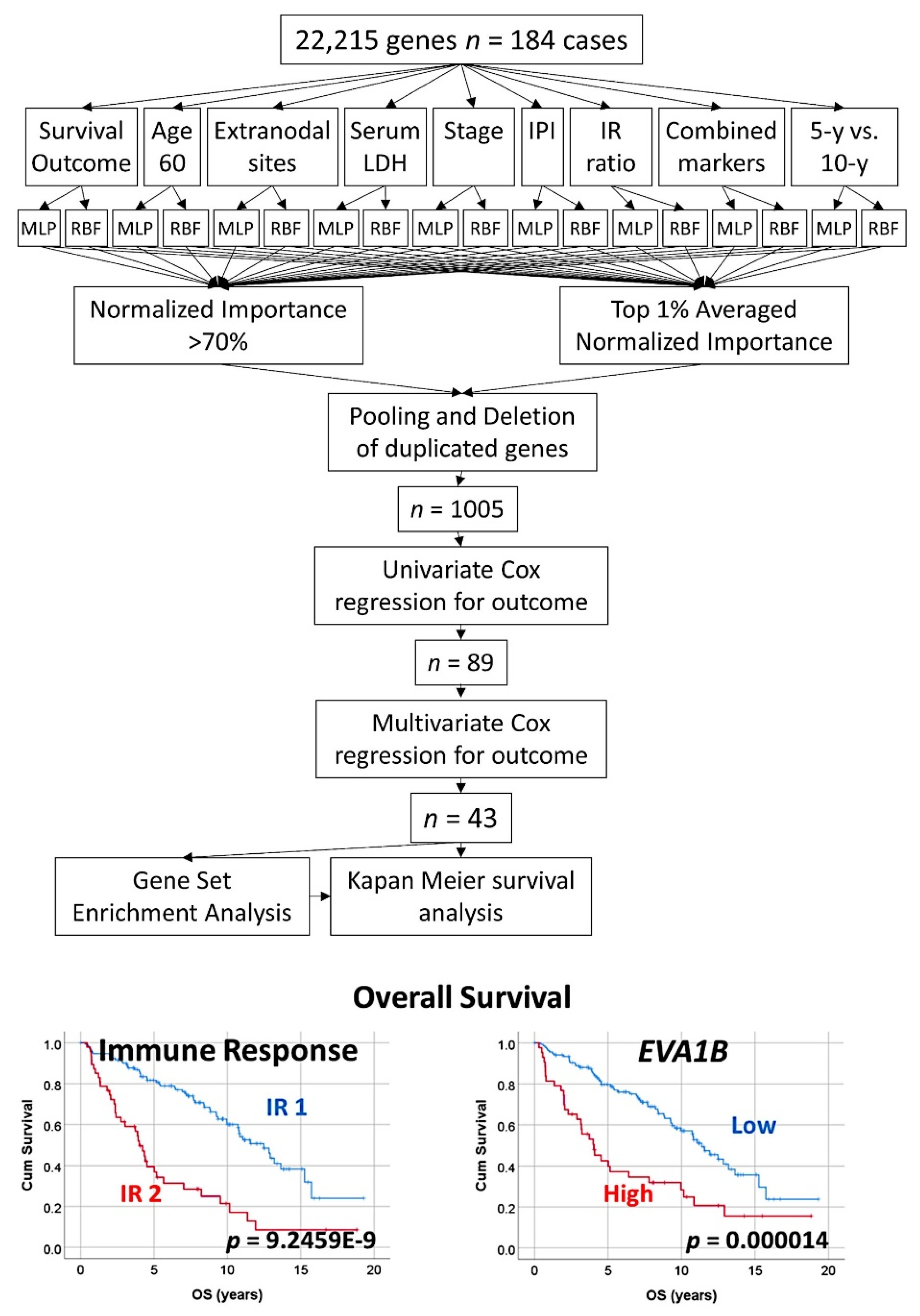
3.3. Follicular Lymphoma, Random Number Generator-Based Strategy
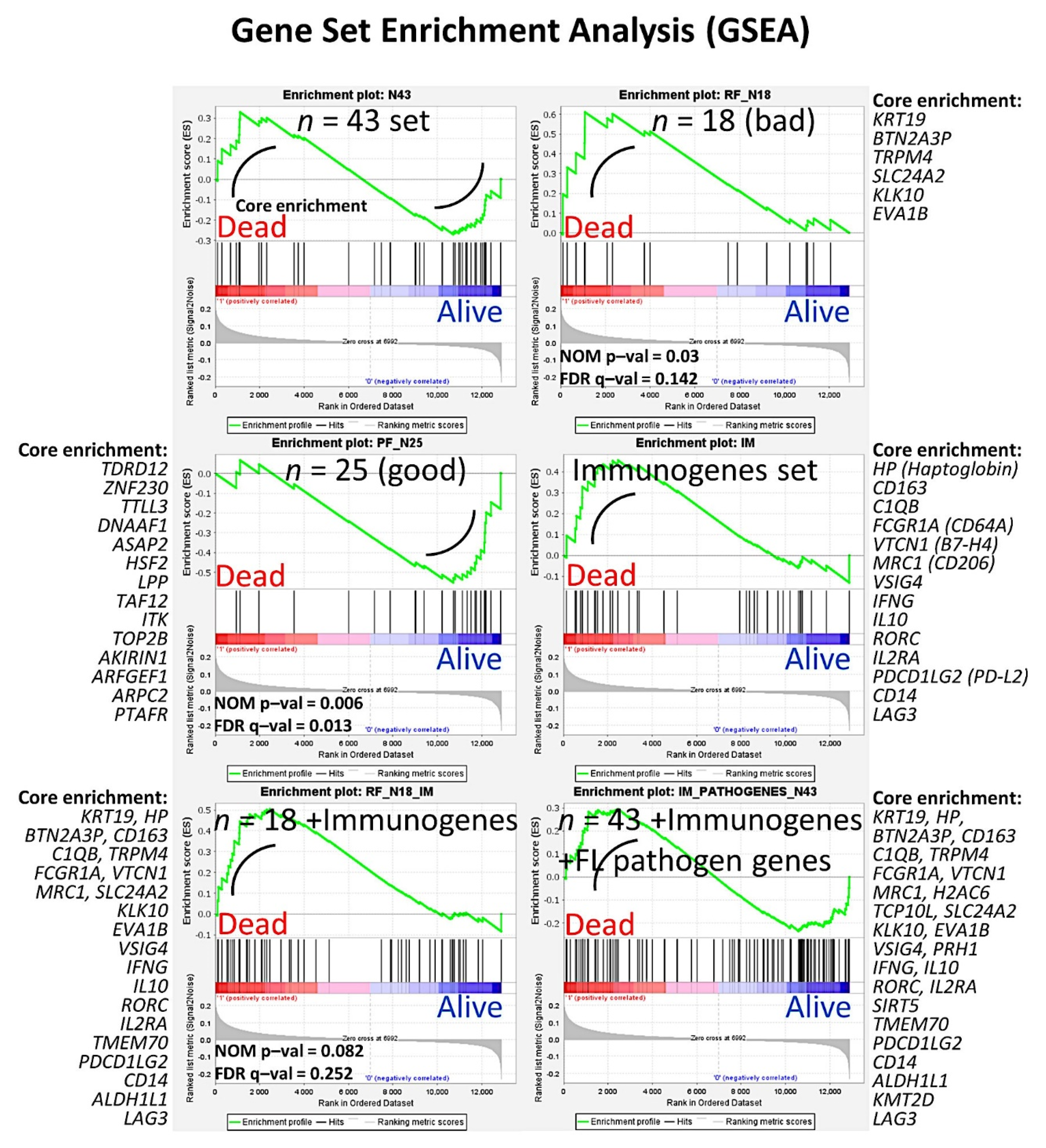
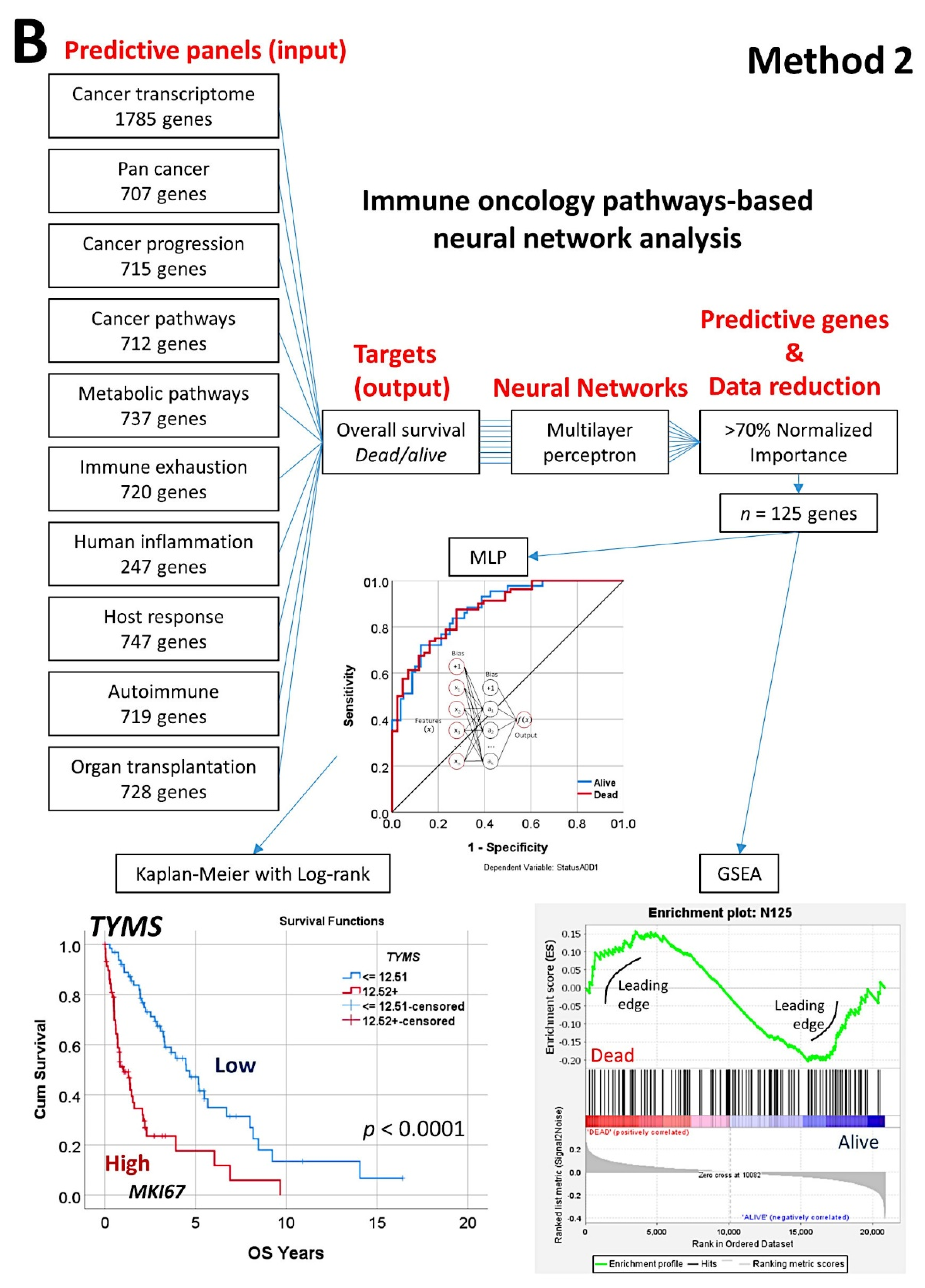
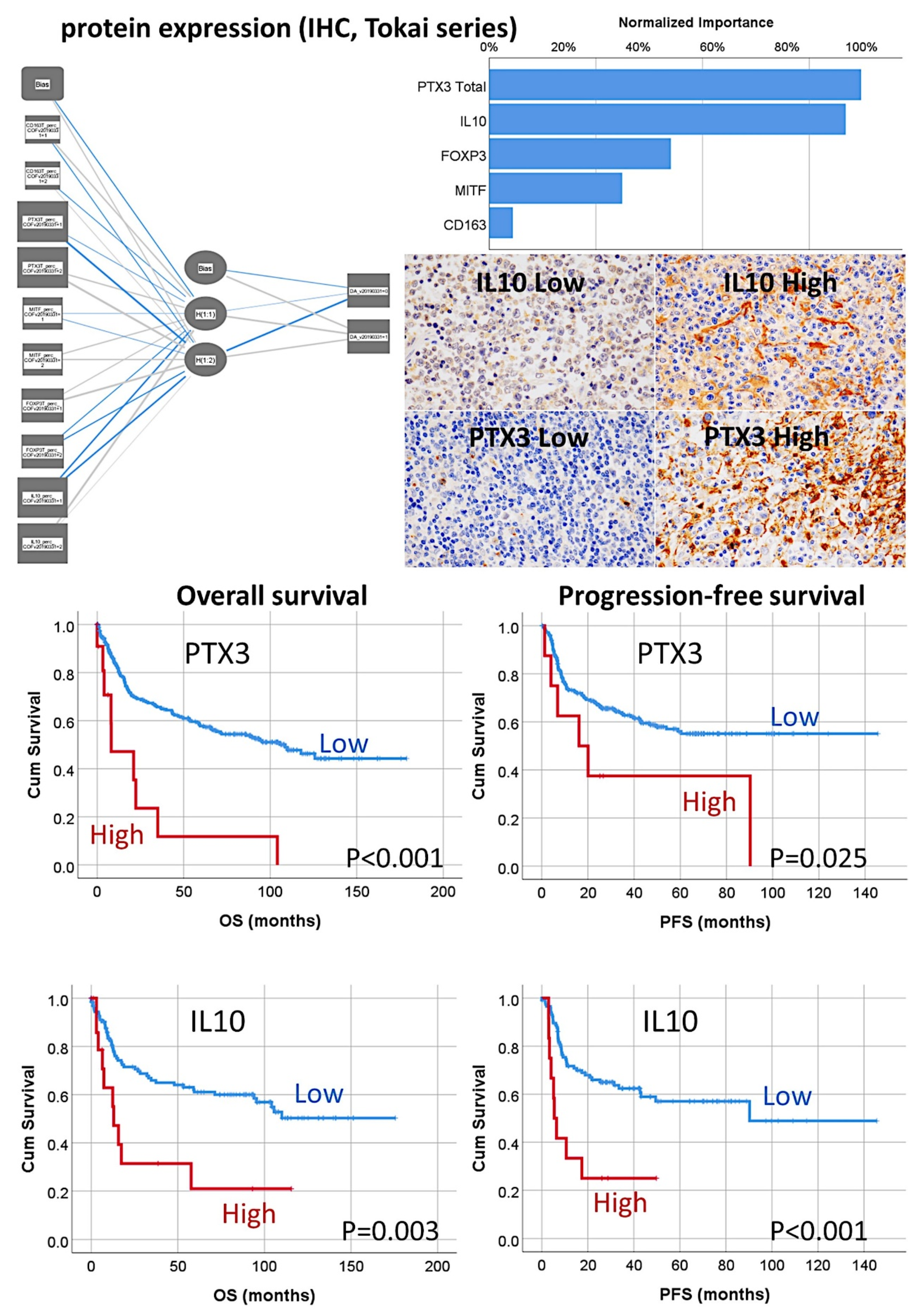
- The random number generation created 120 independent multilayer perceptron solutions and 22,215 gene probes were ranked according to their averaged normalized importance for predicting the overall survival [35].
- The analysis identified new predictor genes, which were related to cell adhesion and migration, cell signaling, and metabolism. These genes were also correlated to the immuno-oncology markers of CD163, CSF1R, FOXP3, PDCD1 (PD-1), TNFRSF14 (HVEM), and IL10 [35].
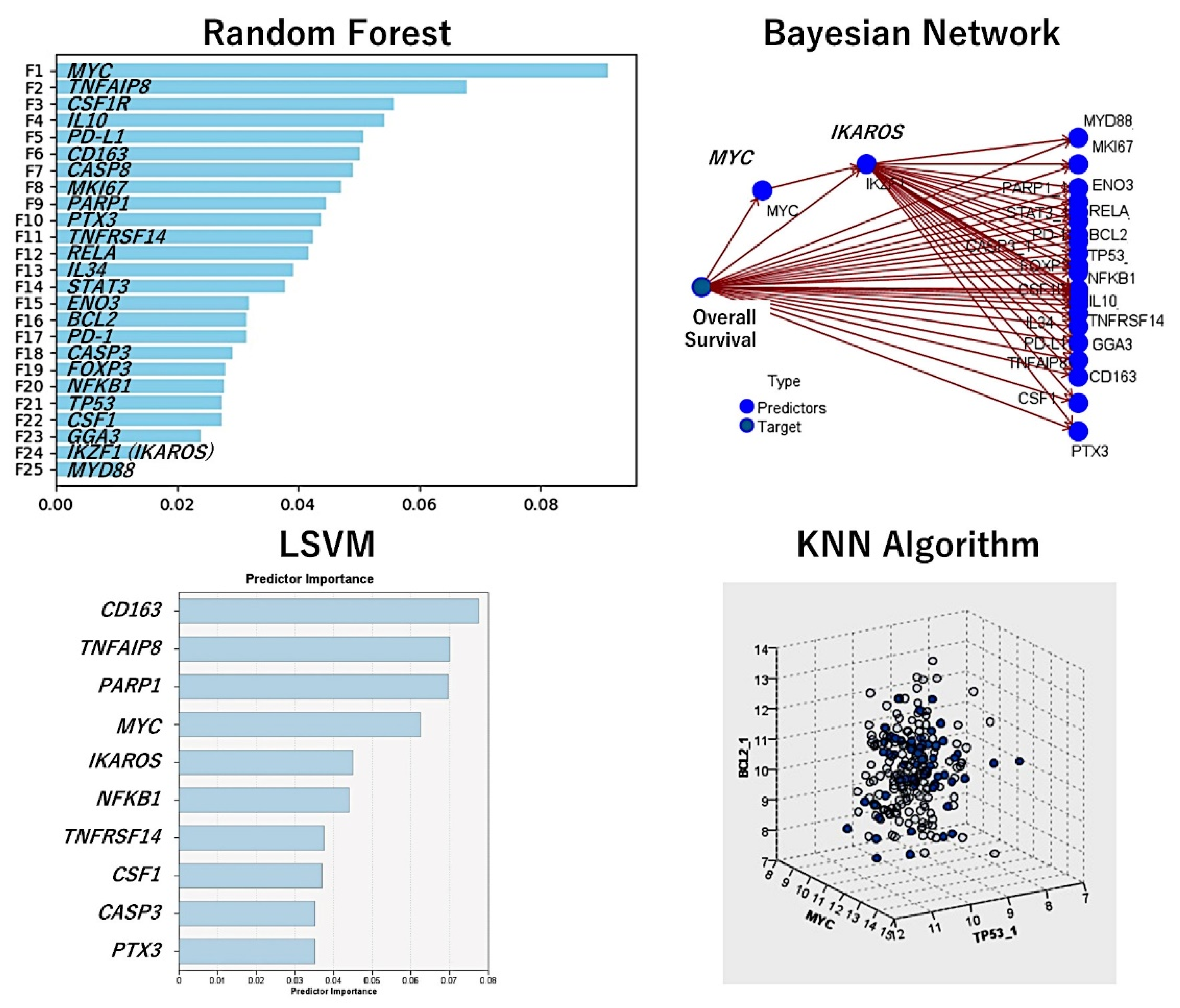
- A comparison with other machine learning techniques was also performed. Machine learning included the following techniques: Bayesian network, C&R tree, C5 tree, CHAID tree, discriminant analysis, KNN algorithms, logistic regression, LSVM, Quest tree, random forest, random trees, SVM, tree-AS, XGBoost linear, and XGBoost tree. A neural network analysis was also made [35] (Figure 8).
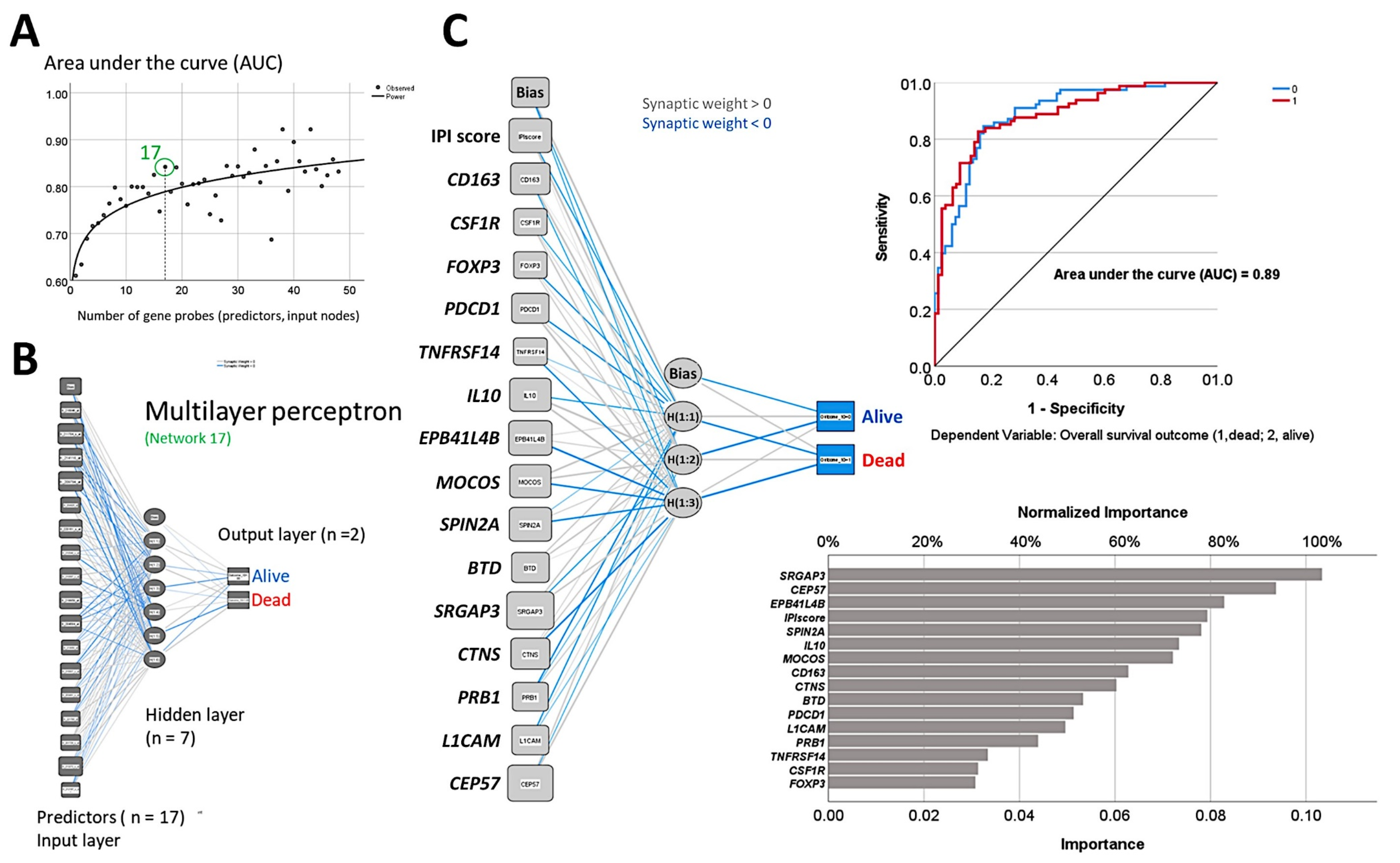
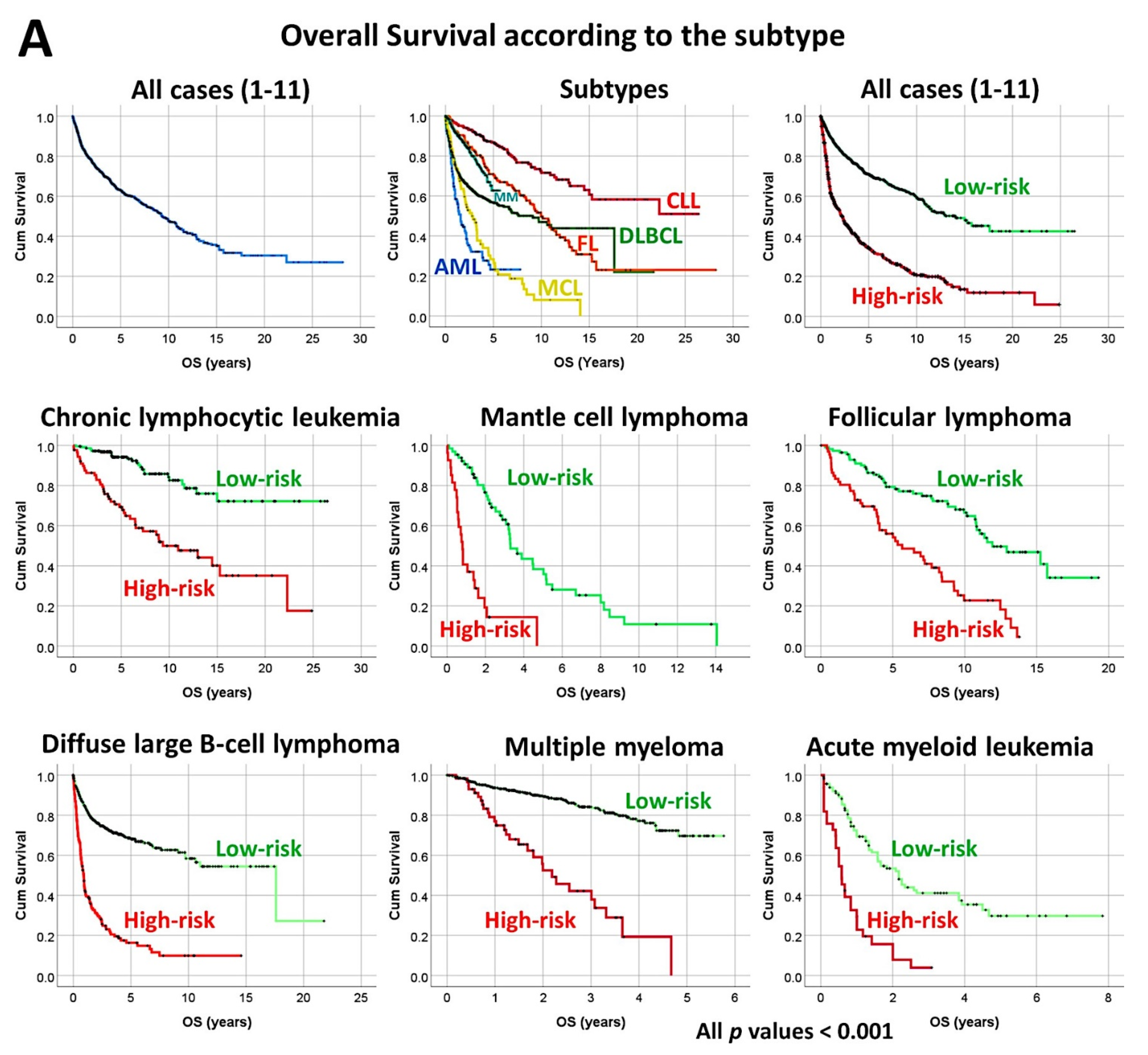
3.4. Mantle Cell Lymphoma, Use of Immuno-Oncology Panels to Predict Survival
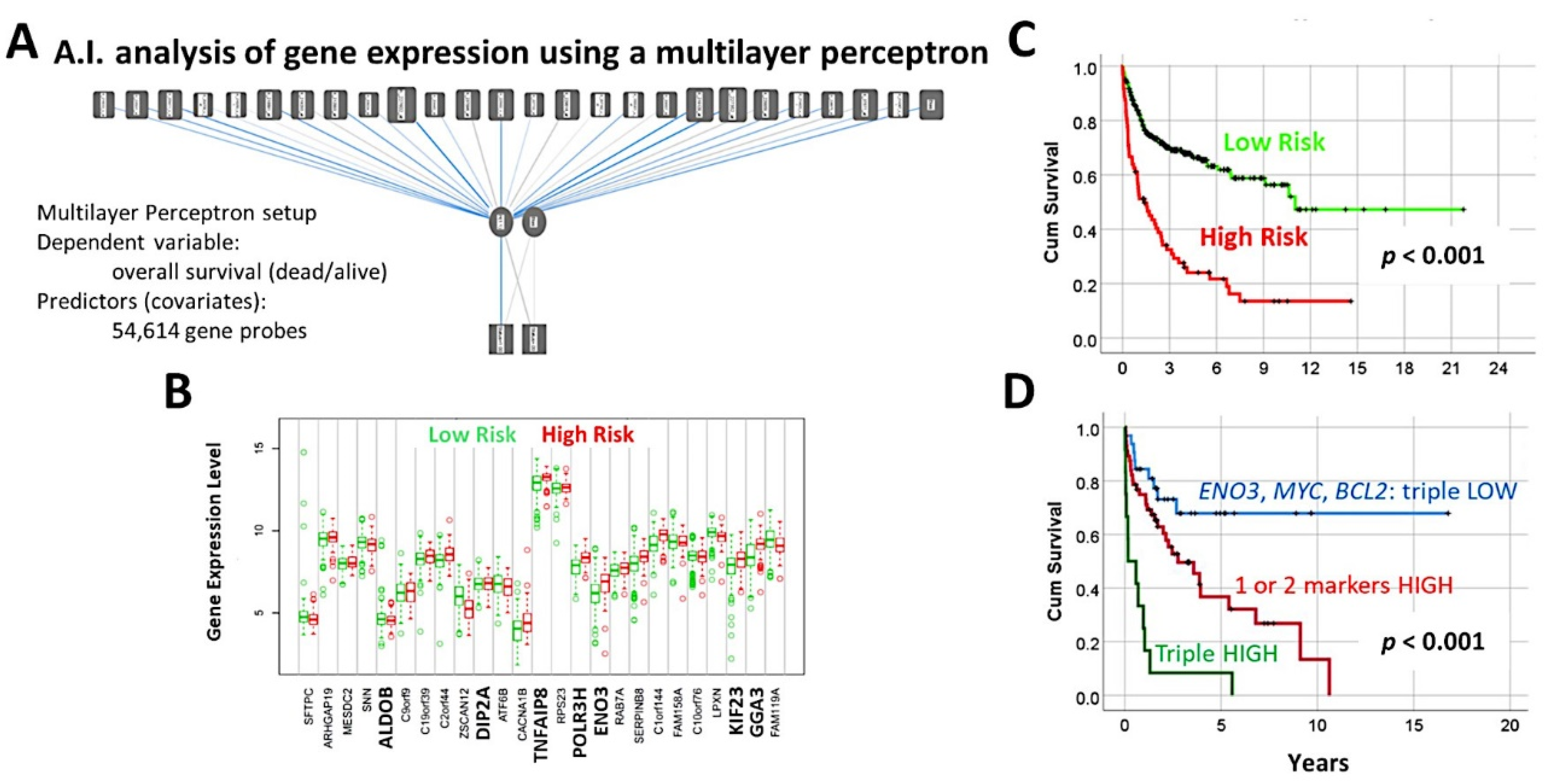
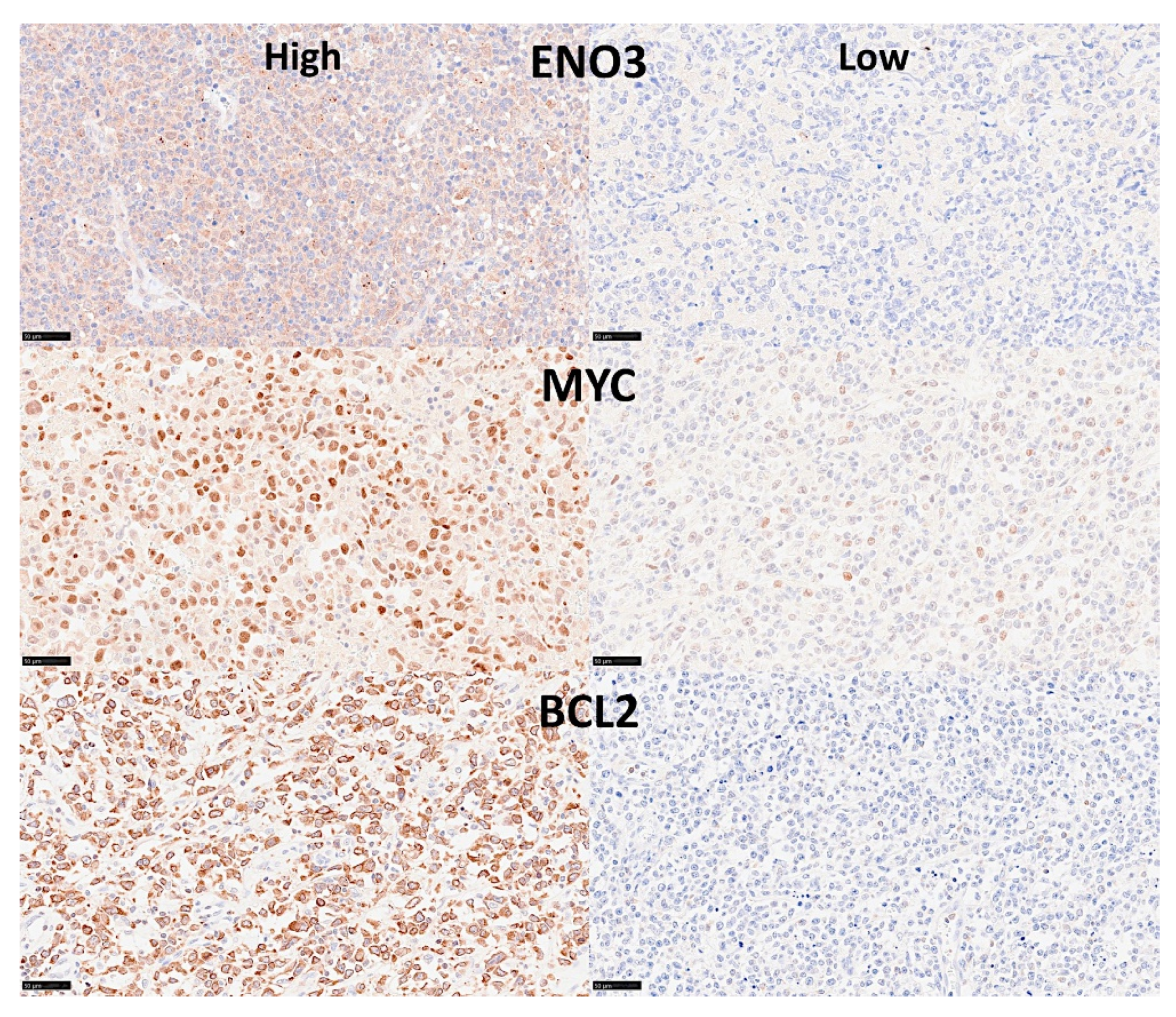
- An analysis algorithm included several analysis techniques such as neural networks (both the multilayer perceptron artificial and radial basis function), GSEA, and conventional statistics. In this analysis, 20,862 genes were correlated with 28 prognostic genes of mantle cell lymphoma. After dimensionality reduction, the patients’ overall survival was predicted, and new markers were highlighted (Figure 9) [34].
- The highlighted genes were related to the cell cycle, apoptosis, and metabolism. The genes not only predicted the survival of mantle cell lymphoma, but also of diffuse large B-cell lymphoma and a large pan-cancer series of the TCGA [34].
3.5. Diffuse Large B-Cell Lymphoma, Identification of the 25 Genes Set
- A multilayer perceptron analysis predicted the overall survival of 100 cases using as input 54,614 gene probes, and highlighted 25 genes with prognostic value [27].
3.6. Diffuse Large B-Cell Lymphoma, Prognostic Value of the 25 Genes in Hematological Neoplasia, and TNFAIP8 Validation
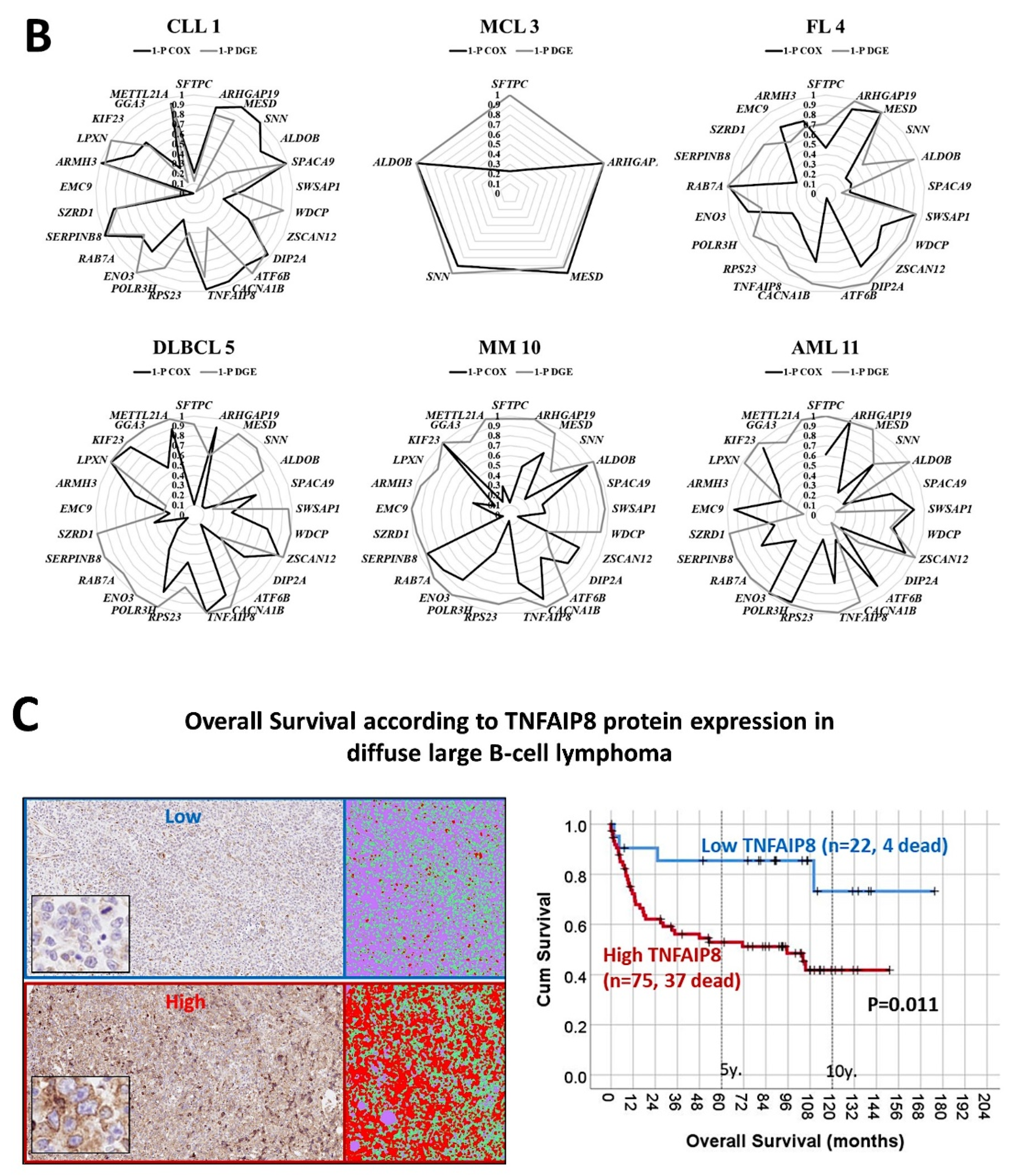
- The previously identified set of 25 genes not only predicted the prognosis of 741 cases of diffuse large B-cell lymphoma, but also predicted other hematological neoplasia, including chronic lymphocytic leukemia (n = 308), mantle cell lymphoma (n = 92), follicular lymphoma (n = 180), multiple myeloma (n = 559), and acute myeloid leukemia (n = 149) [28].
- The TNFAIP8 marker was highlighted in this analysis. Because of TNFAIP8’s importance in the apoptotic pathway, it was validated by immunohistochemistry (i.e., at protein level) in an independent series of 97 cases from Tokai University. Digital image quantification of TNFAIP8 was performed using an AI-based method. Correlations with the prognosis of the patients showed that high TNFAIP8 is associated with poor survival [28].
3.7. Diffuse Large B-Cell Lymphoma, Prediction of Survival by Caspase-8
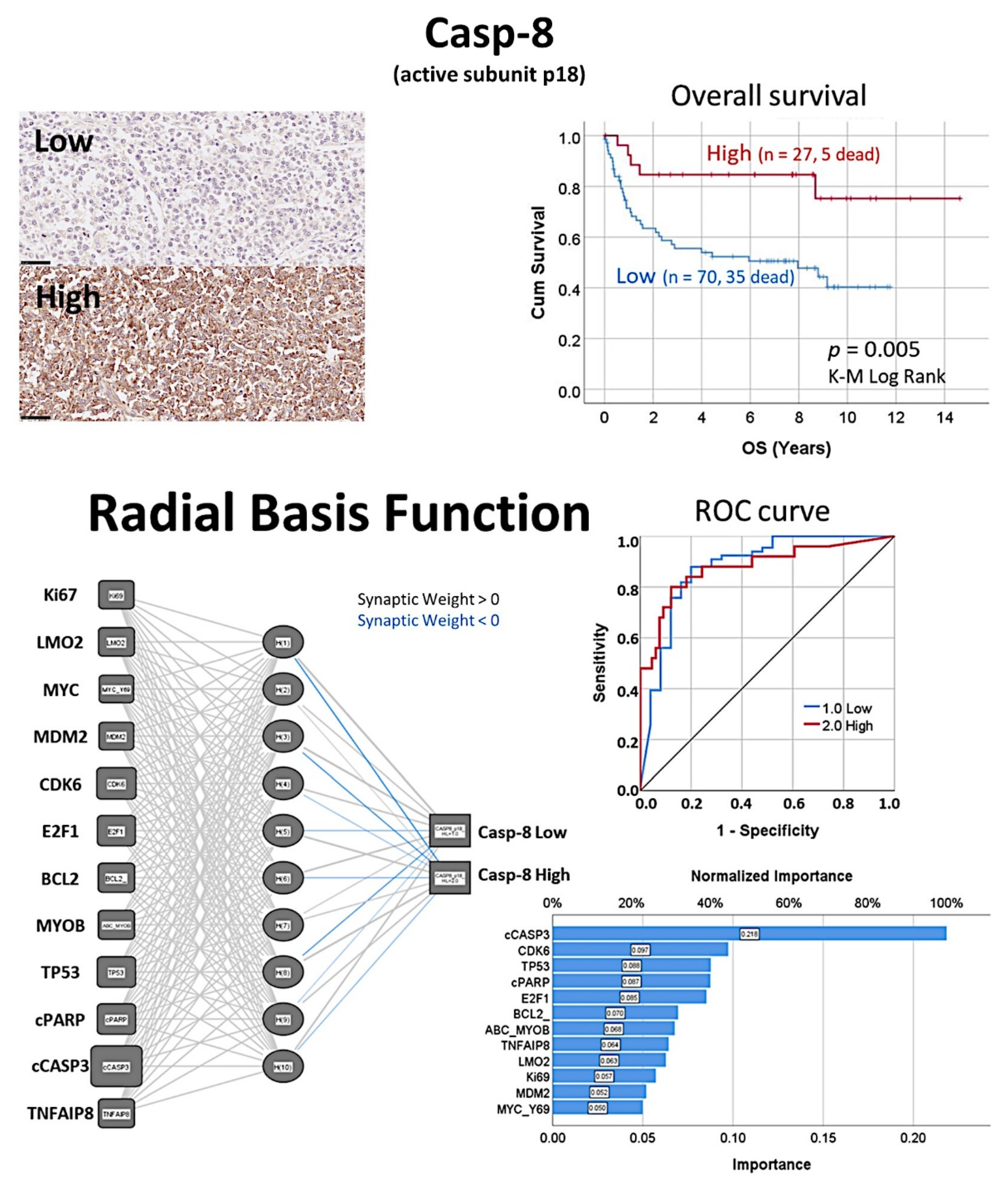
- The protein expression of caspase-8 (which is inhibited by TNFAIP8) was analyzed by immunohistochemistry in a series of 97 cases of diffuse large B-cell lymphoma, and high expression correlated with a favorable overall and progression-free survival [31].
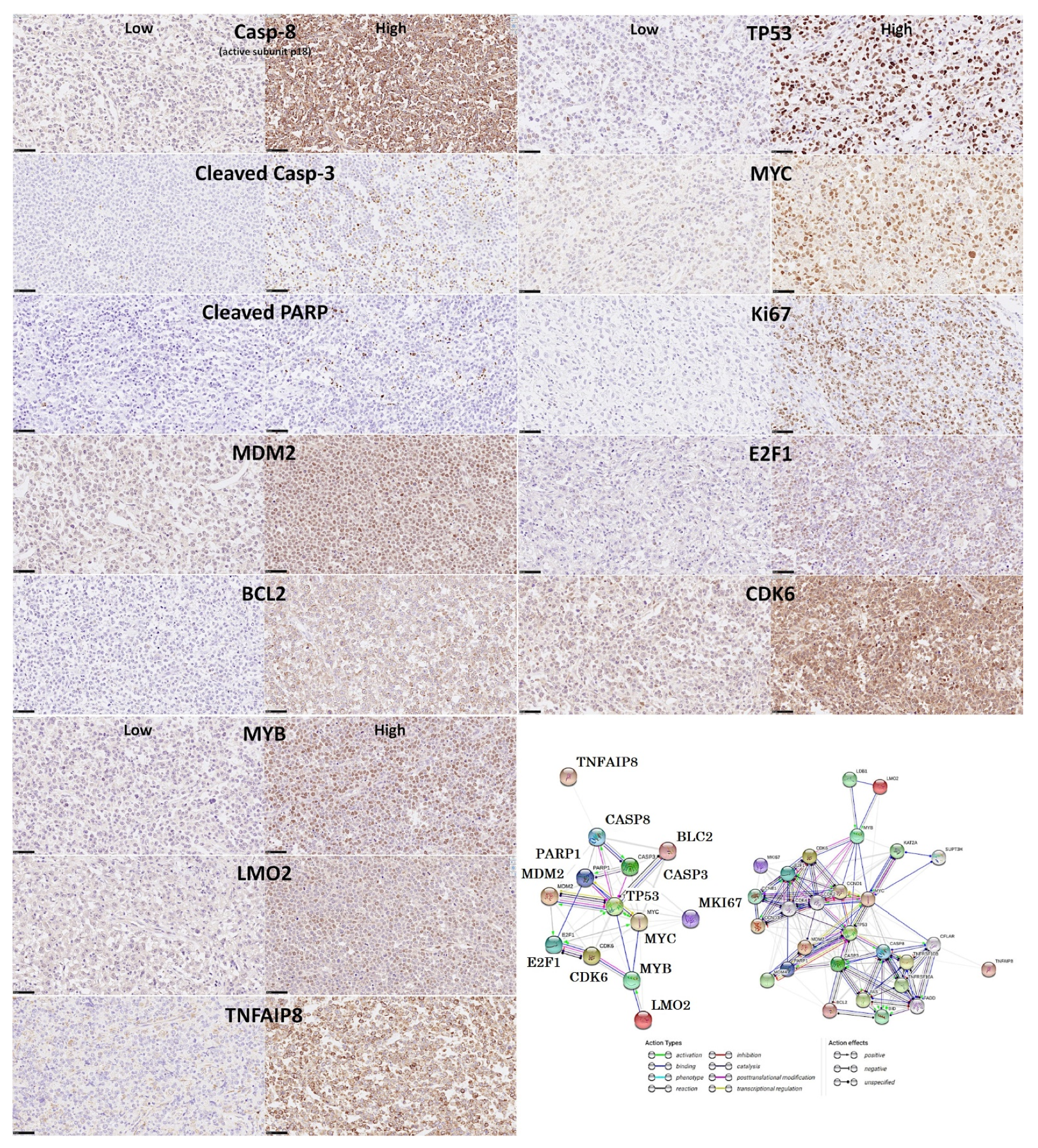
- Based on an immunohistochemical analysis, caspase-8 was correlated with other markers of its pathway, including BCL2, caspase-3, CDK6, cleaved PARP, E2F1, Ki67, LMO2, MDM2, MYB, MYC, TNFAIP8, and TP53 [31].
3.8. Diffuse Large B-Cell Lymphoma, CD274 (PD-L1) and IKAROS
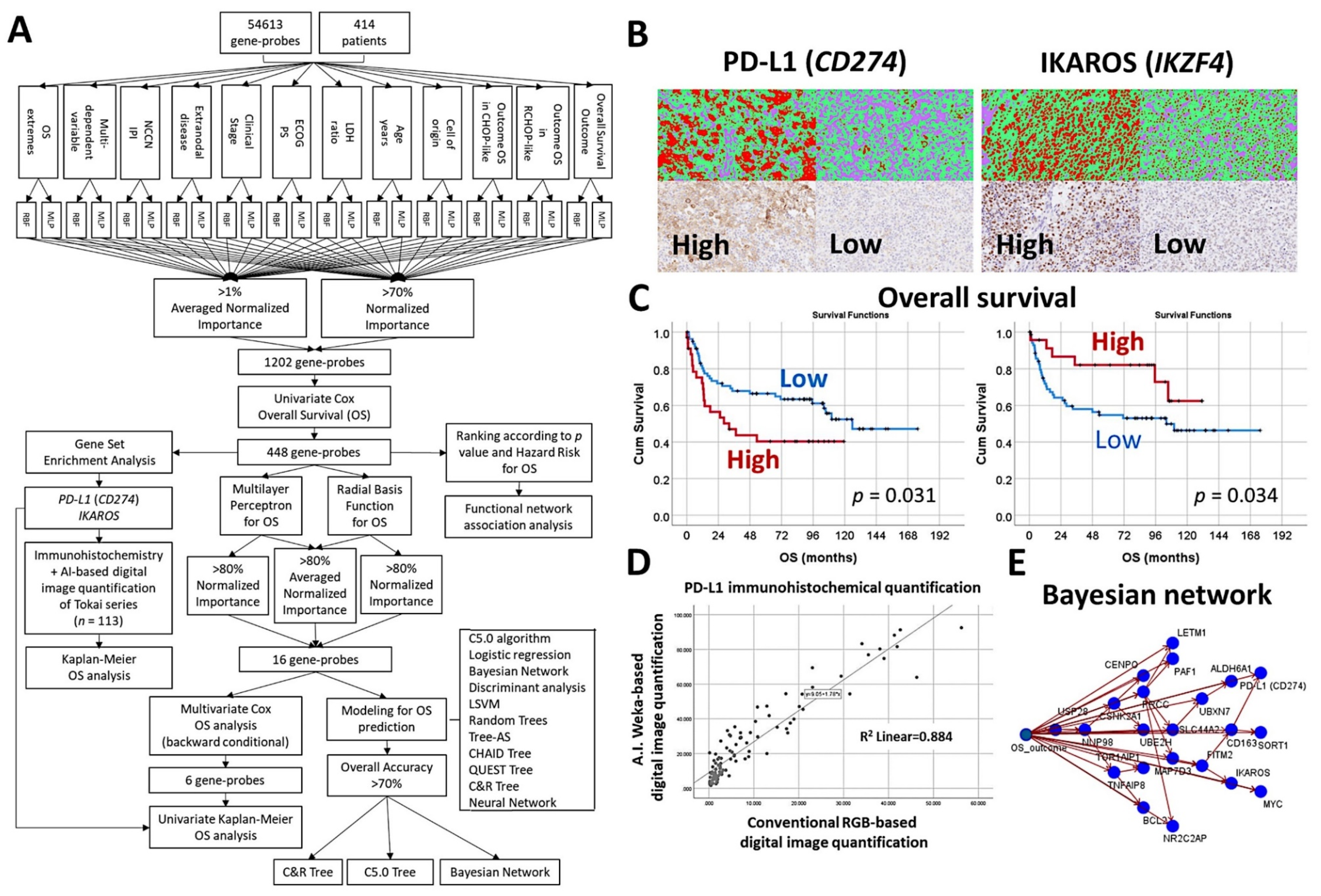
- An algorithm included multilayer perceptron, radial basis function, GSEA, COX regression, and several machine learning techniques to predict the overall survival of 414 cases of diffuse large B-cell lymphoma [30].
- The machine learning techniques were Bayesian network, C5.0 algorithm, chi-squared automatic interaction detection CHAID tree, classification and regression (C&R) tree, discriminant analysis, logistic regression, Quest tree, random trees, and tree-AS. The neural network was the multilayer perceptron [30].
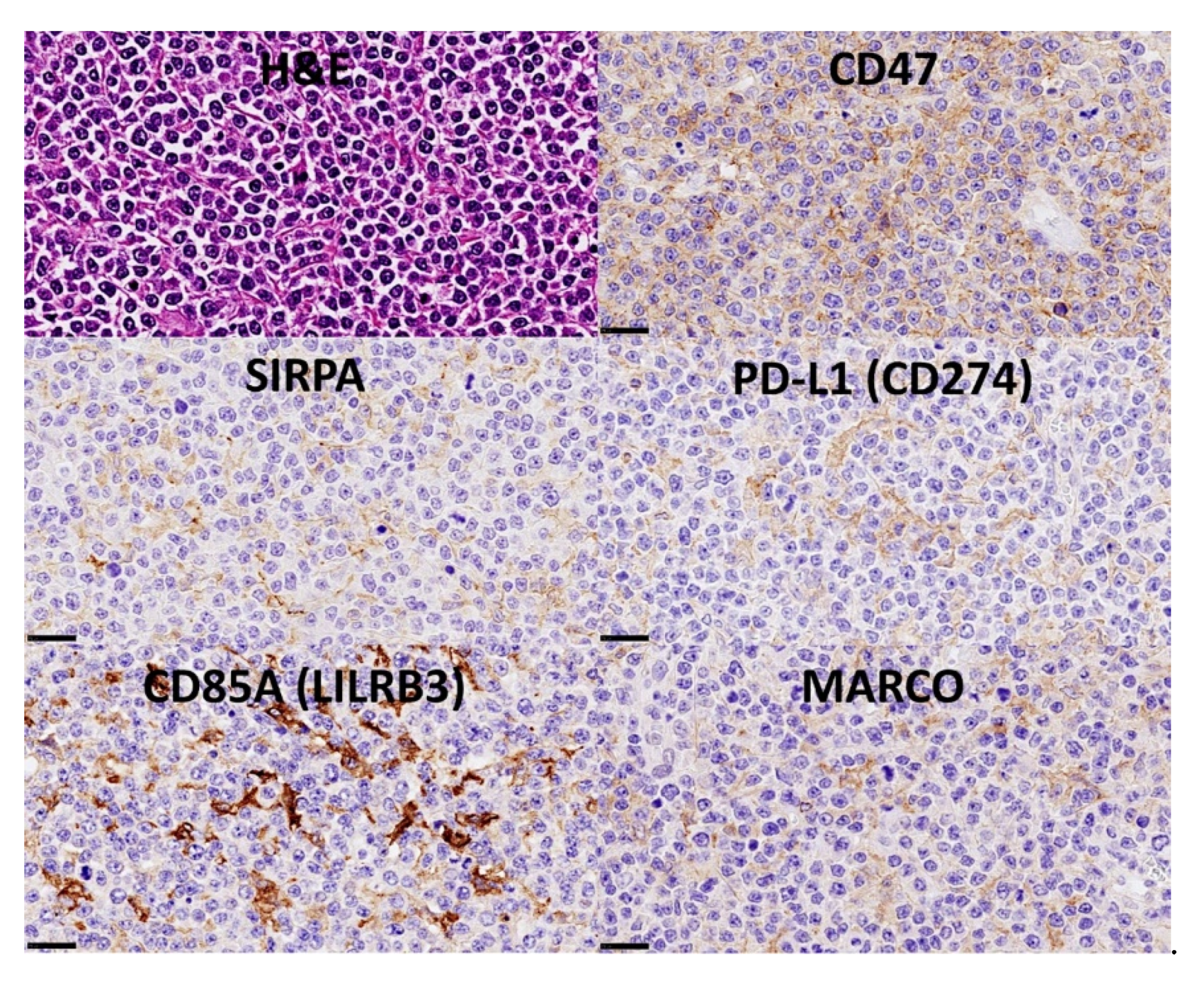
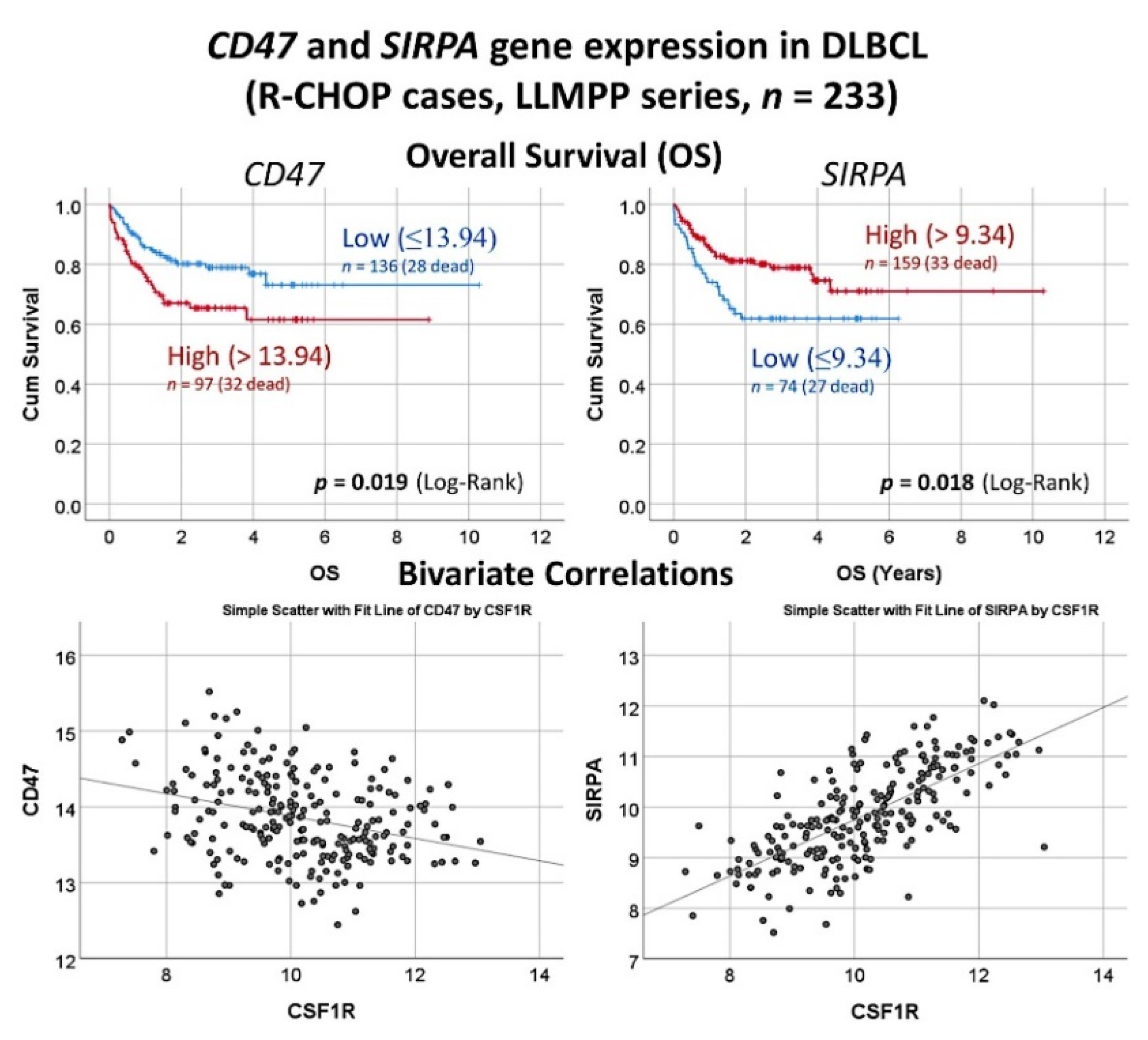
3.9. Diffuse Large B-Cell Lymphoma, CSF1R
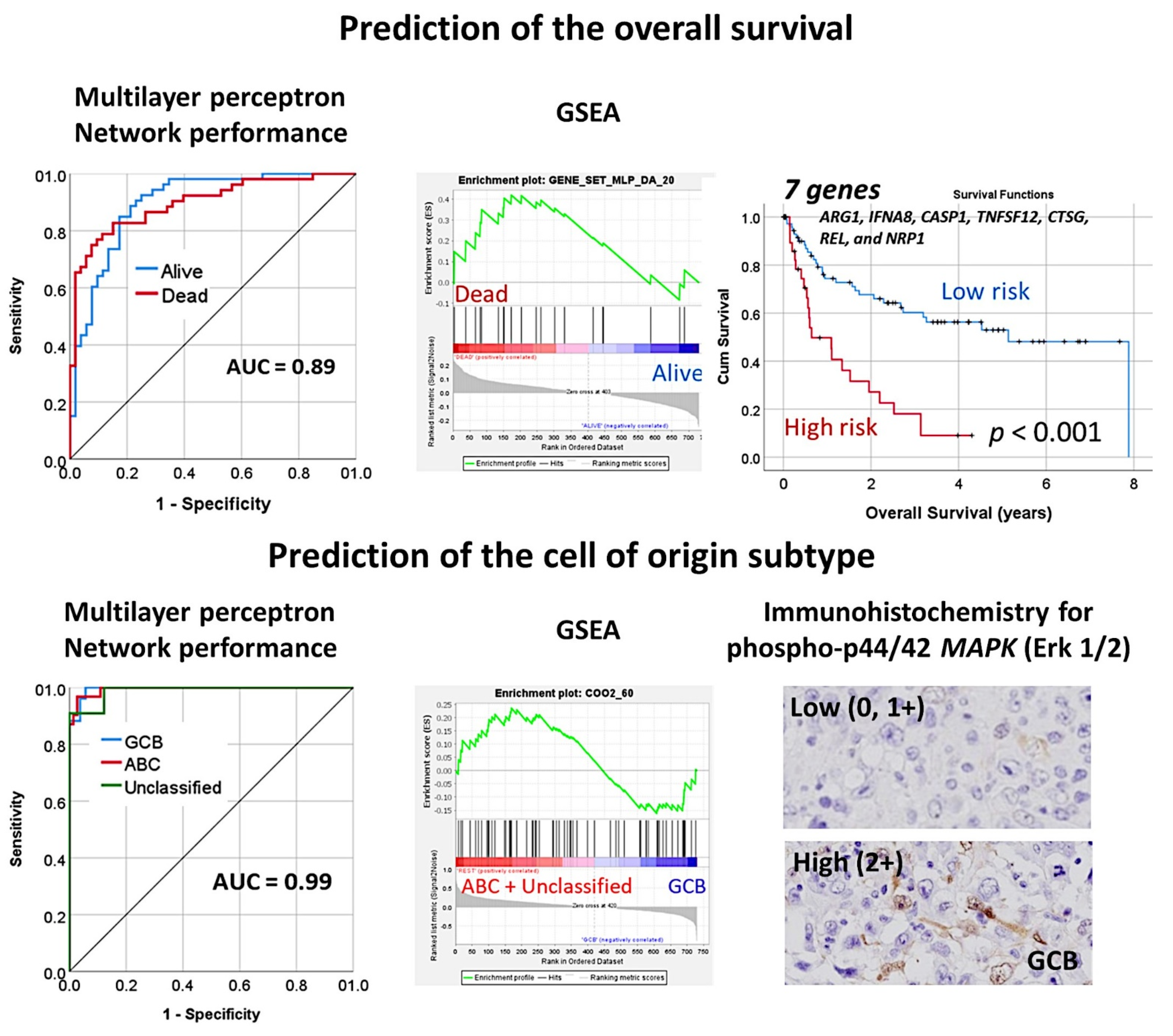
3.10. Diffuse Large B-Cell Lymphoma, Pan-Cancer Immuno-Oncology Panel
- An immuno-oncology panel of 730 genes predicted the overall survival and cell-of-origin phenotype (Lymph2Cx assay) of a series of 106 diffuse large B-cell lymphoma cases, using artificial neural networks and machine learning [33].
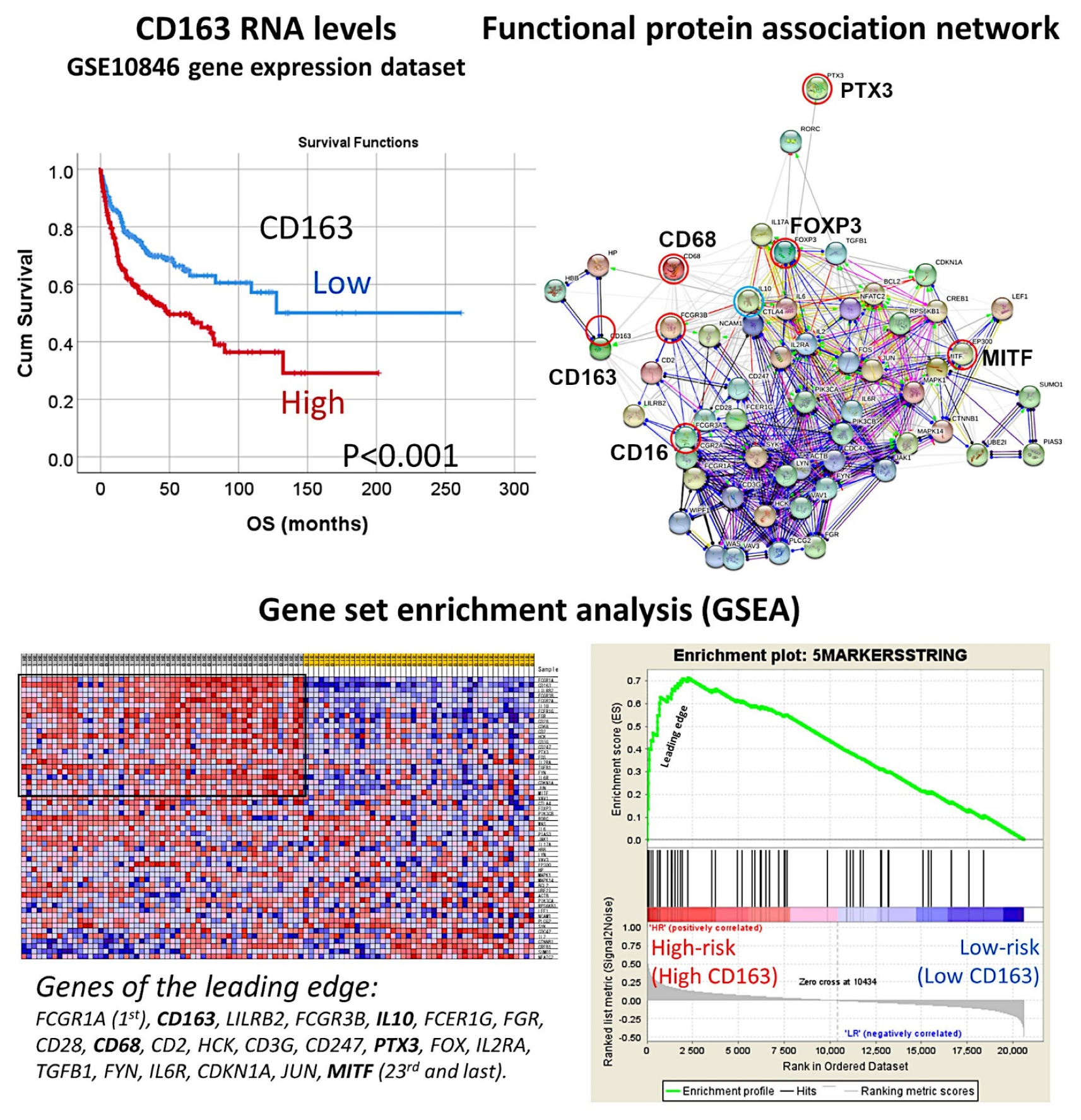
3.11. Diffuse Large B-Cell Lymphoma, Integrative Analysis of Macrophage Markers
4. Discussion
- (1)
- Decision process. Based on the labeled or unlabeled input data, an estimated pattern is produced by the algorithm.
- (2)
- Error function, which evaluates the prediction of the model.
- (3)
- Model optimization process. During the fitting, the weights are adjusted to reduce discrepancy between the known and the estimates, and weights are updated autonomously until a threshold of accuracy is met.
- (1)
- Supervised, which use labeled datasets, such as linear regression, logistic regression, random forest, and support vector machine (SVM).
- (2)
- Unsupervised, which use unlabeled datasets and discover hidden patterns or data groupings without the need of human intervention, such as principal component analysis (PCA), singular value decomposition (SVD), and k-means clustering.
5. Review of the Literature and Future Perspective in Hematological Neoplasia Using AI
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diagnosis | Dataset | No. of Cases | Reference |
|---|---|---|---|
| Non-Hodgkin lymphomas | GSE132929 | 290 | [40] |
| Follicular lymphoma | 65 | ||
| Mantle cell lymphoma | 43 | ||
| Diffuse large B-cell lymphoma | 100 | ||
| Burkitt lymphoma | 59 | ||
| Marginal zone lymphoma | 23 | ||
| Chronic lymphocytic leukemia | GSE22762 | 107 | [41,42] |
| ICGC CLLE-ES | 201 | ||
| Diffuse large B-cell lymphoma | GSE10846 | 414 | [43,44] |
| GSE23501 | 69 | [45] | |
| GSE4475 | 159 | [46,47] | |
| TCGA-DLBCL v.2016 | 47 | ||
| E-TABM-346 | 52 | [48] | |
| Follicular lymphoma | GSE16131 | 180 | [49] |
| Mantle cell lymphoma | LLMPP Rosenwald 2003 | 92 | [50] |
| GSE93291 | 123 | [51] | |
| Multiple myeloma | GSE2658 | 559 | [52,53,54,55,56,57] |
| Acute Myeloid Leukemia | TCGA-AML v.2016 | 149 |
Appendix C. Comments and Analysis Of breast Cancer Detection Using Deep Neural Networks
References
- Harris, N.L.; Jaffe, E.S.; Diebold, J.; Flandrin, G.; Muller-Hermelink, H.K.; Vardiman, J. Lymphoma classification—From controversy to consensus: The R.E.A.L. and WHO Classification of lymphoid neoplasms. Ann. Oncol. 2000, 11 (Suppl. 1), 3–10. [Google Scholar] [CrossRef] [PubMed]
- Campo, E.; Swerdlow, S.H.; Harris, N.L.; Pileri, S.; Stein, H.; Jaffe, E.S. The 2008 WHO classification of lymphoid neoplasms and beyond: Evolving concepts and practical applications. Blood 2011, 117, 5019–5032. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, E.S. Diagnosis and classification of lymphoma: Impact of technical advances. Semin. Hematol. 2019, 56, 30–36. [Google Scholar] [CrossRef]
- De Leval, L.; Jaffe, E.S. Lymphoma Classification. Cancer J. 2020, 26, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Campo, E.; Jaffe, E.S.; Cook, J.R.; Quintanilla-Martinez, L.; Swerdlow, S.H.; Anderson, K.C.; Brousset, P.; Cerroni, L.; de Leval, L.; Dirnhofer, S.; et al. The International Consensus Classification of Mature Lymphoid Neoplasms: A Report from the Clinical Advisory Committee. Blood 2022, 140, 1229–1250. [Google Scholar] [CrossRef]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef]
- Chiorazzi, N.; Rai, K.R.; Ferrarini, M. Chronic lymphocytic leukemia. N. Engl. J. Med. 2005, 352, 804–815. [Google Scholar] [CrossRef]
- Mozas, P.; Sorigué, M.; López-Guillermo, A. Follicular lymphoma: An update on diagnosis, prognosis, and management. Med. Clin. 2021, 157, 440–448. [Google Scholar] [CrossRef]
- Randall, C.; Fedoriw, Y. Pathology and diagnosis of follicular lymphoma and related entities. Pathology 2020, 52, 30–39. [Google Scholar] [CrossRef]
- Donzel, M.; Baseggio, L.; Fontaine, J.; Pesce, F.; Ghesquières, H.; Bachy, E.; Verney, A.; Traverse-Glehen, A. New Insights into the Biology and Diagnosis of Splenic Marginal Zone Lymphomas. Curr. Oncol. 2021, 28, 50297. [Google Scholar] [CrossRef] [PubMed]
- Vilarrasa-Blasi, R.; Verdaguer-Dot, N.; Belver, L.; Soler-Vila, P.; Beekman, R.; Chapaprieta, V.; Kulis, M.; Queirós, A.C.; Parra, M.; Calasanz, M.J.; et al. Insights into the mechanisms underlying aberrant SOX11 oncogene expression in mantle cell lymphoma. Leukemia 2022, 36, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.; Beà, S.; Jares, P.; Campo, E. Molecular Pathogenesis of Mantle Cell Lymphoma. Hematol. Oncol. Clin. N. Am. 2020, 34, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.W.; Mottok, A.; Ennishi, D.; Wright, G.W.; Farinha, P.; Ben-Neriah, S.; Kridel, R.; Barry, G.S.; Hother, C.; Abrisqueta, P.; et al. Prognostic Significance of Diffuse Large B-Cell Lymphoma Cell of Origin Determined by Digital Gene Expression in Formalin-Fixed Paraffin-Embedded Tissue Biopsies. J. Clin. Oncol. 2015, 33, 2848–2856. [Google Scholar] [CrossRef] [PubMed]
- Robetorye, R.S.; Ramsower, C.A.; Rosenthal, A.C.; Yip, T.K.; Wendel Spiczka, A.J.; Glinsmann-Gibson, B.J.; Rimsza, L.M. Incorporation of Digital Gene Expression Profiling for Cell-of-Origin Determination (Lymph2Cx Testing) into the Routine Work-Up of Diffuse Large B-Cell Lymphoma. J. Hematop. 2019, 12, 3–10. [Google Scholar] [CrossRef]
- Ferry, J.A. Burkitt’s lymphoma: Clinicopathologic features and differential diagnosis. Oncologist 2006, 11, 375–383. [Google Scholar] [CrossRef]
- Molyneux, E.M.; Rochford, R.; Griffin, B.; Newton, R.; Jackson, G.; Menon, G.; Harrison, C.J.; Israels, T.; Bailey, S. Burkitt’s lymphoma. Lancet 2012, 379, 1234–1244. [Google Scholar] [CrossRef]
- Dunleavy, K.; Little, R.F.; Wilson, W.H. Update on Burkitt Lymphoma. Hematol. Oncol. Clin. N. Am. 2016, 30, 1333–1343. [Google Scholar] [CrossRef] [PubMed]
- Carreras, J.; Hamoudi, R. Artificial Neural Network Analysis of Gene Expression Data Predicted Non-Hodgkin Lymphoma Subtypes with High Accuracy. Mach. Learn. Knowl. Extr. 2021, 3, 36. [Google Scholar] [CrossRef]
- IBM Cloud Education. IBM Cloud Learn Hub. Machine Learning. July, 2020. Available online: https://www.ibm.com/cloud/learn/machine-learning (accessed on 22 July 2022).
- Kavlakoglu, E. AI vs. Machine Learning vs. Deep Learning vs. Neural Networks: What’s the Difference? Available online: https://www.ibm.com/cloud/blog/ai-vs-machine-learning-vs-deep-learning-vs-neural-networks (accessed on 22 July 2022).
- IBM Cloud Education. Deep Learning. 1 May 2020. Available online: https://www.ibm.com/cloud/learn/deep-learning (accessed on 22 July 2022).
- Delua, J. Supervised vs. Unsupervised Learning: What’s the Difference? IBM Analytics. 12 March 2021. Available online: https://www.ibm.com/cloud/blog/supervised-vs-unsupervised-learning (accessed on 22 July 2022).
- Blais, A.; Mertz, D. An Introduction to Neural Networks. Pattern Learning with the Back-Propagation Algorithm. 19 August 2018. Available online: https://developer.ibm.com/articles/l-neural/ (accessed on 22 July 2022).
- IBM Corporation. Introduction to Neural Networks. 28 February 2021. Available online: https://www.ibm.com/docs/en/spss-statistics/27.0.0?topic=networks-introduction-neural (accessed on 22 July 2022).
- IBM Corporation. IBM SPSS Neural Networks V27; IBM Corporation: Armonk, NY, USA, 2020; pp. 10504–11785. [Google Scholar]
- Carreras, J.; Hamoudi, R.; Nakamura, N. Artificial Intelligence Analysis of Gene Expression Data Predicted the Prognosis of Patients with Diffuse Large B-Cell Lymphoma. Tokai J. Exp. Clin. Med. 2020, 45, 37–48. [Google Scholar]
- Carreras, J.; Kikuti, Y.Y.; Miyaoka, M.; Hiraiwa, S.; Tomita, S.; Ikoma, H.; Kondo, Y.; Ito, A.; Shiraiwa, S.; Hamoudi, R.; et al. A Single Gene Expression Set Derived from Artificial Intelligence Predicted the Prognosis of Several Lymphoma Subtypes; and High Immunohistochemical Expression of TNFAIP8 Associated with Poor Prognosis in Diffuse Large B-Cell Lymphoma. AI 2020, 1, 23. [Google Scholar] [CrossRef]
- Carreras, J.; Kikuti, Y.Y.; Miyaoka, M.; Hiraiwa, S.; Tomita, S.; Ikoma, H.; Kondo, Y.; Ito, A.; Nakamura, N.; Hamoudi, R. Artificial Intelligence Analysis of the Gene Expression of Follicular Lymphoma Predicted the Overall Survival and Correlated with the Immune Microenvironment Response Signatures. Mach. Learn. Knowl. Extr. 2020, 2, 35. [Google Scholar] [CrossRef]
- Carreras, J.; Kikuti, Y.Y.; Miyaoka, M.; Hiraiwa, S.; Tomita, S.; Ikoma, H.; Kondo, Y.; Ito, A.; Nakamura, N.; Hamoudi, R. A Combination of Multilayer Perceptron, Radial Basis Function Artificial Neural Networks and Machine Learning Image Segmentation for the Dimension Reduction and the Prognosis Assessment of Diffuse Large B-Cell Lymphoma. AI 2021, 2, 8. [Google Scholar] [CrossRef]
- Carreras, J.; Kikuti, Y.Y.; Roncador, G.; Miyaoka, M.; Hiraiwa, S.; Tomita, S.; Ikoma, H.; Kondo, Y.; Ito, A.; Shiraiwa, S.; et al. High Expression of Caspase-8 Associated with Improved Survival in Diffuse Large B-Cell Lymphoma: Machine Learning and Artificial Neural Networks Analyses. BioMedInformatics 2021, 1, 3. [Google Scholar] [CrossRef]
- Carreras, J.; Kikuti, Y.Y.; Miyaoka, M.; Roncador, G.; Garcia, J.F.; Hiraiwa, S.; Tomita, S.; Ikoma, H.; Kondo, Y.; Ito, A.; et al. Integrative Statistics, Machine Learning and Artificial Intelligence Neural Network Analysis Correlated CSF1R with the Prognosis of Diffuse Large B-Cell Lymphoma. Hemato 2021, 2, 11. [Google Scholar] [CrossRef]
- Carreras, J.; Hiraiwa, S.; Kikuti, Y.Y.; Miyaoka, M.; Tomita, S.; Ikoma, H.; Ito, A.; Kondo, Y.; Roncador, G.; Garcia, J.F.; et al. Artificial Neural Networks Predicted the Overall Survival and Molecular Subtypes of Diffuse Large B-Cell Lymphoma Using a Pancancer Immune-Oncology Panel. Cancers 2021, 13, 6384. [Google Scholar] [CrossRef] [PubMed]
- Carreras, J.; Nakamura, N.; Hamoudi, R. Artificial Intelligence Analysis of Gene Expression Predicted the Overall Survival of Mantle Cell Lymphoma and a Large Pan-Cancer Series. Healthcare 2022, 10, 155. [Google Scholar] [CrossRef]
- Carreras, J.; Kikuti, Y.Y.; Miyaoka, M.; Hiraiwa, S.; Tomita, S.; Ikoma, H.; Kondo, Y.; Ito, A.; Hamoudi, R.; Nakamura, N. The Use of the Random Number Generator and Artificial Intelligence Analysis for Dimensionality Reduction of Follicular Lymphoma Transcriptomic Data. BioMedInformatics 2022, 2, 17. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Cheson, B.D.; Pfistner, B.; Juweid, M.E.; Gascoyne, R.D.; Specht, L.; Horning, S.J.; Coiffier, B.; Fisher, R.I.; Hagenbeek, A.; Zucca, E.; et al. International Harmonization Project on Lymphoma. Revised response criteria for malignant lymphoma. J. Clin. Oncol. 2007, 25, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Gamboa, R.; Gomez-Rueda, H.; Martínez-Ledesma, E.; Martínez-Torteya, A.; Chacolla-Huaringa, R.; Rodriguez-Barrientos, A.; Tamez-Peña, J.G.; Treviño, V. SurvExpress: An online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS ONE 2013, 8, e74250. [Google Scholar] [CrossRef]
- Ma, M.C.J.; Tadros, S.; Bouska, A.; Heavican, T.; Yang, H.; Deng, Q.; Moore, D.; Akhter, A.; Hartert, K.; Jain, N.; et al. Subtype-specific and co-occurring genetic alterations in B-cell non-Hodgkin lymphoma. Haematologica 2022, 107, 690–701. [Google Scholar] [CrossRef]
- Herold, T.; Jurinovic, V.; Metzeler, K.H.; Boulesteix, A.L.; Bergmann, M.; Seiler, T.; Mulaw, M.; Thoene, S.; Dufour, A.; Pasalic, Z.; et al. An eight-gene expression signature for the prediction of survival and time to treatment in chronic lymphocytic leukemia. Leukemia 2011, 25, 1639–1645. [Google Scholar] [CrossRef]
- Herold, T.; Mulaw, M.A.; Jurinovic, V.; Seiler, T.; Metzeler, K.H.; Dufour, A.; Schneider, S.; Kakadia, P.M.; Spiekermann, K.; Mansmann, U.; et al. High expression of MZB1 predicts adverse prognosis in chronic lymphocytic leukemia, follicular lymphoma and diffuse large B-cell lymphoma and is associated with a unique gene expression signature. Leuk. Lymphoma 2013, 54, 1652–1657. [Google Scholar] [CrossRef] [PubMed]
- Lenz, G.; Wright, G.; Dave, S.S.; Xiao, W.; Powell, J.; Zhao, H.; Xu, W.; Tan, B.; Goldschmidt, N.; Iqbal, J.; et al. Lymphoma/Leukemia Molecular Profiling Project. Stromal gene signatures in large-B-cell lymphomas. N. Engl. J. Med. 2008, 359, 2313–2323. [Google Scholar] [CrossRef] [PubMed]
- Cardesa-Salzmann, T.M.; Colomo, L.; Gutierrez, G.; Chan, W.C.; Weisenburger, D.; Climent, F.; González-Barca, E.; Mercadal, S.; Arenillas, L.; Serrano, S.; et al. High microvessel density determines a poor outcome in patients with diffuse large B-cell lymphoma treated with rituximab plus chemotherapy. Haematologica 2011, 96, 996–1001. [Google Scholar] [CrossRef]
- Shaknovich, R.; Geng, H.; Johnson, N.A.; Tsikitas, L.; Cerchietti, L.; Greally, J.M.; Gascoyne, R.D.; Elemento, O.; Melnick, A. DNA methylation signatures define molecular subtypes of diffuse large B-cell lymphoma. Blood 2010, 116, e81–e89. [Google Scholar] [CrossRef]
- Hummel, M.; Bentink, S.; Berger, H.; Klapper, W.; Wessendorf, S.; Barth, T.F.; Bernd, H.W.; Cogliatti, S.B.; Dierlamm, J.; Feller, A.C.; et al. Molecular Mechanisms in Malignant Lymphomas Network Project of the Deutsche Krebshilfe. A biologic definition of Burkitt’s lymphoma from transcriptional and genomic profiling. N. Engl. J. Med. 2006, 354, 2419–2430. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.; Schlesner, M.; Hoffmann, S.; Kreuz, M.; Leich, E.; Burkhardt, B.; Rosolowski, M.; Ammerpohl, O.; Wagener, R.; Bernhart, S.H.; et al. ICGC MMML-Seq Project. Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat. Genet. 2012, 44, 1316–1320. [Google Scholar] [CrossRef] [PubMed]
- Jais, J.P.; Haioun, C.; Molina, T.J.; Rickman, D.S.; de Reynies, A.; Berger, F.; Gisselbrecht, C.; Brière, J.; Reyes, F.; Gaulard, P.; et al. Groupe d’Etude des Lymphomes de l’Adulte. The expression of 16 genes related to the cell of origin and immune response predicts survival in elderly patients with diffuse large B-cell lymphoma treated with CHOP and rituximab. Leukemia 2008, 22, 1917–1924. [Google Scholar] [CrossRef]
- Leich, E.; Salaverria, I.; Bea, S.; Zettl, A.; Wright, G.; Moreno, V.; Gascoyne, R.D.; Chan, W.C.; Braziel, R.M.; Rimsza, L.M.; et al. Follicular lymphomas with and without translocation t(14;18) differ in gene expression profiles and genetic alterations. Blood 2009, 114, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Rosenwald, A.; Wright, G.; Wiestner, A.; Chan, W.C.; Connors, J.M.; Campo, E.; Gascoyne, R.D.; Grogan, T.M.; Muller-Hermelink, H.K.; Smeland, E.B.; et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell 2003, 3, 185–197. [Google Scholar] [CrossRef]
- Scott, D.W.; Abrisqueta, P.; Wright, G.W.; Slack, G.W.; Mottok, A.; Villa, D.; Jares, P.; Rauert-Wunderlich, H.; Royo, C.; Clot, G.; et al. Lymphoma/Leukemia Molecular Profiling Project. New Molecular Assay for the Proliferation Signature in Mantle Cell Lymphoma Applicable to Formalin-Fixed Paraffin-Embedded Biopsies. J. Clin. Oncol. 2017, 35, 1668–1677. [Google Scholar] [CrossRef]
- Hanamura, I.; Huang, Y.; Zhan, F.; Barlogie, B.; Shaughnessy, J. Prognostic value of cyclin D2 mRNA expression in newly diagnosed multiple myeloma treated with high-dose chemotherapy and tandem autologous stem cell transplantations. Leukemia 2006, 20, 1288–1290. [Google Scholar] [CrossRef] [PubMed]
- Zhan, F.; Huang, Y.; Colla, S.; Stewart, J.P.; Hanamura, I.; Gupta, S.; Epstein, J.; Yaccoby, S.; Sawyer, J.; Burington, B.; et al. The molecular classification of multiple myeloma. Blood 2006, 108, 2020–2028. [Google Scholar] [CrossRef]
- Zhan, F.; Barlogie, B.; Arzoumanian, V.; Huang, Y.; Williams, D.R.; Hollmig, K.; Pineda-Roman, M.; Tricot, G.; van Rhee, F.; Zangari, M.; et al. Gene-expression signature of benign monoclonal gammopathy evident in multiple myeloma is linked to good prognosis. Blood 2007, 109, 1692–1700. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, S.; Zhou, Y.; Wu, X.; Entin, I.; Epstein, J.; Yaccoby, S.; Xiong, W.; Barlogie, B.; Shaughnessy, J.D., Jr.; et al. Identification of early growth response protein 1 (EGR-1) as a novel target for JUN-induced apoptosis in multiple myeloma. Blood 2010, 115, 61–70. [Google Scholar] [CrossRef]
- Qiang, Y.W.; Ye, S.; Huang, Y.; Chen, Y.; Van Rhee, F.; Epstein, J.; Walker, B.A.; Morgan, G.J.; Davies, F.E. MAFb protein confers intrinsic resistance to proteasome inhibitors in multiple myeloma. BMC Cancer 2018, 18, 724. [Google Scholar] [CrossRef]
- Went, M.; Sud, A.; Försti, A.; Halvarsson, B.M.; Weinhold, N.; Kimber, S.; van Duin, M.; Thorleifsson, G.; Holroyd, A.; Johnson, D.C.; et al. PRACTICAL consortium. Identification of multiple risk loci and regulatory mechanisms influencing susceptibility to multiple myeloma. Nat. Commun. 2018, 9, 3707. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, L.R.; Young, K.H. New agents and regimens for diffuse large B cell lymphoma. J. Hematol. Oncol. 2020, 13, 175. [Google Scholar] [CrossRef] [PubMed]
- Carreras, J. Artificial Intelligence Analysis of Celiac Disease Using an Autoimmune Discovery Transcriptomic Panel Highlighted Pathogenic Genes including BTLA. Healthcare 2022, 10, 1550. [Google Scholar] [CrossRef] [PubMed]
- Carreras, J. Artificial Intelligence Analysis of Ulcerative Colitis Using an Autoimmune Discovery Transcriptomic Panel. Healthcare 2022, 10, 1476. [Google Scholar] [CrossRef] [PubMed]
- Lisson, C.S.; Lisson, C.G.; Mezger, M.F.; Wolf, D.; Schmidt, S.A.; Thaiss, W.M.; Tausch, E.; Beer, A.J.; Stilgenbauer, S.; Beer, M. Deep Neural Networks and Machine Learning Radiomics mode for Prediction of Relapse in Mantle Cell Lymphoma. Cancers 2022, 14, 2008. [Google Scholar] [CrossRef]
- Sadik, M.; López-Urdaneta, J.; Ulén, J.; Enqvist, O.; Krupic, A.; Kumar, R.; Andersson, P.O.; Trägårdh, E. Artificial intelligence could alert for focal skeleton/bone marrow uptake in Hodgkin’s lymphoma patients staged with FDG-PET/CT. Sci. Rep. 2021, 11, 10382. [Google Scholar] [CrossRef]
- Wang, Y.J.; Baratto, L.; Hawk, K.E.; Theruvath, A.J.; Pribnow, A.; Thakor, A.S.; Gatidis, S.; Lu, R.; Gummidipundi, S.E.; Garcia-Diaz, J.; et al. Artificial intelligence enables whole-body positron emission tomography scans with minimal radiation exposure. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 2771–2781. [Google Scholar] [CrossRef] [PubMed]
- Pinochet, P.; Eude, F.; Becker, S.; Shah, V.; Sibille, L.; Toledano, M.N.; Modzelewski, R.; Vera, P.; Decazes, P. Evaluation of an Automatic Classification Algorithm Using Convolutional Neural Networks in Oncological Positron Emission Tomography. Front. Med. 2021, 8, 628179. [Google Scholar] [CrossRef]
- El Hussein, S.; Chen, P.; Medeiros, L.J.; Wistuba, I.I.; Jaffray, D.; Wu, J.; Khoury, J.D. Artificial intelligence strategy integrating morphologic and architectural biomarkers provides robust diagnostic accuracy for disease progression in chronic lymphocytic leukemia. J. Pathol. 2022, 256, 4–14. [Google Scholar] [CrossRef]
- Swiderska-Chadaj, Z.; Hebeda, K.M.; van den Brand, M.; Litjens, G. Artificial intelligence to detect MYC translocation in slides of diffuse large B-cell lymphoma. Virchows Arch. 2021, 479, 617–621. [Google Scholar] [CrossRef]
- Steinbuss, G.; Kriegsmann, M.; Zgorzelski, C.; Brobeil, A.; Goeppert, B.; Dietrich, S.; Mechtersheimer, G.; Kriegsmann, K. Deep Learning for the Classification of Non-Hodgkin Lymphoma on Histopathological Images. Cancers 2021, 13, 2419. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, K.; Jiang, M.; Yang, L. Research on the classification of lymphoma pathological images based on deep residual neural network. Technol. Health Care 2021, 29, 335–344. [Google Scholar] [CrossRef]
- Tang, G.; Fu, X.; Wang, Z.; Chen, M. A Machine Learning Tool Using Digital Microscopy (Morphogo) for the Identification of Abnormal Lymphocytes in the Bone Marrow. Acta Cytol. 2021, 65, 354–357. [Google Scholar] [CrossRef]
- Yu, W.H.; Li, C.H.; Wang, R.C.; Yeh, C.Y.; Chuang, S.S. Machine Learning Based on Morphological Features Enables Classification of Primary Intestinal T-Cell Lymphomas. Cancers 2021, 13, 5463. [Google Scholar] [CrossRef]
- Zhou, M.; Wu, K.; Yu, L.; Xu, M.; Yang, J.; Shen, Q.; Liu, B.; Shi, L.; Wu, S.; Dong, B.; et al. Development and Evaluation of a Leukemia Diagnosis System Using Deep Learning in Real Clinical Scenarios. Front. Pediatr. 2021, 9, 693676. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cui, W.; Guo, X.; Wang, B.; Wang, Z. Classification of digital pathological images of non-Hodgkin’s lymphoma subtypes based on the fusion of transfer learning and principal component analysis. Med. Phys. 2020, 47, 4241–4253. [Google Scholar] [CrossRef]
- Mohlman, J.S.; Leventhal, S.D.; Hansen, T.; Kohan, J.; Pascucci, V.; Salama, M.E. Improving Augmented Human Intelligence to Distinguish Burkitt Lymphoma from Diffuse Large B-Cell Lymphoma Cases. Am. J. Clin. Pathol. 2020, 153, 743–759. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Bledsoe, J.R.; Zeng, Y.; Liu, W.; Hu, Y.; Bi, K.; Liang, A.; Li, S. A deep learning diagnostic platform for diffuse large B-cell lymphoma with high accuracy across multiple hospitals. Nat. Commun. 2020, 11, 6004. [Google Scholar] [CrossRef]
- Miyoshi, H.; Sato, K.; Kabeya, Y.; Yonezawa, S.; Nakano, H.; Takeuchi, Y.; Ozawa, I.; Higo, S.; Yanagida, E.; Yamada, K.; et al. Deep learning shows the capability of high-level computer-aided diagnosis in malignant lymphoma. Lab. Investig. 2020, 100, 1300–1310. [Google Scholar] [CrossRef] [PubMed]
- Zorman, M.; de la Rosa, J.L.S.; Dinevski, D. Classification of follicular lymphoma images: A holistic approach with symbol-based machine learning methods. Wien. Klin. Wochenschr. 2011, 123, 700–709. [Google Scholar] [CrossRef]
- Zhao, M.; Mallesh, N.; Höllein, A.; Schabath, R.; Haferlach, C.; Haferlach, T.; Elsner, F.; Lüling, H.; Krawitz, P.; Kern, W. Hematologist-Level Classification of Mature B-Cell Neoplasm Using Deep Learning on Multiparameter Flow Cytometry Data. Cytom. A 2020, 97, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Gaidano, V.; Tenace, V.; Santoro, N.; Varvello, S.; Cignetti, A.; Prato, G.; Saglio, G.; De Rosa, G.; Geuna, M. A Clinically Applicable Approach to the Classification of B-Cell Non-Hodgkin Lymphomas with Flow Cytometry and Machine Learning. Cancers 2020, 12, 1684. [Google Scholar] [CrossRef] [PubMed]
- Zhan, M.; Chen, Z.B.; Ding, C.C.; Qu, Q.; Wang, G.Q.; Liu, S.; Wen, F.Q. Machine learning to predict high-dose methotrexate-related neutropenia and fever in children with B-cell acute lymphoblastic leukemia. Leuk. Lymphoma 2021, 62, 2502–2513. [Google Scholar] [CrossRef] [PubMed]
- Buciński, A.; Marszałł, M.P.; Krysiński, J.; Lemieszek, A.; Załuski, J. Contribution of artificial intelligence to the knowledge of prognostic factors in Hodgkin’s lymphoma. Eur. J. Cancer Prev. 2010, 19, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Xu-Monette, Z.Y.; Zhang, H.; Zhu, F.; Tzankov, A.; Bhagat, G.; Visco, C.; Dybkaer, K.; Chiu, A.; Tam, W.; Zu, Y.; et al. A refined cell-of-origin classifier with targeted NGS and artificial intelligence shows robust predictive value in DLBCL. Blood Adv. 2020, 4, 3391–3404. [Google Scholar] [CrossRef]
- Zhang, W.; Yang, L.; Guan, Y.Q.; Shen, K.F.; Zhang, M.L.; Cai, H.D.; Wang, J.C.; Wang, Y.; Huang, L.; Cao, Y.; et al. Novel bioinformatic classification system for genetic signatures identification in diffuse large B-cell lymphoma. BMC Cancer 2020, 20, 714. [Google Scholar] [CrossRef] [PubMed]
- Parodi, S.; Manneschi, C.; Verda, D.; Ferrari, E.; Muselli, M. Logic Learning Machine and standard supervised methods for Hodgkin’s lymphoma prognosis using gene expression data and clinical variables. Health Inform. J. 2018, 24, 54–65. [Google Scholar] [CrossRef]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef]
- Carreras, J.; Kikuti, Y.Y.; Hiraiwa, S.; Miyaoka, M.; Tomita, S.; Ikoma, H.; Ito, A.; Kondo, Y.; Itoh, J.; Roncador, G.; et al. High PTX3 expression is associated with a poor prognosis in diffuse large B-cell lymphoma. Cancer Sci. 2022, 113, 334–348. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Loibl, S.; Poortmans, P.; Morrow, M.; Denkert, C.; Curigliano, G. Breast cancer. Lancet 2021, 397, 1750–1769. [Google Scholar] [CrossRef]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef]
- Chen, S.; Parmigiani, G. Meta-analysis of BRCA1 and BRCA2 penetrance. J. Clin. Oncol. 2007, 25, 1329–1333. [Google Scholar] [CrossRef]
- Basurto-Hurtado, J.A.; Cruz-Albarran, I.A.; Toledano-Ayala, M.; Ibarra-Manzano, M.A.; Morales-Hernandez, L.A.; Perez-Ramirez, C.A. Diagnostic Strategies for Breast Cancer Detection: From Image Generation to Classification Strategies Using Artificial Intelligence Algorithms. Cancers 2022, 14, 3442. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, M.; Wang, H.; Jiang, H.; Yao, Y.; Zhang, H.; Xin, J. Breast Cancer Detection Using Extreme Learning Machine Based on Feature Fusion with CNN Deep Features. IEEE Access 2019, 7, 105146–105158. [Google Scholar] [CrossRef]
- Yap, M.H.; Pons, G.; Marti, J.; Ganau, S.; Sentis, M.; Zwiggelaar, R.; Davison, A.K.; Marti, R. Automated Breast Ultrasound Lesions Detection Using Convolutional Neural Networks. IEEE J. Biomed. Health Inform. 2018, 22, 1218–1226. [Google Scholar] [CrossRef]
- Teare, P.; Fishman, M.; Benzaquen, O.; Toledano, E.; Elnekave, E. Malignancy Detection on Mammography Using Dual Deep Convolutional Neural Networks and Genetically Discovered False Color Input Enhancement. J. Digit. Imaging 2017, 30, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Margolies, L.R.; Rothstein, J.H.; Fluder, E.; McBride, R.; Sieh, W. Deep Learning to Improve Breast Cancer Detection on Screening Mammography. Sci. Rep. 2019, 9, 12495. [Google Scholar] [CrossRef] [PubMed]
- Gamage, T.P.B.; Malcolm, D.T.K.; Talou, G.D.M.; Mîra, A.; Doyle, A.; Nielsen, P.M.F.; Nash, M.P. An automated computational biomechanics workflow for improving breast cancer diagnosis and treatment. Interface Focus 2019, 9, 20190034. [Google Scholar] [CrossRef]
- Bouron, C.; Mathie, C.; Seegers, V.; Morel, O.; Jézéquel, P.; Lasla, H.; Guillerminet, C.; Girault, S.; Lacombe, M.; Sher, A.; et al. Prognostic Value of Metabolic, Volumetric and Textural Parameters of Baseline [18F]FDG PET/CT in Early Triple-Negative Breast Cancer. Cancers 2022, 14, 637. [Google Scholar] [CrossRef] [PubMed]
- Mughal, B.; Sharif, M.; Muhammad, N. Bi-model processing for early detection of breast tumor in CAD system. Eur. Phys. J. Plus 2017, 132, 266. [Google Scholar] [CrossRef]
- Wang, S.; Rao, R.V.; Chen, P.; Zhang, Y.; Liu, A.; Wei, L. Abnormal Breast Detection in Mammogram Images by Feed-forward Neural Network Trained by Jaya Algorithm. Fundam. Inform. 2017, 151, 191–211. [Google Scholar] [CrossRef]
- Muduli, D.; Dash, R.; Majhi, B. Automated breast cancer detection in digital mammograms: A moth flame optimization based ELM approach. Biomed. Signal Process. Control 2020, 59, 101912. [Google Scholar] [CrossRef]
- Shiji, T.P.; Remya, S.; Lakshmanan, R.; Pratab, T.; Thomas, V. Evolutionary intelligence for breast lesion detection in ultrasound images: A wavelet modulus maxima and SVM based approach. J. Intell. Fuzzy Syst. 2020, 38, 6279–6290. [Google Scholar] [CrossRef]
- Chakraborty, J.; Midya, A.; Rabidas, R. Computer-aided detection and diagnosis of mammographic masses using multi-resolution analysis of oriented tissue patterns. Expert Syst. Appl. 2018, 99, 168–179. [Google Scholar] [CrossRef]
- Zahoor, S.; Shoaib, U.; Lali, I.U. Breast Cancer Mammograms Classification Using Deep Neural Network and Entropy-Controlled Whale Optimization Algorithm. Diagnostics 2022, 12, 557. [Google Scholar] [CrossRef]
- Jagtap, A.D.; Shin, Y.; Kawaguchi, K.; Em Karniadakis, G. Deep Kronecker neural networks: A general framework for neural networks with adaptive activation functions. Neurocomputing 2022, 468, 165–180. [Google Scholar] [CrossRef]
- Ali, M.J.; Raza, B.; Shahid, A.R. Multi-level Kronecker Convolutional Neural Network (ML-KCNN) for Glioma Segmentation from Multi-modal MRI Volumetric Data. J. Digit. Imaging 2021, 34, 905–921. [Google Scholar] [CrossRef]

























| Marker | Target/Pathway | Primary Antibody | Company |
|---|---|---|---|
| BCL2 | Apoptosis | bcl2/100/D5 | Novocastra |
| BCL6 | Germinal center | LN22 | Novocastra |
| cCASP3 | Apoptosis | Asp175, #9661 | Cell Signaling |
| CASP8 | Apoptosis | active subunit p18, 11B6 | Novocastra |
| CD3 | T lymphocytes | CD3 epsilon, LN10 | Novocastra |
| CD5 | T lymphocytes | 4C7 | Novocastra |
| CD10 | Germinal center | 56C6 | Novocastra |
| CD16 | M1-like macrophages | 2H7 | Novocastra |
| CD20 | B lymphocytes | L26 | Novocastra |
| CD47 | B lymphocytes | D3O7P | Cell Signaling |
| CD68 | Pan-macrophages | 514H12 | Novocastra |
| CD85A/LILRB3 | M2-like macrophages | FRAS92B | CNIO |
| CD163 | M2-like macrophages | 10D6 | Novocastra |
| CDK6 | Cell cycle | 98D | CNIO |
| CSF1 | CSF1R pathway | 2D10 | LSBio |
| CSF1R | M2-like macrophages | 2D10 | LSBio |
| Cyclin D1 | Cell cycle | P2D11F11 | Novocastra |
| E2F1 | Cell cycle | Agro368V | CNIO |
| EBER | Epstein-Barr virus | #PB0589, #AR0833 | Novocastra |
| IKAROS | Cytokine signaling | D6N9Y | Cell Signaling |
| IL10 | M2c-like macrophages | LS-B7432 | Lifespan Bioscience |
| Ki67 | Cell cycle | MM1 | Novocastra |
| LMO2 | Proto-oncogene | 299B | CNIO |
| MARCO | Macrophages | HPA063793 | Atlas antibodies |
| MDM2 | p53 signaling | IF2 | Invitrogen |
| MITF | M2-like macrophages | C5/D5/MAB10775 | Abnova |
| MUM1 | Plasma cells | IRF4, EAU32 | Novocastra |
| MYC | Proto-oncogene | Y69 | Abcam |
| NFKB p105/p50 | NFKB pathway | #3035 | Cell Signaling |
| cPARP | Apoptosis | Asp214, D64E10 | Cell Signaling |
| PD-L1 | Immune checkpoint | E1J2J | Cell Signaling |
| p-p44/42 MAPK | MAPK pathway | Thr202/Tyr204, #4370 | Cell Signaling |
| pSTAT3 | STAT3 pathway | Tyr705, D3A7 | Cell Signaling |
| PTX3 | M2c-like macrophages | PPZ1228 | Perseus Proteomics |
| RGS1 | Signal transduction | Rabbit polyclonal | Thermo Fisher |
| SIRPA | M2-like macrophages | D6I3M | Cell Signaling |
| TNFAIP8 | Apoptosis | #14559-MM01 | Sino Biological |
| TP53 | Cell cycle, apoptosis | DO-7 | Novocastra |
| RGS1 | Signal transduction | Rabbit polyclonal | Thermo Fisher |
| Model | No. of Predictors | Overall Accuracy (%) |
|---|---|---|
| XGBoost Tree | 25 | 100 |
| Random Forest | 25 | 98.3 |
| Random Trees | 25 | 97.1 |
| Bayesian Network | 25 | 89.3 |
| SVM | 25 | 84.5 |
| KNN Algorithm | 25 | 81.9 |
| CHAID | 6 | 79.8 |
| LSVM | 25 | 78.5 |
| Logistic Regression | 25 | 78.1 |
| C5 Tree | 3 | 75.9 |
| Tree-AS | 2 | 74.3 |
| XGBoost Linear | 25 | 74.3 |
| Quest | 25 | 74.3 |
| C&R Tree | 25 | 74.3 |
| Neural Net | 25 | 74.3 |
| Discriminant | 25 | 72.9 |
| Marker | Target Cell/Pathway | Function/Prognostic Association |
|---|---|---|
| FOXP3 | Tregs | Immune tolerance and homeostasis of the immune system. High frequency associated with a favorable prognosis of DLBCL. |
| PD-1 | T lymphocytes | Co-inhibition |
| BTLA | B and T lymphocytes | Co-inhibition |
| CD163 | M2-like TAMs | Pro-tumoral. High frequency is associated with poor prognosis of DLBCL and FL. |
| CSF1R | M2-like TAMs | Pro-tumoral. High CSF1R + TAMs associated with poor prognosis, but high CSF1R + B-cells of DLBCL with favorable prognosis. |
| CSF1 | B lymphocytes | Ligand of CSF1R |
| PD-L1 | M2c-like TAMs | Pro-tumoral, immune regulatory macrophages (M2c-like). High expression associated with poor prognosis of DLBCL. |
| SIRPA | M2-like TAMs | Limit phagocytosis |
| CD47 | B lymphocytes | Limit phagocytosis |
| IL10 | M2c-like TAMs | Pro-tumoral, immune regulatory macrophages (M2c-like). High expression associated with poor prognosis of DLBCL and FL. |
| TNFRSF14 | Antigen-presenting cells | Ligand of BTLA, co-inhibitory pathway |
| IKAROS | Pathway-related | Transcription factor, chromatin remodeling, hemolymphopoietic system. High expression associated with a favorable prognosis of DLBCL. |
| STAT3 | Pathway-related | Cell growth and apoptosis |
| NFKB1 | Pathway-related | Activated by cytokines, oxidant-free radicals, ultraviolet irradiation, and bacterial or viral products. Activated NFKB translocates into the nucleus and stimulates expression multiple genes of wide variety of biological functions. |
| MAPK | Pathway-related | p44/42 MAPK (Erk1/2) signaling pathway. High expression associated with GCB phenotype of DLBCL (and a favorable prognosis). |
| TNFAIP8 | Pathway-related | Anti-apoptosis. High expression associated with poor prognosis of DLBCL. |
| BCL2 | Pathway-related | Anti-apoptosis |
| CASP8 | Pathway-related | Pro-apoptosis. High expression associated with a favorable prognosis of DLBCL. |
| CASP3 | Pathway-related | Pro-apoptosis |
| PARP | Pathway-related | Pro-apoptosis |
| MDM2 | Pathway-related | TP53 in inhibitor |
| E2F1 | Pathway-related | Transcription factor, cell cycle, tumor suppressor |
| CDK6 | Pathway-related | Cell cycle |
| MYB | Germinal center B-cells | Transcriptional transactivator |
| LMO2 | Germinal center B-cells | Hematopoietic development |
| ENO3 | Pathway-related | Glycolysis and glycosaminoglycan metabolism. High expression associated with a poor prognosis of DLBCL. |
| GGA3 | Pathway-related | Positive regulation of protein catabolic processes |
| Authors (Year) | Journal | Research Title | Summary | Technique Used | Reference |
|---|---|---|---|---|---|
| (1) PET/CT scan-based AI | |||||
| Lisson CS et al. (2022) | Cancers (Basel) | Deep neural networks and machine learning radiomics modeling for the prediction of relapse in mantle cell lymphoma | This research predicted the relapse of mantle cell lymphoma (MCL) using baseline CT scans. The accuracies of predictions ranged from 64% to 70%. | 3D SEResNet50, 3D DenseNet, optimized 3D CNN, K-nearest Neighbor (KNN), and Random Forest (RF) | [61] |
| Sadik M et al. (2021) | Sci Rep. | Artificial intelligence could alert for focal skeleton/bone marrow uptake in patients with Hodgkin’s lymphoma staged with FDG-PET/CT | Detection of focal skeleton/bone marrow uptake (BMU) in patients with Hodgkin’s lymphoma (HL) undergoing staging with FDG-PET/CT. Training set, n = 153; validation set, n = 48. | Convolutional neural network (CNN) | [62] |
| Wang YJ et al. (2021) | Eur J Nucl Med Mol Imaging | Artificial intelligence enables whole-body positron emission tomography scans with minimal radiation exposure | Thirty-three diagnostic 18F-FDG PET images of patients with pediatric cancer were generated from ultra-low dose 18F-FDG PET input images using an AI algorithm. Then, the AI-generated PET scans were compared with clinical standard PET scans. | Convolutional neural network (CNN) | [63] |
| Pinochet P et al. (2021) | Front Med (Lausanne) | Evaluation of an automatic classification algorithm using convolutional neural networks in oncological positron emission tomography | This research measured the efficiency and performance in both clinical and research environments of a system called positron emission tomography (PET)-assisted reporting system (PARS) (Siemens Healthineers). The method was based on a convolutional neural network (CNN) that identified suspected cancer sites in fluorine-18 fluorodeoxyglucose (18F-FDG) PET/computed tomography. These data were correlated with the survival of the patients. Two cohorts were evaluated: 119 cases of DLBCL, and 430 cases of DLBCL and other tumors. | Dice score | [64] |
| (2) Histological images-based AI | |||||
| El Hussein S et al. (2022) | J Pathol. | Artificial intelligence strategy integrating morphologic and architectural biomarkers provide robust diagnostic accuracy for disease progression in chronic lymphocytic leukemia | Cytologic and architectural features obtained from whole-slides images were used to classify 125 samples into three subtypes: chronic lymphocytic leukemia (CLL, n = 69), progression to accelerated CLL (aCLL, n = 44), and transformation to diffuse large B-cell lymphoma (Richter transformation; RT, n = 80). | Hover-Net | [65] |
| Swiderska-Chadaj Z et al. (2021) | Virchows Arch. | Artificial intelligence to detect MYC translocation in slides of diffuse large B-cell lymphoma | The H&E slides of 287 cases were evaluated using a deep learning algorithm to identify MYC rearrangement by DNA in situ hybridization (FISH). | Deep learning neural network (U-Net) and classical machine learning (random forest classification) | [66] |
| Steinbuss G et al. (2021) | Cancers (Basel) | Deep learning for the classification of non-Hodgkin lymphoma on histopathological images | In this research, the training set included 84,139 image patches from 629 patients that were classified as reactive lymph nodes, nodal small lymphocytic lymphoma/chronic lymphocytic leukemia, and nodal diffuse large B-cell lymphoma. The validation set included 16,960 image patches from 125 patients. The final model had an accuracy of 96%. | EfficientNet convolutional neuronal network (CNN) | [67] |
| Zhang X et al. (2021) | Technol Health Care | Research on the classification of lymphoma pathological images-based on deep residual neural networks | The analysis used 374 pathological images, including chronic lymphocytic leukemia, follicular lymphoma, and mantle cell lymphoma. | BP neural network and BP neural network optimized by genetic algorithm (GA-BP), deep residual neural network model (ResNet50), softmax layer | [68] |
| Tang G et al. (2021) | Acta Cytol. | A machine learning tool using digital microscopy (Morphogo) for the identification of abnormal lymphocytes in the bone marrow | Morphological differentiation of abnormal lymphocytes in bone marrow was evaluated in 53 cases of different subtypes of B-cell lymphomas, using automated digital images. | “Morphogo” system | [69] |
| Yu WH et al. (2021) | Cancers (Basel) | Machine learning based on morphological features enables the classification of primary intestinal T-cell lymphomas. | A total of 40 primary intestinal T-cell lymphomas (PITL), including 26 monomorphic epitheliotropic intestinal T-cell lymphoma (MEITL), 10 intestinal T-cell lymphoma, not otherwise specified (ITCL-NOS), and 4 borderline cases were analyzed. The inputs were the morphological features and the immunophenotypes (CD8 and CD56). | XGBoost and CNN (HTC-RCNN with ResNet50) | [70] |
| Zhou M et al. (2021) | Front Pediatr. | Development and evaluation of a leukemia diagnosis system using deep learning in real clinical scenarios | A total of 1732 bone marrow, raw images of 89 children with leukemia were analyzed with convolutional neural networks, with a performance accuracy of 89%. Apart from detecting leukocytes, the system also detected bone marrow metastasis of lymphoma and neuroblastomas. | RetinaNet, VGG, Feature Pyramid Network, ResNet, convolutional neural network (CNN) | [71] |
| Zhang J et al. (2020) | Med Phys. | Classification of digital pathological images of non-Hodgkin’s lymphoma subtypes based on the fusion of transfer learning and principal component analysis | Digital pathology images of non-Hodgkin lymphoma, including chronic lymphocytic leukemia (CLL), follicular lymphoma (FL), and mantle cell lymphoma (MCL) tumor were analyzed and classified. The model had an overall accuracy of 98.9%. | Transfer learning (TL) and principal component analysis (PCA) | [72] |
| Mohlman JS et al. (2020) | Am J Clin Pathol. | Improving augmented human intelligence to distinguish Burkitt lymphoma from diffuse large B-cell lymphoma cases | A total of 10,818 H&E images from 34 cases of Burkitt lymphoma and 36 cases of diffuse large B-cell lymphoma were used to train and differentiate the two lymphoma subtypes. | Convolutional neural network (CNN) | [73] |
| Li D et al. (2020) | Nat Commun. | A deep learning diagnostic platform for diffuse large B-cell lymphoma with high accuracy across multiple hospitals | This research used histological images of H&E to classify diffuse large B-cell lymphoma (DLBCL) vs non-DLBCL. Non-DLBCL included metastatic carcinoma, melanoma, and other lymphomas including small lymphocytic lymphoma/chronic lymphocytic leukemia, mantle cell lymphoma, follicular lymphoma, and classical Hodgkin lymphoma. The GOTDP-MP-CNNs (with combined 17 CNNs) model had an accuracy of 99.7% to 100%. | 17 types of CNN: AlexNet, GoogLeNet (ImageNet), GoogLeNet (Places365), ResNet18, ResNet50, ResNet101, Vgg16, Vgg19, Inceptionv3, InceptionResNetv2, SqueezeNet, DenseNet201, MobileNetv2, ShuffleNet, Xception, NasNetmobile, Nasnetlarge | [74] |
| Miyoshi H et al. (2020) | Lab Invest. | Deep learning shows the capability of high-level computer-aided diagnosis of malignant lymphoma. | The H&E images of 388 cases, including 259 with diffuse large B-cell lymphoma, 89 with follicular lymphoma, and 40 with reactive lymphoid hyperplasia, were analyzed using deep learning. The accuracy of the model was 97%. | Convolutional neural network (CNN) | [75] |
| Zorman M et al. (2011) | Wien Klin Wochenschr. | Classification of follicular lymphoma images: a holistic approach with symbol-based machine learning methods. | Analysis of follicular lymphoma images, focusing on the identification of follicles. | Decision trees (MtDeciT 3.1, RSES 2.2, and Weka 3) and artificial neural networks (multilayer perceptron) | [76] |
| (3) Immunophenotype-based AI | |||||
| Zhao M et al. (2020) | Cytometry A. | Hematologist-level classification of mature B-cell neoplasms using deep learning on multiparameter flow cytometry data | Information captured by multiparameter flow cytometry (MFC) of 18,274 cases, including chronic lymphocytic leukemia and its precursor monoclonal B-cell lymphocytosis, marginal zone lymphoma, mantle cell lymphoma, prolymphocytic leukemia, follicular lymphoma, hairy cell leukemia, lymphoplasmacytic lymphoma were analyzed; the model was tested on a set of 2346 cases. The model performance had an F1 score of 0.94. | Self-organizing maps and convolutional neural networks | [77] |
| Gaidano V et al. (2020) | Cancers (Basel) | A clinically applicable approach to the classification of B-cell non-Hodgkin lymphomas with flow cytometry and machine learning | The immunophenotype data from flow cytometry of 1465 B-cell non-Hodgkin lymphoma (NHL) cases were analyzed. The cases included chronic lymphocytic leukemia (CLL), diffuse large B-cell lymphoma (DLBCL), Burkitt lymphoma (BL), follicular cell lymphoma (FCL), hairy cell leukemia (HCL), splenic lymphoma (SL), mantle cell lymphoma (MCL), marginal zone lymphoma (MZL), and lymphoplasmacytic lymphoma (LPL). The accuracy of the classification ranged from 92% to 100%. | Classification trees | [78] |
| (4) Clinicopathological variables-based AI | |||||
| Zhan M et al. (2021) | Leuk Lymphoma | Machine learning to predict high-dose methotrexate-related neutropenia and fever in children with B-cell acute lymphoblastic leukemia | A model included 57 SNPs of 16 genes and clinical variables to predict neutropenia and fever in 139 pediatric cases of acute lymphoblastic leukemia treated with high-dose methotrexate (MTX). | Random forest | [79] |
| Buciński A et al. (2010) | Eur J Cancer Prev. | Contribution of artificial intelligence to the knowledge of prognostic factors in Hodgkin’s lymphoma | A total of 31 variables from 114 patients with Hodgkin’s lymphoma were used to predict the prognosis of the patients. | Artificial neural network (ANN) | [80] |
| (5) Gene expression, mutational, and integrative analysis-based AI | |||||
| Carreras J et al. (2022) | Healthcare (Basel) | Artificial intelligence analysis of gene expression predicted the overall survival of mantle cell lymphoma and a large pan-cancer Series | The gene expression data of 123 cases of mantle cell lymphoma (MCL) were analyzed with artificial neural networks to predict the overall survival of the patients with high accuracy. The survival of diffuse large B-cell lymphoma (DLBCL), and a pan-cancer series was also predicted. | Several machine learning techniques, and artificial neural networks | [34] |
| Carreras J et al. (2021) | Cancers (Basel) | Artificial neural networks predicted the overall survival and molecular subtypes of diffuse large B-cell lymphoma using a pan-cancer immune-oncology panel | The gene expression of an immuno-oncology panel of a series of 106 cases of diffuse large B-cell lymphoma was analyzed using artificial intelligence to predict the overall survival and the cell of origin molecular subtypes. The model had a high accuracy of classification. | Several machine learning techniques, and artificial neural networks | [33] |
| Carreras J et al. (2021) | Tokai J Exp Clin Med. | Artificial intelligence analysis of gene expression data predicted the prognosis of patients with diffuse large B-cell lymphoma | The gene expression of a series of 414 cases of diffuse large B-cell lymphoma (DLBCL) was analyzed to predict the overall survival, and was correlated with other known pathogenic genes such as BCL2 and MYC. | Artificial neural networks (ANN) | [27] |
| Xu-Monette ZY et al. (2020) | Blood Adv. | A refined cell of origin classifier with targeted NGS and artificial intelligence showed robust predictive value in DLBCL | The series of diffuse large B-cell lymphoma of 418 cases included immunohistochemical, gene expression, DNA in situ hybridization, array CGH, and NGS sequencing. Using an autoencoder, the cases were classified according to the cell of origin and the survival (overall survival and progression-free survival). | Autoencoder, logistic regression, and CPH models | [81] |
| Zhang W et al. (2020) | BMC Cancer | Novel bioinformatic classification system for genetic signature identification in diffuse large B-cell lymphoma | A total of 342 cases of diffuse large B-cell lymphoma were analyzed using mutational data from a panel of 46 genes by NGS. | Random forest | [82] |
| Parodi S et al. (2018) | Health Informatics J. | Logic learning machine and standard supervised methods for Hodgkin’s lymphoma prognosis using gene expression data and clinical variables | The data of 130 patients diagnosed with Hodgkin’s lymphoma, including a small set of clinical variables and more than 54,000 gene features, were used to predict the prognosis. | K-nearest neighbor (KNN), artificial neural network (ANN), support vector machine (SVM), decision tree, and the innovative logic learning machine method | [83] |
| Schmitz R et al. (2018) | N Engl J Med. | Genetics and pathogenesis of diffuse large B-cell lymphoma | The data of 574 diffuse large B-cell lymphoma cases, which included exome and transcriptome sequencing, array-based DNA copy-number analysis, and targeted amplicon resequencing of 372 genes, were used to identify genetic subtypes. | Random forest | [84] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carreras, J.; Roncador, G.; Hamoudi, R. Artificial Intelligence Predicted Overall Survival and Classified Mature B-Cell Neoplasms Based on Immuno-Oncology and Immune Checkpoint Panels. Cancers 2022, 14, 5318. https://doi.org/10.3390/cancers14215318
Carreras J, Roncador G, Hamoudi R. Artificial Intelligence Predicted Overall Survival and Classified Mature B-Cell Neoplasms Based on Immuno-Oncology and Immune Checkpoint Panels. Cancers. 2022; 14(21):5318. https://doi.org/10.3390/cancers14215318
Chicago/Turabian StyleCarreras, Joaquim, Giovanna Roncador, and Rifat Hamoudi. 2022. "Artificial Intelligence Predicted Overall Survival and Classified Mature B-Cell Neoplasms Based on Immuno-Oncology and Immune Checkpoint Panels" Cancers 14, no. 21: 5318. https://doi.org/10.3390/cancers14215318
APA StyleCarreras, J., Roncador, G., & Hamoudi, R. (2022). Artificial Intelligence Predicted Overall Survival and Classified Mature B-Cell Neoplasms Based on Immuno-Oncology and Immune Checkpoint Panels. Cancers, 14(21), 5318. https://doi.org/10.3390/cancers14215318