
Next-Generation Antisense Oligonucleotide of TGF-β2 Enhances T Cell-Mediated Anticancer Efficacy of Anti-PD-1 Therapy in a Humanized Mouse Model of Immune-Excluded Melanoma

 ,
, 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Oligonucleotide
2.2. Cell Cultures
2.3. qRT-PCR and Western Blot
2.4. Experimental Animals
2.5. Generation of a Melanoma-Bearing Human Immune System Mouse Model
2.6. ngTASO and Anti-PD-1 Antibody Treatment
2.7. Fluorescence-Activated Cell Sorting (FACS) Analysis
2.8. Immunohistochemistry (IHC)
2.9. Statistical Analysis
3. Results
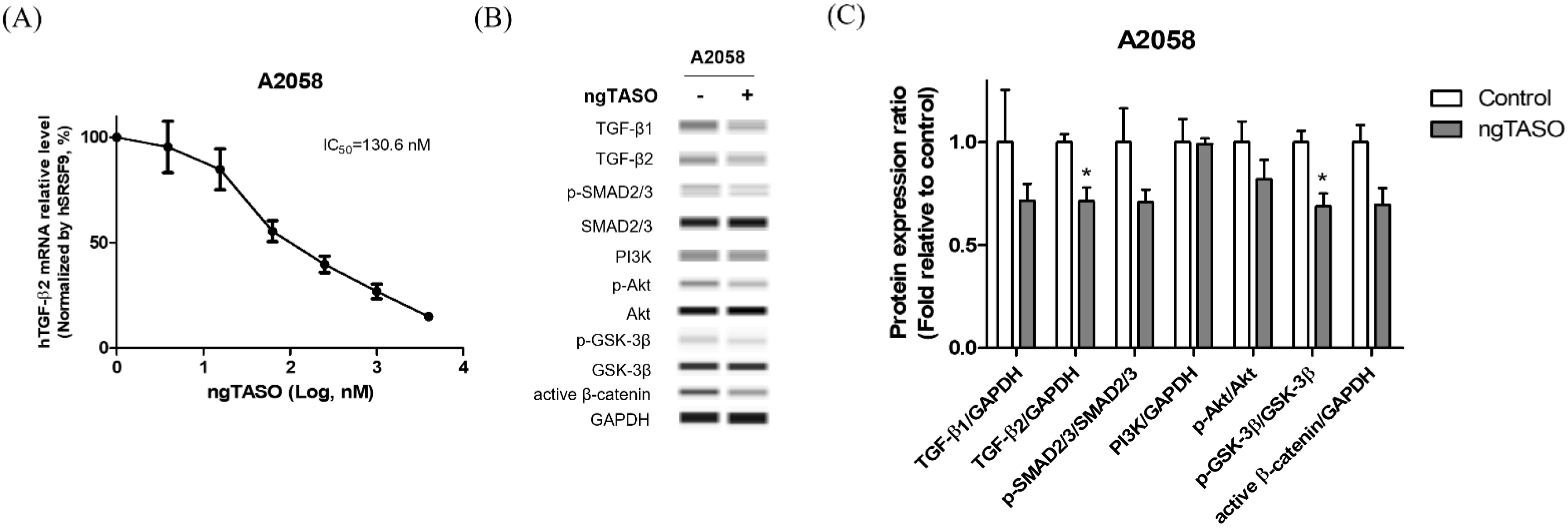
3.1. ngTASO Downregulates TGF-β2 and Its Downstream Signaling
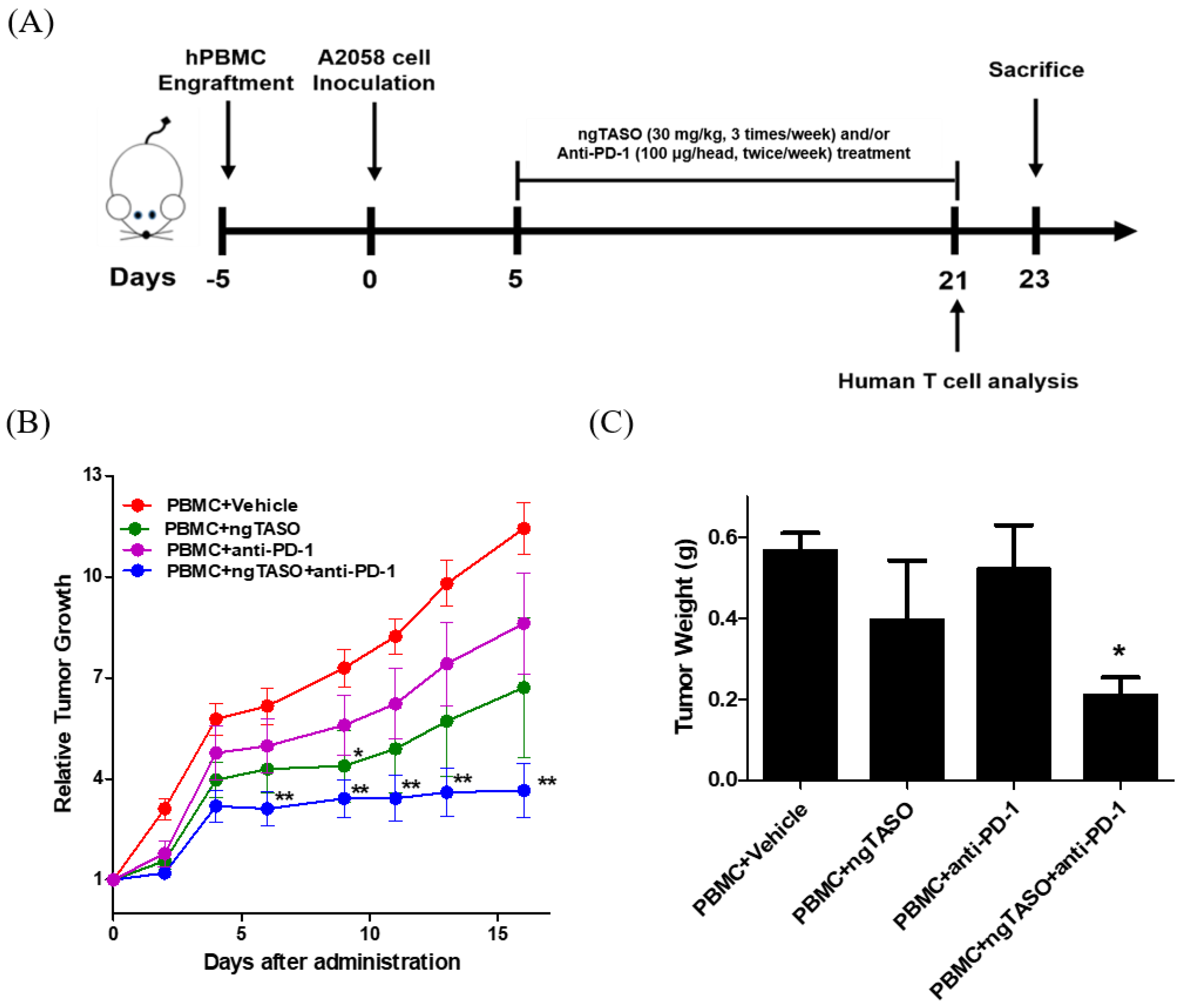
3.2. TGF-β2 Blockade by ngTASO in Combination with PD-1 Inhibition Delays Tumor Growth
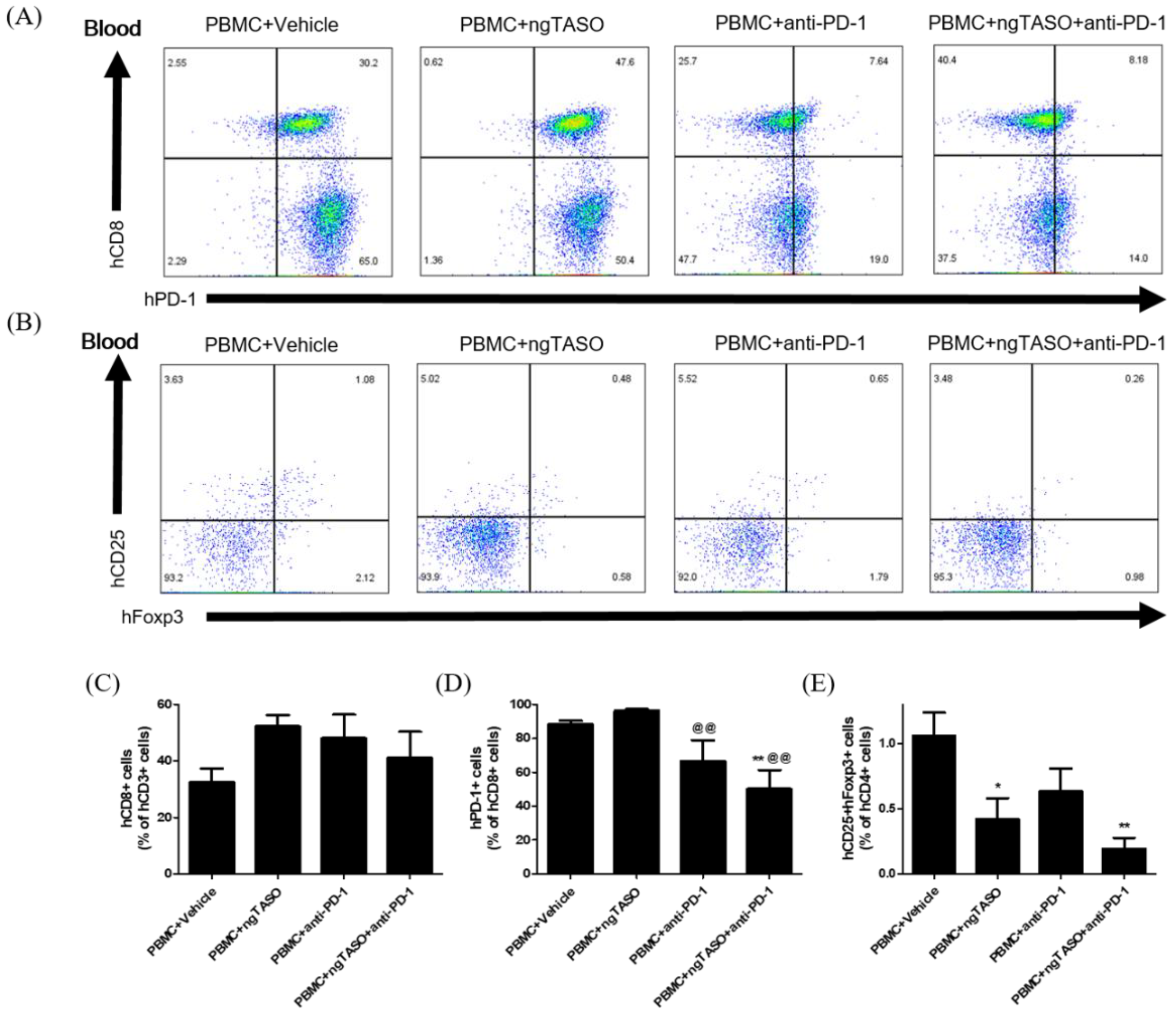
3.3. ngTASO and Anti-PD-1 Administration Modifies the Subpopulation of Human CD8+ T Cells and Tregs in Peripheral Blood
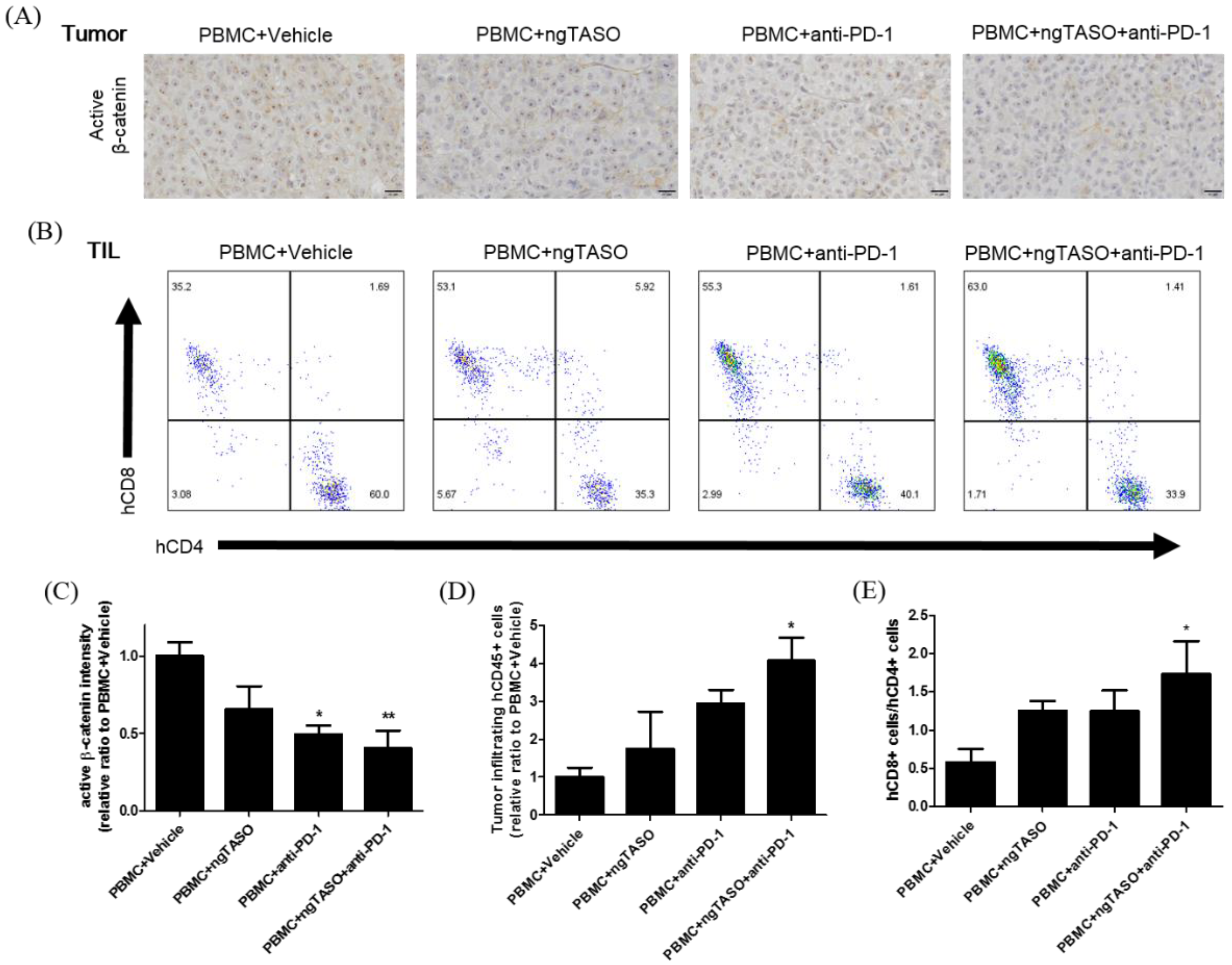
3.4. TGF-β2 Blockade by ngTASO Combined with PD-1 Inhibition Downregulates the β-Catenin Activation and Facilitates Human CD8+ T Cell Infiltration in Tumors
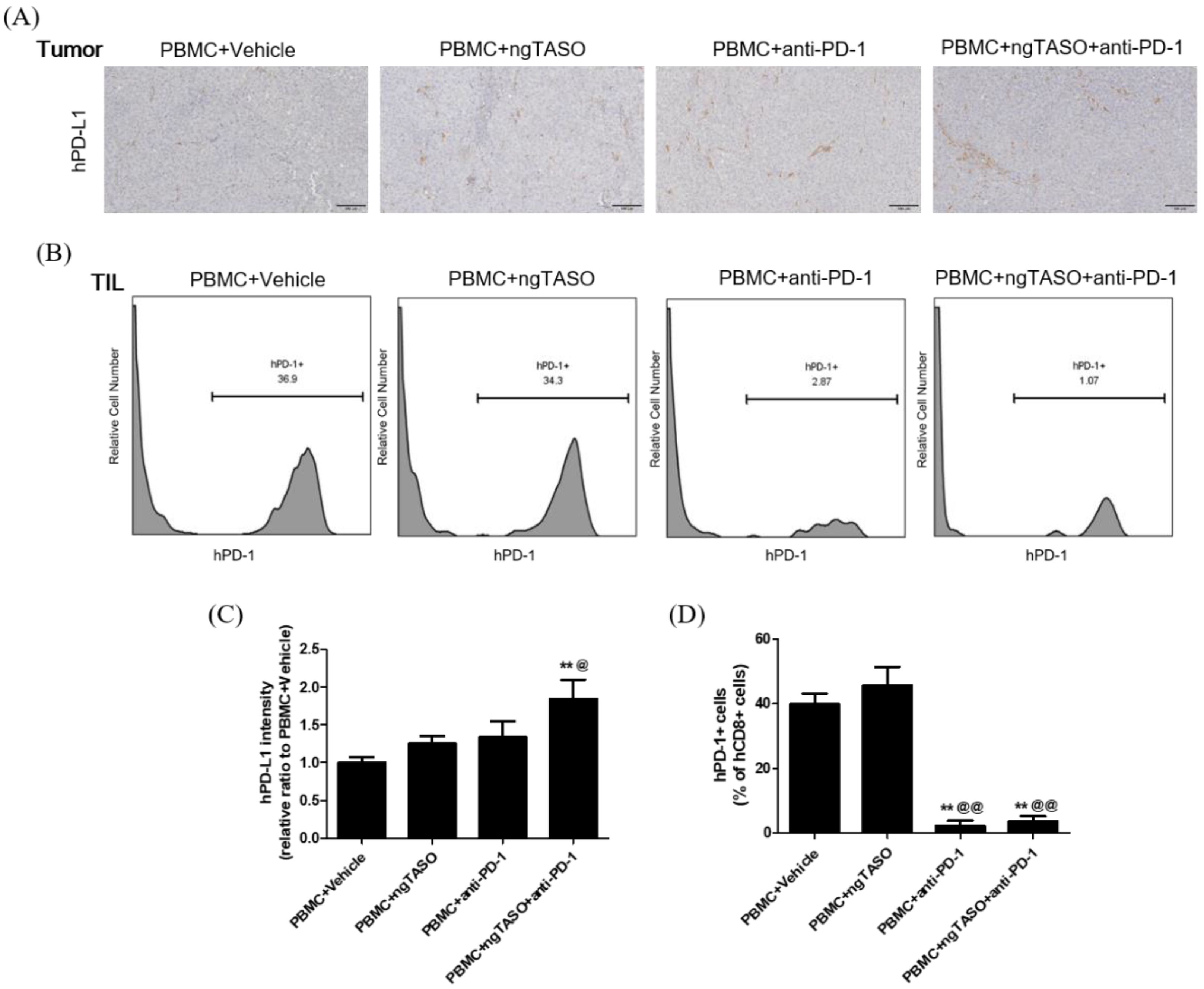
3.5. TGF-β2 Blockade by ngTASO Combined with PD-1 Inhibition Upregulates PD-L1 Expression in Tumors
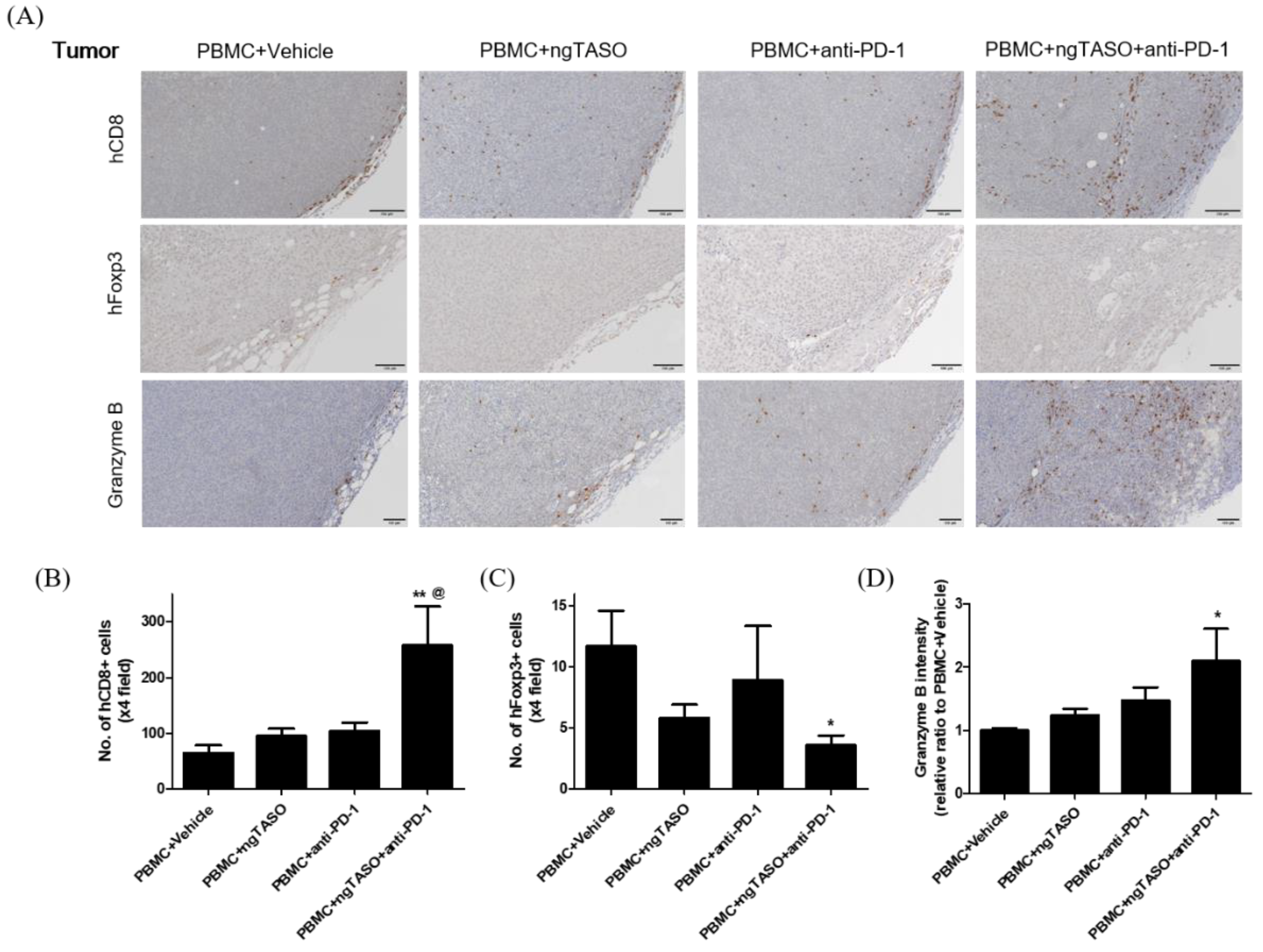
3.6. TGF-β2 Blockade by ngTASO Combined with PD-1 Inhibition Regulates Infiltration of the T Cell Subpopulation and Their Activation in Tumors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hamanishi, J.; Mandai, M.; Matsumura, N.; Abiko, K.; Baba, T.; Konishi, I. Pd-1/pd-l1 blockade in cancer treatment: Perspectives and issues. Int. J. Clin. Oncol. 2016, 21, 462–473. [Google Scholar] [CrossRef] [PubMed]
- Bennett, F.; Luxenberg, D.; Ling, V.; Wang, I.M.; Marquette, K.; Lowe, D.; Khan, N.; Veldman, G.; Jacobs, K.A.; Valge-Archer, V.E.; et al. Program death-1 engagement upon tcr activation has distinct effects on costimulation and cytokine-driven proliferation: Attenuation of icos, il-4, and il-21, but not cd28, il-7, and il-15 responses. J. Immunol. 2003, 170, 711–718. [Google Scholar] [CrossRef]
- Chikuma, S.; Terawaki, S.; Hayashi, T.; Nabeshima, R.; Yoshida, T.; Shibayama, S.; Okazaki, T.; Honjo, T. Pd-1-mediated suppression of il-2 production induces cd8+ t cell anergy in vivo. J. Immunol. 2009, 182, 6682–6689. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral cd4+cd25- naive t cells to cd4+cd25+ regulatory t cells by tgf-beta induction of transcription factor foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.N.; Sarkissian, S.; Chao, J.; Klempner, S.J. Pd-1 and pd-l1 as emerging therapeutic targets in gastric cancer: Current evidence. Gastrointest. Cancer 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. Pd-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Barone, A.; Hazarika, M.; Theoret, M.R.; Mishra-Kalyani, P.; Chen, H.; He, K.; Sridhara, R.; Subramaniam, S.; Pfuma, E.; Wang, Y.; et al. Fda approval summary: Pembrolizumab for the treatment of patients with unresectable or metastatic melanoma. Clin. Cancer Res. 2017, 23, 5661–5665. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations associated with acquired resistance to pd-1 blockade in melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Ren, D.; Hua, Y.; Yu, B.; Ye, X.; He, Z.; Li, C.; Wang, J.; Mo, Y.; Wei, X.; Chen, Y.; et al. Predictive biomarkers and mechanisms underlying resistance to pd1/pd-l1 blockade cancer immunotherapy. Mol. Cancer 2020, 19, 19. [Google Scholar] [CrossRef]
- Terabe, M.; Robertson, F.C.; Clark, K.; De Ravin, E.; Bloom, A.; Venzon, D.J.; Kato, S.; Mirza, A.; Berzofsky, J.A. Blockade of only tgf-beta 1 and 2 is sufficient to enhance the efficacy of vaccine and pd-1 checkpoint blockade immunotherapy. Oncoimmunology 2017, 6, e1308616. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Akhurst, R.J.; Balmain, A. Tgf-beta signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Hussein, M.R. Transforming growth factor-beta and malignant melanoma: Molecular mechanisms. J. Cutan. Pathol. 2005, 32, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Lebrun, J.J. The dual role of tgfbeta in human cancer: From tumor suppression to cancer metastasis. ISRN Mol. Biol. 2012, 2012, 381428. [Google Scholar] [PubMed]
- Zonneville, J.; Safina, A.; Truskinovsky, A.M.; Arteaga, C.L.; Bakin, A.V. Tgf-beta signaling promotes tumor vasculature by enhancing the pericyte-endothelium association. BMC Cancer 2018, 18, 670. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Baker, D.; Ten Dijke, P. Tgf-beta-mediated epithelial-mesenchymal transition and cancer metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. Tgf-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Bierie, B.; Moses, H.L. Transforming growth factor beta (tgf-beta) and inflammation in cancer. Cytokine Growth Factor Rev. 2010, 21, 49–59. [Google Scholar] [CrossRef]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Canellas, A.; Hernando-Momblona, X.; et al. Tgfbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef]
- Thomas, D.A.; Massague, J. Tgf-beta directly targets cytotoxic t cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef]
- Moo-Young, T.A.; Larson, J.W.; Belt, B.A.; Tan, M.C.; Hawkins, W.G.; Eberlein, T.J.; Goedegebuure, P.S.; Linehan, D.C. Tumor-derived tgf-beta mediates conversion of cd4+foxp3+ regulatory t cells in a murine model of pancreas cancer. J. Immunother. 2009, 32, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Ji, H.J.; Shin, S.K.; Koo, J.; Kim, T.H.; Kim, C.W.; Seong, Y.H.; Park, J.E.; Choi, K.C. Targeting transforming growth factor-beta2 by antisense oligodeoxynucleotide accelerates t cell-mediated tumor rejection in a humanized mouse model of triple-negative breast cancer. Cancer Immunol. Immunother. 2022, 71, 2213–2226. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Shin, H.J.; Koo, J.; Kim, T.H.; Kim, C.W.; Go, R.E.; Seong, Y.H.; Park, J.E.; Choi, K.C. Blockade of transforming growth factor beta2 by anti-sense oligonucleotide improves immunotherapeutic potential of il-2 against melanoma in a humanized mouse model. Cytotherapy 2021, 23, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Ahn, D.; Kim, C.W.; Go, R.E.; Choi, K.C. Evaluation of mitochondrial oxidative toxicity in mammalian cardiomyocytes by determining the highly reproducible and reliable increase in mitochondrial superoxides after exposure to therapeutic drugs. Toxicol. In Vitro 2022, 83, 105393. [Google Scholar] [CrossRef] [PubMed]
- Crowe, A.R.; Yue, W. Semi-quantitative determination of protein expression using immunohistochemistry staining and analysis: An integrated protocol. Bio-Protocol 2019, 9, e3465. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yao, L.C.; Cheng, M.; Cai, D.; Martinek, J.; Pan, C.X.; Shi, W.; Ma, A.H.; De Vere White, R.W.; Airhart, S.; et al. Humanized mice in studying efficacy and mechanisms of pd-1-targeted cancer immunotherapy. FASEB J. 2018, 32, 1537–1549. [Google Scholar] [CrossRef]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Nsengimana, J.; Laye, J.; Filia, A.; O′Shea, S.; Muralidhar, S.; Pozniak, J.; Droop, A.; Chan, M.; Walker, C.; Parkinson, L.; et al. Beta-catenin-mediated immune evasion pathway frequently operates in primary cutaneous melanomas. J. Clin. Investig. 2018, 128, 2048–2063. [Google Scholar] [CrossRef]
- Pantaleo, M.A.; Tarantino, G.; Agostinelli, C.; Urbini, M.; Nannini, M.; Saponara, M.; Castelli, C.; Stacchiotti, S.; Fumagalli, E.; Gatto, L.; et al. Immune microenvironment profiling of gastrointestinal stromal tumors (gist) shows gene expression patterns associated to immune checkpoint inhibitors response. Oncoimmunology 2019, 8, e1617588. [Google Scholar] [CrossRef]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. Pd-1 and pd-l1 checkpoint signaling inhibition for cancer immunotherapy: Mechanism, combinations, and clinical outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef]
- Liu, F.; Lang, R.; Zhao, J.; Zhang, X.; Pringle, G.A.; Fan, Y.; Yin, D.; Gu, F.; Yao, Z.; Fu, L. Cd8(+) cytotoxic t cell and foxp3(+) regulatory t cell infiltration in relation to breast cancer survival and molecular subtypes. Breast Cancer Res. Treat. 2011, 130, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Meslin, F.; Thiery, J.; Richon, C.; Jalil, A.; Chouaib, S. Granzyme b-induced cell death involves induction of p53 tumor suppressor gene and its activation in tumor target cells. J. Biol. Chem. 2007, 282, 32991–32999. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Massague, J. Transforming growth factor-beta signaling in immunity and cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef] [PubMed]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.K.; Flavell, R.A. Transforming growth factor-beta regulation of immune responses. Annu. Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef]
- Ciardiello, D.; Elez, E.; Tabernero, J.; Seoane, J. Clinical development of therapies targeting tgfbeta: Current knowledge and future perspectives. Ann. Oncol. 2020, 31, 1336–1349. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. Tgfbeta attenuates tumour response to pd-l1 blockade by contributing to exclusion of t cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Akhmetshina, A.; Palumbo, K.; Dees, C.; Bergmann, C.; Venalis, P.; Zerr, P.; Horn, A.; Kireva, T.; Beyer, C.; Zwerina, J.; et al. Activation of canonical wnt signalling is required for tgf-beta-mediated fibrosis. Nat. Commun. 2012, 3, 735. [Google Scholar] [CrossRef]
- Amini Nik, S.; Ebrahim, R.P.; Van Dam, K.; Cassiman, J.J.; Tejpar, S. Tgf-beta modulates beta-catenin stability and signaling in mesenchymal proliferations. Exp. Cell Res. 2007, 313, 2887–2895. [Google Scholar] [CrossRef]
- Ruiz de Galarreta, M.; Bresnahan, E.; Molina-Sanchez, P.; Lindblad, K.E.; Maier, B.; Sia, D.; Puigvehi, M.; Miguela, V.; Casanova-Acebes, M.; Dhainaut, M.; et al. Beta-catenin activation promotes immune escape and resistance to anti-pd-1 therapy in hepatocellular carcinoma. Cancer Discov. 2019, 9, 1124–1141. [Google Scholar] [CrossRef]
- Freudenberg, K.; Lindner, N.; Dohnke, S.; Garbe, A.I.; Schallenberg, S.; Kretschmer, K. Critical role of tgf-beta and il-2 receptor signaling in foxp3 induction by an inhibitor of DNA methylation. Front. Immunol. 2018, 9, 125. [Google Scholar] [CrossRef]
- Collison, L.W.; Workman, C.J.; Kuo, T.T.; Boyd, K.; Wang, Y.; Vignali, K.M.; Cross, R.; Sehy, D.; Blumberg, R.S.; Vignali, D.A. The inhibitory cytokine il-35 contributes to regulatory t-cell function. Nature 2007, 450, 566–569. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, V.; Di Rella, F.; Di Giacomo, A.; Matarese, G. Regulatory t cells as suppressors of anti-tumor immunity: Role of metabolism. Cytokine Growth Factor Rev. 2017, 35, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.L.; Betts, G.J.; Plant, A.; Wright, K.L.; El-Shanawany, T.M.; Harrop, R.; Torkington, J.; Rees, B.I.; Williams, G.T.; Gallimore, A.M.; et al. Cd4+cd25+foxp3+ regulatory t cells suppress anti-tumor immune responses in patients with colorectal cancer. PLoS ONE 2006, 1, e129. [Google Scholar] [CrossRef]
- Linette, G.P.; Carreno, B.M. Tumor-infiltrating lymphocytes in the checkpoint inhibitor era. Curr. Hematol. Malig. Rep. 2019, 14, 286–291. [Google Scholar] [CrossRef]
- Ballot, E.; Ladoire, S.; Routy, B.; Truntzer, C.; Ghiringhelli, F. Tumor infiltrating lymphocytes signature as a new pan-cancer predictive biomarker of anti pd-1/pd-l1 efficacy. Cancers 2020, 12, 2418. [Google Scholar] [CrossRef] [PubMed]
- Wouters, M.C.A.; Nelson, B.H. Prognostic significance of tumor-infiltrating b cells and plasma cells in human cancer. Clin. Cancer Res. 2018, 24, 6125–6135. [Google Scholar] [CrossRef] [PubMed]
- Gide, T.N.; Wilmott, J.S.; Scolyer, R.A.; Long, G.V. Primary and acquired resistance to immune checkpoint inhibitors in metastatic melanoma. Clin. Cancer Res. 2018, 24, 1260–1270. [Google Scholar] [CrossRef] [PubMed]
- Principe, D.R.; Park, A.; Dorman, M.J.; Kumar, S.; Viswakarma, N.; Rubin, J.; Torres, C.; McKinney, R.; Munshi, H.G.; Grippo, P.J.; et al. Tgfbeta blockade augments pd-1 inhibition to promote t-cell-mediated regression of pancreatic cancer. Mol. Cancer Ther. 2019, 18, 613–620. [Google Scholar] [CrossRef]
- Spranger, S.; Spaapen, R.M.; Zha, Y.; Williams, J.; Meng, Y.; Ha, T.T.; Gajewski, T.F. Up-regulation of pd-l1, ido, and t(regs) in the melanoma tumor microenvironment is driven by cd8(+) t cells. Sci. Transl. Med. 2013, 5, 200ra116. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the tumor microenvironment in pd-l1/pd-1-mediated tumor immune escape. Mol. Cancer 2019, 18, 10. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Species | Forward/Reverse | Sequence (5′-3′) | Tm (°C) |
|---|---|---|---|---|
| TGFB2 | Human | F | CAGCACACTCGATATGGACCA | 57 |
| R | CCTCGGGCTCAGGATAGTCT | 61 | ||
| SRSF9 | Human | F | TGTGCAGAAGGATGGAGT | 55 |
| R | CTGGTGCTTCTCTCAGGATA | 54 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.K.; Kim, C.-W.; Ahn, D.; Go, R.-E.; Choi, Y.; Choi, K.-C. Next-Generation Antisense Oligonucleotide of TGF-β2 Enhances T Cell-Mediated Anticancer Efficacy of Anti-PD-1 Therapy in a Humanized Mouse Model of Immune-Excluded Melanoma. Cancers 2022, 14, 5220. https://doi.org/10.3390/cancers14215220
Lee HK, Kim C-W, Ahn D, Go R-E, Choi Y, Choi K-C. Next-Generation Antisense Oligonucleotide of TGF-β2 Enhances T Cell-Mediated Anticancer Efficacy of Anti-PD-1 Therapy in a Humanized Mouse Model of Immune-Excluded Melanoma. Cancers. 2022; 14(21):5220. https://doi.org/10.3390/cancers14215220
Chicago/Turabian StyleLee, Hong Kyu, Cho-Won Kim, Dohee Ahn, Ryeo-Eun Go, Youngdong Choi, and Kyung-Chul Choi. 2022. "Next-Generation Antisense Oligonucleotide of TGF-β2 Enhances T Cell-Mediated Anticancer Efficacy of Anti-PD-1 Therapy in a Humanized Mouse Model of Immune-Excluded Melanoma" Cancers 14, no. 21: 5220. https://doi.org/10.3390/cancers14215220
APA StyleLee, H. K., Kim, C.-W., Ahn, D., Go, R.-E., Choi, Y., & Choi, K.-C. (2022). Next-Generation Antisense Oligonucleotide of TGF-β2 Enhances T Cell-Mediated Anticancer Efficacy of Anti-PD-1 Therapy in a Humanized Mouse Model of Immune-Excluded Melanoma. Cancers, 14(21), 5220. https://doi.org/10.3390/cancers14215220