
Molecular Classification of Colorectal Cancer by microRNA Profiling: Correlation with the Consensus Molecular Subtypes (CMS) and Validation of miR-30b Targets
 ,
,  , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and RNA
2.2. MicroRNA Expression Analysis and Tumor Classification
2.3. Identification of miRs Targets and Correlation with mRNA Expression
2.4. External Dataset Validation
2.5. MicroRNAs Differentially Expressed between Tumor-Epithelia and Tumor-Stroma
2.6. Evaluation of miRs-Subtypes Using miRaCL20 Classifier
2.7. Cell Lines, Transformation, Transfection and Luciferase Assay
2.8. Site-Directed Mutagenesis
2.9. Statistical Analysis
3. Results
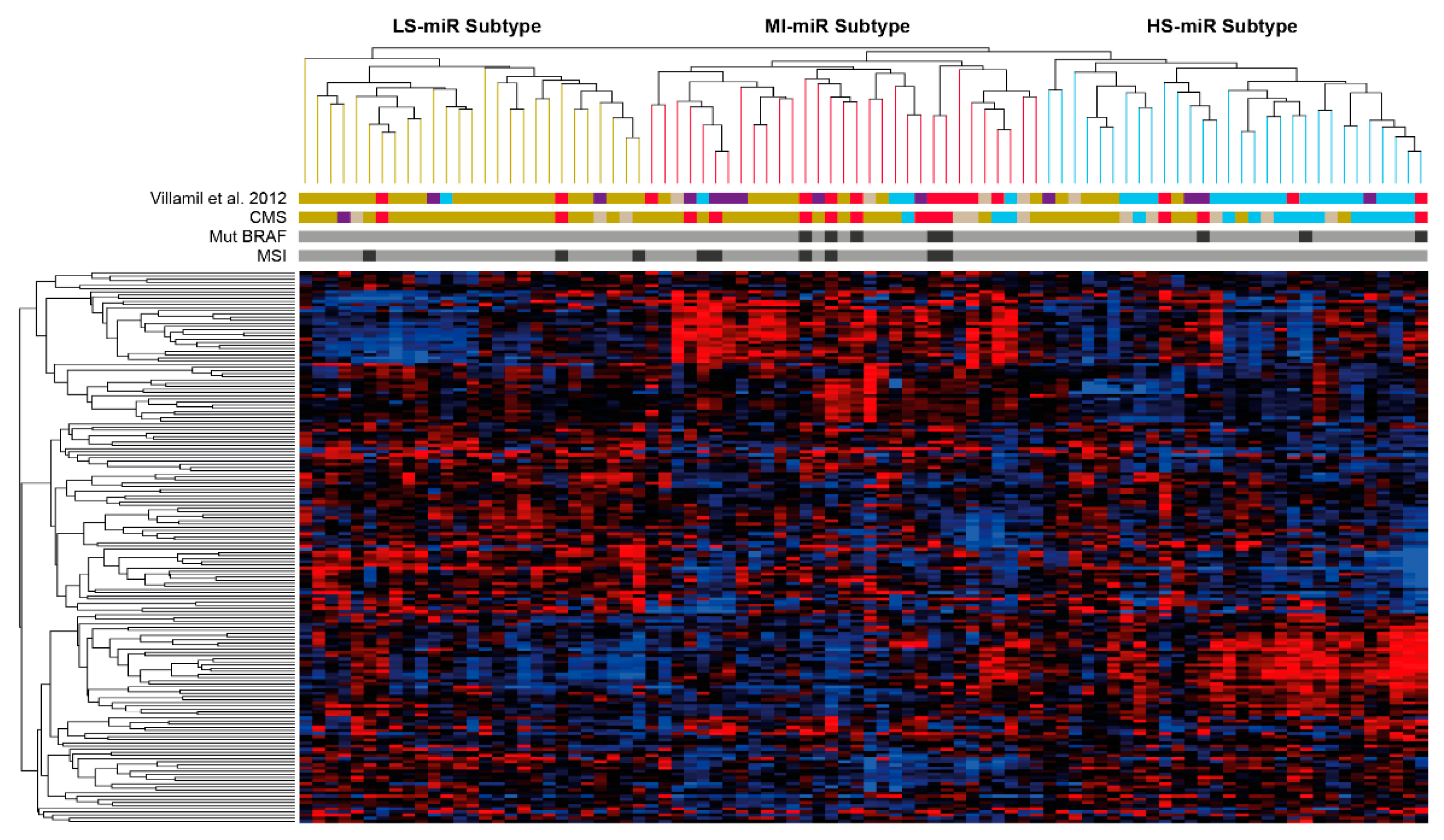
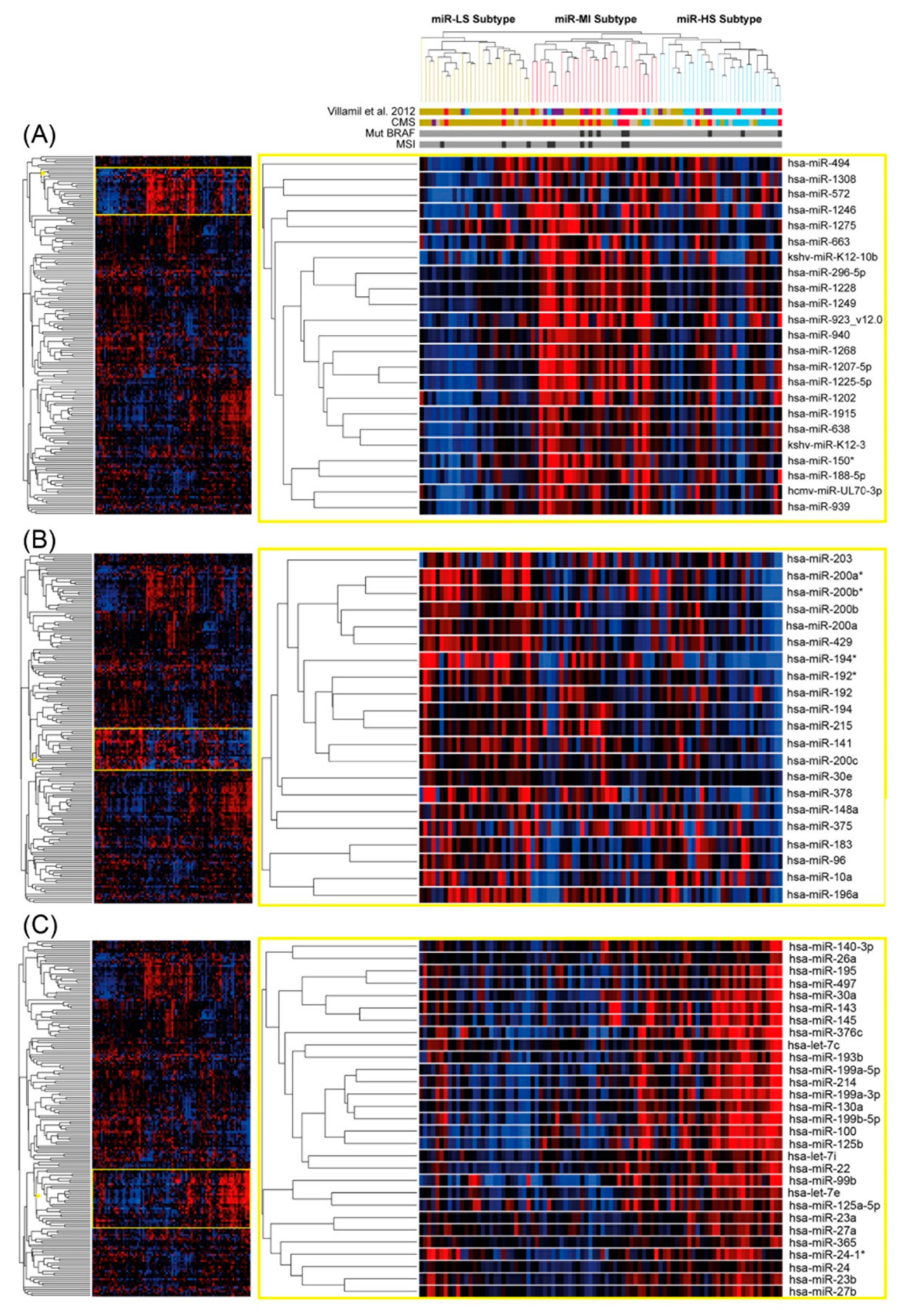
3.1. Tumor Classification Based on miR Expression Patterns and Association to mRNA Subtypes
3.2. External Dataset Validation
3.3. Comparison between miR-LS, miR-HS and miR-MI with the miRCL20 Classifier Subtypes
3.4. Stromal or Epithelial Localization of the miRs Differentially Expressed between Subtypes
3.5. Identification of miRs Targets, Selection of Relevant Interactions Associated to Subtypes and Altered Pathways
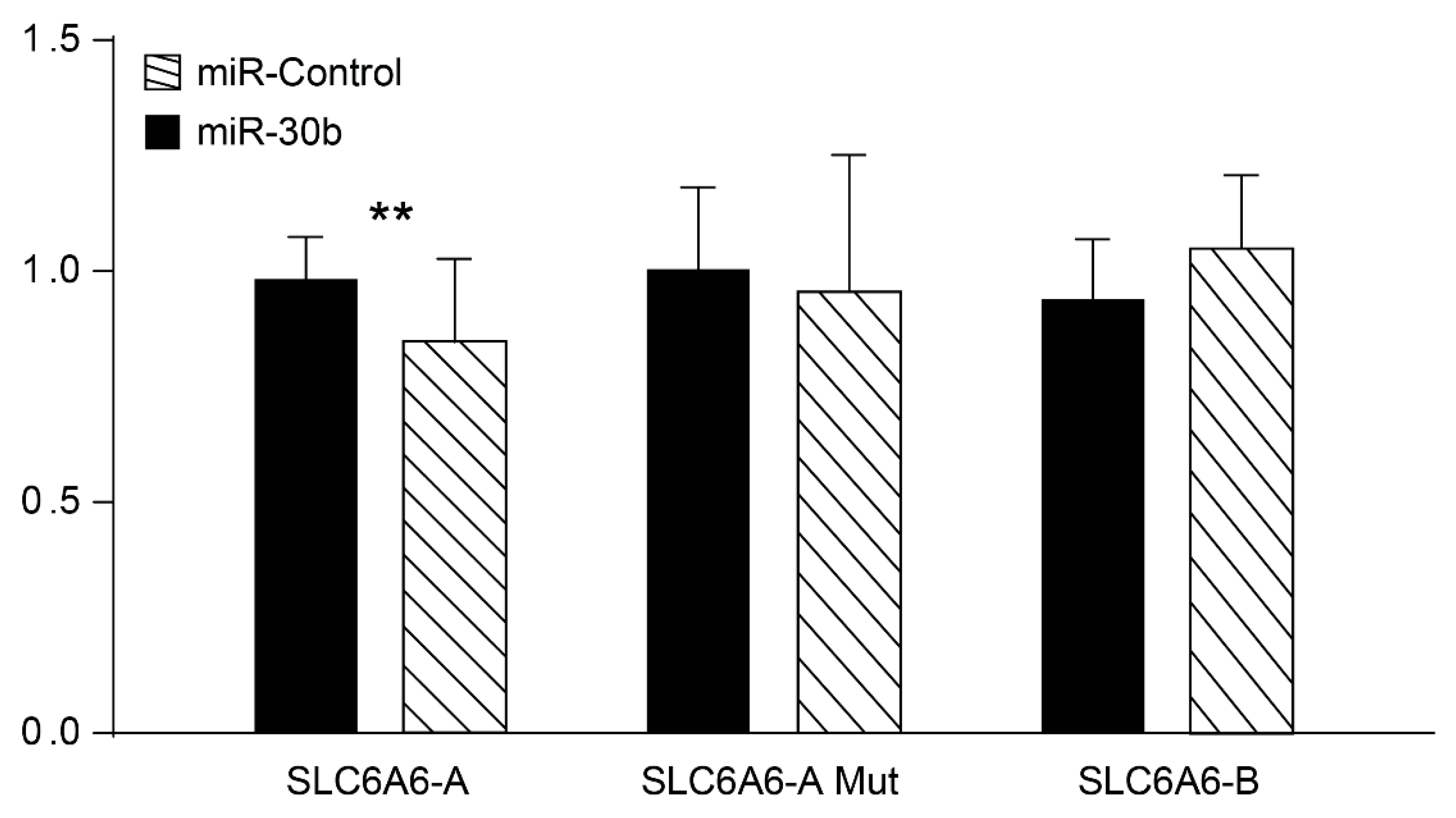
3.6. SCL6A6 Up-Regulated in the High-Stroma/CMS4 Subtype Shows Specific Interaction with miR-30b In Vitro
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Perez-Villamil, B.; Romera-Lopez, A.; Hernandez-Prieto, S.; Lopez-Campos, G.; Calles, A.; Lopez-Asenjo, J.; Sanz-Ortega, J.; Fernandez-Perez, C.; Sastre, J.; Alfonso, R.; et al. Colon cancer molecular subtypes identified by expression profiling and associated to stroma, mucinous type and different clinical behaviour. BMC Cancer 2012, 12, 260. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniés, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Hoorn, S.T.; de Back, T.R.; Sommeijer, D.W.; Vermeulen, L. Clinical Value of Consensus Molecular Subtypes in Colorectal Cancer: A Systematic Review and Meta-Analysis. J. Natl. Cancer Inst. 2021, 144, 503–516. [Google Scholar] [CrossRef]
- Mooi, J.K.; Wirapati, P.; Asher, R.; Lee, C.K.; Savas, P.; Price, T.J.; Townsend, A.; Hardingham, J.; Buchanan, D.; Williams, D.; et al. The prognostic impact of consensus molecular subtypes (CMS) and its predictive effects for bevacizumab benefit in metastatic colorectal cancer: Molecular analysis of the AGITG MAX clinical trial. Ann. Oncol. 2018, 29, 2240–2246. [Google Scholar] [CrossRef] [PubMed]
- Lenz, H.; Ou, F.; Venook, A.P.; Hochster, H.S.; Niedzwiecki, D.; Goldberg, R.M.; Mayer, R.J.; Bertagnolli, M.M.; Blanke, C.D.; Zemla, T.; et al. Impact of consensus molecular subtype on survival in patients with metastatic colorectal cancer: Results from CALGB/SWOG 80405 (Alliance). J. Clin. Oncol. 2019, 37, 1876–1885. [Google Scholar] [CrossRef] [PubMed]
- Al-Akhrass, H.; Christou, N. The clinical assessment of microrna diagnostic, prognostic, and theranostic value in colorectal cancer. Cancers 2021, 13, 2916. [Google Scholar] [CrossRef] [PubMed]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–221. [Google Scholar] [CrossRef] [PubMed]
- Fessler, E.; Jansen, M.; De Sousa E Melo, F.; Zhao, L.; Prasentyanti, P.R.; Rodermond, H.; Kandimalla, R.; Linnekamp, J.F.; Franitza, M.; van Hoof, S.R.; et al. A multidimensional network approach reveals microRNAs as determinants of the mesenchymal colorectal cancer subtype. Oncogene 2016, 35, 6026–6037. [Google Scholar] [CrossRef] [PubMed]
- Cantini, L.; Isella, C.; Petti, C.; Picco, G.; Chiola, S.; Ficarra, E.; Caselle, M.; Medico, E. MicroRNA-mRNA interactions underlying colorectal cancer molecular subtypes. Nat. Commun. 2015, 6, 8878. [Google Scholar] [CrossRef] [PubMed]
- Kandimalla, R.; Shimura, T.; Mallik, S.; Sonohara, F.; Tsai, S.; Evans, D.B.; Kim, S.C.; Baba, H.; Kodera, Y.; Von Hoff, D.; et al. Identification of Serum miRNA Signature and Establishment of a Nomogram for Risk Stratification in Patients with Pancreatic Ductal Adenocarcinoma. Ann. Surg. 2022, 275, E229–E237. [Google Scholar] [CrossRef] [PubMed]
- Adam, R.S.; Poel, D.; Ferreira, L.M.; Spronck, J.M.A.; de Back, T.R.; Torang, A.; Gomez, P.M.B.; ten Hoorn, S.; Markowetz, F.; Wang, X.; et al. Development of a miRNA-based classifier for detection of colorectal cancer molecular subtypes. Mol. Oncol. 2022, 16, 2693–2709. [Google Scholar] [CrossRef] [PubMed]
- Muniategui, A.; Nogales-Cadenas, R.; Vázquez, M.; Aranguren, X.L.; Aguirre, X.; Luttun, A.; Prosper, F.; Pascual-Montano, A.; Rubio, A. Quantification of miRNA-mRNA interactions. PLoS ONE 2012, 7, e30766. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Del Rio, G.; Koschützki, D.; Coello, G. How to identify essential genes from molecular networks? BMC Syst. Biol. 2009, 3, 102. [Google Scholar] [CrossRef]
- Tabas-Madrid, D.; Muniategui, A.; Sánchez-Caballero, I.; Martínez-Herrera, D.J.; Sorzano, C.O.S.; Rubio, A.; Pascual-Montano, A. Improving miRNA-mRNA interaction predictions. BMC Genomics 2014, 15 (Suppl. 10), S2. [Google Scholar] [CrossRef] [PubMed]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Nishida, N.; Magahara, M.; Sato, T.; Mimori, K.; Sudo, T.; Tanaka, F.; Shibata, K.; Ishii, H.; Sugihara, K.; Doki, Y.; et al. Microarray analysis of colorectal cancer stromal tissue reveals upregulation of two oncogenic miRNA clusters. Clin. Cancer Res. 2012, 18, 3054–3070. [Google Scholar] [CrossRef]
- Ogando, J.; Tardáguila, M.; Díaz-Alderete, A.; Usategui, A.; Miranda-Ramos, V.; Martínez-Herrera, D.J.; de la Fuente, L.; García-León, M.J.; Moreno, M.C.; Escudero, S.; et al. Notch-regulated MIR-223 targets the aryl hydrocarbon receptor pathway and increases cytokine production in macrophages from rheumatoid arthritis patients. Sci. Rep. 2016, 6, 20223. [Google Scholar] [CrossRef]
- Wang, S.; He, X.; Zhou, R.; Jia, G.; Qiao, Q. STAT3 induces colorectal carcinoma progression through a novel miR-572-MOAP-1 pathway. Onco. Targets Ther. 2018, 11, 3475–3484. [Google Scholar] [CrossRef]
- Wang, S.; Zeng, Y.; Zhou, J.; Nie, S.; Peng, Q.; Gong, J.; Huo, J. MicroRNA-1246 promotes growth and metastasis of colorectal cancer cells involving CCNG2 reduction. Mol. Med. Rep. 2016, 13, 273–280. [Google Scholar] [CrossRef]
- Sun, H.; Chen, X.; Ji, H.; Wu, T.; Lu, H.; Zhang, Y.; Li, H.; Li, Y. MiR-494 is an independent prognostic factor and promotes cell migration and invasion in colorectal cancer by directly targeting PTEN. Int. J. Oncol. 2014, 45, 2486–2494. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, D.R.; Chinnaiyan, A.M. Integrative analysis of the cancer transcriptome. Nat. Genet. 2005, 37, S31–S37. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lin, Y.; Guo, Y.; Pu, X.; Li, M. Functional dissection of human targets for KSHV-encoded miRNAs using network analysis. Sci. Rep. 2017, 7, 3159. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, A.R.; Shango, J.; Seal, A.; Shukla, D.; Nares, S. Viral miRNAs alter host cell miRNA profiles and modulate innate immune responses. Front. Immunol. 2018, 9, 433. [Google Scholar] [CrossRef]
- Zeng, M.; Zhu, L.; Li, L.; Kang, C. miR-378 suppresses the proliferation, migration and invasion of colon cancer cells by inhibiting SDAD1. Cell. Mol. Biol. Lett. 2017, 22, 12. [Google Scholar] [CrossRef]
- Chang, H.; Ye, S.; Pan, S.; Kuo, T.; Liu, B.C.; Chen, Y.; Huang, T. Overexpression of miR-194 Reverses HMGA2-driven Signatures in Colorectal Cancer. Theranostics 2017, 7, 3889–3900. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhao, X.; Liu, Q.; Li, C.; Graves-Deal, R.; Cao, Z.; Singh, B.; Franklin, J.L.; Wang, J.; Hu, H.; et al. LncRNA MIR100HG-derived miR-100 and miR-125b mediate cetuximab resistance via Wnt/β-catenin signaling. Nat. Med. 2017, 23, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
- Michael, M.Z.; O’Connor, S.M.; van Holst Pellekaan, N.G.; Young, G.P.; James, R.J. Reduced Accumulation of Specific MicroRNAs in Colorectal Neoplasia. Mol. Cancer Res. 2003, 1, 882–891. [Google Scholar]
- Chivukula, R.R.; Shi, G.; Acharya, A.; Mills, E.W.; Zeitels, L.R.; Anandam, J.L.; Abdelnaby, A.A.; Balck, G.C.; Mansour, J.C.; Yopp, A.C.; et al. An essential mesenchymal function for miR-143/145 in intestinal epithelial regeneration. Cell 2014, 157, 1104–1116. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Qi, L.; Liu, B.; Liu, J.; Zhang, H.; Che, D.; Cao, J.; Shen, J.; Geng, J.; Bi, Y.; et al. Fibroblast activation protein overexpression and clinical implications in solid tumours: A meta-analysis. PLoS ONE 2015, 10, e0116683. [Google Scholar] [CrossRef]
- Yasunaga, M.; Matsumura, Y. Role of SLC6A6 in promoting the survival and multidrug resistance of colorectal cancer. Sci. Rep. 2014, 4, 4852. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miR-LS (n = 27) | miR-MI (n = 30) | miR-HS (n = 31) | p Value | ||
|---|---|---|---|---|---|
| Villamil et al., 2012 Subtypes | Low Stroma | 22 | 7 | 6 | 0.000 χ2 |
| Immunoglobul | 2 | 6 | 4 | ||
| High Stroma | 1 | 4 | 17 | ||
| Mucinous-MSI | 2 | 9 | 3 | ||
| Unclassified | 0 | 4 | 1 | ||
| RF | CMS1 | 1 | 7 | 2 | 0.000 χ2 |
| CMS2 | 15 | 9 | 10 | ||
| CMS3 | 4 | 0 | 0 | ||
| CMS4 | 0 | 3 | 13 | ||
| NA | 7 | 11 | 6 | ||
| SSP | CMS1 | 2 | 8 | 3 | 0.001 χ2 |
| CMS2 | 21 | 16 | 11 | ||
| CMS3 | 1 | 0 | 0 | ||
| CMS4 | 0 | 3 | 12 | ||
| NA | 3 | 3 | 5 | ||
| Microsatellite | MSS | 24 | 24 | 31 | 0.036 χ2 |
| MSI | 3 | 6 | 0 | ||
| Histologic type | Conventional | 26 | 24 | 28 | 0.144 χ2 |
| Mucinous | 1 | 6 | 3 | ||
| BRAF | WT | 27 | 25 | 28 | 0.091 χ2 |
| Mut | 0 | 5 | 3 | ||
| FF Stroma | Range | (5–28) | (5–40) | (8–65) | 0.000 KW |
| Median | 7.5 | 13.75 | 22.5 | ||
| FFPE Stroma | Range | (5–20) | (5–40) | (5–60) | 0.004 KW |
| Median | 10 | 10 | 20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paz-Cabezas, M.; Calvo-López, T.; Romera-Lopez, A.; Tabas-Madrid, D.; Ogando, J.; Fernández-Aceñero, M.-J.; Sastre, J.; Pascual-Montano, A.; Mañes, S.; Díaz-Rubio, E.; et al. Molecular Classification of Colorectal Cancer by microRNA Profiling: Correlation with the Consensus Molecular Subtypes (CMS) and Validation of miR-30b Targets. Cancers 2022, 14, 5175. https://doi.org/10.3390/cancers14215175
Paz-Cabezas M, Calvo-López T, Romera-Lopez A, Tabas-Madrid D, Ogando J, Fernández-Aceñero M-J, Sastre J, Pascual-Montano A, Mañes S, Díaz-Rubio E, et al. Molecular Classification of Colorectal Cancer by microRNA Profiling: Correlation with the Consensus Molecular Subtypes (CMS) and Validation of miR-30b Targets. Cancers. 2022; 14(21):5175. https://doi.org/10.3390/cancers14215175
Chicago/Turabian StylePaz-Cabezas, Mateo, Tania Calvo-López, Alejandro Romera-Lopez, Daniel Tabas-Madrid, Jesus Ogando, María-Jesús Fernández-Aceñero, Javier Sastre, Alberto Pascual-Montano, Santos Mañes, Eduardo Díaz-Rubio, and et al. 2022. "Molecular Classification of Colorectal Cancer by microRNA Profiling: Correlation with the Consensus Molecular Subtypes (CMS) and Validation of miR-30b Targets" Cancers 14, no. 21: 5175. https://doi.org/10.3390/cancers14215175
APA StylePaz-Cabezas, M., Calvo-López, T., Romera-Lopez, A., Tabas-Madrid, D., Ogando, J., Fernández-Aceñero, M.-J., Sastre, J., Pascual-Montano, A., Mañes, S., Díaz-Rubio, E., & Perez-Villamil, B. (2022). Molecular Classification of Colorectal Cancer by microRNA Profiling: Correlation with the Consensus Molecular Subtypes (CMS) and Validation of miR-30b Targets. Cancers, 14(21), 5175. https://doi.org/10.3390/cancers14215175