



Association between Dysfunction of the Nucleolar Stress Response and Multidrug Resistance in Pediatric Acute Lymphoblastic Leukemia

 , , , and
, , , and 
Abstract
Simple Summary
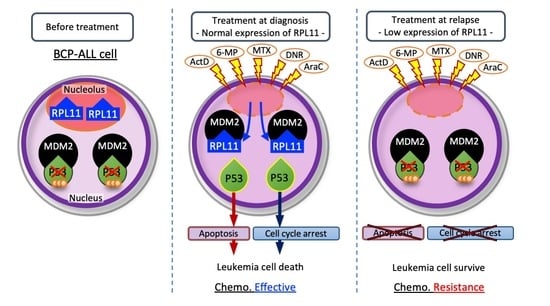
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Patients and Samples
2.2. Cell Culture and Reagents
2.3. Quantitative Real-Time Polymerase Chain Reaction (PCR)
2.4. Immunoblotting Analysis
2.5. Transfection with siRNA
2.6. Drug Resistance and Apoptosis
2.7. Immunoprecipitation
2.8. Immunofluorescence
2.9. Statistical Analyses
3. Results
3.1. The Nucleolar Stress Response as an Anti-Leukemia Mechanism
3.2. Sensitivity to 6-Mercaptopurine, Methotrexate, Daunorubicin, and Cytarabine
3.3. P53 Pathway Activation via RPL11
3.4. Suppression of 45S pre-rRNA mRNA Expression Followed by Disassembly of the Nucleolar and Protein Binding
3.5. RPL11 mRNA Expression in Leukemia Cells of Patients
3.6. Resistance to 6-Mercaptopurine, Methotrexate, Daunorubicin, and Cytarabine
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Levine, A.J.; Oren, M. The First 30 Years of P53: Growing ever More Complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Jin, A.; Itahana, K.; O’Keefe, K.; Zhang, Y. Inhibition of HDM2 and Activation of P53 by Ribosomal Protein L23. Mol. Cell Biol. 2004, 24, 7669–7680. [Google Scholar] [CrossRef] [PubMed]
- Lohrum, M.A.E.; Ludwig, R.L.; Kubbutat, M.H.G.; Hanlon, M.; Vousden, K.H. Regulation of HDM2 Activity by the Ribosomal Protein L11. Cancer Cell 2003, 3, 577–587. [Google Scholar] [CrossRef]
- Dai, M.S.; Lu, H. Inhibition of MDM2-Mediated P53 Ubiquitination and Degradation by Ribosomal Protein L5. J. Biol. Chem. 2004, 279, 44475–44482. [Google Scholar] [CrossRef]
- Suzuki, A.; Kogo, R.; Kawahara, K.; Sasaki, M.; Nishio, M.; Maehama, T.; Sasaki, T.; Mimori, K.; Mori, M. A New PICTure of Nucleolar Stress. Cancer Sci. 2012, 103, 632–637. [Google Scholar] [CrossRef]
- Sasaki, M.; Kawahara, K.; Nishio, M.; Mimori, K.; Kogo, R.; Hamada, K.; Itoh, B.; Wang, J.; Komatsu, Y.; Yang, Y.R.; et al. Regulation of the MDM2-P53 Pathway and Tumor Growth by PICT1 via Nucleolar RPL11. Nat. Med. 2011, 17, 944–951. [Google Scholar] [CrossRef]
- Uchi, R.; Kogo, R.; Kawahara, K.; Sudo, T.; Yokobori, T.; Eguchi, H.; Sugimachi, K.; Maehama, T.; Mori, M.; Suzuki, A.; et al. PICT1 Regulates TP53 via RPL11 and Is Involved in Gastric Cancer Progression. Br. J. Cancer 2013, 109, 2199–2206. [Google Scholar] [CrossRef]
- Kawahata, T.; Kawahara, K.; Shimokawa, M.; Sakiyama, A.; Shiraishi, T.; Minami, K.; Yamamoto, M.; Shinsato, Y.; Arima, K.; Hamada, T.; et al. Involvement of Ribosomal Protein L11 Expression in Sensitivity of Gastric Cancer against 5–FU. Oncol. Lett. 2020, 19, 2258–2264. [Google Scholar] [CrossRef]
- Okamura, K.; Takayama, K.; Kawahara, K.; Harada, T.; Nishio, M.; Otsubo, K.; Ijichi, K.; Kohno, M.; Iwama, E.; Fujii, A.; et al. PICT1 Expression Is a Poor Prognostic Factor in Non-Small Cell Lung Cancer. Oncoscience 2014, 25, 375–382. [Google Scholar] [CrossRef]
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 15, 1541–1552. [Google Scholar] [CrossRef]
- Toledo, F.; Wahl, G.M. Regulating the P53 Pathway: In Vitro Hypotheses, in Vivo Veritas. Nat. Rev. Cancer 2006, 6, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Petitjean, A.; Achatz, M.I.W.; Borresen-Dale, A.L.; Hainaut, P.; Olivier, M. TP53 Mutations in Human Cancers: Functional Selection and Impact on Cancer Prognosis and Outcomes. Oncogene 2007, 26, 2157–2165. [Google Scholar] [CrossRef]
- Ueno, H.; Yoshida, K.; Shiozawa, Y.; Nannya, Y.; Iijima-Yamashita, Y.; Kiyokawa, N.; Shiraishi, Y.; Chiba, K.; Tanaka, H.; Isobe, T.; et al. Landscape of Driver Mutations and Their Clinical Impacts in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia. Blood Adv. 2020, 4, 5165–5173. [Google Scholar] [CrossRef] [PubMed]
- Holleman, A.; Cheok, M.H.; den Boer, M.L.; Yang, W.; Veerman, A.J.P.; Kazemier, K.M.; Pei, D.; Cheng, C.; Pui, C.-H.; Relling, M.V.; et al. Gene-Expression Patterns in Drug-Resistant Acute Lymphoblastic Leukemia Cells and Response to Treatment. N. Engl. J. Med. 2004, 351, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wolf, G.W.; Bhat, K.; Jin, A.; Allio, T.; Burkhart, W.A.; Xiong, Y. Ribosomal Protein L11 Negatively Regulates Oncoprotein MDM2 and Mediates a P53-Dependent Ribosomal-Stress Checkpoint Pathway. Mol. Cell Biol. 2003, 23, 8902–8912. [Google Scholar] [CrossRef] [PubMed]
- Gilkes, D.M.; Chen, L.; Chen, J. MDMX Regulation of P53 Response to Ribosomal Stress. EMBO J. 2006, 25, 5614–5625. [Google Scholar] [CrossRef] [PubMed]
- Tzoneva, G.; Dieck, C.L.; Oshima, K.; Ambesi-Impiombato, A.; Sánchez-Martín, M.; Madubata, C.J.; Khiabanian, H.; Yu, J.; Waanders, E.; Iacobucci, I.; et al. Clonal Evolution Mechanisms in NT5C2 Mutant-Relapsed Acute Lymphoblastic Leukaemia. Nature 2018, 553, 511–514. [Google Scholar] [CrossRef]
- Hryniuk, W.M.; Henderson, J.F.; Tamaoki, T. Consequences of Methotrexate Inhibition of Purine Biosynthesis in L5178Y Cells. Cancer Res. 1975, 35, 1427–1432. [Google Scholar]
- Tewey, K.M.; Rowe, T.C.; Yang, L.; Halligan, B.D.; Liu, L.F. Adriamycin-Induced DNA Damage Mediated by Mammalian DNA Topoisomerase II. Science 1984, 26, 466–468. [Google Scholar] [CrossRef]
- di Marco, A.; Silvestrini, R.; di Marco, S.; Dasdia, T. Inhibiting Effect of the New Cytotoxic Antibiotic Daunomycin on Nucleic Acids and Mitotic Activity of HeLa Cells. J. Cell Biol. 1965, 27, 545–550. [Google Scholar] [CrossRef]
- Yang, F.; Teves, S.S.; Kemp, C.J.; Henikoff, S. Doxorubicin, DNA Torsion, and Chromatin Dynamics. Biochim. Biophys. Acta Rev. Cancer 2014, 1845, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Feinstein, E.; Canaani, E.; Weiner, L.M. Dependence of Nucleic Acid Degradation on in Situ Free-Radical Production by Adriamycin. Biochemistry 1993, 32, 13156–13161. [Google Scholar] [CrossRef] [PubMed]
- Bökkerink, J.P.M.; Stet, E.H.; de Abreu, R.A.; Damen, F.J.M.; Hulscher, T.W.; Bakker, M.A.H.; van Baal, J.A. 6-Mercaptopurine: Cytotoxicity and Biochemical Pharmacology in Human Malignant T-Lymphoblasts. Biochem. Pharmacol. 1993, 45, 1455–1463. [Google Scholar] [CrossRef]
- Holmberg, C.A.; Osbum, B.I.; Chuang, R.Y. Inhibition of Human Lymphoma dna-Dependent rna Polymerase Activity by 6-Mercaptopurine Ribonucleoside Triphosphate. Cancer Res. 1983, 43, 3655–3659. [Google Scholar]
- Burger, K.; Mühl, B.; Harasim, T.; Rohrmoser, M.; Malamoussi, A.; Orban, M.; Kellner, M.; Gruber-Eber, A.; Kremmer, E.; Hölzel, M.; et al. Chemotherapeutic Drugs Inhibit Ribosome Biogenesis at Various Levels. J. Biol. Chem. 2010, 285, 12416–12425. [Google Scholar] [CrossRef]
- Dano, K.; Frederiksen, S.; Hellung-Larsen, P. Inhibition of DNA and RNA Synthesis by Daunorubicin in Sensitive and Resistant Ehrlich Ascites Tumor Cells in vitro. Cancer Res. 1972, 32, 1307–1314. [Google Scholar]
- Meriwether, W.D.; Bachur, N.R. Inhibition of DNA and RNA Metabolism by Daunorubicin and Adriamycin in L1210 Mouse Leukemia. Cancer Res. 1972, 32, 1137–1142. [Google Scholar]
- Jones, R.E.; Moscona, A.A. Effects of Cytosine Arabinoside on Differential Gene Expression in Embryonic Neural Retina. II. Immunochemical Studies on the Accumulation of Glutamine Synthetase. J. Cell Biol. 1977, 74, 30–42. [Google Scholar] [CrossRef]
- Zdunski, D.; Ilan, J. The Effect of Cytosine Arabinoside on the Synthesis of Rapidly Labeled RNA during DNA Replicating and Non-DNA Replicating Periods of the Cell Cycle. Cell Differ. 1980, 9, 181–191. [Google Scholar] [CrossRef]
- Chuang, R.Y.; Chuang, L.F. Inhibition of RNA Polymerase as a Possible Anti-Leukaemic Action of Cytosine Arabinoside. Nature 1976, 260, 549–550. [Google Scholar] [CrossRef]
- Li, B.; Brady, S.W.; Ma, X.; Shen, S.; Zhang, Y.; Li, Y.; Szlachta, K.; Dong, L.; Liu, Y.; Yang, F.; et al. Therapy-Induced Mutations Drive the Genomic Landscape of Relapsed Acute Lymphoblastic Leukemia. Blood 2020, 135, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Gu, L.; Abshire, T.C.; Homans, A.; Billett, A.L.; Yeager, A.M.; Findley, H.W.; Findley, H. Incidence and Prognostic Significance of MDM2 Oncoprotein Overexpression in Relapsed Childhood Acute Lymphoblastic Leukemia. Leukemia 2000, 14, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Morishita, N.; Tsukahara, H.; Chayama, K.; Ishida, T.; Washio, K.; Miyamura, T.; Yamashita, N.; Oda, M.; Morishima, T. Activation of Akt Is Associated with Poor Prognosis and Chemotherapeutic Resistance in Pediatric B-Precursor Acute Lymphoblastic Leukemia. Pediatr. Blood Cancer 2012, 59, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.; Handgretinger, R.; Klingebiel, T.; Dopfer, R.; Schaich, M.; Ehninger, G.; Niethammer, D.; Gekeler, V. Expression of PKC Isozyme and MDR-Associated Genes in Primary and Relapsed State AML. Leukemia 1996, 10, 426–433. [Google Scholar]
- den Boer, M.L.; Pieters, R.; Kazemier, K.M.; Rottier, M.M.A.; Zwaan, C.M.; Kaspers, G.J.L.; Janka-Schaub, G.; Henze, G.; Creutzig, U.; Scheper, R.J.; et al. Relationship between Major Vault Protein/Lung Resistance Protein, Multidrug Resistance-Associated Protein, P-Glycoprotein Expression, and Drug Resistance in Childhood Leukemia. Blood 1998, 91, 2092–2098. [Google Scholar] [CrossRef]
- Olson, D.P.; Taylor, B.J.; La, M.; Sather, H.; Reaman, G.H.; Ivy, S.P. The Prognostic Significance of P-Glycoprotein, Multidrug Resistance-Related Protein 1 and Lung Resistance Protein in Pediatric Acute Lymphoblastic Leukemia: A Retrospective Study of 295 Newly Diagnosed Patients by the Children’s Oncology Group. Leuk. Lymphoma 2005, 46, 681–691. [Google Scholar] [CrossRef]
- Stam, R.W.; van den Heuvel-Eibrink, M.M.; den Boer, M.L.; Ebus, M.E.G.; Janka-Schaub, G.E.; Allen, J.D.; Pieters, R. Multidrug Resistance Genes in Infant Acute Lymphoblastic Leukemia: Ara-C Is Not a Substrate for the Breast Cancer Resistance Protein. Leukemia 2004, 18, 78–83. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Toyoda, Y.; Manabe, A.; Tsuchida, M.; Hanada, R.; Ikuta, K.; Okimoto, Y.; Ohara, A.; Ohkawa, Y.; Mori, T.; Ishimoto, K.; et al. Six Months of Maintenance Chemotherapy after Intensified Treatment for Acute Lymphoblastic Leukemia of Childhood. J. Clin. Oncol. 2000, 18, 1508–1516. [Google Scholar] [CrossRef] [PubMed]
- Gionfriddo, I.; Brunetti, L.; Mezzasoma, F.; Milano, F.; Cardinali, V.; Ranieri, R.; Venanzi, A.; Pierangeli, S.; Vetro, C.; Spinozzi, G.; et al. Dactinomycin Induces Complete Remission Associated with Nucleolar Stress Response in Relapsed/Refractory NPM1-Mutated AML. Leukemia 2021, 35, 2552–2562. [Google Scholar] [CrossRef]
- Chen, J.; Stark, L.A. Insights into the Relationship between Nucleolar Stress and the NF-ΚB Pathway. Trends Genet. 2019, 35, 768–780. [Google Scholar] [CrossRef]
- Pagliara, V.; Saide, A.; Mitidieri, E.; di Bianca, R.d.V.; Sorrentino, R.; Russo, G.; Russo, A. 5-FU Targets RpL3 to Induce Mitochondrial Apoptosis via Cystathionine-β-Synthase in Colon Cancer Cells Lacking P53. Oncotarget 2016, 7, 50333–50348. [Google Scholar] [CrossRef] [PubMed]
- Donati, G.; Montanaro, L.; Derenzini, M. Ribosome Biogenesis and Control of Cell Proliferation: P53 Is Not Alone. Cancer Res. 2012, 72, 1602–1607. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakagawa, S.; Kawahara, K.; Okamoto, Y.; Kodama, Y.; Nishikawa, T.; Kawano, Y.; Furukawa, T. Association between Dysfunction of the Nucleolar Stress Response and Multidrug Resistance in Pediatric Acute Lymphoblastic Leukemia. Cancers 2022, 14, 5127. https://doi.org/10.3390/cancers14205127
Nakagawa S, Kawahara K, Okamoto Y, Kodama Y, Nishikawa T, Kawano Y, Furukawa T. Association between Dysfunction of the Nucleolar Stress Response and Multidrug Resistance in Pediatric Acute Lymphoblastic Leukemia. Cancers. 2022; 14(20):5127. https://doi.org/10.3390/cancers14205127
Chicago/Turabian StyleNakagawa, Shunsuke, Kohichi Kawahara, Yasuhiro Okamoto, Yuichi Kodama, Takuro Nishikawa, Yoshifumi Kawano, and Tatsuhiko Furukawa. 2022. "Association between Dysfunction of the Nucleolar Stress Response and Multidrug Resistance in Pediatric Acute Lymphoblastic Leukemia" Cancers 14, no. 20: 5127. https://doi.org/10.3390/cancers14205127
APA StyleNakagawa, S., Kawahara, K., Okamoto, Y., Kodama, Y., Nishikawa, T., Kawano, Y., & Furukawa, T. (2022). Association between Dysfunction of the Nucleolar Stress Response and Multidrug Resistance in Pediatric Acute Lymphoblastic Leukemia. Cancers, 14(20), 5127. https://doi.org/10.3390/cancers14205127