
Decitabine-Mediated Upregulation of CSPG4 in Ovarian Carcinoma Cells Enables Targeting by CSPG4-Specific CAR-T Cells

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Reagents
2.2. Treatment with Decitabine
2.3. T-Cell Expansion
2.4. Flow Cytometry
2.5. mRNA-Electroporation
2.6. Cytokine Secretion
2.7. Cytotoxicity Assay
2.8. Statistical Analysis
3. Results
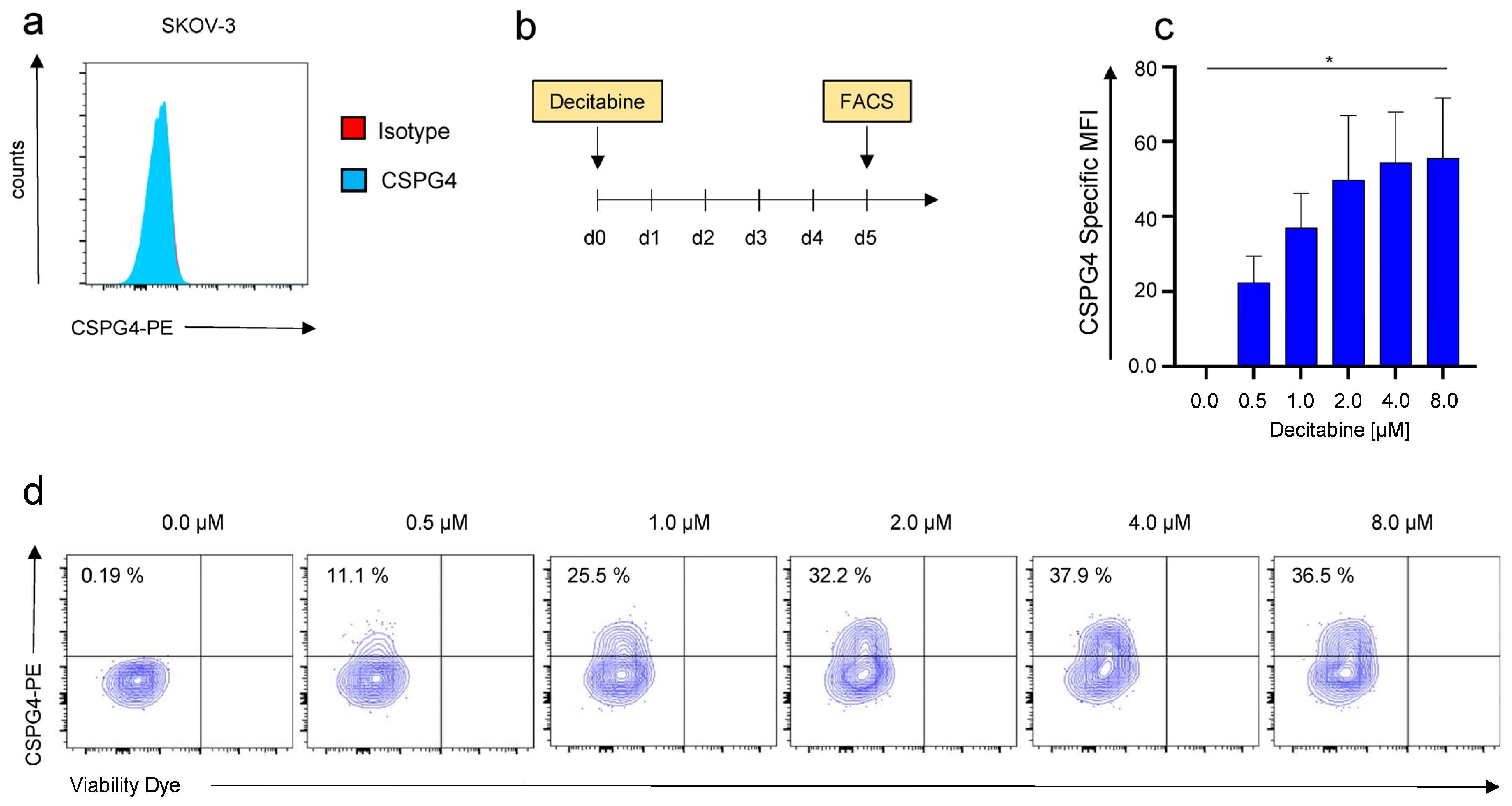
3.1. Decitabine Mediates Dose-Dependent Upregulation of CSPG4 on SKOV-3 Ovarian Cancer Cells
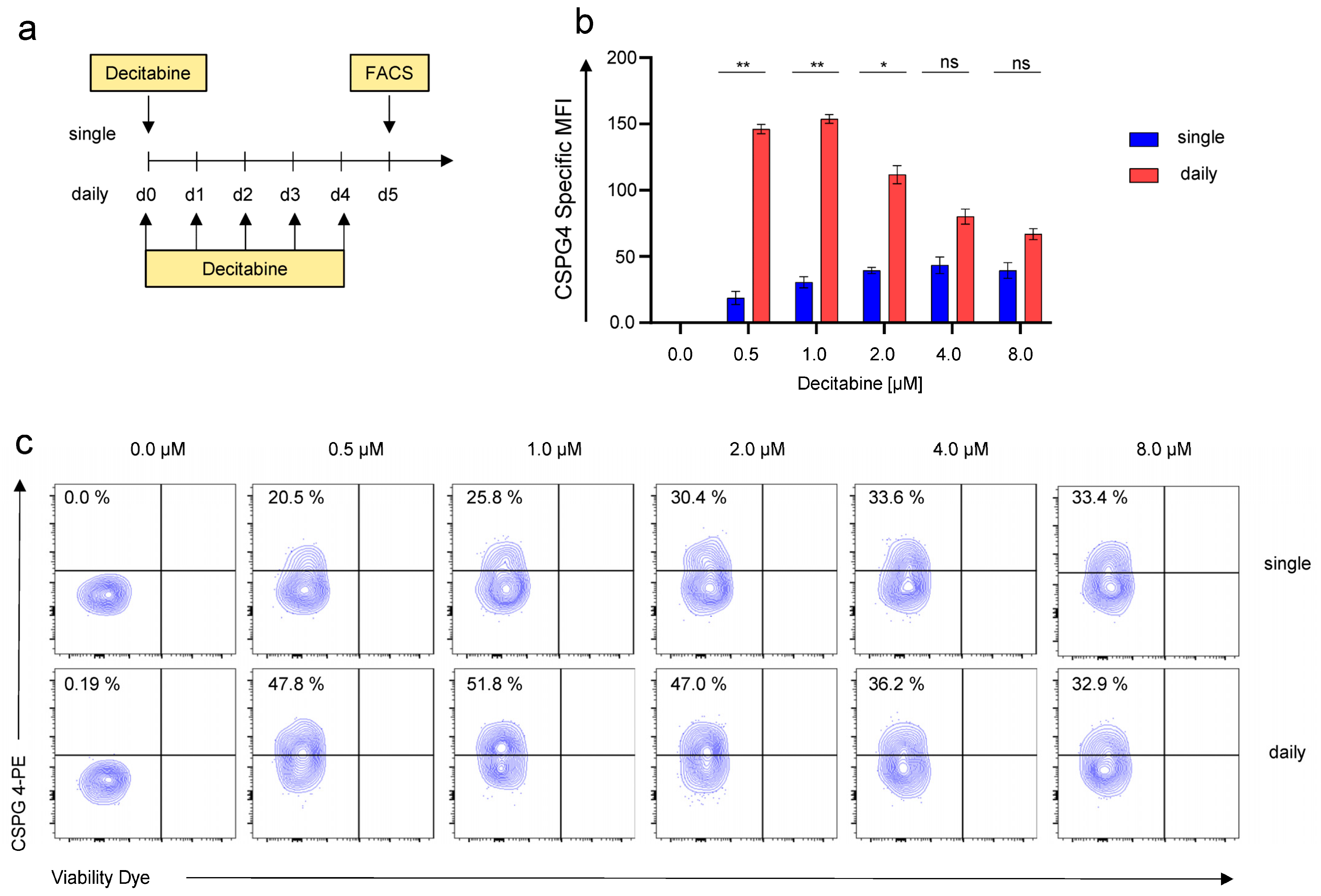
3.2. Daily Application of Decitabine Mediates Superior Upregulation of CSPG4 on SKOV-3 Ovarian Cancer Cells
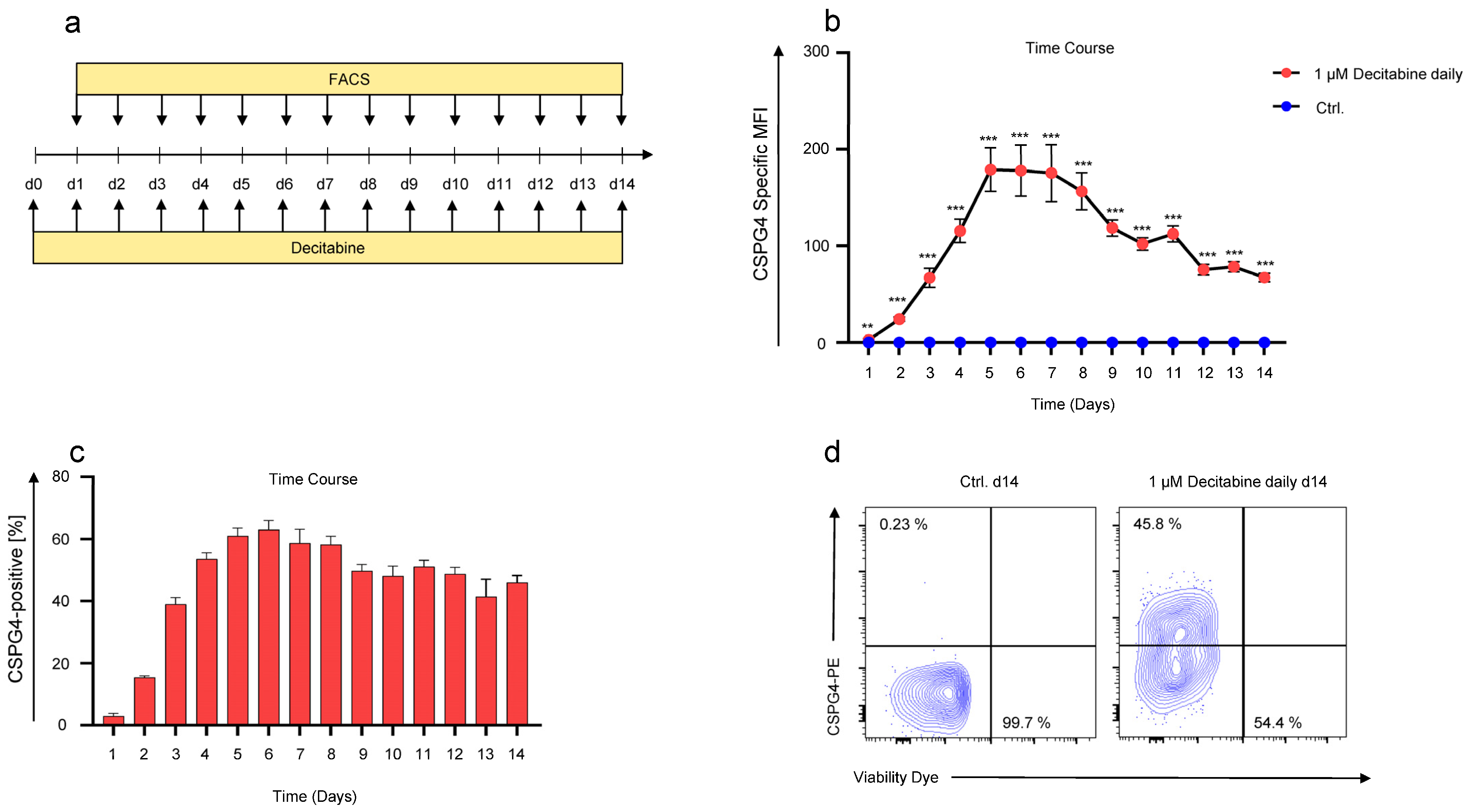
3.3. Kinetics of Decitabine-Mediated CSPG4-Upregulation on SKOV-3 Ovarian Cancer Cells
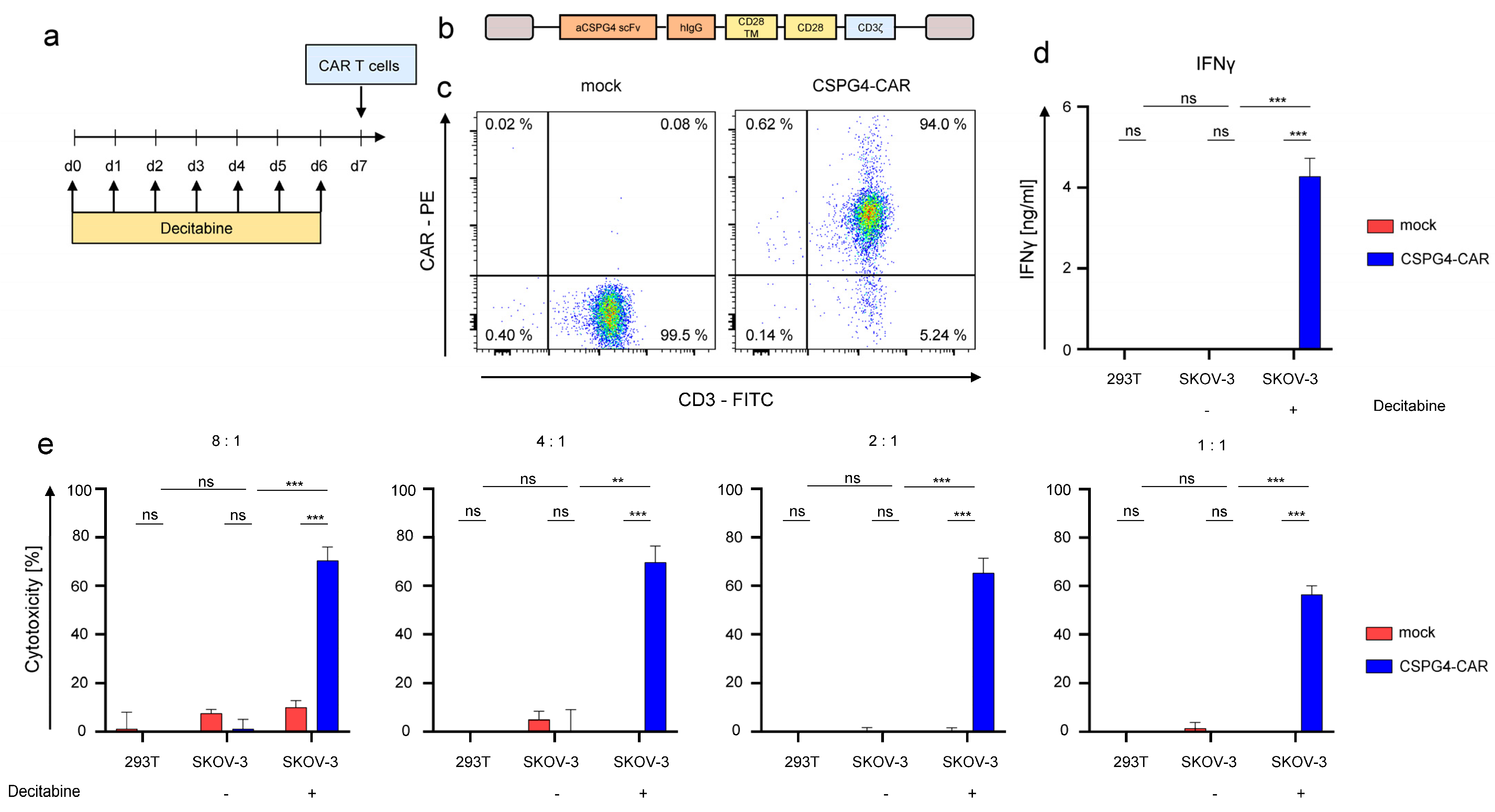
3.4. Decitabine-Mediated Upregulation of CSPG4 on SKOV-3 Ovarian Cancer Cells Mediates Recognition by CSPG4-Specific CAR-T Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Holzinger, A.; Abken, H. Treatment with Living Drugs: Pharmaceutical Aspects of CAR T Cells. Pharmacology 2022, 107, 446–463. [Google Scholar] [CrossRef] [PubMed]
- Schaft, N. The Landscape of CAR-T Cell Clinical Trials against Solid Tumors-A Comprehensive Overview. Cancers 2020, 12, 2567. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, S.; Brentjens, R.J. Tumors evading CARs-the chase is on. Nat. Med. 2018, 24, 1492–1493. [Google Scholar] [CrossRef] [PubMed]
- Harrer, D.C.; Dörrie, J.; Schaft, N. CSPG4 as Target for CAR-T-Cell Therapy of Various Tumor Entities-Merits and Challenges. Int. J. Mol. Sci. 2019, 20, 5942. [Google Scholar] [CrossRef] [PubMed]
- Chekenya, M.; Enger, P.O.; Thorsen, F.; Tysnes, B.B.; Al-Sarraj, S.; Read, T.A.; Furmanek, T.; Mahesparan, R.; Levine, J.M.; Butt, A.M.; et al. The glial precursor proteoglycan, NG2, is expressed on tumour neovasculature by vascular pericytes in human malignant brain tumours. Neuropathol. Appl. Neurobiol. 2002, 28, 367–380. [Google Scholar] [CrossRef]
- Ilieva, K.M.; Cheung, A.; Mele, S.; Chiaruttini, G.; Crescioli, S.; Griffin, M.; Nakamura, M.; Spicer, J.F.; Tsoka, S.; Lacy, K.E.; et al. Chondroitin Sulfate Proteoglycan 4 and Its Potential As an Antibody Immunotherapy Target across Different Tumor Types. Front. Immunol. 2017, 8, 1911. [Google Scholar] [CrossRef]
- Price, M.A.; Colvin Wanshura, L.E.; Yang, J.; Carlson, J.; Xiang, B.; Li, G.; Ferrone, S.; Dudek, A.Z.; Turley, E.A.; McCarthy, J.B. CSPG4, a potential therapeutic target, facilitates malignant progression of melanoma. Pigment Cell Melanoma Res. 2011, 24, 1148–1157. [Google Scholar] [CrossRef]
- Luo, W.; Wang, X.; Kageshita, T.; Wakasugi, S.; Karpf, A.R.; Ferrone, S. Regulation of high molecular weight-melanoma associated antigen (HMW-MAA) gene expression by promoter DNA methylation in human melanoma cells. Oncogene 2006, 25, 2873–2884. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Leick, M.B.; Silva, H.; Scarfò, I.; Larson, R.; Choi, B.D.; Bouffard, A.A.; Gallagher, K.; Schmidts, A.; Bailey, S.R.; Kann, M.C.; et al. Non-cleavable hinge enhances avidity and expansion of CAR-T cells for acute myeloid leukemia. Cancer Cell 2022, 40, 494–508.e5. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, S.; Braunstein, M.; Oza, A.M. Epithelial ovarian cancer: Evolution of management in the era of precision medicine. CA Cancer J. Clin. 2019, 69, 280–304. [Google Scholar] [CrossRef]
- Wiesinger, M.; März, J.; Kummer, M.; Schuler, G.; Dörrie, J.; Schuler-Thurner, B.; Schaft, N. Clinical-Scale Production of CAR-T Cells for the Treatment of Melanoma Patients by mRNA Transfection of a CSPG4-Specific CAR under Full GMP Compliance. Cancers 2019, 11, 1198. [Google Scholar] [CrossRef] [PubMed]
- Harrer, D.C.; Simon, B.; Fujii, S.-I.; Shimizu, K.; Uslu, U.; Schuler, G.; Gerer, K.F.; Hoyer, S.; Dörrie, J.; Schaft, N. RNA-transfection of γ/δ T cells with a chimeric antigen receptor or an α/β T-cell receptor: A safer alternative to genetically engineered α/β T cells for the immunotherapy of melanoma. BMC Cancer 2017, 17, 551. [Google Scholar] [CrossRef] [PubMed]
- Dörrie, J.; Babalija, L.; Hoyer, S.; Gerer, K.F.; Schuler, G.; Heinzerling, L.; Schaft, N. BRAF and MEK Inhibitors Influence the Function of Reprogrammed T Cells: Consequences for Adoptive T-Cell Therapy. Int. J. Mol. Sci. 2018, 19, 289. [Google Scholar] [CrossRef] [PubMed]
- Krug, C.; Birkholz, K.; Paulus, A.; Schwenkert, M.; Schmidt, P.; Hoffmann, N.; Hombach, A.; Fey, G.; Abken, H.; Schuler, G.; et al. Stability and activity of MCSP-specific chimeric antigen receptors (CARs) depend on the scFv antigen-binding domain and the protein backbone. Cancer Immunol. Immunother. 2015, 64, 1623–1635. [Google Scholar] [CrossRef] [PubMed]
- Krug, C.; Wiesinger, M.; Abken, H.; Schuler-Thurner, B.; Schuler, G.; Dörrie, J.; Schaft, N. A GMP-compliant protocol to expand and transfect cancer patient T cells with mRNA encoding a tumor-specific chimeric antigen receptor. Cancer Immunol. Immunother. 2014, 63, 999–1008. [Google Scholar] [CrossRef]
- Gerer, K.F.; Hoyer, S.; Dörrie, J.; Schaft, N. Electroporation of mRNA as Universal Technology Platform to Transfect a Variety of Primary Cells with Antigens and Functional Proteins. Methods Mol. Biol. 2017, 1499, 165–178. [Google Scholar] [CrossRef]
- Yang, J.; Liao, Q.; Price, M.; Moriarity, B.; Wolf, N.; Felices, M.; Miller, J.S.; Geller, M.A.; Bendzick, L.; Hopps, R.; et al. Chondroitin sulfate proteoglycan 4, a targetable oncoantigen that promotes ovarian cancer growth, invasion, cisplatin resistance and spheroid formation. Transl. Oncol. 2022, 16, 101318. [Google Scholar] [CrossRef]
- Montesinos, P.; Roboz, G.J.; Bulabois, C.-E.; Subklewe, M.; Platzbecker, U.; Ofran, Y.; Papayannidis, C.; Wierzbowska, A.; Shin, H.J.; Doronin, V.; et al. Safety and efficacy of talacotuzumab plus decitabine or decitabine alone in patients with acute myeloid leukemia not eligible for chemotherapy: Results from a multicenter, randomized, phase 2/3 study. Leukemia 2021, 35, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Filì, C.; Candoni, A.; Zannier, M.E.; Olivieri, J.; Imbergamo, S.; Caizzi, M.; Nadali, G.; Di Bona, E.; Ermacora, A.; Gottardi, M.; et al. Efficacy and toxicity of Decitabine in patients with acute myeloid leukemia (AML): A multicenter real-world experience. Leuk. Res. 2019, 76, 33–38. [Google Scholar] [CrossRef] [PubMed]
- de Castro, F.; Benedetti, M.; Antonaci, G.; Del Coco, L.; de Pascali, S.A.; Muscella, A.; Marsigliante, S.; Fanizzi, F.P. Response of Cisplatin Resistant Skov-3 Cells to Pt(O,O’-Acac)(γ-Acac)(DMS) Treatment Revealed by a Metabolomic ¹H-NMR Study. Molecules 2018, 23, 2301. [Google Scholar] [CrossRef]
- Chmielewski, M.; Hombach, A.; Heuser, C.; Adams, G.P.; Abken, H. T cell activation by antibody-like immunoreceptors: Increase in affinity of the single-chain fragment domain above threshold does not increase T cell activation against antigen-positive target cells but decreases selectivity. J. Immunol. 2004, 173, 7647–7653. [Google Scholar] [CrossRef] [PubMed]
- Schoutrop, E.; El-Serafi, I.; Poiret, T.; Zhao, Y.; Gultekin, O.; He, R.; Moyano-Galceran, L.; Carlson, J.W.; Lehti, K.; Hassan, M.; et al. Mesothelin-Specific CAR T Cells Target Ovarian Cancer. Cancer Res. 2021, 81, 3022–3035. [Google Scholar] [CrossRef] [PubMed]
- Shu, R.; Evtimov, V.J.; Hammett, M.V.; Nguyen, N.-Y.N.; Zhuang, J.; Hudson, P.J.; Howard, M.C.; Pupovac, A.; Trounson, A.O.; Boyd, R.L. Engineered CAR-T cells targeting TAG-72 and CD47 in ovarian cancer. Mol. Ther. Oncolytics 2021, 20, 325–341. [Google Scholar] [CrossRef]
- Uslu, U.; Schuler, G.; Dorrie, J.; Schaft, N. Combining a chimeric antigen receptor and a conventional T-cell receptor to generate T cells expressing two additional receptors (TETARs) for a multi-hit immunotherapy of melanoma. Exp. Dermatol. 2016, 25, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Abken, H. TRUCKs: The fourth generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef]
- Chmielewski, M.; Kopecky, C.; Hombach, A.A.; Abken, H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011, 71, 5697–5706. [Google Scholar] [CrossRef]
- Beard, R.E.; Abate-Daga, D.; Rosati, S.F.; Zheng, Z.; Wunderlich, J.R.; Rosenberg, S.A.; Morgan, R.A. Gene expression profiling using nanostring digital RNA counting to identify potential target antigens for melanoma immunotherapy. Clin. Cancer Res. 2013, 19, 4941–4950. [Google Scholar] [CrossRef]
- Beard, R.E.; Zheng, Z.; Lagisetty, K.H.; Burns, W.R.; Tran, E.; Hewitt, S.M.; Abate-Daga, D.; Rosati, S.F.; Fine, H.A.; Ferrone, S.; et al. Multiple chimeric antigen receptors successfully target chondroitin sulfate proteoglycan 4 in several different cancer histologies and cancer stem cells. J. Immunother. Cancer 2014, 2, 25. [Google Scholar] [CrossRef] [PubMed]
- Geldres, C.; Savoldo, B.; Hoyos, V.; Caruana, I.; Zhang, M.; Yvon, E.; Del Vecchio, M.; Creighton, C.J.; Ittmann, M.; Ferrone, S.; et al. T lymphocytes redirected against the chondroitin sulfate proteoglycan-4 control the growth of multiple solid tumors both in vitro and in vivo. Clin. Cancer Res. 2014, 20, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Schlingemann, R.O.; Rietveld, F.J.; de Waal, R.M.; Ferrone, S.; Ruiter, D.J. Expression of the high molecular weight melanoma-associated antigen by pericytes during angiogenesis in tumors and in healing wounds. Am. J. Pathol. 1990, 136, 1393–1405. [Google Scholar] [PubMed]
- Titov, A.; Kaminskiy, Y.; Ganeeva, I.; Zmievskaya, E.; Valiullina, A.; Rakhmatullina, A.; Petukhov, A.; Miftakhova, R.; Rizvanov, A.; Bulatov, E. Knowns and Unknowns about CAR-T Cell Dysfunction. Cancers 2022, 14, 1078. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harrer, D.C.; Schenkel, C.; Berking, C.; Herr, W.; Abken, H.; Dörrie, J.; Schaft, N. Decitabine-Mediated Upregulation of CSPG4 in Ovarian Carcinoma Cells Enables Targeting by CSPG4-Specific CAR-T Cells. Cancers 2022, 14, 5033. https://doi.org/10.3390/cancers14205033
Harrer DC, Schenkel C, Berking C, Herr W, Abken H, Dörrie J, Schaft N. Decitabine-Mediated Upregulation of CSPG4 in Ovarian Carcinoma Cells Enables Targeting by CSPG4-Specific CAR-T Cells. Cancers. 2022; 14(20):5033. https://doi.org/10.3390/cancers14205033
Chicago/Turabian StyleHarrer, Dennis Christoph, Charlotte Schenkel, Carola Berking, Wolfgang Herr, Hinrich Abken, Jan Dörrie, and Niels Schaft. 2022. "Decitabine-Mediated Upregulation of CSPG4 in Ovarian Carcinoma Cells Enables Targeting by CSPG4-Specific CAR-T Cells" Cancers 14, no. 20: 5033. https://doi.org/10.3390/cancers14205033
APA StyleHarrer, D. C., Schenkel, C., Berking, C., Herr, W., Abken, H., Dörrie, J., & Schaft, N. (2022). Decitabine-Mediated Upregulation of CSPG4 in Ovarian Carcinoma Cells Enables Targeting by CSPG4-Specific CAR-T Cells. Cancers, 14(20), 5033. https://doi.org/10.3390/cancers14205033