

Hepcidin Upregulation in Colorectal Cancer Associates with Accumulation of Regulatory Macrophages and Epithelial–Mesenchymal Transition and Correlates with Progression of the Disease
 , ,
, ,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Samples
2.2. Cell Culture
2.3. Protein Extraction and Western Blotting
2.4. Immunohistochemistry and Immunofluorescence
2.5. RNA Extraction and RT-PCR
2.6. Flow Cytometry
2.7. UALCAN
2.8. Gene Expression Profiling Interactive Analysis (GEPIA)
2.9. Tumor Immune Estimation Resource (TIMER)
2.10. Statistical Analysis
3. Results
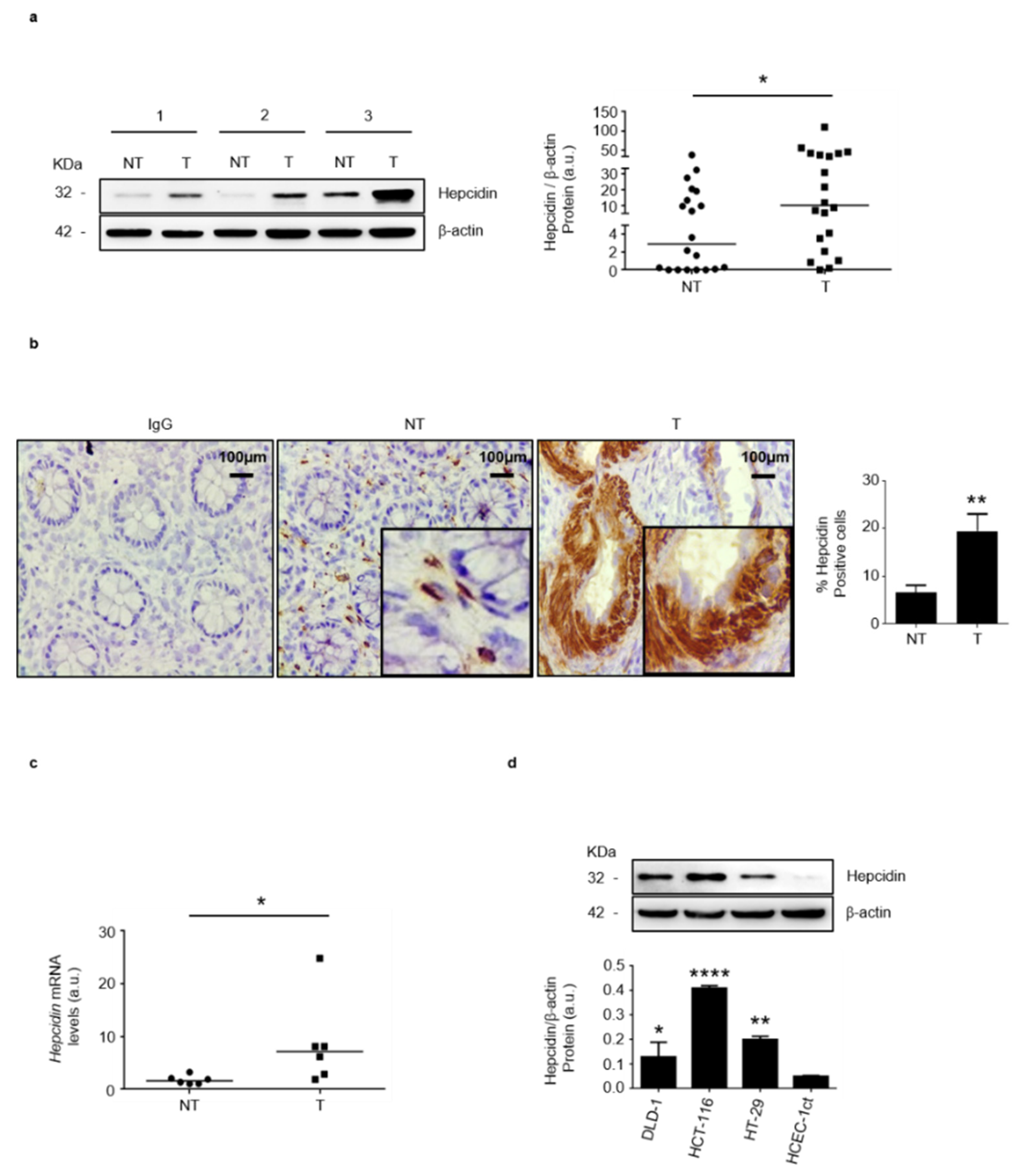
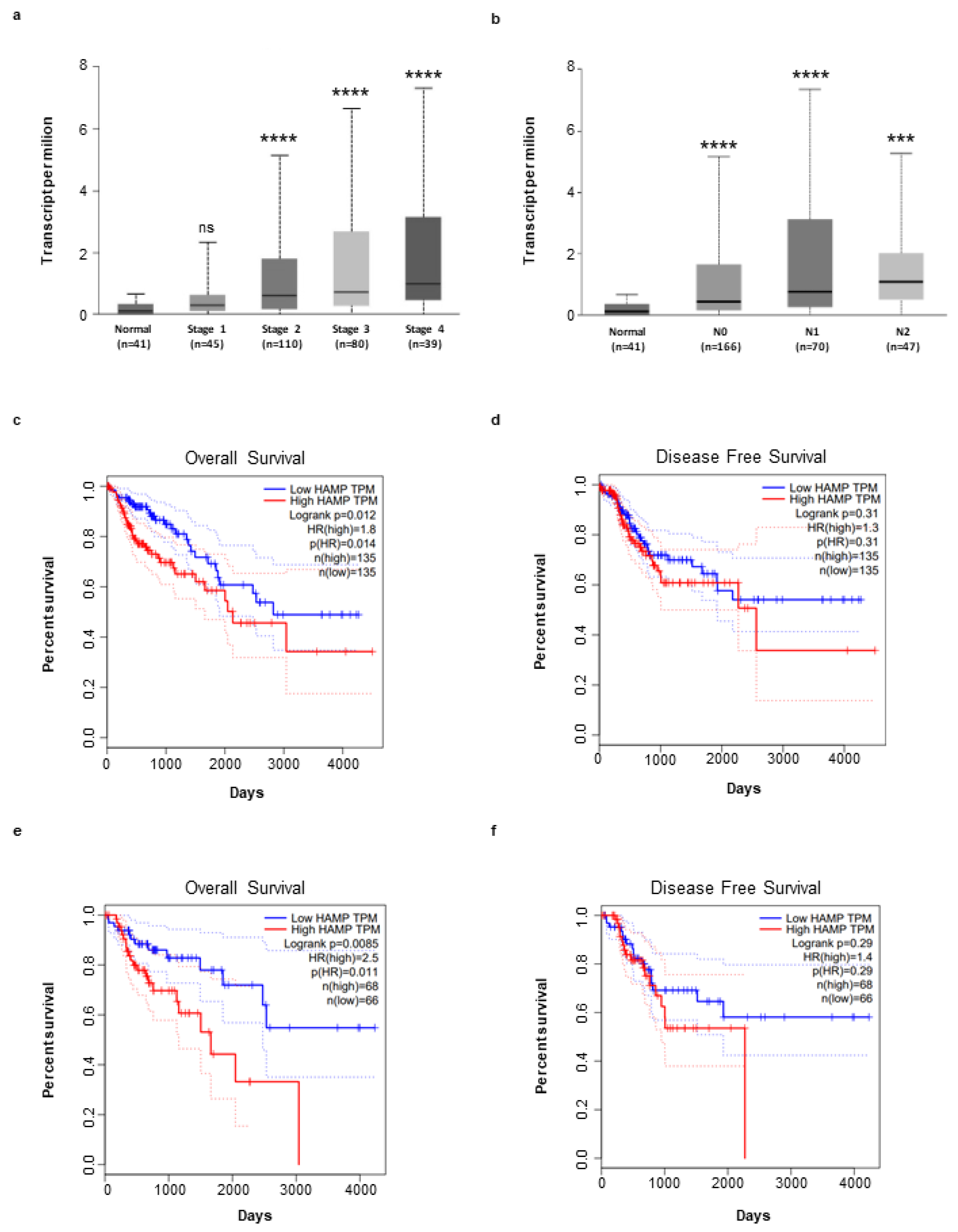
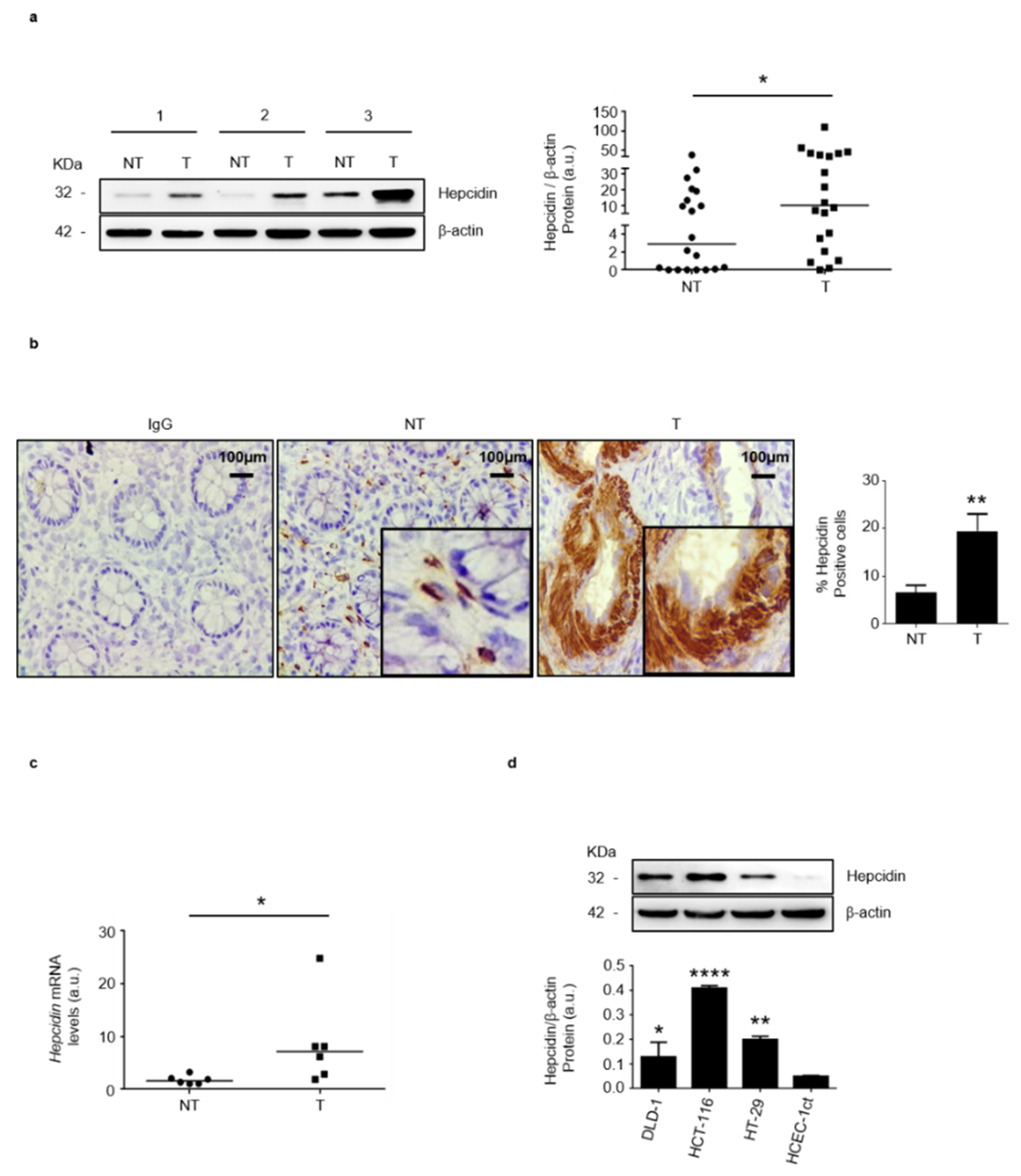
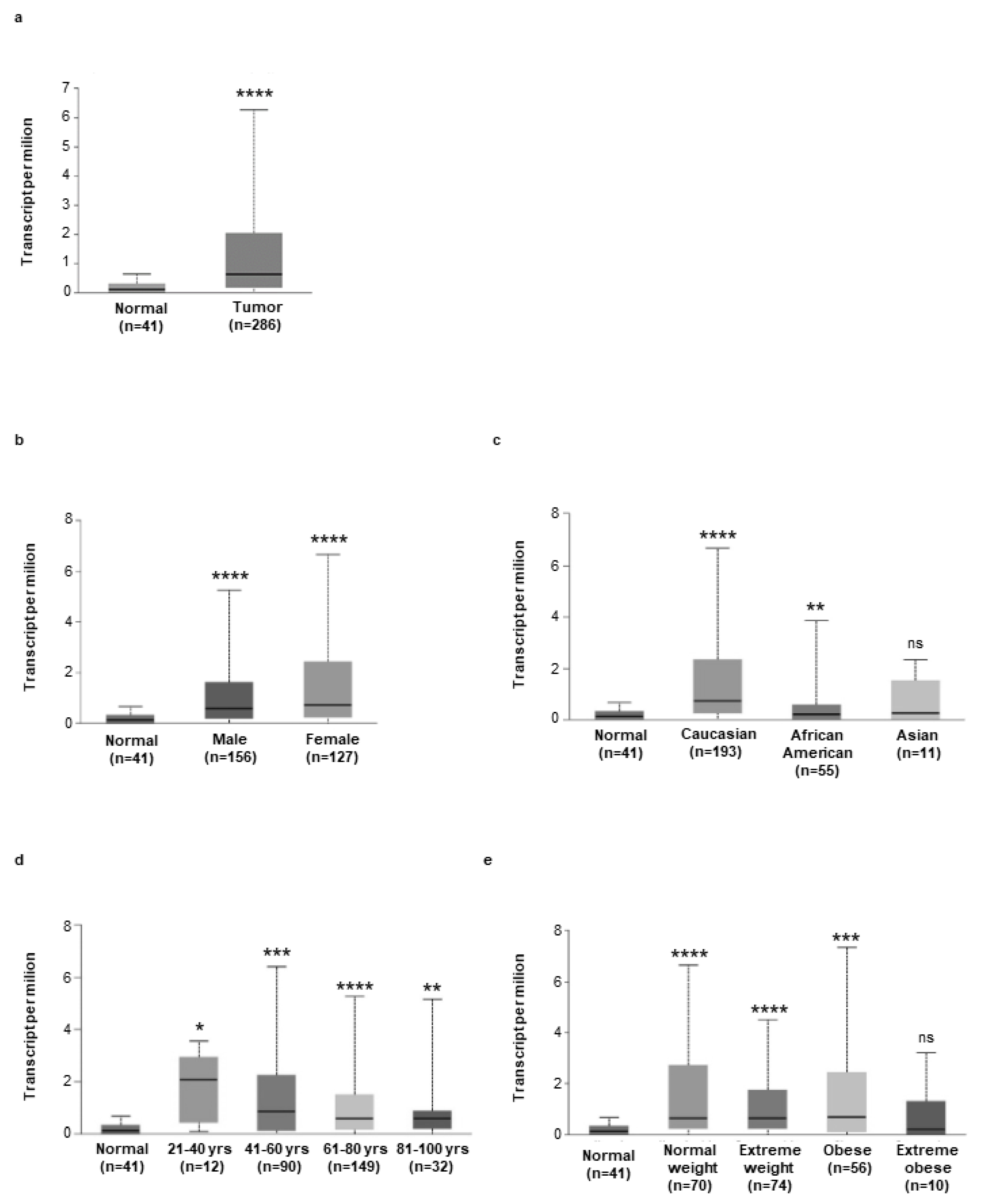
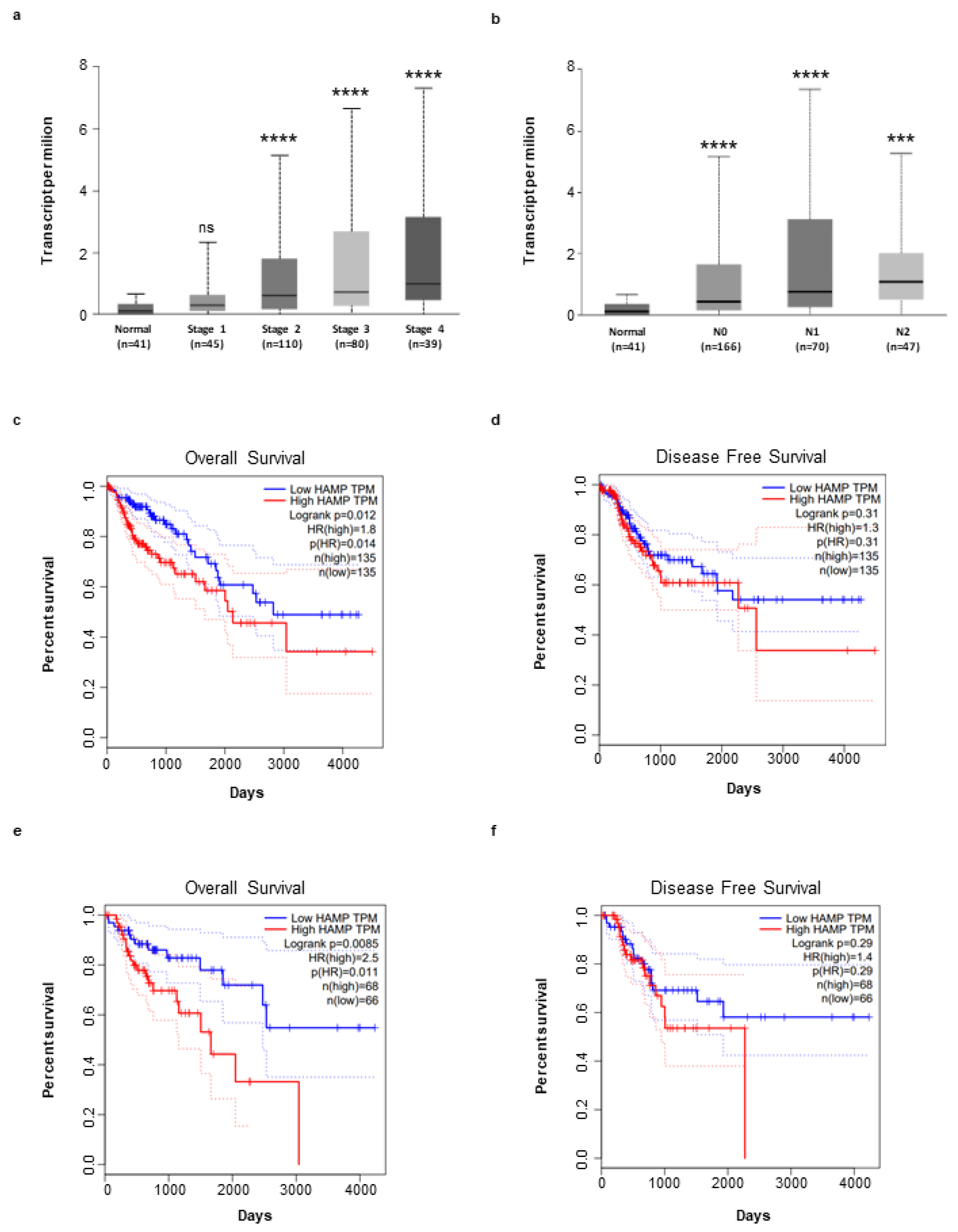
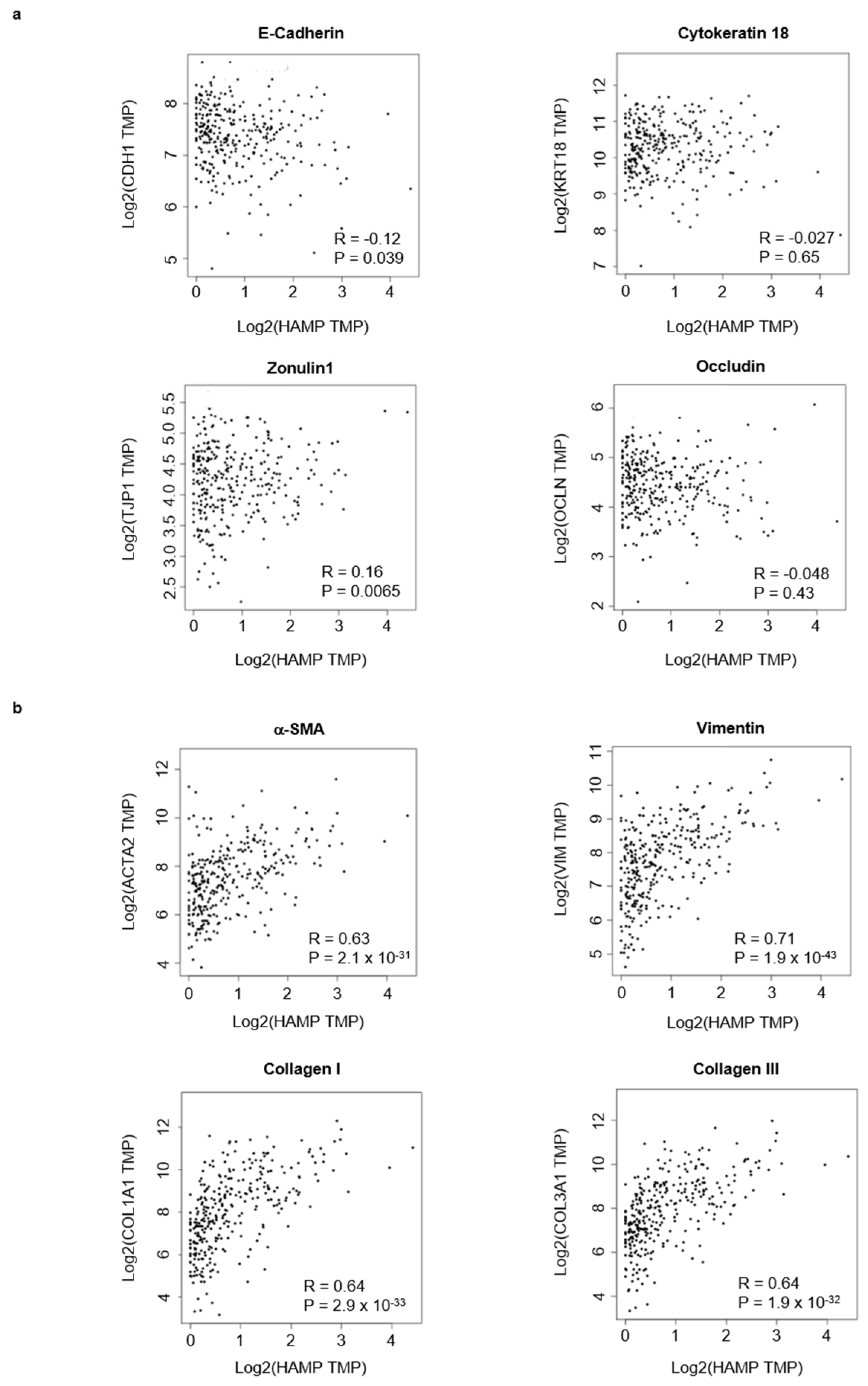
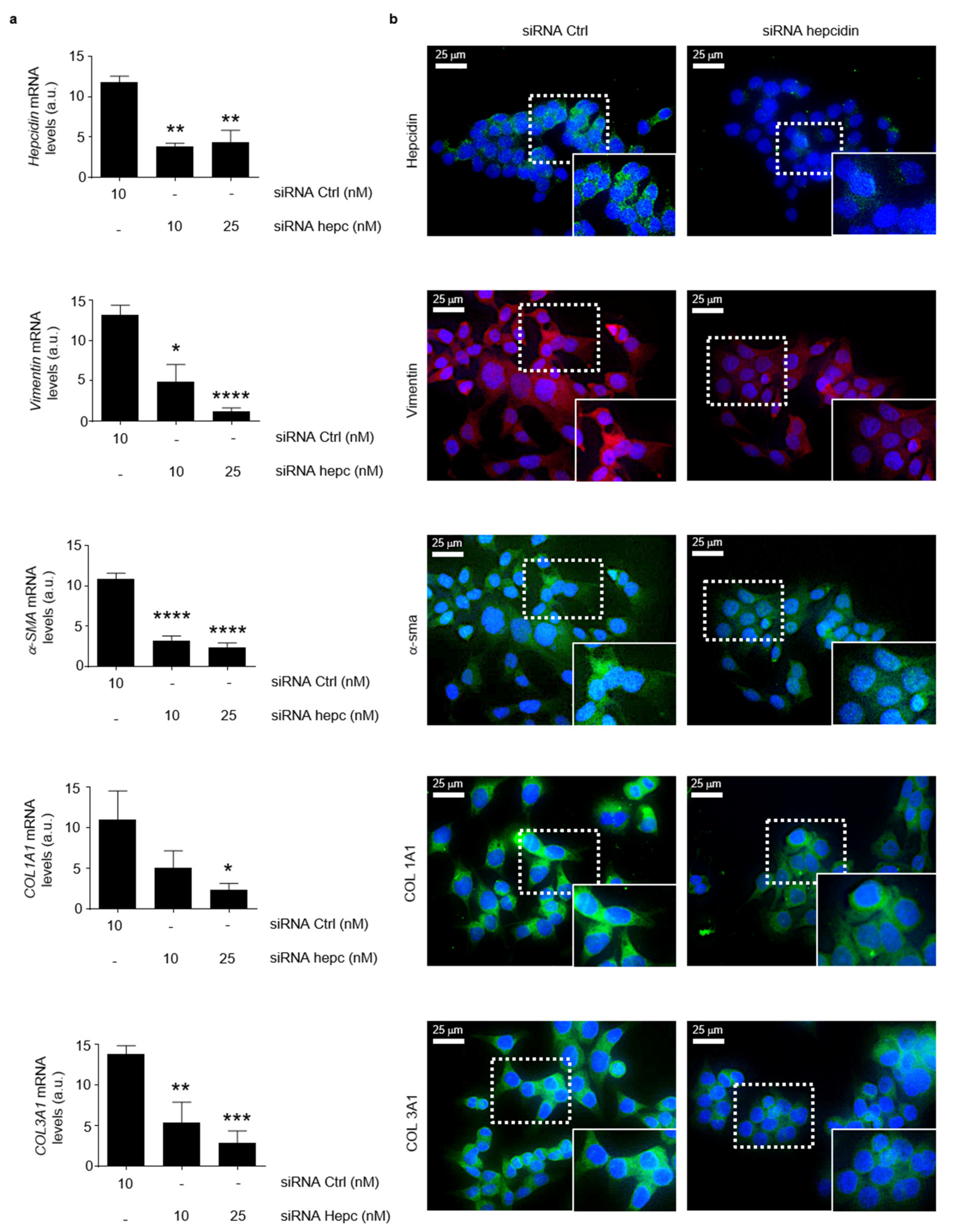
3.1. Hepcidin Is Up-Regulated in Human Sporadic CRC
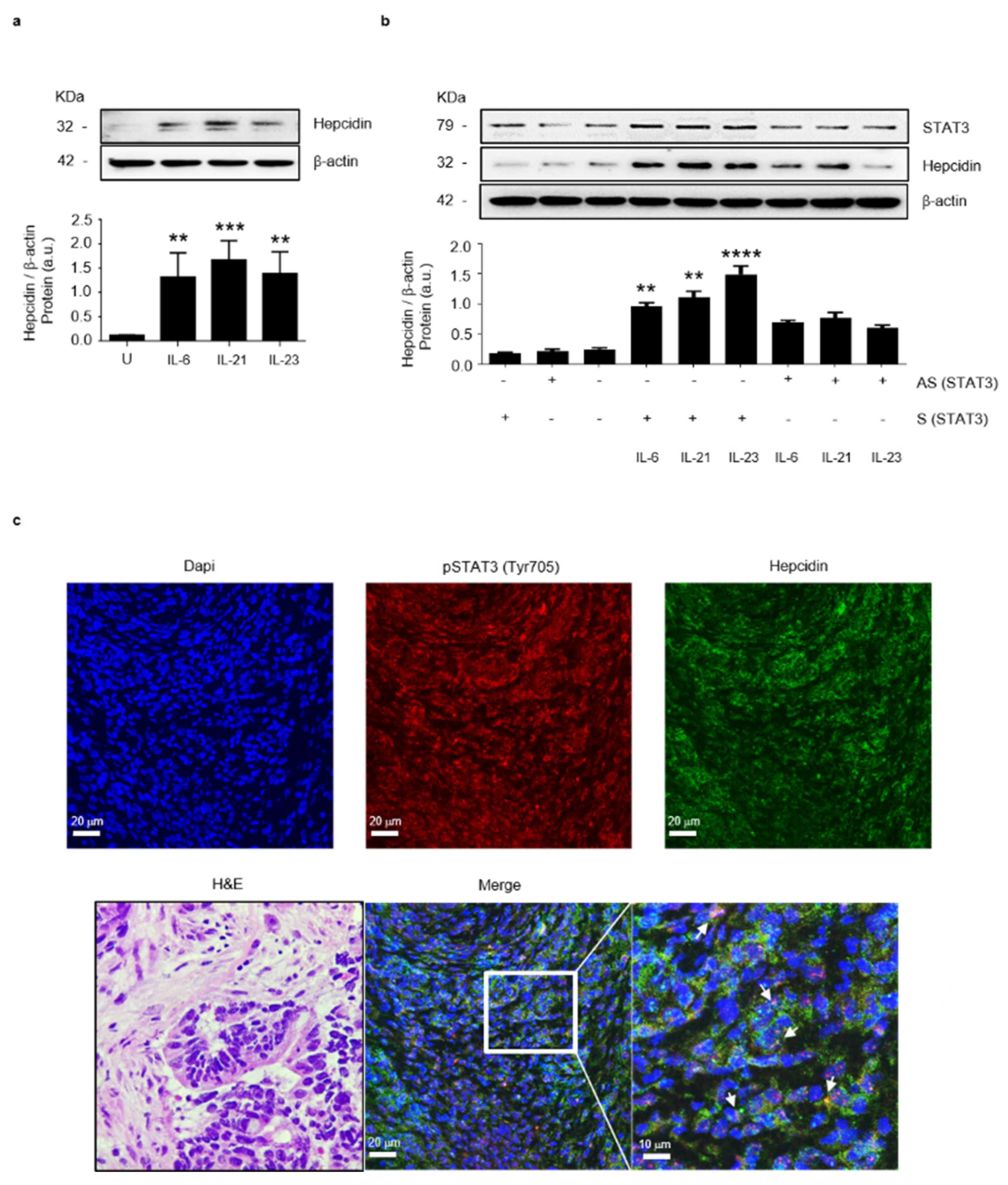
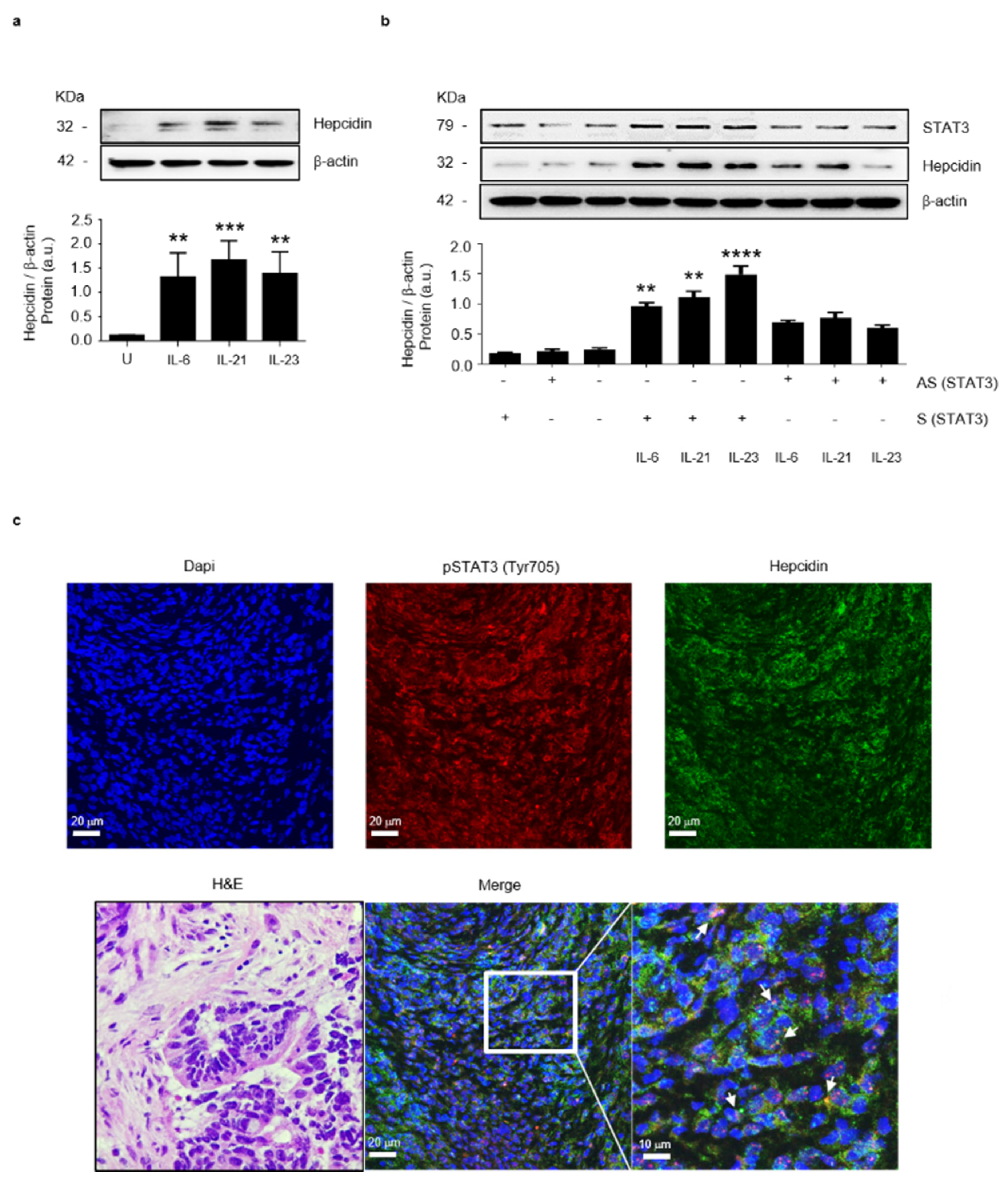
3.2. Stat3 Regulates Positively Hepcidin Production
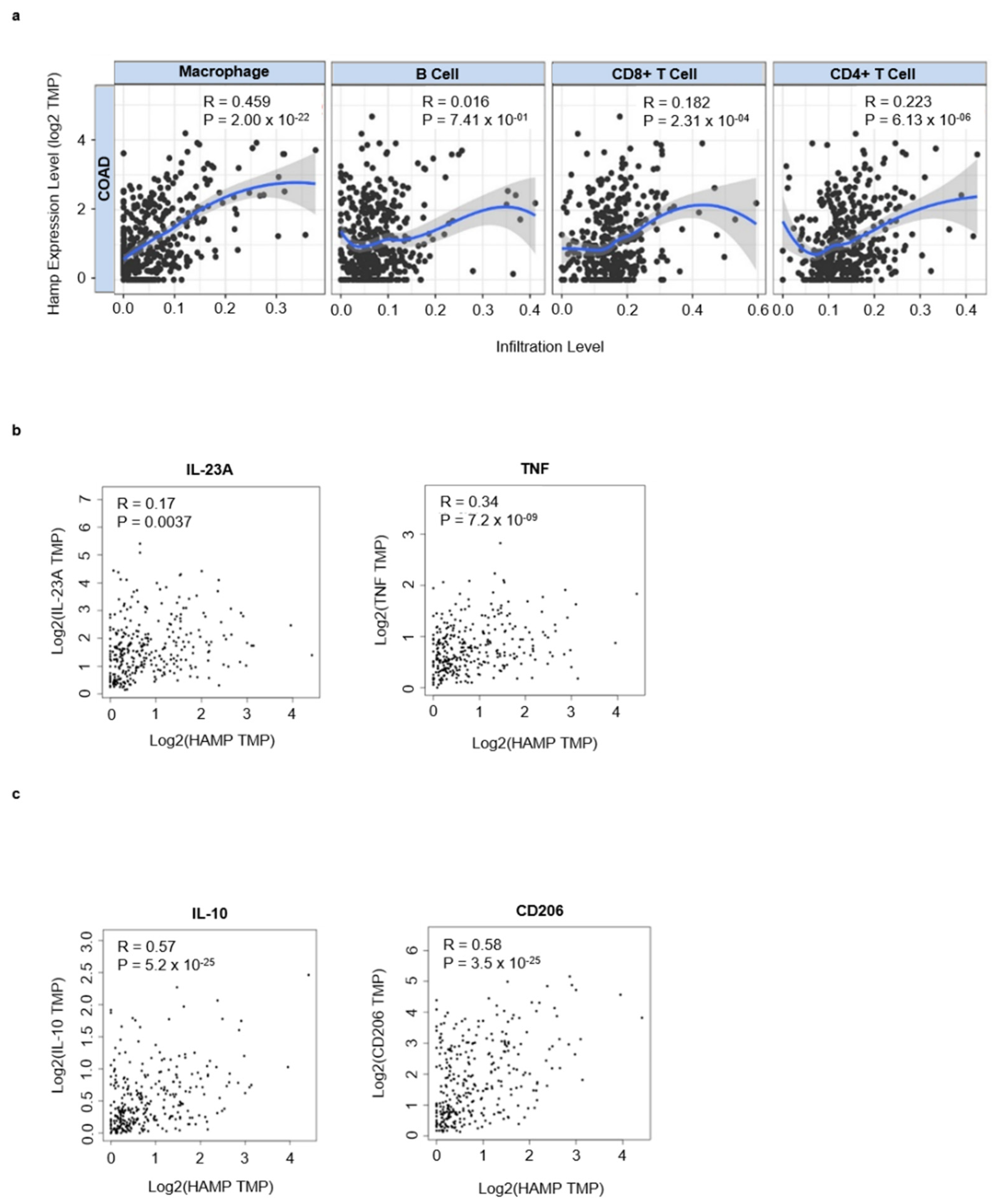
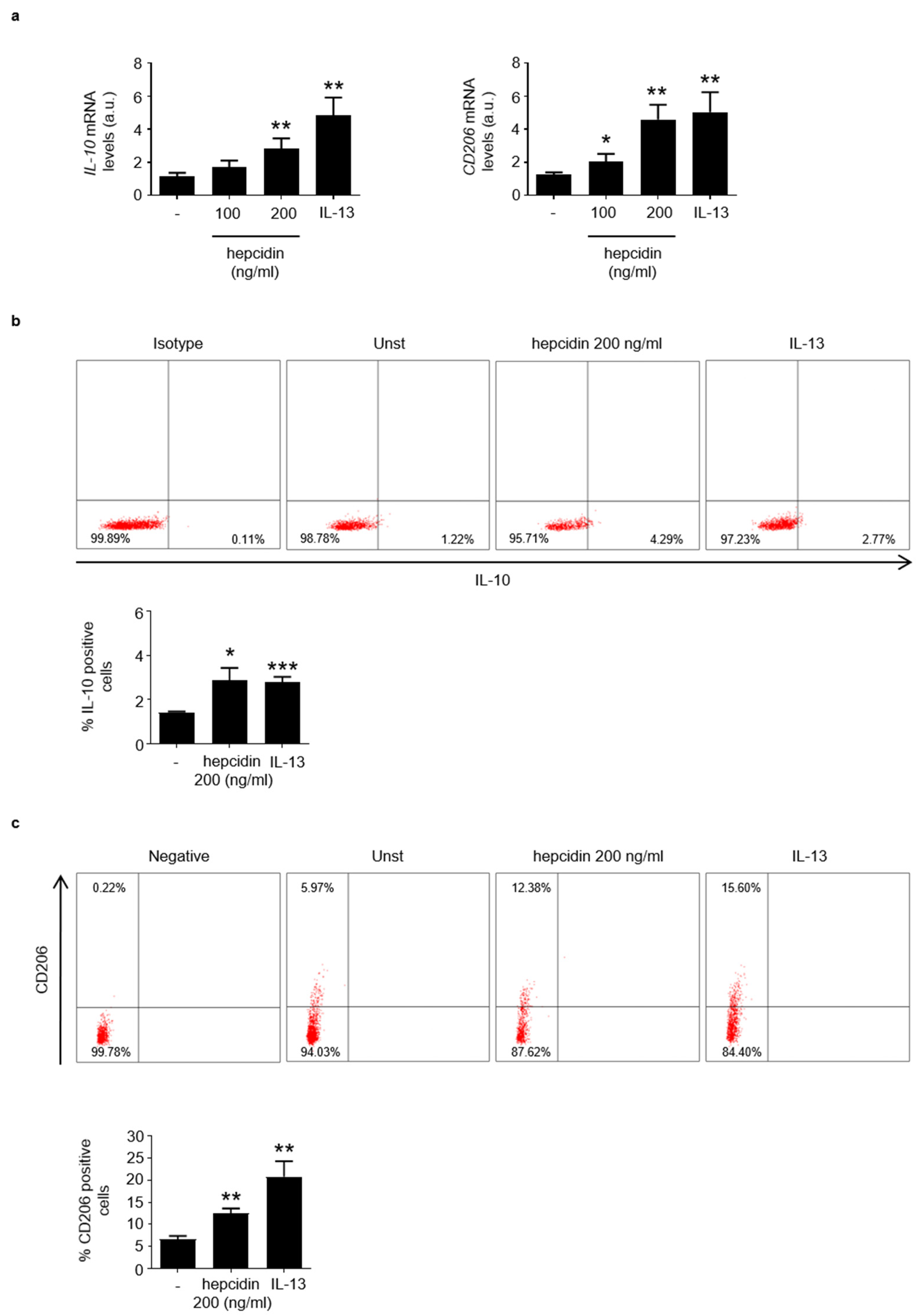
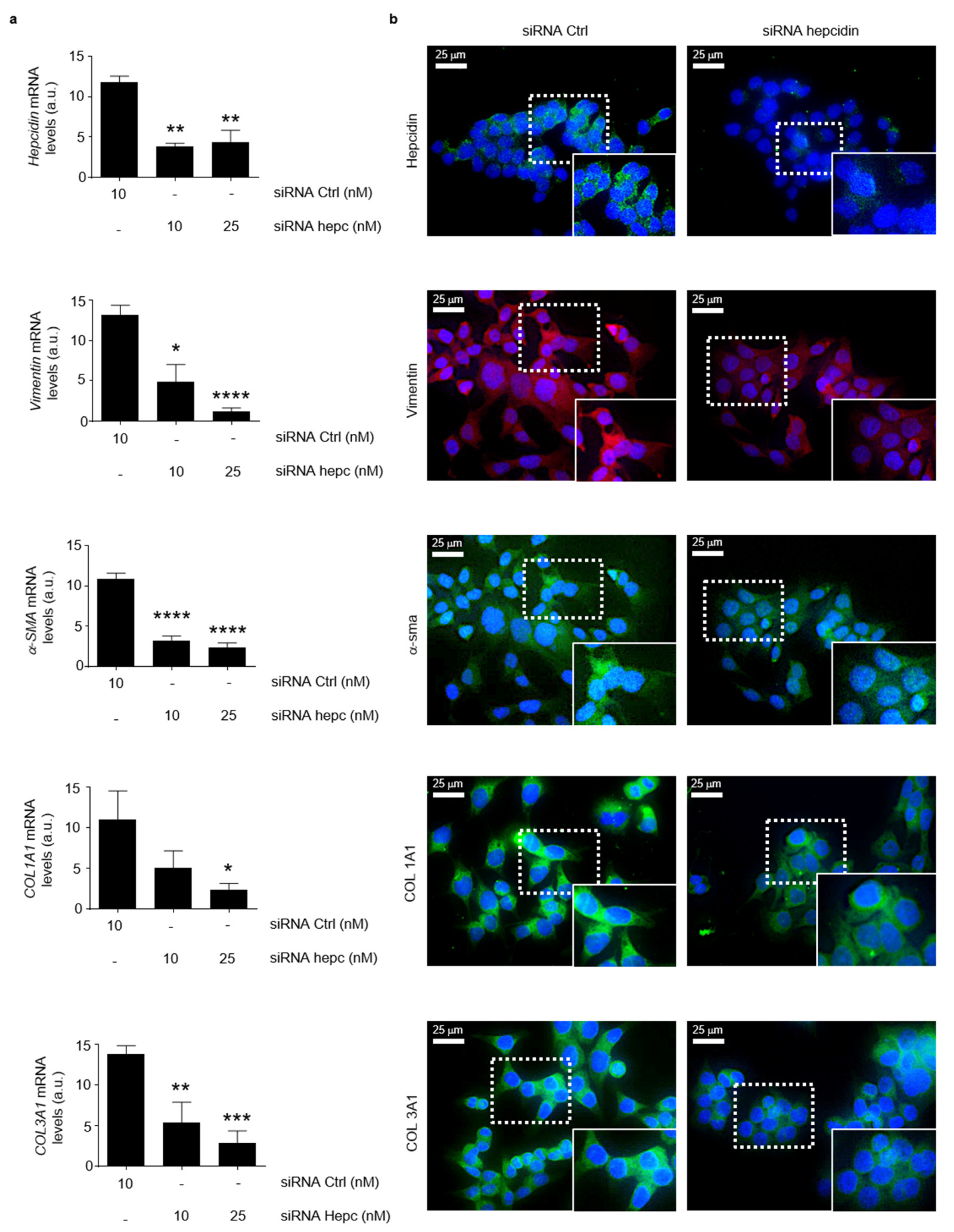
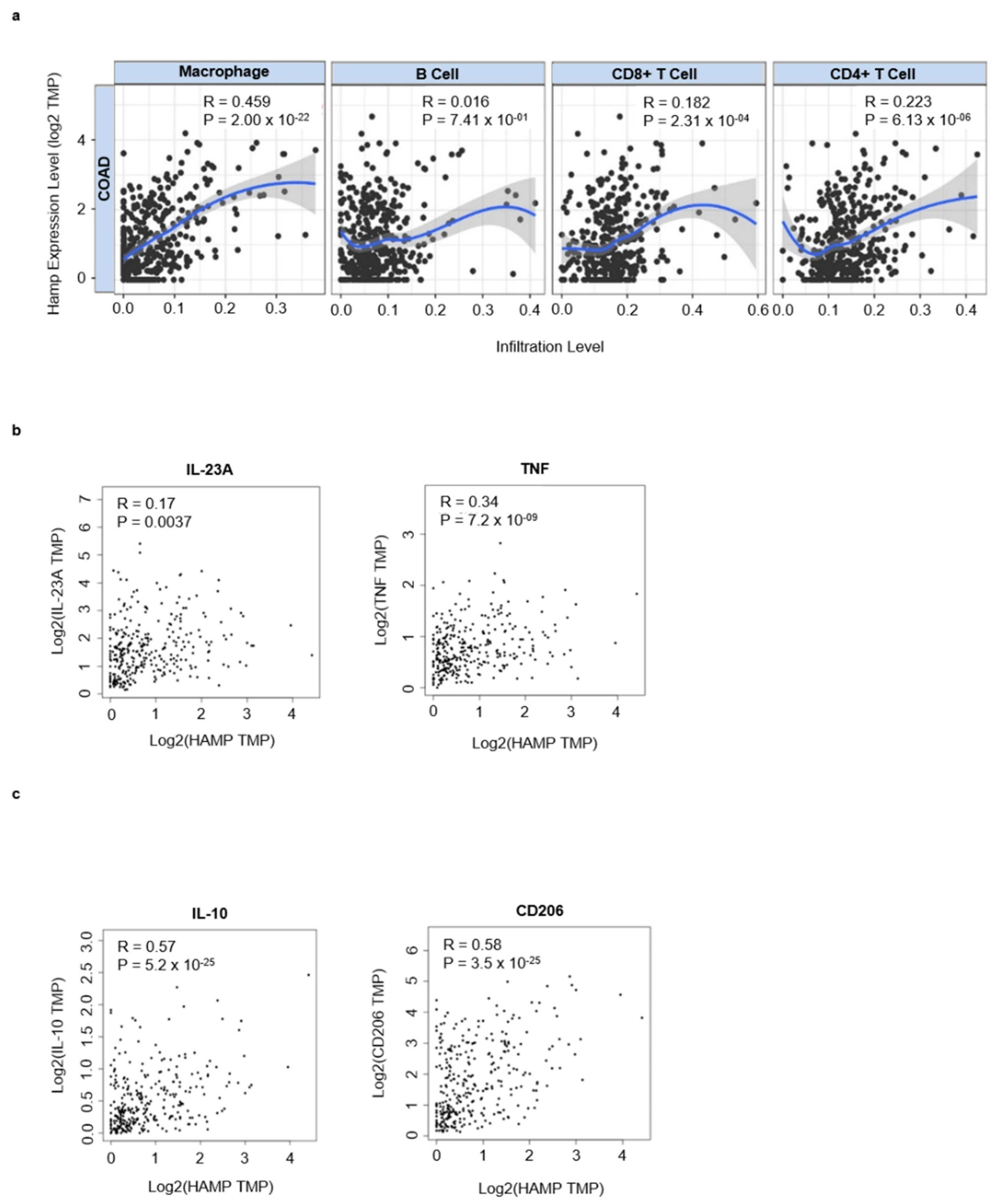
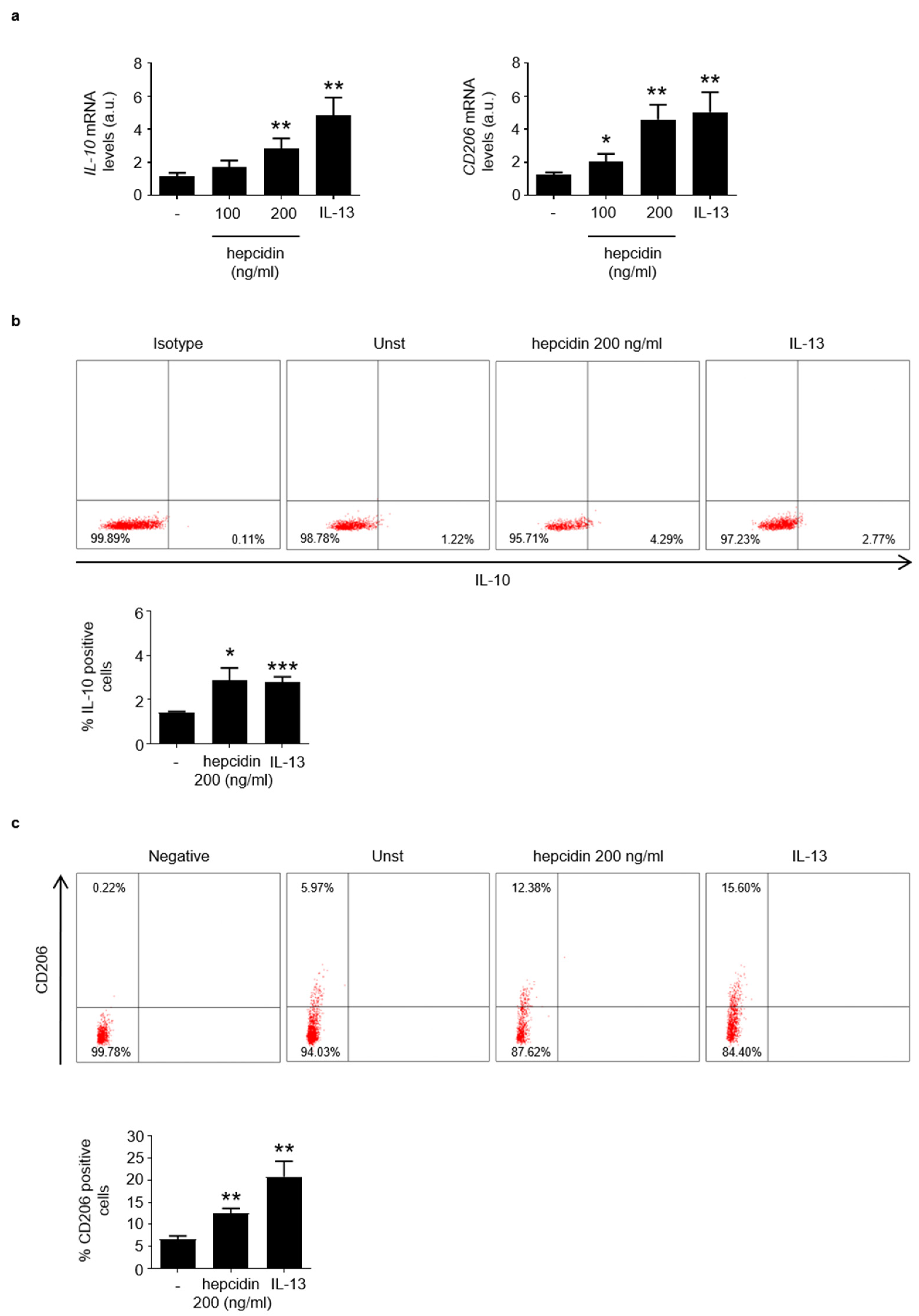
3.3. Hepcidin Promotes Induction of Regulatory Macrophage-Associated Markers
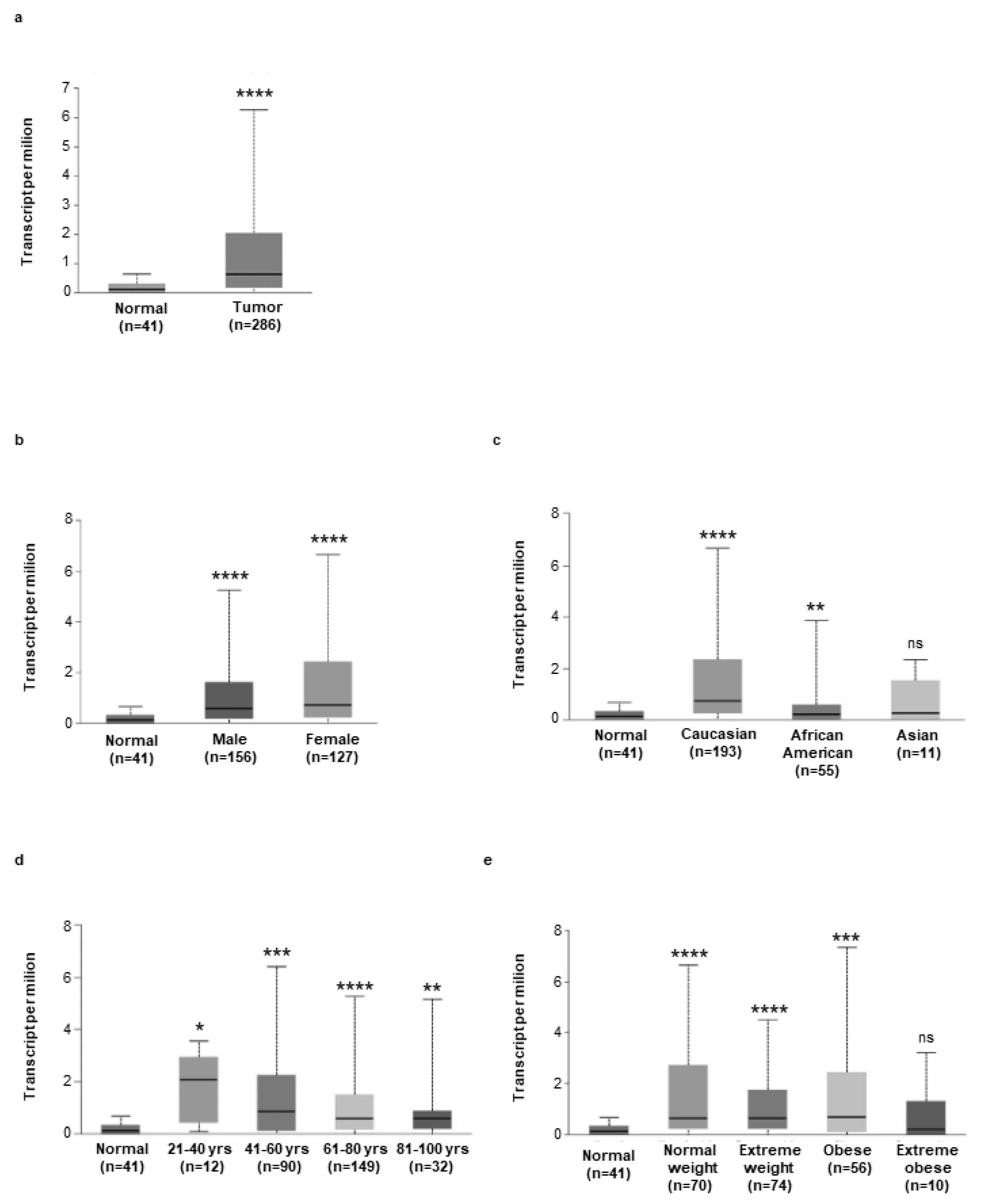
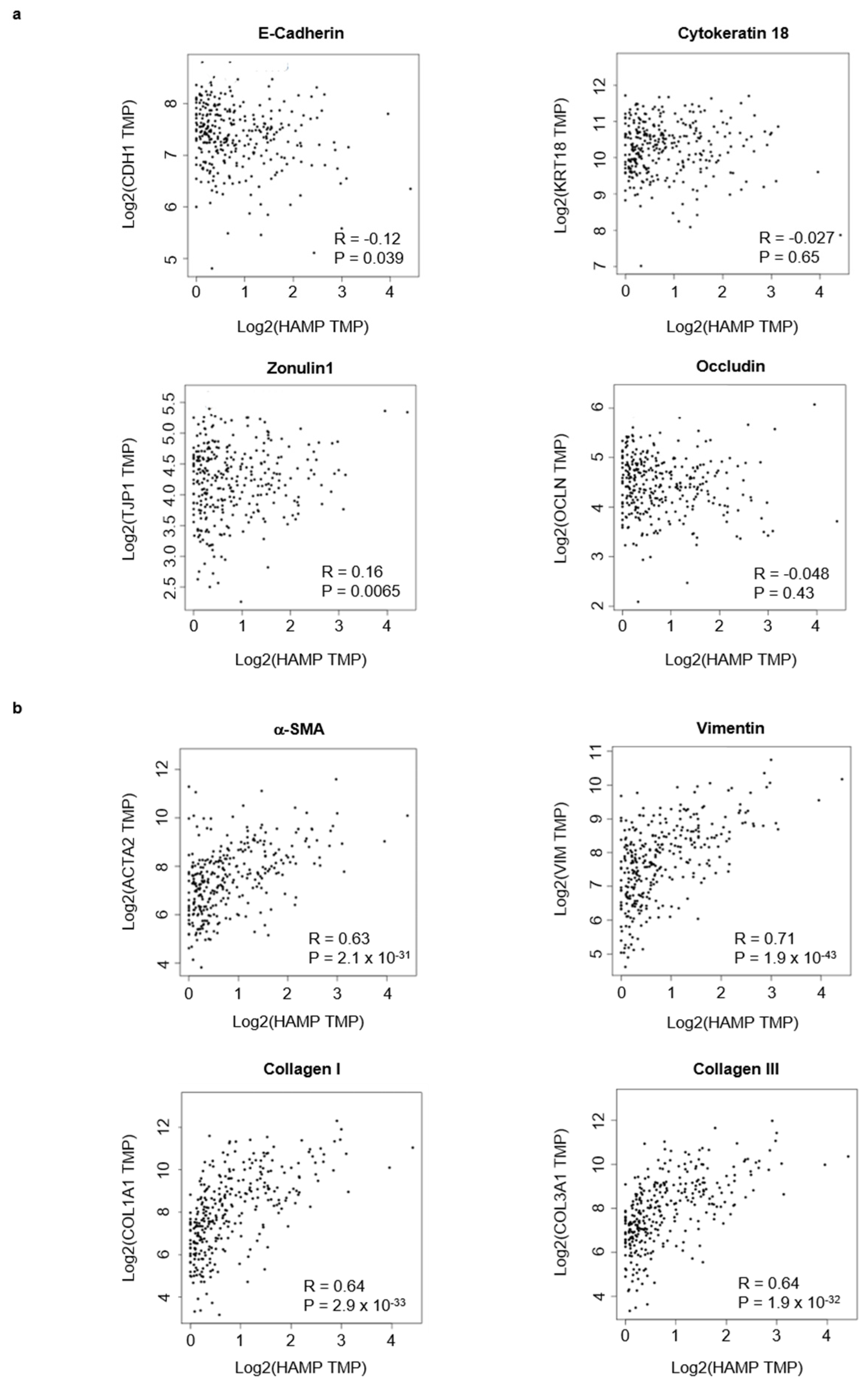
3.4. High Hepcidin Expression Associates with Low Survival of CRC Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017, 66, 683–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradbury, K.E.; Murphy, N.; Key, T.J. Diet and colorectal cancer in UK Biobank: A prospective study. Int. J. Epidemiol. 2020, 49, 246–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Chen, Y.; Ma, S.; Zheng, R.; Zhao, P.; Zhang, L.; Liu, Y.; Yu, Q.; Deng, Q.; Zhang, K. Dietary fibre intake and risk of breast cancer: A systematic review and meta-analysis of epidemiological studies. Oncotarget 2016, 7, 80980–80989. [Google Scholar] [CrossRef] [Green Version]
- Fedirko, V.; Tramacere, I.; Bagnardi, V.; Rota, M.; Scotti, L.; Islami, F.; Negri, E.; Straif, K.; Romieu, I.; La Vecchia, C.; et al. Alcohol drinking and colorectal cancer risk: An overall and dose-response meta-analysis of published studies. Ann. Oncol. 2011, 22, 1958–1972. [Google Scholar] [CrossRef] [PubMed]
- Giovannucci, E.; Willett, W.C. Dietary factors and risk of colon cancer. Ann. Med. 1994, 26, 443–452. [Google Scholar] [CrossRef]
- Maida, M.; Macaluso, F.S.; Ianiro, G.; Mangiola, F.; Sinagra, E.; Hold, G.; Maida, C.; Cammarota, G.; Gasbarrini, A.; Scarpulla, G. Screening of colorectal cancer: Present and future. Expert Rev. Anticancer Ther. 2017, 17, 1131–1146. [Google Scholar] [CrossRef]
- McQuade, R.M.; Stojanovska, V.; Bornstein, J.C.; Nurgali, K. Colorectal Cancer Chemotherapy: The Evolution of Treatment and New Approaches. Curr. Med. Chem. 2017, 24, 1537–1557. [Google Scholar] [CrossRef]
- Ganz, T. Systemic iron homeostasis. Physiol. Rev. 2013, 93, 1721–1741. [Google Scholar] [CrossRef] [Green Version]
- Vogt, A.-C.; Arsiwala, T.; Mohsen, M.; Vogel, M.; Manolova, V.; Bachmann, M. On Iron Metabolism and Its Regulation. Int. J. Mol. Sci. 2021, 22, 4591. [Google Scholar] [CrossRef]
- Miseta, A.; Nagy, J.; Nagy, T.; Poor, V.; Fekete, Z.; Sipos, K. Hepcidin and its potential clinical utility. Cell Biol. Int. 2015, 39, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.V.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, H.N.; Fulton, D.B.; Ganz, T.; Vogel, H.J. The solution structure of human hepcidin, a peptide hormone with antimicrobial activity that is involved in iron uptake and hereditary hemochromatosis. J. Biol. Chem. 2002, 277, 37597–37603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, J.B.; Poppe, L.; Haniu, M.; Arvedson, T.; Syed, R.; Li, V.; Kohno, H.; Kim, H.; Schnier, P.D.; Harvey, T.S.; et al. Hepcidin revisited, disulfide connectivity, dynamics, and structure. J. Biol. Chem. 2009, 284, 24155–24167. [Google Scholar] [CrossRef] [Green Version]
- Krause, A.; Neitz, S.; Mägert, H.-J.; Schulz, A.; Forssmann, W.-G.; Schulz-Knappe, P.; Adermann, K. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett. 2000, 480, 147–150. [Google Scholar] [CrossRef] [Green Version]
- Hawula, Z.J.; Wallace, D.F.; Subramaniam, V.N.; Rishi, G. Therapeutic Advances in Regulating the Hepcidin/Ferroportin Axis. Pharmaceuticals 2019, 12, 170. [Google Scholar] [CrossRef] [Green Version]
- Joachim, J.H.; Mehta, K.J. Hepcidin in hepatocellular carcinoma. Br. J. Cancer 2022, 127, 185–192. [Google Scholar] [CrossRef]
- Udali, S.; Castagna, A.; Corbella, M.; Ruzzenente, A.; Moruzzi, S.; Mazzi, F.; Campagnaro, T.; De Santis, D.; Franceschi, A.; Pattini, P.; et al. Hepcidin and DNA promoter methylation in hepatocellular carcinoma. Eur. J. Clin. Investig. 2018, 48, e12870. [Google Scholar] [CrossRef]
- Shibabaw, T.; Teferi, B.; Molla, M.D.; Ayelign, B. Inflammation Mediated Hepcidin-Ferroportin Pathway and Its Therapeutic Window in Breast Cancer. Breast Cancer 2020, 12, 165–180. [Google Scholar] [CrossRef]
- Pan, X.; Lu, Y.; Cheng, X.; Wang, J. Hepcidin and ferroportin expression in breast cancer tissue and serum and their relationship with anemia. Curr. Oncol. 2016, 23, e24–e26. [Google Scholar] [CrossRef]
- Scimeca, M.; Bonanno, E. New highlight in breast cancer development: The key role of hepcidin and iron metabolism. Ann. Transl. Med. 2018, 6 (Suppl. S1), S56. [Google Scholar] [CrossRef] [PubMed]
- Yalovenko, T.M.; Todor, I.M.; Lukianova, N.Y.; Chekhun, V.F. Hepcidin as a possible marker in determination of malignancy degree and sensitivity of breast cancer cells to cytostatic drugs. Exp. Oncol. 2016, 38, 84–88. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, L.; Ma, Y.; Wu, X.; Jin, L.; Yu, F. Increased hepcidin expression in non-small cell lung cancer tissue and serum is associated with clinical stage. Thorac. Cancer 2014, 5, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Tesfay, L.; Clausen, K.A.; Kim, J.W.; Hegde, P.; Wang, X.; Miller, L.D.; Deng, Z.; Blanchette, N.; Arvedson, T.; Miranti, C.K.; et al. Hepcidin regulation in prostate and its disruption in prostate cancer. Cancer Res. 2015, 75, 2254–2263. [Google Scholar] [CrossRef] [Green Version]
- Toshiyama, R.; Konno, M.; Eguchi, H.; Asai, A.; Noda, T.; Koseki, J.; Asukai, K.; Ohashi, T.; Matsushita, K.; Iwagami, Y.; et al. Association of iron metabolic enzyme hepcidin expression levels with the prognosis of patients with pancreatic cancer. Oncol. Lett. 2018, 15, 8125–8133. [Google Scholar] [CrossRef] [PubMed]
- Sornjai, W.; Van Long, F.N.; Pion, N.; Pasquer, A.; Saurin, J.-C.; Marcel, V.; Diaz, J.J.; Mertani, H.C.; Smith, D.R. Iron and hepcidin mediate human colorectal cancer cell growth. Chem.-Biol. Interact. 2020, 319, 109021. [Google Scholar] [CrossRef]
- Schwartz, A.J.; Goyert, J.W.; Solanki, S.; Kerk, S.A.; Chen, B.; Castillo, C.; Hsu, P.P.; Do, B.T.; Singhal, R.; Dame, M.K.; et al. Hepcidin sequesters iron to sustain nucleotide metabolism and mitochondrial function in colorectal cancer epithelial cells. Nat. Metab. 2021, 3, 969–982. [Google Scholar] [CrossRef]
- Gineste, A.; Martin, P.; Oswald, E.; Coppin, H.; Roth, M.P. Evidence for IL-6/STAT3-independent induction of lipocalin-2 in the liver of mice infected with Escherichia coli. Hepatology 2016, 63, 673–674. [Google Scholar] [CrossRef] [PubMed]
- Allavena, P.; Sica, A.; Solinas, G.; Porta, C.; Mantovani, A. The inflammatory micro-environment in tumor progression: The role of tumor-associated macrophages. Crit. Rev. Oncol./Hematol. 2008, 66, 1–9. [Google Scholar] [CrossRef]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef]
- Hao, N.B.; Lu, M.H.; Fan, Y.H.; Cao, Y.L.; Zhang, Z.R.; Yang, S.M. Macrophages in tumor microenvironments and the progression of tumors. Clin. Dev. Immunol. 2012, 2012, 948098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, H.; Xu, E.; Liu, H.; Wan, L.; Lai, M. Epithelial-mesenchymal transition in colorectal cancer metastasis: A system review. Pathol. Res. Pract. 2015, 211, 557–569. [Google Scholar] [CrossRef]
- Choi, J.E.; Bae, J.S.; Kang, M.J.; Chung, M.J.; Jang, K.Y.; Park, H.S.; Moon, W.S. Expression of epithelial-mesenchymal transition and cancer stem cell markers in colorectal adenocarcinoma: Clinicopathological significance. Oncol. Rep. 2017, 38, 1695–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niknami, Z.; Muhammadnejad, A.; Ebrahimi, A.; Harsani, Z.; Shirkoohi, R. Significance of E-cadherin and Vimentin as epithelial-mesenchymal transition markers in colorectal carcinoma prognosis. EXCLI J. 2020, 19, 917–926. [Google Scholar] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Xu, P. Global colorectal cancer burden in 2020 and projections to 2040. Transl. Oncol. 2021, 14, 101174. [Google Scholar] [CrossRef]
- Ward, D.G.; Roberts, K.; Brookes, M.J.; Joy, H.; Martin, A.; Ismail, T.; Spychal, R.; Iqbal, T.; Tselepis, C. Increased hepcidin expression in colorectal carcinogenesis. World J. Gastroenterol. 2008, 14, 1339–1345. [Google Scholar] [CrossRef]
- Laudisi, F.; Cherubini, F.; Monteleone, G.; Stolfi, C. STAT3 Interactors as Potential Therapeutic Targets for Cancer Treatment. Int. J. Mol. Sci. 2018, 19, 1787. [Google Scholar] [CrossRef] [Green Version]
- De Simone, V.; Franzè, E.; Ronchetti, G.; Colantoni, A.; Fantini, M.C.; Di Fusco, D.; Sica, G.S.; Sileri, P.; Macdonald, T.T.; Pallone, F.; et al. Th17-type cytokines, IL-6 and TNF-alpha synergistically activate STAT3 and NF-kB to promote colorectal cancer cell growth. Oncogene 2015, 34, 3493–3503. [Google Scholar] [CrossRef]
- De Simone, V.; Ronchetti, G.; Franzè, E.; Colantoni, A.; Ortenzi, A.; Fantini, M.C.; Rizzo, A.; Sica, G.S.; Sileri, P.; Rossi, P.; et al. Interleukin-21 sustains inflammatory signals that contribute to sporadic colon tumorigenesis. Oncotarget 2015, 6, 9908–9923. [Google Scholar] [CrossRef]
- Stolfi, C.; Rizzo, A.; Franzè, E.; Rotondi, A.; Fantini, M.C.; Sarra, M.; Caruso, R.; Monteleone, I.; Sileri, P.; Franceschilli, L.; et al. Involvement of interleukin-21 in the regulation of colitis-associated colon cancer. J. Exp. Med. 2011, 208, 2279–2290. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.; Chauvet, C.; Viatte, L.; Danan, J.L.; Bigard, X.; Devaux, I.; Beaumont, C.; Kahn, A.; Vaulont, S. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J. Clin. Investig. 2002, 110, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Peng, H.; Gelbart, T.; Wang, L.; Beutler, E. Regulation of hepcidin transcription by interleukin-1 and interleukin-6. Proc. Natl. Acad. Sci. USA 2005, 102, 1906–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteleone, G.; Franze, E.; Troncone, E.; Maresca, C.; Marfini, I. Interleukin-34 Mediates Cross-Talk Between Stromal Cells and Immune Cells in the Gut. Front. Immunol. 2022, 13, 873332. [Google Scholar] [CrossRef]
- Monteleone, G.; Maresca, C.; Colella, M.; Pacifico, T.; Congiu, D.; Troncone, E.; Marafini, I. Targeting IL-34/MCSF-1R Axis in Colon Cancer. Front. Immunol. 2022, 13, 917955. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Wynn, T.A. Myeloid-cell differentiation redefined in cancer. Nat. Immunol. 2013, 14, 197–199. [Google Scholar] [CrossRef] [Green Version]
- De Domenico, I.; Zhang, T.Y.; Koening, C.L.; Branch, R.W.; London, N.; Lo, E.; Daynes, R.A.; Kushner, J.P.; Li, D.; Ward, D.M.; et al. Hepcidin mediates transcriptional changes that modulate acute cytokine-induced inflammatory responses in mice. J. Clin. Investig. 2010, 120, 2395–2405. [Google Scholar] [CrossRef] [Green Version]
- Kamai, T.; Tomosugi, N.; Abe, H.; Arai, K.; Yoshida, K. Increased serum hepcidin-25 level and increased tumor expression of hepcidin mRNA are associated with metastasis of renal cell carcinoma. BMC Cancer 2009, 9, 270. [Google Scholar] [CrossRef]
- Sugai, T.; Yamada, N.; Osakabe, M.; Hashimoto, M.; Uesugi, N.; Eizuka, M.; Tanaka, Y.; Sugimoto, R.; Yanagawa, N.; Matsumoto, T. Microenvironmental markers are correlated with lymph node metastasis in invasive submucosal colorectal cancer. Histopathology 2021, 79, 584–598. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Ng, A.S.; Cai, S.; Li, Q.; Yang, L.; Kerr, D. Novel therapeutic strategies: Targeting epithelial-mesenchymal transition in colorectal cancer. Lancet Oncol. 2021, 22, e358–e368. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Grazia, A.; Di Fusco, D.; Franzè, E.; Colella, M.; Strimpakos, G.; Salvatori, S.; Formica, V.; Laudisi, F.; Maresca, C.; Colantoni, A.; et al. Hepcidin Upregulation in Colorectal Cancer Associates with Accumulation of Regulatory Macrophages and Epithelial–Mesenchymal Transition and Correlates with Progression of the Disease. Cancers 2022, 14, 5294. https://doi.org/10.3390/cancers14215294
Di Grazia A, Di Fusco D, Franzè E, Colella M, Strimpakos G, Salvatori S, Formica V, Laudisi F, Maresca C, Colantoni A, et al. Hepcidin Upregulation in Colorectal Cancer Associates with Accumulation of Regulatory Macrophages and Epithelial–Mesenchymal Transition and Correlates with Progression of the Disease. Cancers. 2022; 14(21):5294. https://doi.org/10.3390/cancers14215294
Chicago/Turabian StyleDi Grazia, Antonio, Davide Di Fusco, Eleonora Franzè, Marco Colella, Georgios Strimpakos, Silvia Salvatori, Vincenzo Formica, Federica Laudisi, Claudia Maresca, Alfredo Colantoni, and et al. 2022. "Hepcidin Upregulation in Colorectal Cancer Associates with Accumulation of Regulatory Macrophages and Epithelial–Mesenchymal Transition and Correlates with Progression of the Disease" Cancers 14, no. 21: 5294. https://doi.org/10.3390/cancers14215294
APA StyleDi Grazia, A., Di Fusco, D., Franzè, E., Colella, M., Strimpakos, G., Salvatori, S., Formica, V., Laudisi, F., Maresca, C., Colantoni, A., Ortenzi, A., Stolfi, C., Monteleone, I., & Monteleone, G. (2022). Hepcidin Upregulation in Colorectal Cancer Associates with Accumulation of Regulatory Macrophages and Epithelial–Mesenchymal Transition and Correlates with Progression of the Disease. Cancers, 14(21), 5294. https://doi.org/10.3390/cancers14215294