

Resistance to Trastuzumab

{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
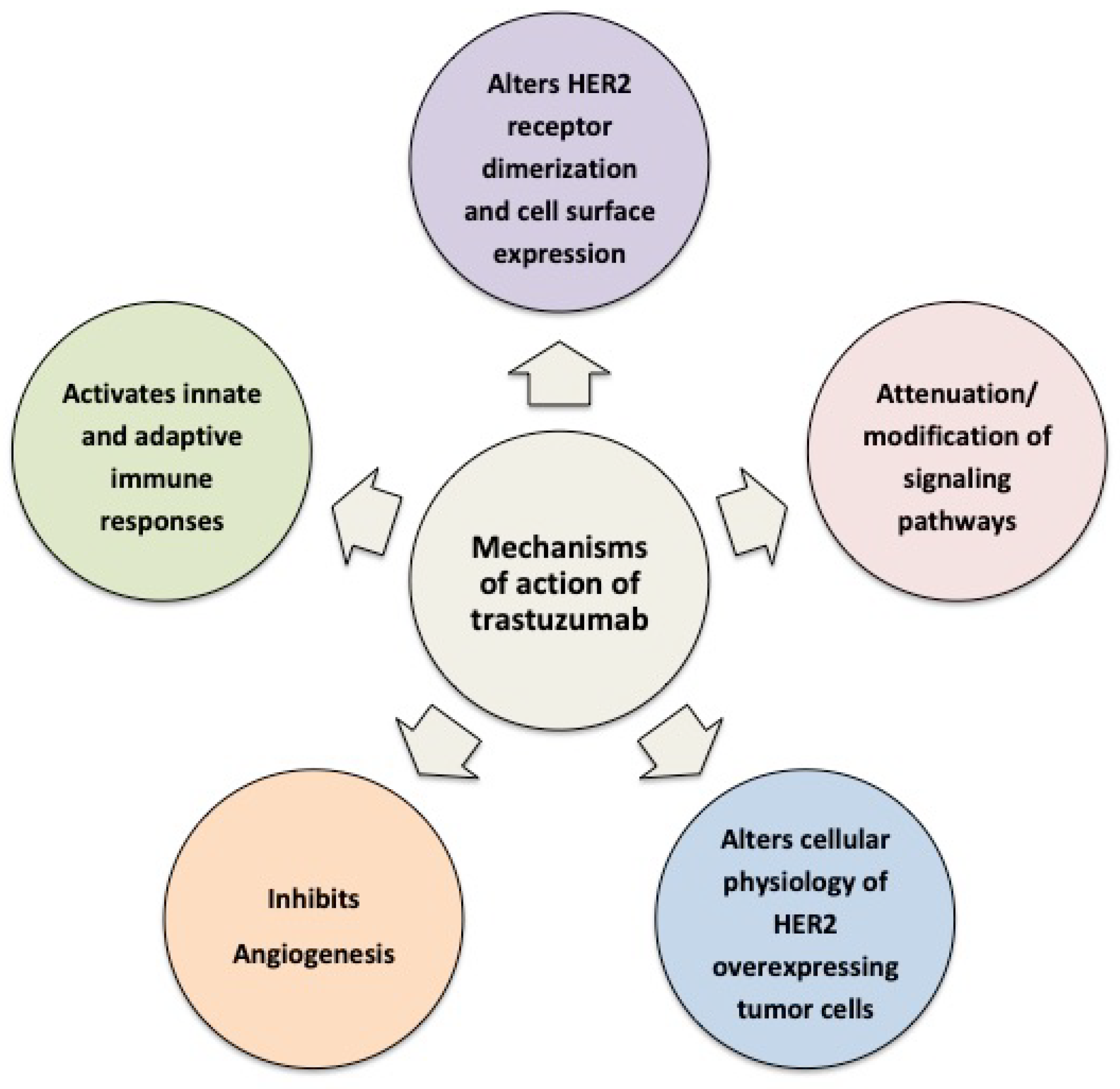
2. Cellular and Molecular Mechanisms of Action of Trastuzumab
2.1. Trastuzumab Alters HER2 Receptor Dimerization and Cell Surface Expression
2.2. Trastuzumab Attenuates/Modifies Proximal HER2 Signaling
2.3. Trastuzumab Alters the Cellular Physiology of HER2-Overexpressing Tumor Cells
2.4. Trastuzumab Limits Formation of Aggressive Tumor Microenvironments
2.5. Trastuzumab Activates Innate and Adaptive Immune Responses
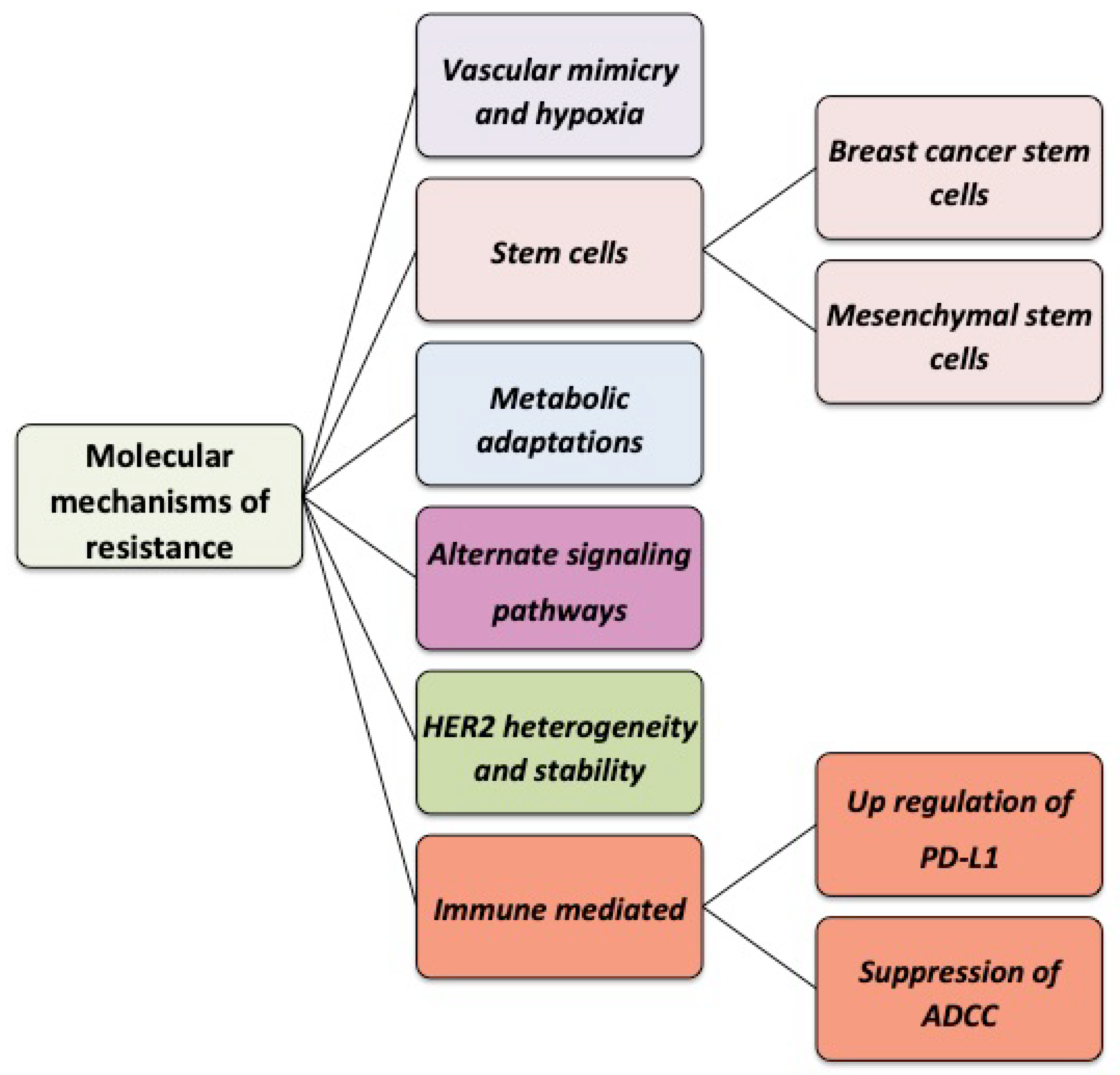
3. Molecular and Cellular Mechanisms of Resistance to Trastuzumab
3.1. Vascular Mimicry and Hypoxia
3.2. The Role of Breast Cancer Stem Cells (BCSCs) in Trastuzumab Resistance
3.3. Trastuzumab Resistance Is Associated with Metabolic Changes
3.4. Activation of Alternative Signaling Pathways
3.5. HER2 Molecular Variants Contribute to Therapeutic Resistance
3.6. HER2 Expression Heterogeneity, Stability, and Molecular Complexing in Tumors Limits Responsiveness to Trastuzumab
3.7. Immune-Mediated Mechanisms of Resistance
4. Resistance to Trastuzumab Drug Conjugates
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- DeSantis, C.E.; Ma, J.; Gaudet, M.M.; Newman, L.A.; Miller, K.D.; Goding Sauer, A.; Jemal, A.; Siegel, R.L. Breast cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Rexer, B.N.; Arteaga, C.L. Intrinsic and acquired resistance to HER2-targeted therapies in HER2 gene-amplified breast cancer: Mechanisms and clinical implications. Crit. Rev. Oncog. 2012, 17, 1–16. [Google Scholar] [CrossRef]
- Kovacs, E.; Zorn, J.A.; Huang, Y.; Barros, T.; Kuriyan, J. A structural perspective on the regulation of the epidermal growth factor receptor. Annu. Rev. Biochem. 2015, 84, 739–764. [Google Scholar] [CrossRef]
- Lu, Y.; Zi, X.; Pollak, M. Molecular mechanisms underlying IGF-I-induced attenuation of the growth-inhibitory activity of trastuzumab (Herceptin) on SKBR3 breast cancer cells. Int. J. Cancer 2004, 108, 334–341. [Google Scholar] [CrossRef]
- Maadi, H.; Soheilifar, M.H.; Choi, W.S.; Moshtaghian, A.; Wang, Z. Trastuzumab Mechanism of Action; 20 Years of Research to Unravel a Dilemma. Cancers 2021, 13, 3540. [Google Scholar] [CrossRef]
- Diermeier, S.; Horvath, G.; Knuechel-Clarke, R.; Hofstaedter, F.; Szollosi, J.; Brockhoff, G. Epidermal growth factor receptor coexpression modulates susceptibility to Herceptin in HER2/neu overexpressing breast cancer cells via specific erbB-receptor interaction and activation. Exp. Cell Res. 2005, 304, 604–619. [Google Scholar] [CrossRef]
- Diwanji, D.; Trenker, R.; Thaker, T.M.; Wang, F.; Agard, D.A.; Verba, K.A.; Jura, N. Structures of the HER2-HER3-NRG1beta complex reveal a dynamic dimer interface. Nature 2021, 600, 339–343. [Google Scholar] [CrossRef]
- Junttila, T.T.; Akita, R.W.; Parsons, K.; Fields, C.; Lewis Phillips, G.D.; Friedman, L.S.; Sampath, D.; Sliwkowski, M.X. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell 2009, 15, 429–440. [Google Scholar] [CrossRef]
- Gaborit, N.; Larbouret, C.; Vallaghe, J.; Peyrusson, F.; Bascoul-Mollevi, C.; Crapez, E.; Azria, D.; Chardes, T.; Poul, M.A.; Mathis, G.; et al. Time-resolved fluorescence resonance energy transfer (TR-FRET) to analyze the disruption of EGFR/HER2 dimers: A new method to evaluate the efficiency of targeted therapy using monoclonal antibodies. J. Biol. Chem. 2011, 286, 11337–11345. [Google Scholar] [CrossRef]
- Zhao, J.; Mohan, N.; Nussinov, R.; Ma, B.; Wu, W.J. Trastuzumab Blocks the Receiver Function of HER2 Leading to the Population Shifts of HER2-Containing Homodimers and Heterodimers. Antibodies 2021, 10, 7. [Google Scholar] [CrossRef]
- Lin, Y.Z.; Clinton, G.M. A soluble protein related to the HER-2 proto-oncogene product is released from human breast carcinoma cells. Oncogene 1991, 6, 639–643. [Google Scholar] [PubMed]
- Pupa, S.M.; Menard, S.; Morelli, D.; Pozzi, B.; De Palo, G.; Colnaghi, M.I. The extracellular domain of the c-erbB-2 oncoprotein is released from tumor cells by proteolytic cleavage. Oncogene 1993, 8, 2917–2923. [Google Scholar] [PubMed]
- Zabrecky, J.R.; Lam, T.; Mckenzie, S.J.; Carney, W. The Extracellular Domain of P185/Neu Is Released from the Surface of Human Breast-Carcinoma Cells, Sk-Br-3. J. Biol. Chem. 1991, 266, 1716–1720. [Google Scholar] [CrossRef]
- Liu, P.C.; Liu, X.; Li, Y.; Covington, M.; Wynn, R.; Huber, R.; Hillman, M.; Yang, G.; Ellis, D.; Marando, C.; et al. Identification of ADAM10 as a major source of HER2 ectodomain sheddase activity in HER2 overexpressing breast cancer cells. Cancer Biol. 2006, 5, 657–664. [Google Scholar] [CrossRef]
- Liu, D.; Zha, L.; Liu, Y.; Zhao, X.; Xu, X.; Liu, S.; Ma, W.; Zheng, J.; Shi, M. beta2-AR activation promotes cleavage and nuclear translocation of Her2 and metastatic potential of cancer cells. Cancer Sci. 2020, 111, 4417–4428. [Google Scholar] [CrossRef]
- Codony-Servat, J.; Albanell, J.; Lopez-Talavera, J.C.; Arribas, J.; Baselga, J. Cleavage of the HER2 ectodomain is a pervanadate-activable process that is inhibited by the tissue inhibitor of metalloproteases-1 in breast cancer cells. Cancer Res. 1999, 59, 1196–1201. [Google Scholar]
- Molina, M.A.; Saez, R.; Ramsey, E.E.; Garcia-Barchino, M.J.; Rojo, F.; Evans, A.J.; Albanell, J.; Keenan, E.J.; Lluch, A.; Garcia-Conde, J.; et al. NH(2)-terminal truncated HER-2 protein but not full-length receptor is associated with nodal metastasis in human breast cancer. Clin. Cancer Res. 2002, 8, 347–353. [Google Scholar]
- Christianson, T.A.; Doherty, J.K.; Lin, Y.J.; Ramsey, E.E.; Holmes, R.; Keenan, E.J.; Clinton, G.M. NH2-terminally truncated HER-2/neu protein: Relationship with shedding of the extracellular domain and with prognostic factors in breast cancer. Cancer Res. 1998, 58, 5123–5129. [Google Scholar]
- Zheng, H.; Zhong, A.; Xie, S.; Wang, Y.; Sun, J.; Zhang, J.; Tong, Y.; Chen, M.; Zhang, G.; Ma, Q.; et al. Elevated serum HER-2 predicts poor prognosis in breast cancer and is correlated to ADAM10 expression. Cancer Med. 2019, 8, 679–685. [Google Scholar] [CrossRef]
- Feldinger, K.; Generali, D.; Kramer-Marek, G.; Gijsen, M.; Ng, T.B.; Wong, J.H.; Strina, C.; Cappelletti, M.; Andreis, D.; Li, J.L.; et al. ADAM10 mediates trastuzumab resistance and is correlated with survival in HER2 positive breast cancer. Oncotarget 2014, 5, 6633–6646. [Google Scholar] [CrossRef] [PubMed]
- Pellikainen, J.M.; Ropponen, K.M.; Kataja, V.V.; Kellokoski, J.K.; Eskelinen, M.J.; Kosma, V.M. Expression of matrix metalloproteinase (MMP)-2 and MMP-9 in breast cancer with a special reference to activator protein-2, HER2, and prognosis. Clin. Cancer Res. 2004, 10, 7621–7628. [Google Scholar] [CrossRef]
- Zhang, B.; Cao, X.; Liu, Y.; Cao, W.; Zhang, F.; Zhang, S.; Li, H.; Ning, L.; Fu, L.; Niu, Y.; et al. Tumor-derived matrix metalloproteinase-13 (MMP-13) correlates with poor prognoses of invasive breast cancer. BMC Cancer 2008, 8, 83. [Google Scholar] [CrossRef]
- Chumsri, S.; Sperinde, J.; Liu, H.; Gligorov, J.; Spano, J.P.; Antoine, M.; Moreno Aspitia, A.; Tan, W.; Winslow, J.; Petropoulos, C.J.; et al. High p95HER2/HER2 Ratio Associated With Poor Outcome in Trastuzumab-Treated HER2-Positive Metastatic Breast Cancer NCCTG N0337 and NCCTG 98-32-52 (Alliance). Clin. Cancer Res. 2018, 24, 3053–3058. [Google Scholar] [CrossRef] [PubMed]
- Kostler, W.J.; Schwab, B.; Singer, C.F.; Neumann, R.; Rucklinger, E.; Brodowicz, T.; Tomek, S.; Niedermayr, M.; Hejna, M.; Steger, G.G.; et al. Monitoring of serum Her-2/neu predicts response and progression-free survival to trastuzumab-based treatment in patients with metastatic breast cancer. Clin. Cancer Res. 2004, 10, 1618–1624. [Google Scholar] [CrossRef] [PubMed]
- Fornier, M.N.; Seidman, A.D.; Schwartz, M.K.; Ghani, F.; Thiel, R.; Norton, L.; Hudis, C. Serum HER2 extracellular domain in metastatic breast cancer patients treated with weekly trastuzumab and paclitaxel: Association with HER2 status by immunohistochemistry and fluorescence in situ hybridization and with response rate. Ann. Oncol. 2005, 16, 234–239. [Google Scholar] [CrossRef]
- Ghedini, G.C.; Ciravolo, V.; Tortoreto, M.; Giuffre, S.; Bianchi, F.; Campiglio, M.; Mortarino, M.; Figini, M.; Coliva, A.; Carcangiu, M.L.; et al. Shed HER2 extracellular domain in HER2-mediated tumor growth and in trastuzumab susceptibility. J. Cell Physiol. 2010, 225, 256–265. [Google Scholar] [CrossRef]
- Valabrega, G.; Montemurro, F.; Aglietta, M. Trastuzumab: Mechanism of action, resistance and future perspectives in HER2-overexpressing breast cancer. Ann. Oncol. 2007, 18, 977–984. [Google Scholar] [CrossRef]
- Klapper, L.N.; Waterman, H.; Sela, M.; Yarden, Y. Tumor-inhibitory antibodies to HER-2/ErbB-2 may act by recruiting c-Cbl and enhancing ubiquitination of HER-2. Cancer Res. 2000, 60, 3384–3388. [Google Scholar]
- Hurwitz, E.; Stancovski, I.; Sela, M.; Yarden, Y. Suppression and promotion of tumor growth by monoclonal antibodies to ErbB-2 differentially correlate with cellular uptake. Proc. Natl. Acad. Sci. USA 1995, 92, 3353–3357. [Google Scholar] [CrossRef] [PubMed]
- Drebin, J.A.; Link, V.C.; Stern, D.F.; Weinberg, R.A.; Greene, M.I. Down-modulation of an oncogene protein product and reversion of the transformed phenotype by monoclonal antibodies. Cell 1985, 41, 697–706. [Google Scholar] [CrossRef]
- Jeong, J.; Shin, J.H.; Li, W.; Hong, J.Y.; Lim, J.; Hwang, J.Y.; Chung, J.J.; Yan, Q.; Liu, Y.; Choi, J.; et al. MAL2 mediates the formation of stable HER2 signaling complexes within lipid raft-rich membrane protrusions in breast cancer cells. Cell Rep. 2021, 37, 110160. [Google Scholar] [CrossRef] [PubMed]
- de Marco, M.C.; Martin-Belmonte, F.; Kremer, L.; Albar, J.P.; Correas, I.; Vaerman, J.P.; Marazuela, M.; Byrne, J.A.; Alonso, M.A. MAL2, a novel raft protein of the MAL family, is an essential component of the machinery for transcytosis in hepatoma HepG2 cells. J. Cell Biol. 2002, 159, 37–44. [Google Scholar] [CrossRef]
- Hommelgaard, A.M.; Lerdrup, M.; van Deurs, B. Association with membrane protrusions makes ErbB2 an internalization-resistant receptor. Mol. Biol. Cell 2004, 15, 1557–1567. [Google Scholar] [CrossRef]
- Wymant, J.M.; Sayers, E.J.; Muir, D.; Jones, A.T. Strategic Trastuzumab Mediated Crosslinking Driving Concomitant HER2 and HER3 Endocytosis and Degradation in Breast Cancer. J. Cancer 2020, 11, 3288–3302. [Google Scholar] [CrossRef]
- Buschenfelde, C.M.Z.; Hermann, C.; Schmidt, B.; Peschel, C.; Bernhard, H. Antihuman epidermal growth factor receptor 2 (HER2) monoclonal antibody trastuzumab enhances cytolytic activity of class I-restricted HER2-specific T lymphocytes against HER2-overexpressing tumor cells. Cancer Res. 2002, 62, 2244–2247. [Google Scholar]
- Valabrega, G.; Montemurro, F.; Sarotto, I.; Petrelli, A.; Rubini, P.; Tacchetti, C.; Aglietta, M.; Comoglio, P.M.; Giordano, S. TGFalpha expression impairs Trastuzumab-induced HER2 downregulation. Oncogene 2005, 24, 3002–3010. [Google Scholar] [CrossRef]
- Scaltriti, M.; Verma, C.; Guzman, M.; Jimenez, J.; Parra, J.L.; Pedersen, K.; Smith, D.J.; Landolfi, S.; Ramon y Cajal, S.; Arribas, J.; et al. Lapatinib, a HER2 tyrosine kinase inhibitor, induces stabilization and accumulation of HER2 and potentiates trastuzumab-dependent cell cytotoxicity. Oncogene 2009, 28, 803–814. [Google Scholar] [CrossRef]
- Cuello, M.; Ettenberg, S.A.; Clark, A.S.; Keane, M.M.; Posner, R.H.; Nau, M.M.; Dennis, P.A.; Lipkowitz, S. Down-regulation of the erbB-2 receptor by trastuzumab (herceptin) enhances tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in breast and ovarian cancer cell lines that overexpress erbB-2. Cancer Res. 2001, 61, 4892–4900. [Google Scholar] [PubMed]
- Longva, K.E.; Pedersen, N.M.; Haslekas, C.; Stang, E.; Madshus, I.H. Herceptin-induced inhibition of ErbB2 signaling involves reduced phosphorylation of Akt but not endocytic down-regulation of ErbB2. Int. J. Cancer 2005, 116, 359–367. [Google Scholar] [CrossRef]
- McLarty, K.; Cornelissen, B.; Cai, Z.; Scollard, D.A.; Costantini, D.L.; Done, S.J.; Reilly, R.M. Micro-SPECT/CT with 111In-DTPA-pertuzumab sensitively detects trastuzumab-mediated HER2 downregulation and tumor response in athymic mice bearing MDA-MB-361 human breast cancer xenografts. J. Nucl. Med. 2009, 50, 1340–1348. [Google Scholar] [CrossRef]
- Ram, S.; Kim, D.; Ober, R.J.; Ward, E.S. The level of HER2 expression is a predictor of antibody-HER2 trafficking behavior in cancer cells. Mabs Austin 2014, 6, 1211–1219. [Google Scholar] [CrossRef]
- Ignatov, T.; Gorbunow, F.; Eggemann, H.; Ortmann, O.; Ignatov, A. Loss of HER2 after HER2-targeted treatment. Breast Cancer Res. Treat 2019, 175, 401–408. [Google Scholar] [CrossRef]
- Barok, M.; Joensuu, H.; Isola, J. Trastuzumab emtansine: Mechanisms of action and drug resistance. Breast Cancer Res. 2014, 16, 209. [Google Scholar] [CrossRef]
- Nagata, Y.; Lan, K.H.; Zhou, X.; Tan, M.; Esteva, F.J.; Sahin, A.A.; Klos, K.S.; Li, P.; Monia, B.P.; Nguyen, N.T.; et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 2004, 6, 117–127. [Google Scholar] [CrossRef]
- Yokoyama, D.; Hisamori, S.; Deguchi, Y.; Nishigori, T.; Okabe, H.; Kanaya, S.; Manaka, D.; Kadokawa, Y.; Hata, H.; Minamiguchi, S.; et al. PTEN is a predictive biomarker of trastuzumab resistance and prognostic factor in HER2-overexpressing gastroesophageal adenocarcinoma. Sci. Rep. UK 2021, 11, 9013. [Google Scholar] [CrossRef]
- Kim, C.; Lee, C.K.; Chon, H.J.; Kim, J.H.; Park, H.S.; Heo, S.J.; Kim, H.J.; Kim, T.S.; Kwon, W.S.; Chung, H.C.; et al. PTEN loss and level of HER2 amplification is associated with trastuzumab resistance and prognosis in HER2-positive gastric cancer. Oncotarget 2017, 8, 113494–113501. [Google Scholar] [CrossRef]
- Deguchi, Y.; Okabe, H.; Oshima, N.; Hisamori, S.; Minamiguchi, S.; Muto, M.; Sakai, Y. PTEN loss is associated with a poor response to trastuzumab in HER2-overexpressing gastroesophageal adenocarcinoma. Gastric Cancer 2017, 20, 416–427. [Google Scholar] [CrossRef]
- Loibl, S.; Darb-Esfahani, S.; Huober, J.; Klimowicz, A.; Furlanetto, J.; Lederer, B.; Hartmann, A.; Eidtmann, H.; Pfitzner, B.; Fasching, P.A.; et al. Integrated Analysis of PTEN and p4EBP1 Protein Expression as Predictors for pCR in HER2-Positive Breast Cancer. Clin. Cancer Res. 2016, 22, 2675–2683. [Google Scholar] [CrossRef]
- Esteva, F.J.; Guo, H.; Zhang, S.; Santa-Maria, C.; Stone, S.; Lanchbury, J.S.; Sahin, A.A.; Hortobagyi, G.N.; Yu, D. PTEN, PIK3CA, p-AKT, and p-p70S6K status: Association with trastuzumab response and survival in patients with HER2-positive metastatic breast cancer. Am. J. Pathol. 2010, 177, 1647–1656. [Google Scholar] [CrossRef]
- Ghosh, R.; Narasanna, A.; Wang, S.E.; Liu, S.; Chakrabarty, A.; Balko, J.M.; Gonzalez-Angulo, A.M.; Mills, G.B.; Penuel, E.; Winslow, J.; et al. Trastuzumab has preferential activity against breast cancers driven by HER2 homodimers. Cancer Res. 2011, 71, 1871–1882. [Google Scholar] [CrossRef]
- Mohsin, S.K.; Weiss, H.L.; Gutierrez, M.C.; Chamness, G.C.; Schiff, R.; Digiovanna, M.P.; Wang, C.X.; Hilsenbeck, S.G.; Osborne, C.K.; Allred, D.C.; et al. Neoadjuvant trastuzumab induces apoptosis in primary breast cancers. J. Clin. Oncol. 2005, 23, 2460–2468. [Google Scholar] [CrossRef]
- Campbell, M.R.; Ruiz-Saenz, A.; Zhang, Y.; Peterson, E.; Steri, V.; Oeffinger, J.; Sampang, M.; Jura, N.; Moasser, M.M. Extensive conformational and physical plasticity protects HER2-HER3 tumorigenic signaling. Cell Rep. 2022, 38, 110285. [Google Scholar] [CrossRef]
- Spector, N.L.; Blackwell, K.L. Understanding the mechanisms behind trastuzumab therapy for human epidermal growth factor receptor 2-positive breast cancer. J. Clin. Oncol. 2009, 27, 5838–5847. [Google Scholar] [CrossRef]
- Fiszman, G.L.; Jasnis, M.A. Molecular Mechanisms of Trastuzumab Resistance in HER2 Overexpressing Breast Cancer. Int. J. Breast Cancer 2011, 2011, 352182. [Google Scholar] [CrossRef]
- Yakes, F.M.; Chinratanalab, W.; Ritter, C.A.; King, W.; Seelig, S.; Arteaga, C.L. Herceptin-induced inhibition of phosphatidylinositol-3 kinase and Akt Is required for antibody-mediated effects on p27, cyclin D1, and antitumor action. Cancer Res. 2002, 62, 4132–4141. [Google Scholar]
- Pereira, P.M.R.; Sharma, S.K.; Carter, L.M.; Edwards, K.J.; Pourat, J.; Ragupathi, A.; Janjigian, Y.Y.; Durack, J.C.; Lewis, J.S. Caveolin-1 mediates cellular distribution of HER2 and affects trastuzumab binding and therapeutic efficacy. Nat. Commun. 2018, 9, 5137. [Google Scholar] [CrossRef]
- Dokmanovic, M.; Wu, Y.; Shen, Y.; Chen, J.; Hirsch, D.S.; Wu, W.J. Trastuzumab-induced recruitment of Csk-homologous kinase (CHK) to ErbB2 receptor is associated with ErbB2-Y1248 phosphorylation and ErbB2 degradation to mediate cell growth inhibition. Cancer Biol. 2014, 15, 1029–1041. [Google Scholar] [CrossRef]
- Lane, H.A.; Beuvink, I.; Motoyama, A.B.; Daly, J.M.; Neve, R.M.; Hynes, N.E. ErbB2 potentiates breast tumor proliferation through modulation of p27(Kip1)-Cdk2 complex formation: Receptor overexpression does not determine growth dependency. Mol. Cell Biol. 2000, 20, 3210–3223. [Google Scholar] [CrossRef]
- Le, X.F.; Claret, F.X.; Lammayot, A.; Tian, L.; Deshpande, D.; LaPushin, R.; Tari, A.M.; Bast, R.C., Jr. The role of cyclin-dependent kinase inhibitor p27Kip1 in anti-HER2 antibody-induced G1 cell cycle arrest and tumor growth inhibition. J. Biol. Chem. 2003, 278, 23441–23450. [Google Scholar] [CrossRef]
- Filipits, M.; Dafni, U.; Gnant, M.; Polydoropoulou, V.; Hills, M.; Kiermaier, A.; de Azambuja, E.; Larsimont, D.; Rojo, F.; Viale, G.; et al. Association of p27 and Cyclin D1 Expression and Benefit from Adjuvant Trastuzumab Treatment in HER2-Positive Early Breast Cancer: A TransHERA Study. Clin. Cancer Res. 2018, 24, 3079–3086. [Google Scholar] [CrossRef]
- Ichikawa, T.; Sato, F.; Terasawa, K.; Tsuchiya, S.; Toi, M.; Tsujimoto, G.; Shimizu, K. Trastuzumab produces therapeutic actions by upregulating miR-26a and miR-30b in breast cancer cells. PLoS ONE 2012, 7, e31422. [Google Scholar] [CrossRef]
- Papadakis, E.; Robson, N.; Yeomans, A.; Bailey, S.; Laversin, S.; Beers, S.; Sayan, A.E.; Ashton-Key, M.; Schwaiger, S.; Stuppner, H.; et al. A combination of trastuzumab and BAG-1 inhibition synergistically targets HER2 positive breast cancer cells. Oncotarget 2016, 7, 18851–18864. [Google Scholar] [CrossRef]
- Carpenter, R.L.; Lo, H.W. Regulation of Apoptosis by HER2 in Breast Cancer. J. Carcinog. Mutagen. 2013, 2013 (Suppl. S7). [Google Scholar] [CrossRef]
- Henson, E.S.; Hu, X.; Gibson, S.B. Herceptin sensitizes ErbB2-overexpressing cells to apoptosis by reducing antiapoptotic Mcl-1 expression. Clin. Cancer Res. 2006, 12, 845–853. [Google Scholar] [CrossRef]
- Pietras, R.J.; Pegram, M.D.; Finn, R.S.; Maneval, D.A.; Slamon, D.J. Remission of human breast cancer xenografts on therapy with humanized monoclonal antibody to HER-2 receptor and DNA-reactive drugs. Oncogene 1998, 17, 2235–2249. [Google Scholar] [CrossRef]
- Pietras, R.J.; Fendly, B.M.; Chazin, V.R.; Pegram, M.D.; Howell, S.B.; Slamon, D.J. Antibody to HER-2/neu receptor blocks DNA repair after cisplatin in human breast and ovarian cancer cells. Oncogene 1994, 9, 1829–1838. [Google Scholar]
- Tagliabue, E.; Campiglio, M.; Pupa, S.M.; Menard, S.; Balsari, A. Activity and resistance of trastuzumab according to different clinical settings. Cancer Treat. Rev. 2012, 38, 212–217. [Google Scholar] [CrossRef]
- Boone, J.J.; Bhosle, J.; Tilby, M.J.; Hartley, J.A.; Hochhauser, D. Involvement of the HER2 pathway in repair of DNA damage produced by chemotherapeutic agents. Mol. Cancer 2009, 8, 3015–3023. [Google Scholar] [CrossRef]
- Pietras, R.J.; Poen, J.C.; Gallardo, D.; Wongvipat, P.N.; Lee, H.J.; Slamon, D.J. Monoclonal antibody to HER-2/neureceptor modulates repair of radiation-induced DNA damage and enhances radiosensitivity of human breast cancer cells overexpressing this oncogene. Cancer Res. 1999, 59, 1347–1355. [Google Scholar] [PubMed]
- Chakraborty, A.K.; Mehra, R.; Digiovanna, M.P. Co-targeting ER and HER family receptors induces apoptosis in HER2-normal or overexpressing breast cancer models. AntiCancer Res. 2015, 35, 1243–1250. [Google Scholar] [PubMed]
- Laughner, E.; Taghavi, P.; Chiles, K.; Mahon, P.C.; Semenza, G.L. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: Novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol. Cell Biol. 2001, 21, 3995–4004. [Google Scholar] [CrossRef] [PubMed]
- Yen, L.; You, X.L.; Al Moustafa, A.E.; Batist, G.; Hynes, N.E.; Mader, S.; Meloche, S.; Alaoui-Jamali, M.A. Heregulin selectively upregulates vascular endothelial growth factor secretion in cancer cells and stimulates angiogenesis. Oncogene 2000, 19, 3460–3469. [Google Scholar] [CrossRef] [PubMed]
- Yen, L.; Benlimame, N.; Nie, Z.R.; Xiao, D.; Wang, T.; Al Moustafa, A.E.; Esumi, H.; Milanini, J.; Hynes, N.E.; Pages, G.; et al. Differential regulation of tumor angiogenesis by distinct ErbB homo- and heterodimers. Mol. Biol. Cell 2002, 13, 4029–4044. [Google Scholar] [CrossRef] [PubMed]
- Viallard, C.; Larrivee, B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis 2017, 20, 409–426. [Google Scholar] [CrossRef]
- Sorace, A.G.; Quarles, C.C.; Whisenant, J.G.; Hanker, A.B.; McIntyre, J.O.; Sanchez, V.M.; Yankeelov, T.E. Trastuzumab improves tumor perfusion and vascular delivery of cytotoxic therapy in a murine model of HER2+ breast cancer: Preliminary results. Breast Cancer Res. Treat. 2016, 155, 273–284. [Google Scholar] [CrossRef]
- Izumi, Y.; Xu, L.; di Tomaso, E.; Fukumura, D.; Jain, R.K. Tumour biology: Herceptin acts as an anti-angiogenic cocktail. Nature 2002, 416, 279–280. [Google Scholar] [CrossRef]
- Musolino, A.; Naldi, N.; Bortesi, B.; Pezzuolo, D.; Capelletti, M.; Missale, G.; Laccabue, D.; Zerbini, A.; Camisa, R.; Bisagni, G.; et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J. Clin. Oncol. 2008, 26, 1789–1796. [Google Scholar] [CrossRef]
- Baerenwaldt, A.; Nimmerjahn, F. Immune regulation: FcgammaRIIB—Regulating the balance between protective and autoreactive immune responses. Immunol. Cell Biol. 2008, 86, 482–484. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Shimizu, C.; Hojo, T.; Akashi-Tanaka, S.; Kinoshita, T.; Yonemori, K.; Kouno, T.; Katsumata, N.; Ando, M.; Aogi, K.; et al. FcgammaR2A and 3A polymorphisms predict clinical outcome of trastuzumab in both neoadjuvant and metastatic settings in patients with HER2-positive breast cancer. Ann. Oncol. 2011, 22, 1302–1307. [Google Scholar] [CrossRef] [PubMed]
- Hurvitz, S.A.; Betting, D.J.; Stern, H.M.; Quinaux, E.; Stinson, J.; Seshagiri, S.; Zhao, Y.; Buyse, M.; Mackey, J.; Driga, A.; et al. Analysis of Fcgamma receptor IIIa and IIa polymorphisms: Lack of correlation with outcome in trastuzumab-treated breast cancer patients. Clin. Cancer Res. 2012, 18, 3478–3486. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Kim, J.H.; Im, S.A.; Kim, Y.J.; Han, H.S.; Kim, J.S.; Han, S.W.; Jeon, Y.K.; Oh, D.Y.; Han, W.; et al. ABCB1, FCGR2A, and FCGR3A polymorphisms in patients with HER2-positive metastatic breast cancer who were treated with first-line taxane plus trastuzumab chemotherapy. Oncology 2012, 83, 218–227. [Google Scholar] [CrossRef]
- Norton, N.; Olson, R.M.; Pegram, M.; Tenner, K.; Ballman, K.V.; Clynes, R.; Knutson, K.L.; Perez, E.A. Association studies of Fcgamma receptor polymorphisms with outcome in HER2+ breast cancer patients treated with trastuzumab in NCCTG (Alliance) Trial N9831. Cancer Immunol. Res. 2014, 2, 962–969. [Google Scholar] [CrossRef]
- Clynes, R.A.; Towers, T.L.; Presta, L.G.; Ravetch, J.V. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat. Med. 2000, 6, 443–446. [Google Scholar] [CrossRef]
- Prager, I.; Watzl, C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J. Leukoc. Biol. 2019, 105, 1319–1329. [Google Scholar] [CrossRef]
- Hubert, P.; Heitzmann, A.; Viel, S.; Nicolas, A.; Sastre-Garau, X.; Oppezzo, P.; Pritsch, O.; Osinaga, E.; Amigorena, S. Antibody-dependent cell cytotoxicity synapses form in mice during tumor-specific antibody immunotherapy. Cancer Res. 2011, 71, 5134–5143. [Google Scholar] [CrossRef]
- Wang, W.; Erbe, A.K.; Hank, J.A.; Morris, Z.S.; Sondel, P.M. NK Cell-Mediated Antibody-Dependent Cellular Cytotoxicity in Cancer Immunotherapy. Front. Immunol. 2015, 6, 368. [Google Scholar] [CrossRef]
- Arnould, L.; Gelly, M.; Penault-Llorca, F.; Benoit, L.; Bonnetain, F.; Migeon, C.; Cabaret, V.; Fermeaux, V.; Bertheau, P.; Garnier, J.; et al. Trastuzumab-based treatment of HER2-positive breast cancer: An antibody-dependent cellular cytotoxicity mechanism? Br. J. Cancer 2006, 94, 259–267. [Google Scholar] [CrossRef]
- Muraro, E.; Comaro, E.; Talamini, R.; Turchet, E.; Miolo, G.; Scalone, S.; Militello, L.; Lombardi, D.; Spazzapan, S.; Perin, T.; et al. Improved Natural Killer cell activity and retained anti-tumor CD8(+) T cell responses contribute to the induction of a pathological complete response in HER2-positive breast cancer patients undergoing neoadjuvant chemotherapy. J. Transl. Med. 2015, 13, 204. [Google Scholar] [CrossRef] [PubMed]
- Treffers, L.W.; van Houdt, M.; Bruggeman, C.W.; Heineke, M.H.; Zhao, X.W.; van der Heijden, J.; Nagelkerke, S.Q.; Verkuijlen, P.; Geissler, J.; Lissenberg-Thunnissen, S.; et al. FcgammaRIIIb Restricts Antibody-Dependent Destruction of Cancer Cells by Human Neutrophils. Front. Immunol. 2018, 9, 3124. [Google Scholar] [CrossRef] [PubMed]
- Grugan, K.D.; McCabe, F.L.; Kinder, M.; Greenplate, A.R.; Harman, B.C.; Ekert, J.E.; van Rooijen, N.; Anderson, G.M.; Nemeth, J.A.; Strohl, W.R.; et al. Tumor-associated macrophages promote invasion while retaining Fc-dependent anti-tumor function. J. Immunol. 2012, 189, 5457–5466. [Google Scholar] [CrossRef]
- Oflazoglu, E.; Stone, I.J.; Gordon, K.A.; Grewal, I.S.; van Rooijen, N.; Law, C.L.; Gerber, H.P. Macrophages contribute to the antitumor activity of the anti-CD30 antibody SGN-30. Blood 2007, 110, 4370–4372. [Google Scholar] [CrossRef]
- Minard-Colin, V.; Xiu, Y.; Poe, J.C.; Horikawa, M.; Magro, C.M.; Hamaguchi, Y.; Haas, K.M.; Tedder, T.F. Lymphoma depletion during CD20 immunotherapy in mice is mediated by macrophage FcgammaRI, FcgammaRIII, and FcgammaRIV. Blood 2008, 112, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Oflazoglu, E.; Stone, I.J.; Brown, L.; Gordon, K.A.; van Rooijen, N.; Jonas, M.; Law, C.L.; Grewal, I.S.; Gerber, H.P. Macrophages and Fc-receptor interactions contribute to the antitumour activities of the anti-CD40 antibody SGN-40. Br. J. Cancer 2009, 100, 113–117. [Google Scholar] [CrossRef]
- Shi, Y.; Fan, X.; Deng, H.; Brezski, R.J.; Rycyzyn, M.; Jordan, R.E.; Strohl, W.R.; Zou, Q.; Zhang, N.; An, Z. Trastuzumab triggers phagocytic killing of high HER2 cancer cells in vitro and in vivo by interaction with Fcgamma receptors on macrophages. J. Immunol. 2015, 194, 4379–4386. [Google Scholar] [CrossRef] [PubMed]
- Tsao, L.C.; Crosby, E.J.; Trotter, T.N.; Agarwal, P.; Hwang, B.J.; Acharya, C.; Shuptrine, C.W.; Wang, T.; Wei, J.; Yang, X.; et al. CD47 blockade augmentation of trastuzumab antitumor efficacy dependent on antibody-dependent cellular phagocytosis. JCI Insight 2019, 4, e131882. [Google Scholar] [CrossRef]
- Bettadapur, A.; Miller, H.W.; Ralston, K.S. Biting Off What Can Be Chewed: Trogocytosis in Health, Infection, and Disease. Infect. Immun. 2020, 88, e00930-19. [Google Scholar] [CrossRef]
- Velmurugan, R.; Challa, D.K.; Ram, S.; Ober, R.J.; Ward, E.S. Macrophage-Mediated Trogocytosis Leads to Death of Antibody-Opsonized Tumor Cells. Mol. Cancer Ther. 2016, 15, 1879–1889. [Google Scholar] [CrossRef]
- Honkanen, T.J.; Tikkanen, A.; Karihtala, P.; Makinen, M.; Vayrynen, J.P.; Koivunen, J.P. Prognostic and predictive role of tumour-associated macrophages in HER2 positive breast cancer. Sci. Rep. Uk 2019, 9, 10961. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, E.; Kataoka, T.R.; Hirata, M.; Kawaguchi, K.; Nishie, M.; Haga, H.; Toi, M. Trogocytosis-mediated expression of HER2 on immune cells may be associated with a pathological complete response to trastuzumab-based primary systemic therapy in HER2-overexpressing breast cancer patients. BMC Cancer 2015, 15, 39. [Google Scholar] [CrossRef] [PubMed]
- Pohlmann, P.R.; Mayer, I.A.; Mernaugh, R. Resistance to Trastuzumab in Breast Cancer. Clin. Cancer Res. 2009, 15, 7479–7491. [Google Scholar] [CrossRef]
- Tsao, L.C.; Crosby, E.J.; Trotter, T.N.; Wei, J.P.; Wang, T.; Yang, X.; Summers, A.N.; Lei, G.J.; Rabiola, C.A.; Chodosh, L.A.; et al. Trastuzumab/pertuzumab combination therapy stimulates antitumor responses through complement-dependent cytotoxicity and phagocytosis. JCI Insight 2022, 7, e155636. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.; Hershman, D.; Shah, N.; Suciu-Foca, N.; Petrylak, D.P.; Taub, R.; Vahdat, L.; Cheng, B.; Pegram, M.; Knutson, K.L.; et al. Augmented HER-2 specific immunity during treatment with trastuzumab and chemotherapy. Clin. Cancer Res. 2007, 13, 5133–5143. [Google Scholar] [CrossRef]
- Norton, N.; Fox, N.; McCarl, C.A.; Tenner, K.S.; Ballman, K.; Erskine, C.L.; Necela, B.M.; Northfelt, D.; Tan, W.W.; Calfa, C.; et al. Generation of HER2-specific antibody immunity during trastuzumab adjuvant therapy associates with reduced relapse in resected HER2 breast cancer. Breast Cancer Res. 2018, 20, 52. [Google Scholar] [CrossRef] [PubMed]
- Knutson, K.L.; Clynes, R.; Shreeder, B.; Yeramian, P.; Kemp, K.P.; Ballman, K.; Tenner, K.S.; Erskine, C.L.; Norton, N.; Northfelt, D.; et al. Improved Survival of HER2+ Breast Cancer Patients Treated with Trastuzumab and Chemotherapy Is Associated with Host Antibody Immunity against the HER2 Intracellular Domain. Cancer Res. 2016, 76, 3702–3710. [Google Scholar] [CrossRef]
- Catenacci, D.V.T.; Kang, Y.K.; Park, H.; Uronis, H.E.; Lee, K.W.; Ng, M.C.H.; Enzinger, P.C.; Park, S.H.; Gold, P.J.; Lacy, J.; et al. Margetuximab plus pembrolizumab in patients with previously treated, HER2-positive gastro-oesophageal adenocarcinoma (CP-MGAH22-05): A single-arm, phase 1b-2 trial. Lancet Oncol. 2020, 21, 1066–1076. [Google Scholar] [CrossRef]
- Gall, V.A.; Philips, A.V.; Qiao, N.; Clise-Dwyer, K.; Perakis, A.A.; Zhang, M.; Clifton, G.T.; Sukhumalchandra, P.; Ma, Q.; Reddy, S.M.; et al. Trastuzumab Increases HER2 Uptake and Cross-Presentation by Dendritic Cells. Cancer Res. 2017, 77, 5374–5383. [Google Scholar] [CrossRef]
- Kono, K.; Sato, E.; Naganuma, H.; Takahashi, A.; Mimura, K.; Nukui, H.; Fujii, H. Trastuzumab (Herceptin) enhances class I-restricted antigen presentation recognized by HER-2/neu-specific T cytotoxic lymphocytes. Clin. Cancer Res. 2004, 10, 2538–2544. [Google Scholar] [CrossRef]
- Park, S.; Jiang, Z.; Mortenson, E.D.; Deng, L.; Radkevich-Brown, O.; Yang, X.; Sattar, H.; Wang, Y.; Brown, N.K.; Greene, M.; et al. The therapeutic effect of anti-HER2/neu antibody depends on both innate and adaptive immunity. Cancer Cell 2010, 18, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Yeo, B.; Kotsori, K.; Mohammed, K.; Walsh, G.; Smith, I.E. Long-term outcome of HER2 positive metastatic breast cancer patients treated with first-line trastuzumab. Breast 2015, 24, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Hori, A.; Shimoda, M.; Naoi, Y.; Kagara, N.; Tanei, T.; Miyake, T.; Shimazu, K.; Kim, S.J.; Noguchi, S. Vasculogenic mimicry is associated with trastuzumab resistance of HER2-positive breast cancer. Breast Cancer Res. 2019, 21, 88. [Google Scholar] [CrossRef] [PubMed]
- Morales-Guadarrama, G.; Garcia-Becerra, R.; Mendez-Perez, E.A.; Garcia-Quiroz, J.; Avila, E.; Diaz, L. Vasculogenic Mimicry in Breast Cancer: Clinical Relevance and Drivers. Cells 2021, 10, 1758. [Google Scholar] [CrossRef]
- Gupta, P.B.; Onder, T.T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009, 138, 645–659. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Santisteban, M.; Reiman, J.M.; Asiedu, M.K.; Behrens, M.D.; Nassar, A.; Kalli, K.R.; Haluska, P.; Ingle, J.N.; Hartmann, L.C.; Manjili, M.H.; et al. Immune-induced epithelial to mesenchymal transition in vivo generates breast cancer stem cells. Cancer Res. 2009, 69, 2887–2895. [Google Scholar] [CrossRef]
- Phillips, T.M.; McBride, W.H.; Pajonk, F. The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. J. Natl. Cancer Inst. 2006, 98, 1777–1785. [Google Scholar] [CrossRef]
- Li, X.; Lewis, M.T.; Huang, J.; Gutierrez, C.; Osborne, C.K.; Wu, M.F.; Hilsenbeck, S.G.; Pavlick, A.; Zhang, X.; Chamness, G.C.; et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J. Natl. Cancer Inst. 2008, 100, 672–679. [Google Scholar] [CrossRef]
- Espinosa-Sanchez, A.; Suarez-Martinez, E.; Sanchez-Diaz, L.; Carnero, A. Therapeutic Targeting of Signaling Pathways Related to Cancer Stemness. Front. Oncol. 2020, 10, 1533. [Google Scholar] [CrossRef]
- Magnifico, A.; Albano, L.; Campaner, S.; Delia, D.; Castiglioni, F.; Gasparini, P.; Sozzi, G.; Fontanella, E.; Menard, S.; Tagliabue, E. Tumor-initiating cells of HER2-positive carcinoma cell lines express the highest oncoprotein levels and are sensitive to trastuzumab. Clin. Cancer Res. 2009, 15, 2010–2021. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, C.E.; Berardi, D.E.; Abrigo, M.; Todaro, L.B.; Bal de Kier Joffe, E.D.; Fiszman, G.L. Breast cancer stem cells are involved in Trastuzumab resistance through the HER2 modulation in 3D culture. J. Cell Biochem. 2018, 119, 1381–1391. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chu, J.; Feng, W.; Yang, M.; Zhang, Y.; Zhang, Y.; Qin, Y.; Xu, J.; Li, J.; Vasilatos, S.N.; et al. EPHA5 mediates trastuzumab resistance in HER2-positive breast cancers through regulating cancer stem cell-like properties. FASEB J. 2019, 33, 4851–4865. [Google Scholar] [CrossRef] [PubMed]
- Grudzien, P.; Lo, S.; Albain, K.S.; Robinson, P.; Rajan, P.; Strack, P.R.; Golde, T.E.; Miele, L.; Foreman, K.E. Inhibition of Notch signaling reduces the stem-like population of breast cancer cells and prevents mammosphere formation. AntiCancer Res. 2010, 30, 3853–3867. [Google Scholar] [CrossRef] [PubMed]
- Sansone, P.; Storci, G.; Giovannini, C.; Pandolfi, S.; Pianetti, S.; Taffurelli, M.; Santini, D.; Ceccarelli, C.; Chieco, P.; Bonafe, M. p66Shc/Notch-3 interplay controls self-renewal and hypoxia survival in human stem/progenitor cells of the mammary gland expanded in vitro as mammospheres. Stem. Cells 2007, 25, 807–815. [Google Scholar] [CrossRef]
- Osipo, C.; Patel, P.; Rizzo, P.; Clementz, A.G.; Hao, L.; Golde, T.E.; Miele, L. ErbB-2 inhibition activates Notch-1 and sensitizes breast cancer cells to a gamma-secretase inhibitor. Oncogene 2008, 27, 5019–5032. [Google Scholar] [CrossRef]
- Lui, G.Y.L.; Grandori, C.; Kemp, C.J. CDK12: An emerging therapeutic target for cancer. J. Clin. Pathol. 2018, 71, 957–962. [Google Scholar] [CrossRef]
- Li, H.; Wang, J.; Yi, Z.; Li, C.; Wang, H.; Zhang, J.; Wang, T.; Nan, P.; Lin, F.; Xu, D.; et al. CDK12 inhibition enhances sensitivity of HER2+ breast cancers to HER2-tyrosine kinase inhibitor via suppressing PI3K/AKT. Eur. J. Cancer 2021, 145, 92–108. [Google Scholar] [CrossRef]
- Choi, H.J.; Jin, S.; Cho, H.; Won, H.Y.; An, H.W.; Jeong, G.Y.; Park, Y.U.; Kim, H.Y.; Park, M.K.; Son, T.; et al. CDK12 drives breast tumor initiation and trastuzumab resistance via WNT and IRS1-ErbB-PI3K signaling. EMBO Rep. 2019, 20, e48058. [Google Scholar] [CrossRef]
- Lamb, R.; Ablett, M.P.; Spence, K.; Landberg, G.; Sims, A.H.; Clarke, R.B. Wnt pathway activity in breast cancer sub-types and stem-like cells. PLoS ONE 2013, 8, e67811. [Google Scholar] [CrossRef]
- Wu, Y.; Ginther, C.; Kim, J.; Mosher, N.; Chung, S.; Slamon, D.; Vadgama, J.V. Expression of Wnt3 activates Wnt/beta-catenin pathway and promotes EMT-like phenotype in trastuzumab-resistant HER2-overexpressing breast cancer cells. Mol. Cancer Res. 2012, 10, 1597–1606. [Google Scholar] [CrossRef]
- Wang, K.; Ma, Q.; Ren, Y.; He, J.; Zhang, Y.; Zhang, Y.; Chen, W. Geldanamycin destabilizes HER2 tyrosine kinase and suppresses Wnt/beta-catenin signaling in HER2 overexpressing human breast cancer cells. Oncol. Rep. 2007, 17, 89–96. [Google Scholar]
- Hartman, Z.C.; Yang, X.Y.; Glass, O.; Lei, G.; Osada, T.; Dave, S.S.; Morse, M.A.; Clay, T.M.; Lyerly, H.K. HER2 overexpression elicits a proinflammatory IL-6 autocrine signaling loop that is critical for tumorigenesis. Cancer Res. 2011, 71, 4380–4391. [Google Scholar] [CrossRef]
- Olayioye, M.A.; Beuvink, I.; Horsch, K.; Daly, J.M.; Hynes, N.E. ErbB receptor-induced activation of stat transcription factors is mediated by Src tyrosine kinases. J. Biol. Chem. 1999, 274, 17209–17218. [Google Scholar] [CrossRef]
- DeArmond, D.; Brattain, M.G.; Jessup, J.M.; Kreisberg, J.; Malik, S.; Zhao, S.; Freeman, J.W. Autocrine-mediated ErbB-2 kinase activation of STAT3 is required for growth factor independence of pancreatic cancer cell lines. Oncogene 2003, 22, 7781–7795. [Google Scholar] [CrossRef]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(-) stem cell-like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef]
- Wei, W.; Tweardy, D.J.; Zhang, M.; Zhang, X.; Landua, J.; Petrovic, I.; Bu, W.; Roarty, K.; Hilsenbeck, S.G.; Rosen, J.M.; et al. STAT3 signaling is activated preferentially in tumor-initiating cells in claudin-low models of human breast cancer. Stem Cells 2014, 32, 2571–2582. [Google Scholar] [CrossRef]
- Dogan, F.; Biray Avci, C. Correlation between telomerase and mTOR pathway in cancer stem cells. Gene 2018, 641, 235–239. [Google Scholar] [CrossRef]
- Goldblatt, E.M.; Erickson, P.A.; Gentry, E.R.; Gryaznov, S.M.; Herbert, B.S. Lipid-conjugated telomerase template antagonists sensitize resistant HER2-positive breast cancer cells to trastuzumab. Breast Cancer Res. Treat. 2009, 118, 21–32. [Google Scholar] [CrossRef]
- Koziel, J.E.; Herbert, B.S. The telomerase inhibitor imetelstat alone, and in combination with trastuzumab, decreases the cancer stem cell population and self-renewal of HER2+ breast cancer cells. Breast Cancer Res. Treat. 2015, 149, 607–618. [Google Scholar] [CrossRef]
- Ridge, S.M.; Sullivan, F.J.; Glynn, S.A. Mesenchymal stem cells: Key players in cancer progression. Mol. Cancer 2017, 16, 31. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Qu, H.; Han, M.; Ding, Y.; Xie, M.; Hu, J.; Chen, Y.; Dong, H. MSC-induced lncRNA AGAP2-AS1 promotes stemness and trastuzumab resistance through regulating CPT1 expression and fatty acid oxidation in breast cancer. Oncogene 2021, 40, 833–847. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, S.; Xu, A.M.; Yuan, X.; Huang, L.; Li, J. Overexpression of GSE1 Related to Trastuzumab Resistance in Gastric Cancer Cells. Biomed. Res. Int. 2021, 2021, 8834923. [Google Scholar] [CrossRef]
- Nami, B.; Wang, Z. HER2 in Breast Cancer Stemness: A Negative Feedback Loop towards Trastuzumab Resistance. Cancers 2017, 9, 40. [Google Scholar] [CrossRef]
- Qiu, Y.; Yang, L.; Liu, H.; Luo, X. Cancer stem cell-targeted therapeutic approaches for overcoming trastuzumab resistance in HER2-positive breast cancer. Stem Cells 2021, 39, 1125–1136. [Google Scholar] [CrossRef]
- Zhao, Y.H.; Zhou, M.; Liu, H.; Ding, Y.; Khong, H.T.; Yu, D.; Fodstad, O.; Tan, M. Upregulation of lactate dehydrogenase A by ErbB2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth. Oncogene 2009, 28, 3689–3701. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, H.; Liu, Z.; Ding, Y.; Ledoux, S.P.; Wilson, G.L.; Voellmy, R.; Lin, Y.; Lin, W.; Nahta, R.; et al. Overcoming trastuzumab resistance in breast cancer by targeting dysregulated glucose metabolism. Cancer Res. 2011, 71, 4585–4597. [Google Scholar] [CrossRef]
- Gu, L.; Waliany, S.; Kane, S.E. Darpp-32 and its truncated variant t-Darpp have antagonistic effects on breast cancer cell growth and herceptin resistance. PLoS ONE 2009, 4, e6220. [Google Scholar] [CrossRef]
- Lenz, G.; Hamilton, A.; Geng, S.; Hong, T.; Kalkum, M.; Momand, J.; Kane, S.E.; Huss, J.M. t-Darpp Activates IGF-1R Signaling to Regulate Glucose Metabolism in Trastuzumab-Resistant Breast Cancer Cells. Clin. Cancer Res. 2018, 24, 1216–1226. [Google Scholar] [CrossRef]
- Gale, M.; Li, Y.; Cao, J.; Liu, Z.Z.; Holmbeck, M.A.; Zhang, M.; Lang, S.M.; Wu, L.; Do Carmo, M.; Gupta, S.; et al. Acquired Resistance to HER2-Targeted Therapies Creates Vulnerability to ATP Synthase Inhibition. Cancer Res. 2020, 80, 524–535. [Google Scholar] [CrossRef]
- Wang, T.; Ma, F.; Qian, H.L. Defueling the cancer: ATP synthase as an emerging target in cancer therapy. Mol. Oncolytics 2021, 23, 82–95. [Google Scholar] [CrossRef]
- Abdel-Wahab, A.F.; Mahmoud, W.; Al-Harizy, R.M. Targeting glucose metabolism to suppress cancer progression: Prospective of anti-glycolytic cancer therapy. Pharm. Res. 2019, 150, 104511. [Google Scholar] [CrossRef]
- Menendez, J.A.; Vellon, L.; Mehmi, I.; Oza, B.P.; Ropero, S.; Colomer, R.; Lupu, R. Inhibition of fatty acid synthase (FAS) suppresses HER2/neu (erbB-2) oncogene overexpression in cancer cells. Proc. Natl. Acad. Sci. USA 2004, 101, 10715–10720. [Google Scholar] [CrossRef]
- Kumar-Sinha, C.; Ignatoski, K.W.; Lippman, M.E.; Ethier, S.P.; Chinnaiyan, A.M. Transcriptome analysis of HER2 reveals a molecular connection to fatty acid synthesis. Cancer Res. 2003, 63, 132–139. [Google Scholar]
- Vazquez-Martin, A.; Fernandez-Real, J.M.; Oliveras-Ferraros, C.; Navarrete, J.M.; Martin-Castillo, B.; Del Barco, S.; Brunet, J.; Menendez, J.A. Fatty acid synthase activity regulates HER2 extracellular domain shedding into the circulation of HER2-positive metastatic breast cancer patients. Int. J. Oncol. 2009, 35, 1369–1376. [Google Scholar] [CrossRef]
- Ferraro, G.B.; Ali, A.; Luengo, A.; Kodack, D.P.; Deik, A.; Abbott, K.L.; Bezwada, D.; Blanc, L.; Prideaux, B.; Jin, X.; et al. Fatty Acid Synthesis Is Required for Breast Cancer Brain Metastasis. Nat. Cancer 2021, 2, 414–428. [Google Scholar] [CrossRef]
- Cordero, A.; Kanojia, D.; Miska, J.; Panek, W.K.; Xiao, A.; Han, Y.; Bonamici, N.; Zhou, W.; Xiao, T.; Wu, M.; et al. FABP7 is a key metabolic regulator in HER2+ breast cancer brain metastasis. Oncogene 2019, 38, 6445–6460. [Google Scholar] [CrossRef]
- Feng, W.W.; Wilkins, O.; Bang, S.; Ung, M.; Li, J.; An, J.; Del Genio, C.; Canfield, K.; DiRenzo, J.; Wells, W.; et al. CD36-Mediated Metabolic Rewiring of Breast Cancer Cells Promotes Resistance to HER2-Targeted Therapies. Cell Rep. 2019, 29, 3405–3420 e3405. [Google Scholar] [CrossRef]
- Ligorio, F.; Di Cosimo, S.; Verderio, P.; Ciniselli, C.M.; Pizzamiglio, S.; Castagnoli, L.; Triulzi, T.; Tagliabue, E.; El-Abed, S.; Izquierdo, M.; et al. High CD36 expression predicts worse event free survival in HER2-positive breast cancer patients treated with neoadjuvant trastuzumab-based therapy: An exploratory analysis of the NeoALTTO study. Cancer Res. 2022, 82 (Suppl. S4). [Google Scholar] [CrossRef]
- Luque-Cabal, M.; Garcia-Teijido, P.; Fernandez-Perez, Y.; Sanchez-Lorenzo, L.; Palacio-Vazquez, I. Mechanisms Behind the Resistance to Trastuzumab in HER2-Amplified Breast Cancer and Strategies to Overcome It. Clin. Med. Insights Oncol. 2016, 10, 21–30. [Google Scholar] [CrossRef]
- Gallardo, A.; Lerma, E.; Escuin, D.; Tibau, A.; Munoz, J.; Ojeda, B.; Barnadas, A.; Adrover, E.; Sanchez-Tejada, L.; Giner, D.; et al. Increased signalling of EGFR and IGF1R, and deregulation of PTEN/PI3K/Akt pathway are related with trastuzumab resistance in HER2 breast carcinomas. Br. J. Cancer 2012, 106, 1367–1373. [Google Scholar] [CrossRef]
- Di Giovanna, M.P.; Stern, D.F.; Edgerton, S.M.; Whalen, S.G.; Moore, D., 2nd; Thor, A.D. Relationship of epidermal growth factor receptor expression to ErbB-2 signaling activity and prognosis in breast cancer patients. J. Clin. Oncol. 2005, 23, 1152–1160. [Google Scholar] [CrossRef] [PubMed]
- Nieto, Y.; Nawaz, F.; Jones, R.B.; Shpall, E.J.; Nawaz, S. Prognostic significance of overexpression and phosphorylation of epidermal growth factor receptor (EGFR) and the presence of truncated EGFRvIII in locoregionally advanced breast cancer. J. Clin. Oncol. 2007, 25, 4405–4413. [Google Scholar] [CrossRef] [PubMed]
- Motoyama, A.B.; Hynes, N.E.; Lane, H.A. The efficacy of ErbB receptor-targeted anticancer therapeutics is influenced by the availability of epidermal growth factor-related peptides. Cancer Res. 2002, 62, 3151–3158. [Google Scholar]
- Ueda, S.; Tsuda, H.; Sato, K.; Takeuchi, H.; Shigekawa, T.; Matsubara, O.; Hiraide, H.; Mochizuki, H. Alternative tyrosine phosphorylation of signaling kinases according to hormone receptor status in breast cancer overexpressing the insulin-like growth factor receptor type 1. Cancer Sci. 2006, 97, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Railo, M.J.; von Smitten, K.; Pekonen, F. The prognostic value of insulin-like growth factor-I in breast cancer patients. Results of a follow-up study on 126 patients. Eur. J. Cancer 1994, 30, 307–311. [Google Scholar] [CrossRef]
- Nahta, R.; Yuan, L.X.; Zhang, B.; Kobayashi, R.; Esteva, F.J. Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res. 2005, 65, 11118–11128. [Google Scholar] [CrossRef]
- Lu, Y.; Zi, X.; Zhao, Y.; Mascarenhas, D.; Pollak, M. Insulin-like growth factor-I receptor signaling and resistance to trastuzumab (Herceptin). J. Natl. Cancer Inst. 2001, 93, 1852–1857. [Google Scholar] [CrossRef]
- Shattuck, D.L.; Miller, J.K.; Carraway, K.L., 3rd; Sweeney, C. Met receptor contributes to trastuzumab resistance of Her2-overexpressing breast cancer cells. Cancer Res. 2008, 68, 1471–1477. [Google Scholar] [CrossRef]
- Zhuang, G.; Brantley-Sieders, D.M.; Vaught, D.; Yu, J.; Xie, L.; Wells, S.; Jackson, D.; Muraoka-Cook, R.; Arteaga, C.; Chen, J. Elevation of receptor tyrosine kinase EphA2 mediates resistance to trastuzumab therapy. Cancer Res. 2010, 70, 299–308. [Google Scholar] [CrossRef]
- Liang, K.; Esteva, F.J.; Albarracin, C.; Stemke-Hale, K.; Lu, Y.; Bianchini, G.; Yang, C.Y.; Li, Y.; Li, X.; Chen, C.T.; et al. Recombinant human erythropoietin antagonizes trastuzumab treatment of breast cancer cells via Jak2-mediated Src activation and PTEN inactivation. Cancer Cell 2010, 18, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Yang, Z.; Hu, M.; Liu, D.; Hu, Y.; Qian, L.; Zhang, W.; Chen, H.; Guo, L.; Yu, M.; et al. Catecholamine-Induced beta2-adrenergic receptor activation mediates desensitization of gastric cancer cells to trastuzumab by upregulating MUC4 expression. J. Immunol. 2013, 190, 5600–5608. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Yang, Z.; Wang, T.; Yang, Z.; Chen, H.; Hu, Y.; Hu, C.; Guo, L.; Deng, Q.; Liu, Y.; et al. beta2-AR signaling controls trastuzumab resistance-dependent pathway. Oncogene 2016, 35, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Morrison, G.; Gillihan, R.; Guo, J.; Ward, R.M.; Fu, X.; Botero, M.F.; Healy, N.A.; Hilsenbeck, S.G.; Phillips, G.L.; et al. Different mechanisms for resistance to trastuzumab versus lapatinib in HER2-positive breast cancers--role of estrogen receptor and HER2 reactivation. Breast Cancer Res. 2011, 13, R121. [Google Scholar] [CrossRef]
- Gianni, L.; Pienkowski, T.; Im, Y.H.; Roman, L.; Tseng, L.M.; Liu, M.C.; Lluch, A.; Staroslawska, E.; de la Haba-Rodriguez, J.; Im, S.A.; et al. Efficacy and safety of neoadjuvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early HER2-positive breast cancer (NeoSphere): A randomised multicentre, open-label, phase 2 trial. Lancet Oncol. 2012, 13, 25–32. [Google Scholar] [CrossRef]
- Kaufman, B.; Mackey, J.R.; Clemens, M.R.; Bapsy, P.P.; Vaid, A.; Wardley, A.; Tjulandin, S.; Jahn, M.; Lehle, M.; Feyereislova, A.; et al. Trastuzumab plus anastrozole versus anastrozole alone for the treatment of postmenopausal women with human epidermal growth factor receptor 2-positive, hormone receptor-positive metastatic breast cancer: Results from the randomized phase III TAnDEM study. J. Clin. Oncol. 2009, 27, 5529–5537. [Google Scholar] [CrossRef]
- Gianni, L.; Bisagni, G.; Colleoni, M.; Del Mastro, L.; Zamagni, C.; Mansutti, M.; Zambetti, M.; Frassoldati, A.; De Fato, R.; Valagussa, P.; et al. Neoadjuvant treatment with trastuzumab and pertuzumab plus palbociclib and fulvestrant in HER2-positive, ER-positive breast cancer (NA-PHER2): An exploratory, open-label, phase 2 study. Lancet Oncol. 2018, 19, 249–256. [Google Scholar] [CrossRef]
- Arribas, J.; Baselga, J.; Pedersen, K.; Parra-Palau, J.L. p95HER2 and breast cancer. Cancer Res. 2011, 71, 1515–1519. [Google Scholar] [CrossRef]
- Sperinde, J.; Jin, X.; Banerjee, J.; Penuel, E.; Saha, A.; Diedrich, G.; Huang, W.; Leitzel, K.; Weidler, J.; Ali, S.M.; et al. Quantitation of p95HER2 in paraffin sections by using a p95-specific antibody and correlation with outcome in a cohort of trastuzumab-treated breast cancer patients. Clin. Cancer Res. 2010, 16, 4226–4235. [Google Scholar] [CrossRef]
- Castiglioni, F.; Tagliabue, E.; Campiglio, M.; Pupa, S.M.; Balsari, A.; Menard, S. Role of exon-16-deleted HER2 in breast carcinomas. Endocr. Relat. Cancer 2006, 13, 221–232. [Google Scholar] [CrossRef]
- Mitra, D.; Brumlik, M.J.; Okamgba, S.U.; Zhu, Y.; Duplessis, T.T.; Parvani, J.G.; Lesko, S.M.; Brogi, E.; Jones, F.E. An oncogenic isoform of HER2 associated with locally disseminated breast cancer and trastuzumab resistance. Mol. Cancer 2009, 8, 2152–2162. [Google Scholar] [CrossRef] [PubMed]
- Marchini, C.; Gabrielli, F.; Iezzi, M.; Zenobi, S.; Montani, M.; Pietrella, L.; Kalogris, C.; Rossini, A.; Ciravolo, V.; Castagnoli, L.; et al. The human splice variant Delta16HER2 induces rapid tumor onset in a reporter transgenic mouse. PLoS ONE 2011, 6, e18727. [Google Scholar] [CrossRef] [PubMed]
- Alajati, A.; Sausgruber, N.; Aceto, N.; Duss, S.; Sarret, S.; Voshol, H.; Bonenfant, D.; Bentires-Alj, M. Mammary tumor formation and metastasis evoked by a HER2 splice variant. Cancer Res. 2013, 73, 5320–5327. [Google Scholar] [CrossRef] [PubMed]
- Castagnoli, L.; Ladomery, M.; Tagliabue, E.; Pupa, S.M. The d16HER2 Splice Variant: A Friend or Foe of HER2-Positive Cancers? Cancers 2019, 11, 902. [Google Scholar] [CrossRef] [PubMed]
- Turpin, J.; Ling, C.; Crosby, E.J.; Hartman, Z.C.; Simond, A.M.; Chodosh, L.A.; Rennhack, J.P.; Andrechek, E.R.; Ozcelik, J.; Hallett, M.; et al. The ErbB2DeltaEx16 splice variant is a major oncogenic driver in breast cancer that promotes a pro-metastatic tumor microenvironment. Oncogene 2016, 35, 6053–6064. [Google Scholar] [CrossRef]
- Huynh, F.C.; Jones, F.E. MicroRNA-7 inhibits multiple oncogenic pathways to suppress HER2Delta16 mediated breast tumorigenesis and reverse trastuzumab resistance. PLoS ONE 2014, 9, e114419. [Google Scholar] [CrossRef]
- Ocana, A.; Amir, E.; Pandiella, A. HER2 heterogeneity and resistance to anti-HER2 antibody-drug conjugates. Breast Cancer Res. 2020, 22, 15. [Google Scholar] [CrossRef]
- Seol, H.; Lee, H.J.; Choi, Y.; Lee, H.E.; Kim, Y.J.; Kim, J.H.; Kang, E.; Kim, S.W.; Park, S.Y. Intratumoral heterogeneity of HER2 gene amplification in breast cancer: Its clinicopathological significance. Mod. Pathol. 2012, 25, 938–948. [Google Scholar] [CrossRef]
- Lee, H.J.; Seo, A.N.; Kim, E.J.; Jang, M.H.; Suh, K.J.; Ryu, H.S.; Kim, Y.J.; Kim, J.H.; Im, S.A.; Gong, G.; et al. HER2 heterogeneity affects trastuzumab responses and survival in patients with HER2-positive metastatic breast cancer. Am. J. Clin. Pathol. 2014, 142, 755–766. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Wu, Y.; Scaltriti, M.; Meric-Bernstam, F.; Hunt, K.K.; Dawood, S.; Esteva, F.J.; Buzdar, A.U.; Chen, H.; Eksambi, S.; et al. Loss of HER2 amplification following trastuzumab-based neoadjuvant systemic therapy and survival outcomes. Clin. Cancer Res. 2009, 15, 7381–7388. [Google Scholar] [CrossRef]
- Citri, A.; Kochupurakkal, B.S.; Yarden, Y. The achilles heel of ErbB-2/HER2: Regulation by the Hsp90 chaperone machine and potential for pharmacological intervention. Cell Cycle 2004, 3, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Wainberg, Z.A.; Anghel, A.; Rogers, A.M.; Desai, A.J.; Kalous, O.; Conklin, D.; Ayala, R.; O’Brien, N.A.; Quadt, C.; Akimov, M.; et al. Inhibition of HSP90 with AUY922 induces synergy in HER2-amplified trastuzumab-resistant breast and gastric cancer. Mol. Cancer 2013, 12, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Scaltriti, M.; Serra, V.; Normant, E.; Guzman, M.; Rodriguez, O.; Lim, A.R.; Slocum, K.L.; West, K.A.; Rodriguez, V.; Prudkin, L.; et al. Antitumor activity of the Hsp90 inhibitor IPI-504 in HER2-positive trastuzumab-resistant breast cancer. Mol. Cancer 2011, 10, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Kim, Y.J.; Park, S.; Park, M.; Farrand, L.; Nguyen, C.T.; Ann, J.; Nam, G.; Park, H.J.; Lee, J.; et al. A novel HSP90 inhibitor targeting the C-terminal domain attenuates trastuzumab resistance in HER2-positive breast cancer. Mol. Cancer 2020, 19, 161. [Google Scholar] [CrossRef] [PubMed]
- Chandarlapaty, S.; Scaltriti, M.; Angelini, P.; Ye, Q.; Guzman, M.; Hudis, C.A.; Norton, L.; Solit, D.B.; Arribas, J.; Baselga, J.; et al. Inhibitors of HSP90 block p95-HER2 signaling in Trastuzumab-resistant tumors and suppress their growth. Oncogene 2010, 29, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Stopeck, A.T.; Gordon, M.S.; Mendelson, D.; Solit, D.B.; Bagatell, R.; Ma, W.; Wheler, J.; Rosen, N.; Norton, L.; et al. Combination of trastuzumab and tanespimycin (17-AAG, KOS-953) is safe and active in trastuzumab-refractory HER-2 overexpressing breast cancer: A phase I dose-escalation study. J. Clin. Oncol. 2007, 25, 5410–5417. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Stopeck, A.; Linden, H.; Solit, D.; Chandarlapaty, S.; Rosen, N.; D’Andrea, G.; Dickler, M.; Moynahan, M.E.; Sugarman, S.; et al. HSP90 Inhibition Is Effective in Breast Cancer: A Phase II Trial of Tanespimycin (17-AAG) Plus Trastuzumab in Patients with HER2-Positive Metastatic Breast Cancer Progressing on Trastuzumab. Clin. Cancer Res. 2011, 17, 5132–5139. [Google Scholar] [CrossRef]
- Sauvage, F.; Messaoudi, S.; Fattal, E.; Barratt, G.; Vergnaud-Gauduchon, J. Heat shock proteins and cancer: How can nanomedicine be harnessed? J. Control. Release 2017, 248, 133–143. [Google Scholar] [CrossRef]
- Hamel, S.; Bouchard, A.; Ferrario, C.; Hassan, S.; Aguilar-Mahecha, A.; Buchanan, M.; Quenneville, L.; Miller, W.; Basik, M. Both t-Darpp and DARPP-32 can cause resistance to trastuzumab in breast cancer cells and are frequently expressed in primary breast cancers. Breast Cancer Res. Treat. 2010, 120, 47–57. [Google Scholar] [CrossRef]
- Belkhiri, A.; Dar, A.A.; Peng, D.F.; Razvi, M.H.; Rinehart, C.; Arteaga, C.L.; El-Rifai, W. Expression of t-DARPP mediates trastuzumab resistance in breast cancer cells. Clin. Cancer Res. 2008, 14, 4564–4571. [Google Scholar] [CrossRef]
- Workman, H.C.; Miller, J.K.; Ingalla, E.Q.; Kaur, R.P.; Yamamoto, D.I.; Beckett, L.A.; Young, L.J.T.; Cardiff, R.D.; Borowsky, A.D.; Carraway, K.L.; et al. The membrane mucin MUC4 is elevated in breast tumor lymph node metastases relative to matched primary tumors and confers aggressive properties to breast cancer cells. Breast Cancer Res. 2009, 11, R70. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.; Friedlander, E.; Tanner, M.; Kapanen, A.I.; Carraway, K.L.; Isola, J.; Jovin, T.M. Decreased accessibility and lack of activation of ErbB2 in JIMT-1, a herceptin-resistant, MUC4-expressing breast cancer cell line. Cancer Res. 2005, 65, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Barok, M.; Isola, J.; Palyi-Krekk, Z.; Nagy, P.; Juhasz, I.; Vereb, G.; Kauraniemi, P.; Kapanen, A.; Tanner, M.; Vereb, G.; et al. Trastuzumab causes antibody-dependent cellular cytotoxicity-mediated growth inhibition of submacroscopic JIMT-1 breast cancer xenografts despite intrinsic drug resistance. Mol. Cancer 2007, 6, 2065–2072. [Google Scholar] [CrossRef] [PubMed]
- Mercogliano, M.F.; De Martino, M.; Venturutti, L.; Rivas, M.A.; Proietti, C.J.; Inurrigarro, G.; Frahm, I.; Allemand, D.H.; Deza, E.G.; Ares, S.; et al. TNFalpha-Induced Mucin 4 Expression Elicits Trastuzumab Resistance in HER2-Positive Breast Cancer. Clin. Cancer Res. 2017, 23, 636–648. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhao, L.; Li, W.; Fan, K.; Qian, W.; Hou, S.; Wang, H.; Dai, J.; Wei, H.; Guo, Y. Feedback activation of STAT3 mediates trastuzumab resistance via upregulation of MUC1 and MUC4 expression. Oncotarget 2014, 5, 8317–8329. [Google Scholar] [CrossRef]
- Palyi-Krekk, Z.; Barok, M.; Isola, J.; Tammi, M.; Szollosi, J.; Nagy, P. Hyaluronan-induced masking of ErbB2 and CD44-enhanced trastuzumab internalisation in trastuzumab resistant breast cancer. Eur. J. Cancer 2007, 43, 2423–2433. [Google Scholar] [CrossRef]
- Chaganty, B.K.R.; Qiu, S.; Gest, A.; Lu, Y.; Ivan, C.; Calin, G.A.; Weiner, L.M.; Fan, Z. Trastuzumab upregulates PD-L1 as a potential mechanism of trastuzumab resistance through engagement of immune effector cells and stimulation of IFNgamma secretion. Cancer Lett. 2018, 430, 47–56. [Google Scholar] [CrossRef]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef]
- Yamashita, K.; Iwatsuki, M.; Yasuda-Yoshihara, N.; Morinaga, T.; Nakao, Y.; Harada, K.; Eto, K.; Kurashige, J.; Hiyoshi, Y.; Ishimoto, T.; et al. Trastuzumab upregulates programmed death ligand-1 expression through interaction with NK cells in gastric cancer. Br. J. Cancer 2021, 124, 595–603. [Google Scholar] [CrossRef]
- Loi, S.; Giobbie-Hurder, A.; Gombos, A.; Bachelot, T.; Hui, R.; Curigliano, G.; Campone, M.; Biganzoli, L.; Bonnefoi, H.; Jerusalem, G.; et al. Pembrolizumab plus trastuzumab in trastuzumab-resistant, advanced, HER2-positive breast cancer (PANACEA): A single-arm, multicentre, phase 1b-2 trial. Lancet Oncol. 2019, 20, 371–382. [Google Scholar] [CrossRef]
- Darwich, A.; Silvestri, A.; Benmebarek, M.R.; Mouries, J.; Cadilha, B.; Melacarne, A.; Morelli, L.; Supino, D.; Taleb, A.; Obeck, H.; et al. Paralysis of the cytotoxic granule machinery is a new cancer immune evasion mechanism mediated by chitinase 3-like-1. J. Immunother. Cancer 2021, 9, e003224. [Google Scholar] [CrossRef] [PubMed]
- Zazo, S.; Gonzalez-Alonso, P.; Martin-Aparicio, E.; Chamizo, C.; Luque, M.; Sanz-Alvarez, M.; Minguez, P.; Gomez-Lopez, G.; Cristobal, I.; Carames, C.; et al. Autocrine CCL5 Effect Mediates Trastuzumab Resistance by ERK Pathway Activation in HER2-Positive Breast Cancer. Mol. Cancer 2020, 19, 1696–1707. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.C.; Goedegebuure, P.S.; Belt, B.A.; Flaherty, B.; Sankpal, N.; Gillanders, W.E.; Eberlein, T.J.; Hsieh, C.S.; Linehan, D.C. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J. Immunol. 2009, 182, 1746–1755. [Google Scholar] [CrossRef]
- Force, J.; Howie, L.J.; Abbott, S.E.; Bentley, R.; Marcom, P.K.; Kimmick, G.; Westbrook, K.; Sammons, S.L.; Parks, M.; Topping, D.L.; et al. Early Stage HER2-Positive Breast Cancers Not Achieving a pCR from Neoadjuvant Trastuzumab- or Pertuzumab-Based Regimens Have an Immunosuppressive Phenotype. Clin. Breast Cancer 2018, 18, 410–417. [Google Scholar] [CrossRef]
- Mamidi, S.; Cinci, M.; Hasmann, M.; Fehring, V.; Kirschfink, M. Lipoplex mediated silencing of membrane regulators (CD46, CD55 and CD59) enhances complement-dependent anti-tumor activity of trastuzumab and pertuzumab. Mol. Oncol. 2013, 7, 580–594. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Teicher, B.A.; Hassan, R. Antibody-drug conjugates for cancer therapy. Lancet Oncol. 2016, 17, e254–e262. [Google Scholar] [CrossRef]
- von Minckwitz, G.; Huang, C.S.; Mano, M.S.; Loibl, S.; Mamounas, E.P.; Untch, M.; Wolmark, N.; Rastogi, P.; Schneeweiss, A.; Redondo, A.; et al. Trastuzumab Emtansine for Residual Invasive HER2-Positive Breast Cancer. N. Engl. J. Med. 2019, 380, 617–628. [Google Scholar] [CrossRef]
- Perez, E.A.; Barrios, C.; Eiermann, W.; Toi, M.; Im, Y.H.; Conte, P.; Martin, M.; Pienkowski, T.; Pivot, X.; Burris, H., 3rd; et al. Trastuzumab Emtansine with or without Pertuzumab Versus Trastuzumab Plus Taxane for Human Epidermal Growth Factor Receptor 2-Positive, Advanced Breast Cancer: Primary Results from the Phase III MARIANNE Study. J. Clin. Oncol. 2017, 35, 141–148. [Google Scholar] [CrossRef]
- Hunter, F.W.; Barker, H.R.; Lipert, B.; Rothe, F.; Gebhart, G.; Piccart-Gebhart, M.J.; Sotiriou, C.; Jamieson, S.M.F. Mechanisms of resistance to trastuzumab emtansine (T-DM1) in HER2-positive breast cancer. Br. J. Cancer 2020, 122, 603–612. [Google Scholar] [CrossRef]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.Y.; Dieras, V.; Guardino, E.; et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef]
- Loganzo, F.; Tan, X.; Sung, M.; Jin, G.; Myers, J.S.; Melamud, E.; Wang, F.; Diesl, V.; Follettie, M.T.; Musto, S.; et al. Tumor cells chronically treated with a trastuzumab-maytansinoid antibody-drug conjugate develop varied resistance mechanisms but respond to alternate treatments. Mol. Cancer 2015, 14, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Hurvitz, S.A.; Martin, M.; Symmans, W.F.; Jung, K.H.; Huang, C.S.; Thompson, A.M.; Harbeck, N.; Valero, V.; Stroyakovskiy, D.; Wildiers, H.; et al. Neoadjuvant trastuzumab, pertuzumab, and chemotherapy versus trastuzumab emtansine plus pertuzumab in patients with HER2-positive breast cancer (KRISTINE): A randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 2018, 19, 115–126. [Google Scholar] [CrossRef]
- Gebhart, G.; Lamberts, L.E.; Wimana, Z.; Garcia, C.; Emonts, P.; Ameye, L.; Stroobants, S.; Huizing, M.; Aftimos, P.; Tol, J.; et al. Molecular imaging as a tool to investigate heterogeneity of advanced HER2-positive breast cancer and to predict patient outcome under trastuzumab emtansine (T-DM1): The ZEPHIR trial. Ann. Oncol. 2016, 27, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Guo, J.; Shen, B.Q.; Yadav, D.B.; Sliwkowski, M.X.; Crocker, L.M.; Lacap, J.A.; Phillips, G.D.L. Mechanisms of Acquired Resistance to Trastuzumab Emtansine in Breast Cancer Cells. Mol. Cancer 2018, 17, 1441–1453. [Google Scholar] [CrossRef] [PubMed]
- Sabbaghi, M.; Gil-Gomez, G.; Guardia, C.; Servitja, S.; Arpi, O.; Garcia-Alonso, S.; Menendez, S.; Arumi-Uria, M.; Serrano, L.; Salido, M.; et al. Defective Cyclin B1 Induction in Trastuzumab-emtansine (T-DM1) Acquired Resistance in HER2-positive Breast Cancer. Clin. Cancer Res. 2017, 23, 7006–7019. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.B.; Wildiers, H.; Krop, I.E.; Smitt, M.; Yu, R.; Lysbet de Haas, S.; Gonzalez-Martin, A. Relationship between tumor biomarkers and efficacy in TH3RESA, a phase III study of trastuzumab emtansine (T-DM1) vs. treatment of physician’s choice in previously treated HER2-positive advanced breast cancer. Int. J. Cancer 2016, 139, 2336–2342. [Google Scholar] [CrossRef]
- Modi, S.; Jacot, W.; Yamashita, T.; Sohn, J.; Vidal, M.; Tokunaga, E.; Tsurutani, J.; Ueno, N.T.; Prat, A.; Chae, Y.S.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. N. Engl. J. Med. 2022, 387, 9–20. [Google Scholar] [CrossRef]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vivekanandhan, S.; Knutson, K.L. Resistance to Trastuzumab. Cancers 2022, 14, 5115. https://doi.org/10.3390/cancers14205115
Vivekanandhan S, Knutson KL. Resistance to Trastuzumab. Cancers. 2022; 14(20):5115. https://doi.org/10.3390/cancers14205115
Chicago/Turabian StyleVivekanandhan, Sneha, and Keith L. Knutson. 2022. "Resistance to Trastuzumab" Cancers 14, no. 20: 5115. https://doi.org/10.3390/cancers14205115
APA StyleVivekanandhan, S., & Knutson, K. L. (2022). Resistance to Trastuzumab. Cancers, 14(20), 5115. https://doi.org/10.3390/cancers14205115