
Changes of Protein Expression after CRISPR/Cas9 Knockout of miRNA-142 in Cell Lines Derived from Diffuse Large B-Cell Lymphoma

 , , , ,
, , , ,  , ,
, ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Generation of Knockout Cells Using CRISPR/Cas 9
2.3. RNA Isolation and Northern Blotting
2.4. Growth and Cell Cycle Analysis of Knockout vs. Wild-Type Cells
2.5. Proteome Analysis
2.6. mRNA-Microarray
2.7. DNA Sequencing and Analysis
2.8. Expression and Reporter Vectors
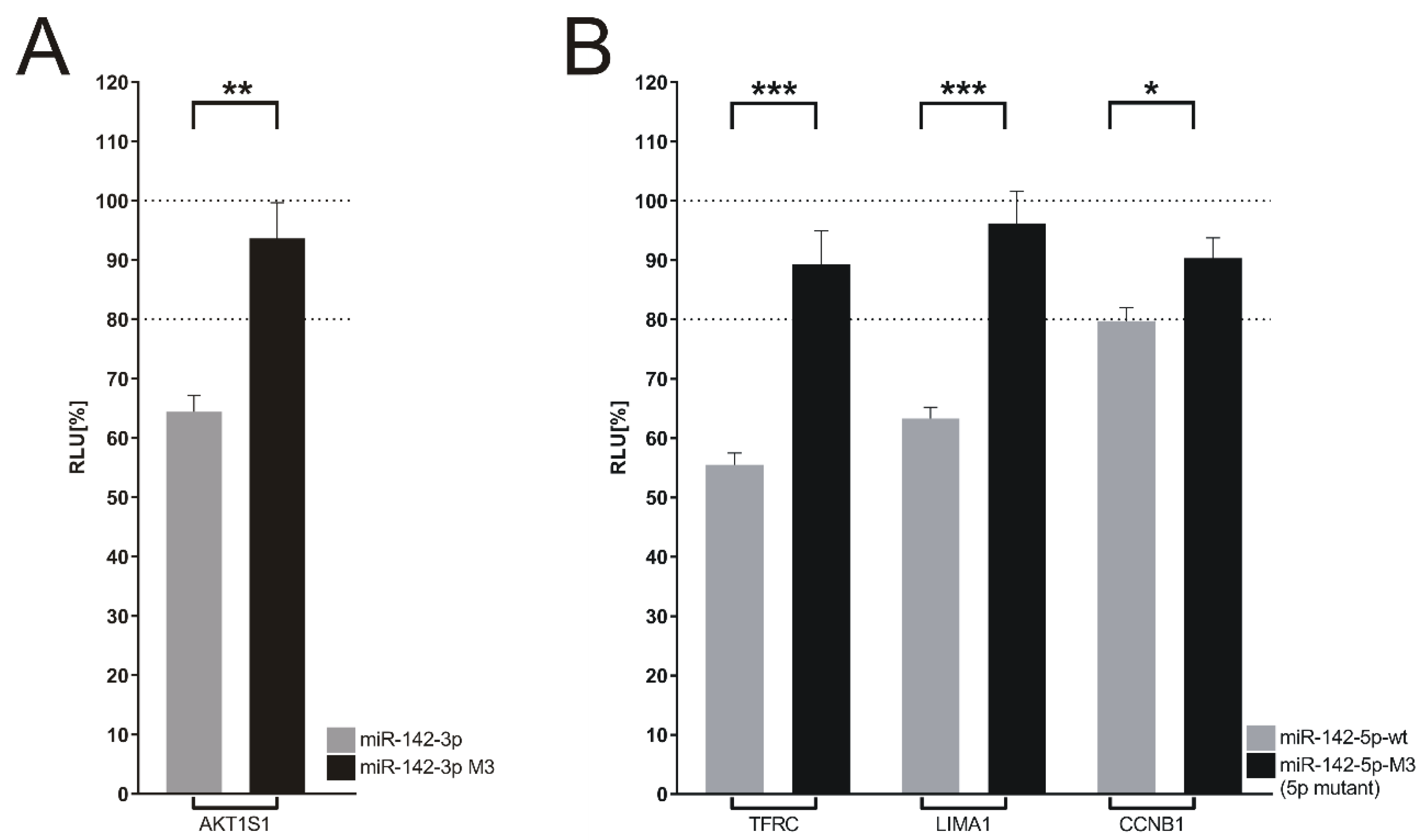
2.9. Dual Luciferase Reporter Assays
2.10. Western Blotting
3. Results
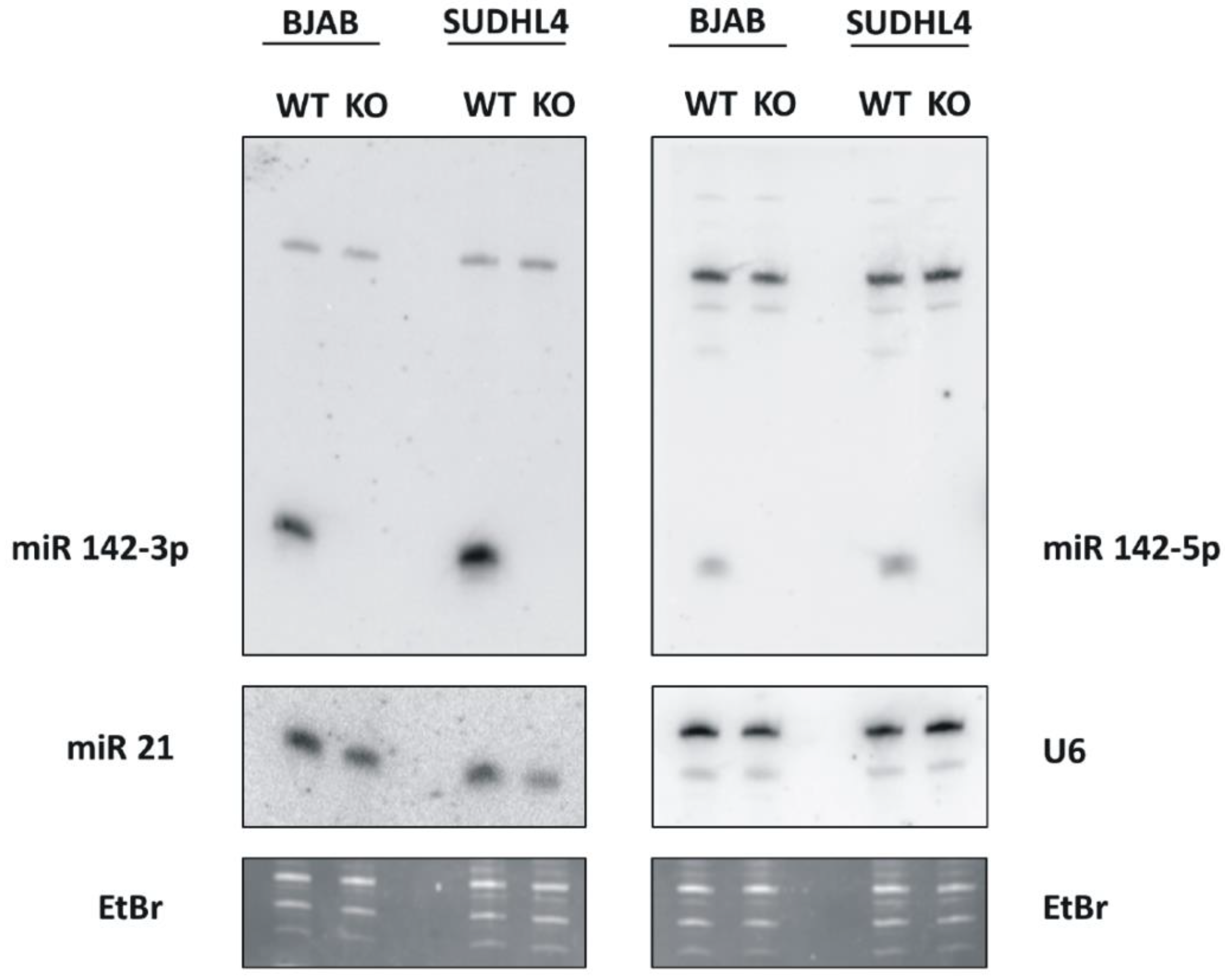
3.1. Inactivation of miR-142 in the GC-DLBCL Lines SUDHL4 and BJAB by CRISPR/Cas9 Knockout
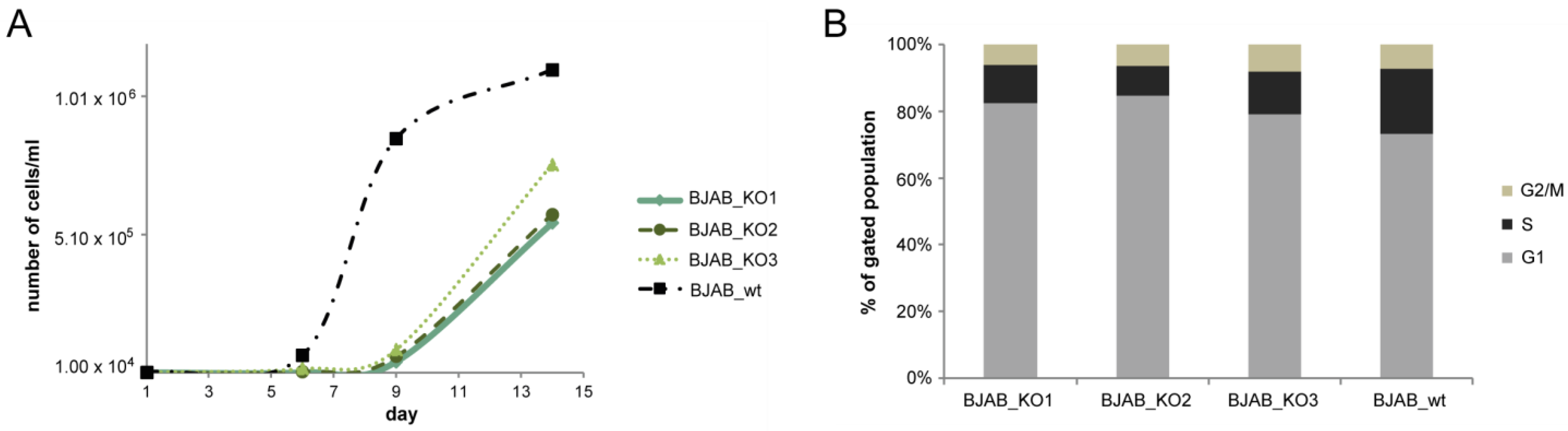
3.2. Growth Properties of miR-142 Knockout Cells
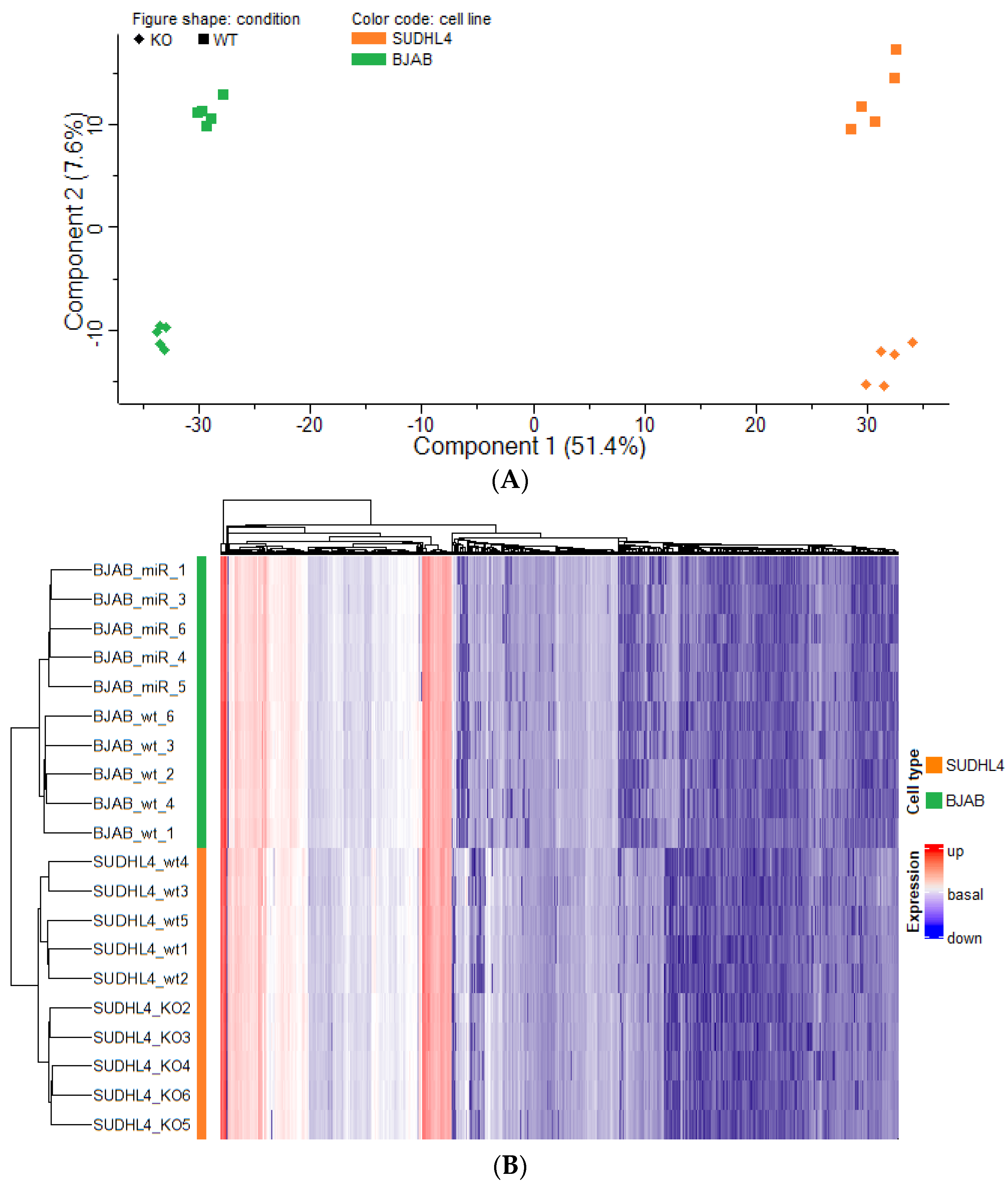
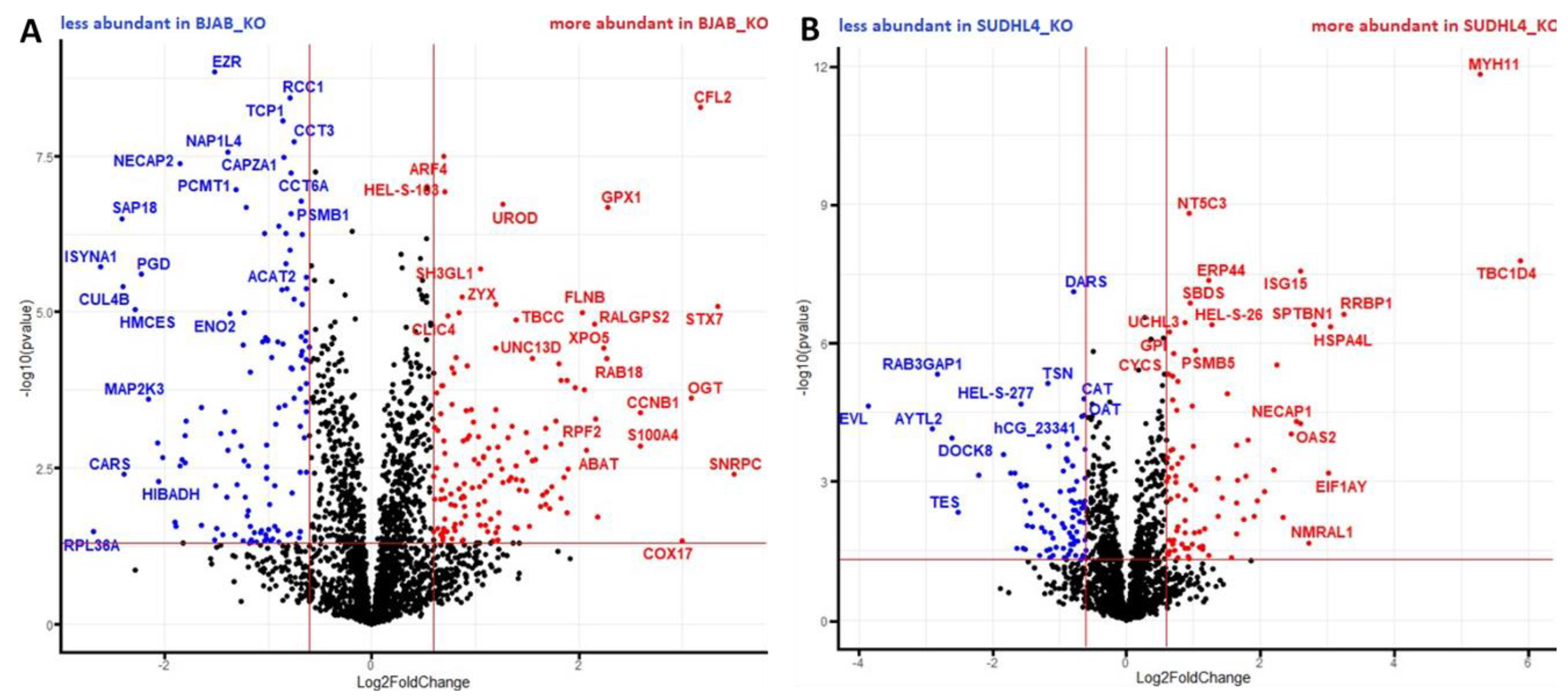
3.3. Analysis of Proteomic Alterations in BJAB and SUDHL4 Cell with Knockout vs. Wild-Type Genotype
3.4. Analysis of Proteomic Alterations in BJAB and SUDHL4 Cells with Knockout vs. Wild-Type Genotype
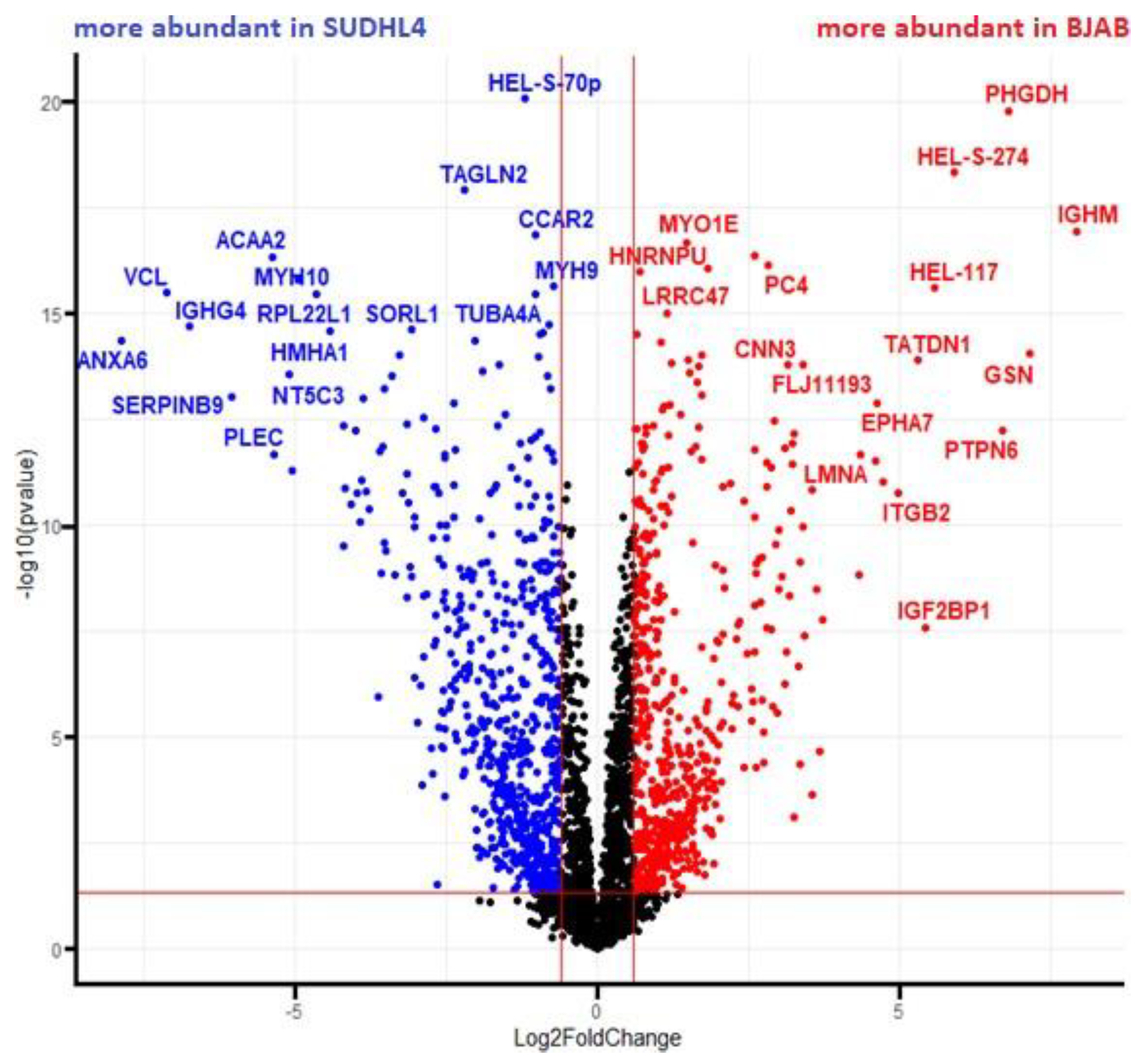
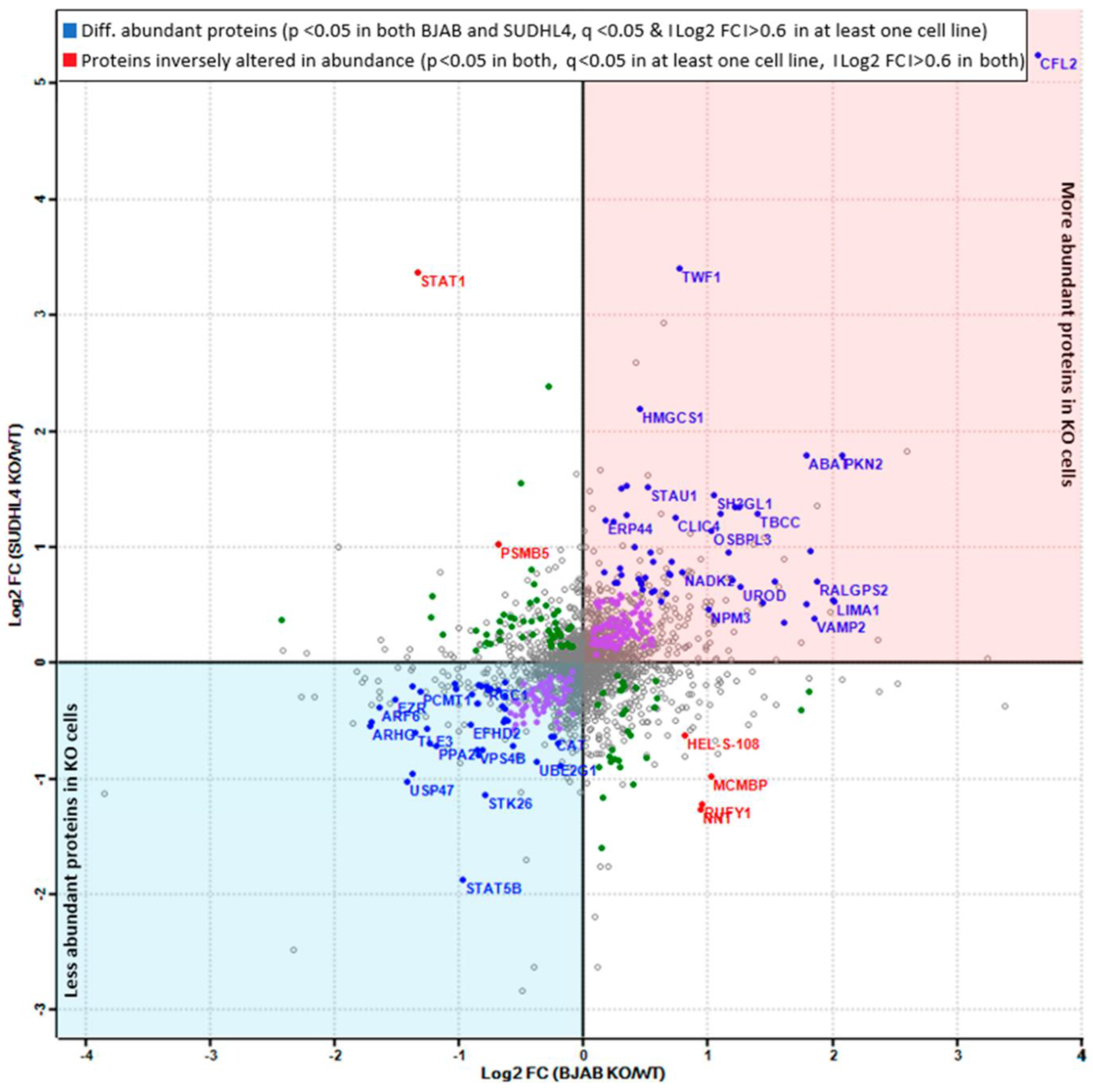
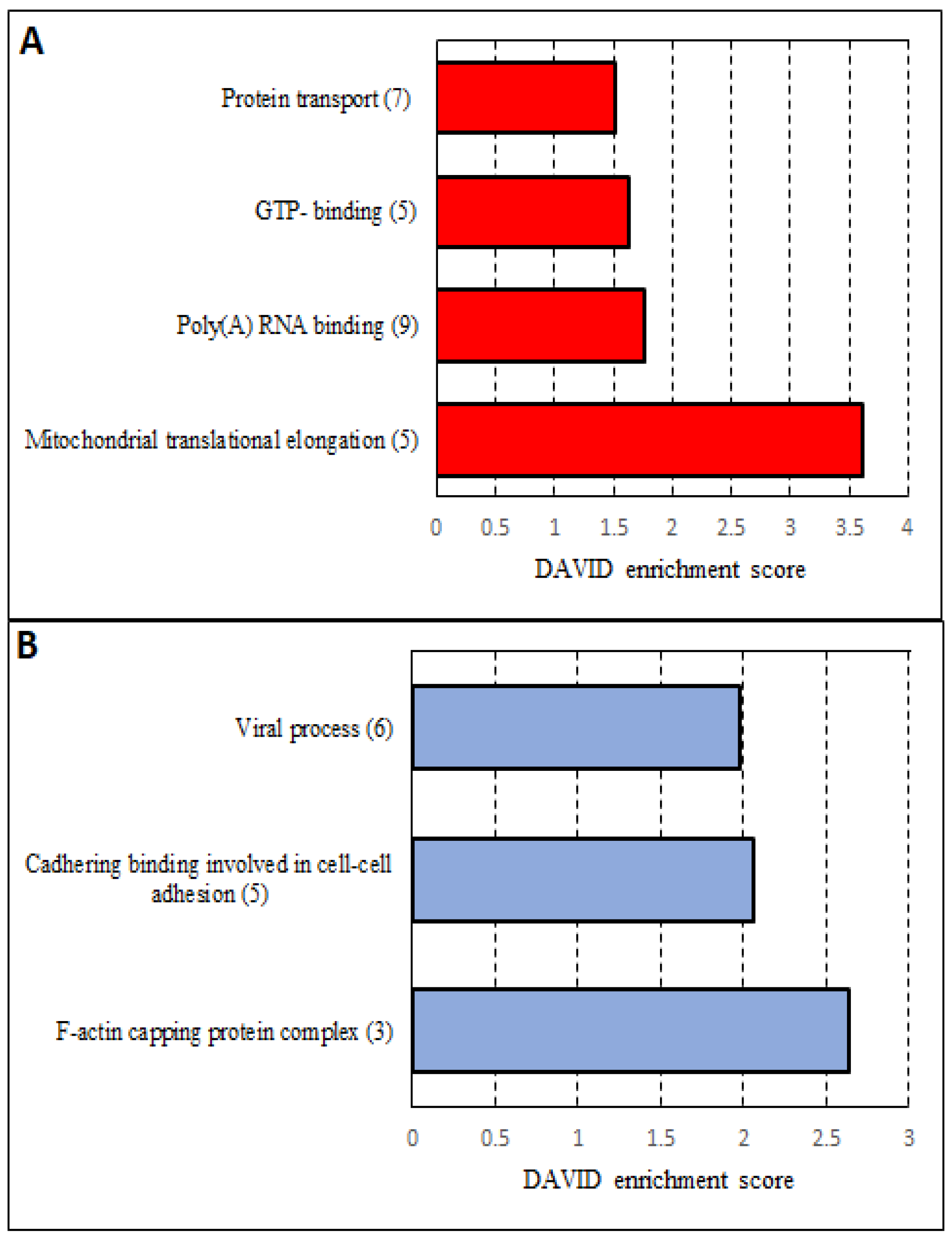
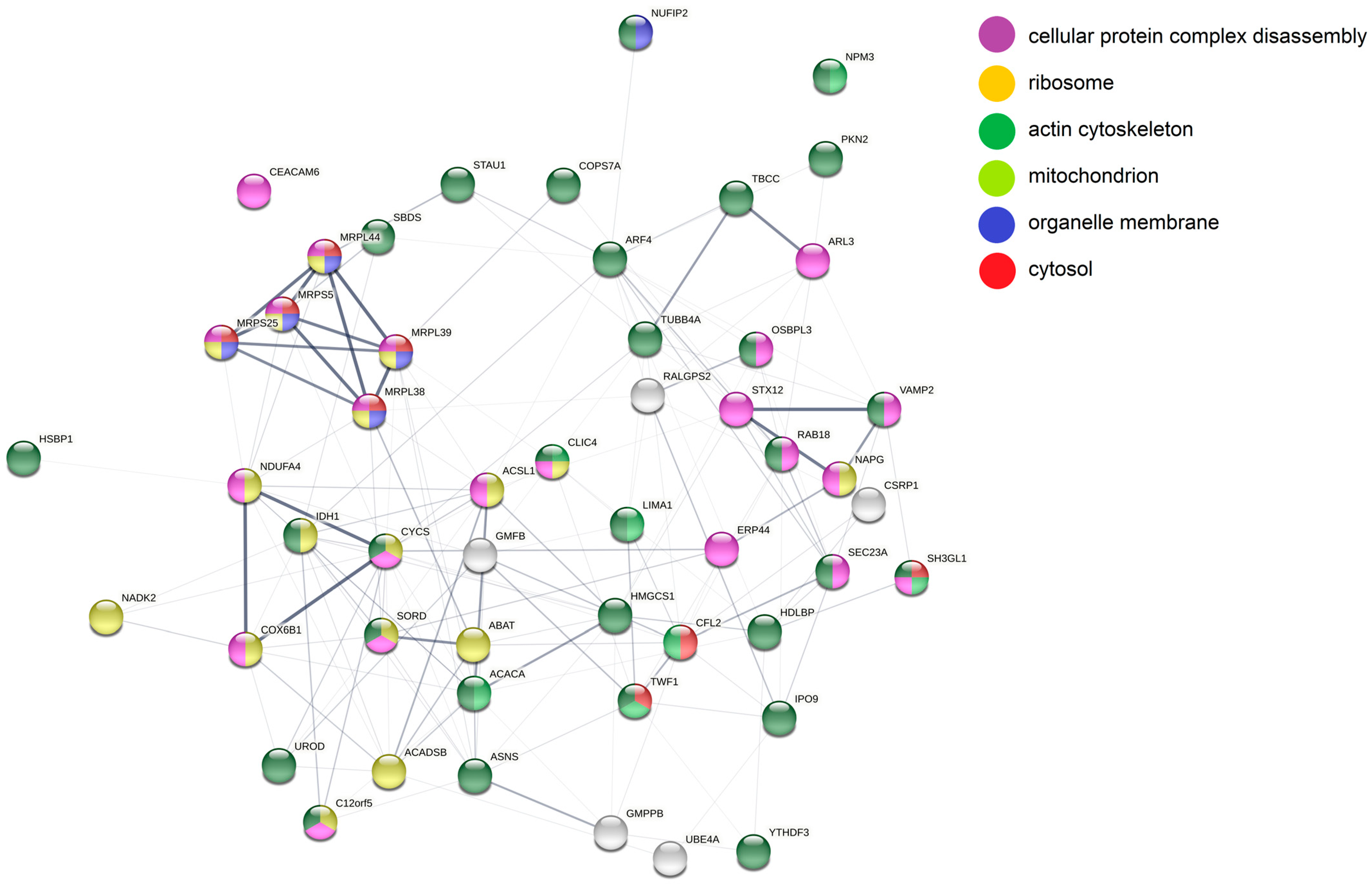
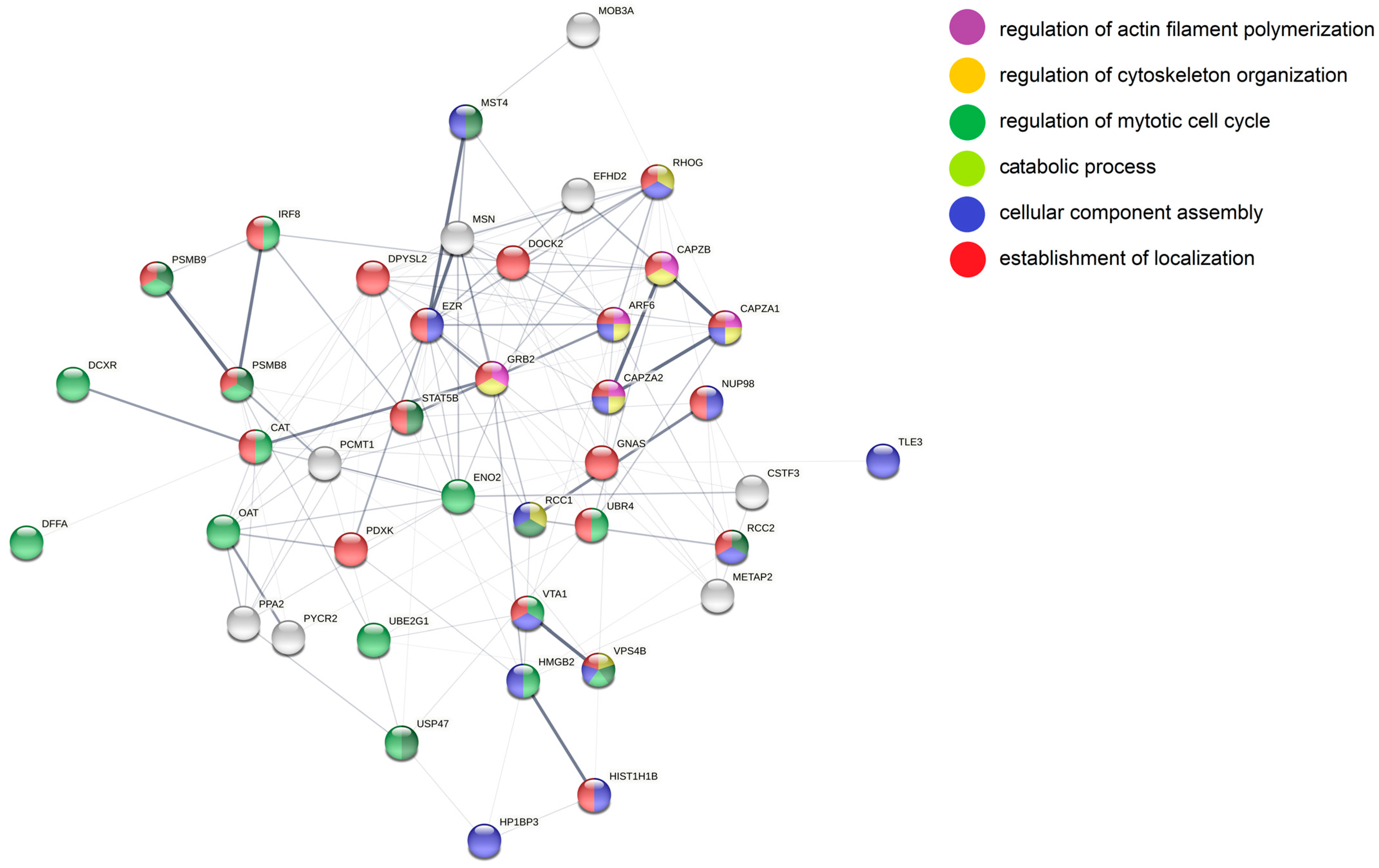
3.5. Analysis of More and Less Abundant Proteins in Both Knockout Cell Lines
3.6. Analysis of mRNA Expression in miR-142 Knockout Cells
3.7. Target Genes of miR-142 Deregulated in DLBCL Knockout Cells
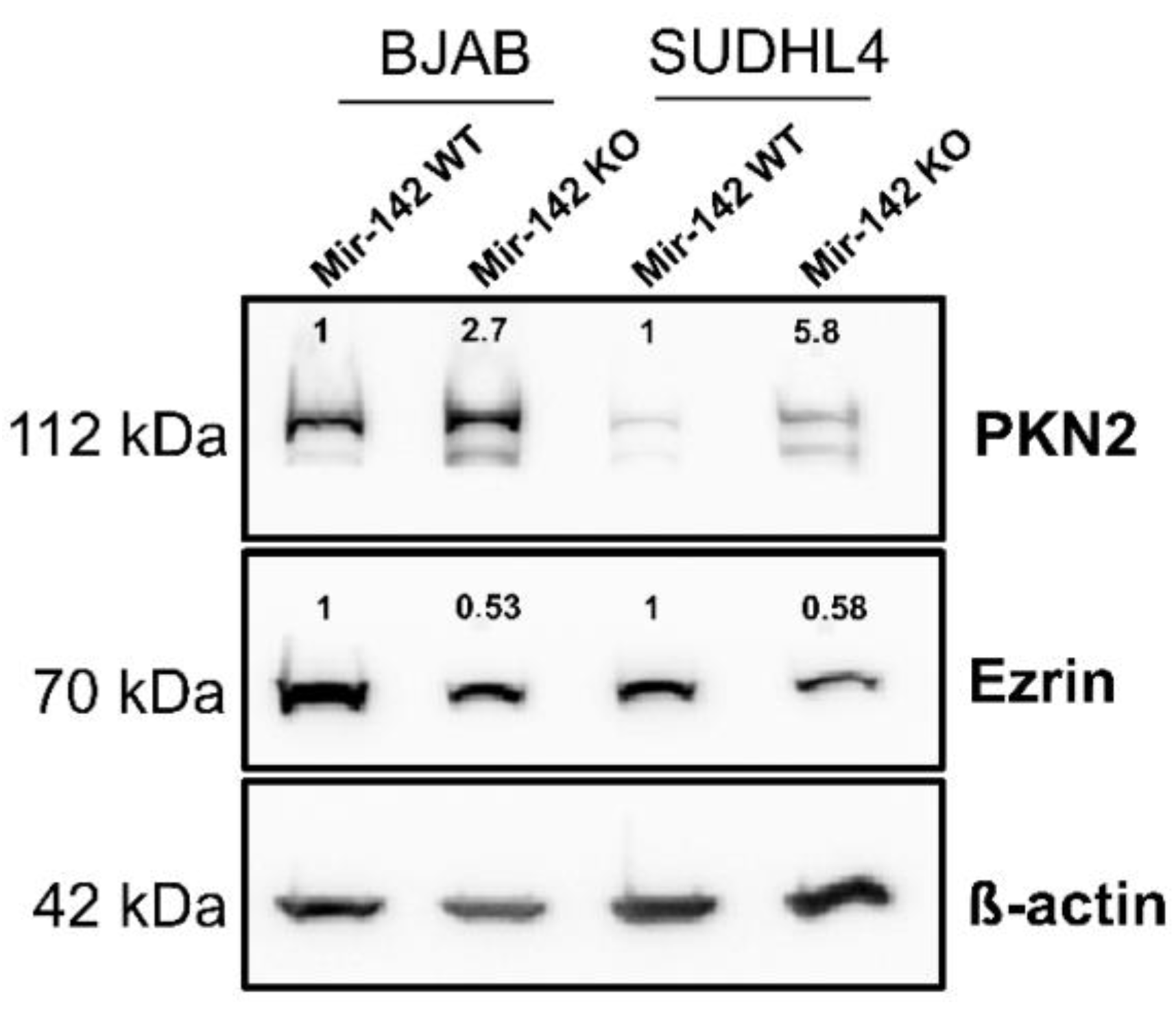
3.8. Western Blot Analysis of PKN2 and Ezrin
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Romano, G.; Veneziano, D.; Acunzo, M.; Croce, C.M. Small non-coding RNA and cancer. Carcinogenesis 2017, 38, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Dueck, A.; Meister, G. Assembly and function of small RNA—Argonaute protein complexes. Biol. Chem. 2014, 395, 611–629. [Google Scholar] [CrossRef]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef]
- Acunzo, M.; Romano, G.; Wernicke, D.; Croce, C.M. MicroRNA and cancer—A brief overview. Adv. Biol. Regul. 2015, 57, 1–9. [Google Scholar] [CrossRef]
- Costinean, S.; Zanesi, N.; Pekarsky, Y.; Tili, E.; Volinia, S.; Heerema, N.; Croce, C.M. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proc. Natl. Acad. Sci. USA 2006, 103, 7024–7029. [Google Scholar] [CrossRef]
- Hermeking, H. The miR-34 family in cancer and apoptosis. Cell Death Differ. 2010, 17, 193–199. [Google Scholar] [CrossRef]
- Welch, C.; Chen, Y.; Stallings, R.L. MicroRNA-34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene 2007, 26, 5017–5022. [Google Scholar] [CrossRef]
- Hayes, J.; Peruzzi, P.P.; Lawler, S. MicroRNAs in cancer: Biomarkers, functions and therapy. Trends Mol. Med. 2014, 20, 460–469. [Google Scholar] [CrossRef]
- Umu, S.U.; Langseth, H.; Bucher-Johannessen, C.; Fromm, B.; Keller, A.; Meese, E.; Lauritzen, M.; Leithaug, M.; Lyle, R.; Rounge, T.B. A comprehensive profile of circulating RNAs in human serum. RNA Biol. 2018, 15, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Backes, C.; Meese, E.; Keller, A. Specific miRNA Disease Biomarkers in Blood, Serum and Plasma: Challenges and Prospects. Mol. Diagn. Ther. 2016, 20, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 2018, 18, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Campo, E.; Swerdlow, S.H.; Harris, N.L.; Pileri, S.; Stein, H.; Jaffe, E.S. The 2008 WHO classification of lymphoid neoplasms and beyond: Evolving concepts and practical applications. Blood 2011, 117, 5019–5032. [Google Scholar] [CrossRef] [PubMed]
- Imig, J.; Motsch, N.; Zhu, J.Y.; Barth, S.; Okoniewski, M.; Reineke, T.; Tinguely, M.; Faggioni, A.; Trivedi, P.; Meister, G.; et al. microRNA profiling in Epstein-Barr virus-associated B-cell lymphoma. Nucleic Acids Res. 2011, 39, 1880–1893. [Google Scholar] [CrossRef]
- Kwanhian, W.; Lenze, D.; Alles, J.; Motsch, N.; Barth, S.; Doll, C.; Imig, J.; Hummel, M.; Tinguely, M.; Trivedi, P.; et al. MicroRNA-142 is mutated in about 20% of diffuse large B-cell lymphoma. Cancer Med. 2012, 1, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Hezaveh, K.; Kloetgen, A.; Bernhart, S.H.; Mahapatra, K.D.; Lenze, D.; Richter, J.; Haake, A.; Bergmann, A.K.; Brors, B.; Burkhardt, B.; et al. Alterations of miRNAs and miRNA-regulated mRNA expression in GC B cell lymphomas determined by integrative sequencing analysis. Haematologica 2016, 101, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Bouska, A.; Zhang, W.; Gong, Q.; Iqbal, J.; Scuto, A.; Vose, J.; Ludvigsen, M.; Fu, K.; Weisenburger, D.D.; Greiner, T.C.; et al. Combined copy number and mutation analysis identifies oncogenic pathways associated with transformation of follicular lymphoma. Leukemia 2017, 31, 83–91. [Google Scholar] [CrossRef]
- Morin, R.D.; Assouline, S.; Alcaide, M.; Mohajeri, A.; Johnston, R.L.; Chong, L.; Grewal, J.; Yu, S.; Fornika, D.; Bushell, K.; et al. Genetic Landscapes of Relapsed and Refractory Diffuse Large B-Cell Lymphomas. Clin. Cancer Res. 2016, 22, 2290–2300. [Google Scholar] [CrossRef] [PubMed]
- Trissal, M.C.; Wong, T.N.; Yao, J.C.; Ramaswamy, R.; Kuo, I.; Baty, J.; Sun, Y.; Jih, G.; Parikh, N.; Berrien-Elliott, M.M.; et al. MIR142 Loss-of-Function Mutations Derepress ASH1L to Increase HOXA Gene Expression and Promote Leukemogenesis. Cancer Res. 2018, 78, 3510–3521. [Google Scholar] [CrossRef] [PubMed]
- Galka-Marciniak, P.; Urbanek-Trzeciak, M.O.; Nawrocka, P.M.; Dutkiewicz, A.; Giefing, M.; Lewandowska, M.A.; Kozlowski, P. Somatic Mutations in miRNA Genes in Lung Cancer-Potential Functional Consequences of Non-Coding Sequence Variants. Cancers 2019, 11, 793. [Google Scholar] [CrossRef] [PubMed]
- Gauwerky, C.E.; Huebner, K.; Isobe, M.; Nowell, P.C.; Croce, C.M. Activation of MYC in a masked t(8;17) translocation results in an aggressive B-cell leukemia. Proc. Natl. Acad. Sci. USA 1989, 86, 8867–8871. [Google Scholar] [CrossRef]
- Robbiani, D.F.; Bunting, S.; Feldhahn, N.; Bothmer, A.; Camps, J.; Deroubaix, S.; McBride, K.M.; Klein, I.A.; Stone, G.; Eisenreich, T.R.; et al. AID produces DNA double-strand breaks in non-Ig genes and mature B cell lymphomas with reciprocal chromosome translocations. Mol. Cell 2009, 36, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Sun, L.Q.; Huang, Y.; Quan, J.; Hu, X.; Tang, D.; Kang, R.; Li, N.; Fan, X.G. miR-142-3p Inhibits the Metastasis of Hepatocellular Carcinoma Cells by Regulating HMGB1 Gene Expression. Curr. Mol. Med. 2018, 18, 135–141. [Google Scholar] [CrossRef]
- Bandres, E.; Cubedo, E.; Agirre, X.; Malumbres, R.; Zarate, R.; Ramirez, N.; Abajo, A.; Navarro, A.; Moreno, I.; Monzo, M.; et al. Identification by Real-time PCR of 13 mature microRNAs differentially expressed in colorectal cancer and non-tumoral tissues. Mol. Cancer 2006, 5, 29. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, Z.; Fang, X.; Yang, H. MiR-142-5p Suppresses Tumorigenesis by Targeting PIK3CA in Non-Small Cell Lung Cancer. Cell Physiol. Biochem. 2017, 43, 2505–2515. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, B.; Mohammadi, A.; Ghasabi, M.; Shirjang, S.; Dehghan, R.; Montazeri, V.; Holmskov, U.; Kazemi, T.; Duijf, P.; Gjerstorff, M.; et al. miR-142-3p as tumor suppressor miRNA in the regulation of tumorigenicity, invasion and migration of human breast cancer by targeting Bach-1 expression. J. Cell. Physiol. 2019, 234, 9816–9825. [Google Scholar] [CrossRef]
- Zhang, X.; Yan, Z.; Zhang, J.; Gong, L.; Li, W.; Cui, J.; Liu, Y.; Gao, Z.; Li, J.; Shen, L.; et al. Combination of hsa-miR-375 and hsa-miR-142-5p as a predictor for recurrence risk in gastric cancer patients following surgical resection. Ann. Oncol. 2011, 22, 2257–2266. [Google Scholar] [CrossRef] [PubMed]
- Dahlhaus, M.; Roolf, C.; Ruck, S.; Lange, S.; Freund, M.; Junghanss, C. Expression and prognostic significance of hsa-miR-142-3p in acute leukemias. Neoplasma 2013, 60, 432–438. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tan, Y.F.; Chen, Z.Y.; Wang, L.; Wang, M.; Liu, X.H. MiR-142-3p functions as an oncogene in prostate cancer by targeting FOXO1. J. Cancer 2020, 11, 1614–1624. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, Z.F.; Wu, H.; Wang, W. miR-142-5p Improves Neural Differentiation and Proliferation of Adipose-Derived Stem Cells. Cell. Physiol. Biochem. 2018, 50, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Kramer, N.J.; Wang, W.L.; Reyes, E.Y.; Kumar, B.; Chen, C.C.; Ramakrishna, C.; Cantin, E.M.; Vonderfecht, S.L.; Taganov, K.D.; Chau, N.; et al. Altered lymphopoiesis and immunodeficiency in miR-142 null mice. Blood 2015, 125, 3720–3730. [Google Scholar] [CrossRef]
- Shrestha, A.; Carraro, G.; El Agha, E.; Mukhametshina, R.; Chao, C.M.; Rizvanov, A.; Barreto, G.; Bellusci, S. Generation and Validation of miR-142 Knock Out Mice. PLoS ONE 2015, 10, e0136913. [Google Scholar] [CrossRef]
- Ayoubian, H.; Ludwig, N.; Fehlmann, T.; Menegatti, J.; Groger, L.; Anastasiadou, E.; Trivedi, P.; Keller, A.; Meese, E.; Grasser, F.A. Epstein-Barr Virus Infection of Cell Lines Derived from Diffuse Large B-Cell Lymphomas Alters MicroRNA Loading of the Ago2 Complex. J. Virol. 2019, 93, e01297-18. [Google Scholar] [CrossRef]
- Flores, O.; Kennedy, E.M.; Skalsky, R.L.; Cullen, B.R. Differential RISC association of endogenous human microRNAs predicts their inhibitory potential. Nucleic Acids Res. 2014, 42, 4629–4639. [Google Scholar] [CrossRef] [PubMed]
- Chapnik, E.; Rivkin, N.; Mildner, A.; Beck, G.; Pasvolsky, R.; Metzl-Raz, E.; Birger, Y.; Amir, G.; Tirosh, I.; Porat, Z.; et al. miR-142 orchestrates a network of actin cytoskeleton regulators during megakaryopoiesis. Elife 2014, 3, e01964. [Google Scholar] [CrossRef] [PubMed]
- Mildner, A.; Chapnik, E.; Varol, D.; Aychek, T.; Lampl, N.; Rivkin, N.; Bringmann, A.; Paul, F.; Boura-Halfon, S.; Hayoun, Y.S.; et al. MicroRNA-142 controls thymocyte proliferation. Eur. J. Immunol. 2017, 47, 1142–1152. [Google Scholar] [CrossRef] [PubMed]
- Charpentier, E.; Doudna, J.A. Biotechnology: Rewriting a genome. Nature 2013, 495, 50–51. [Google Scholar] [CrossRef]
- Deeb, S.J.; D’Souza, R.C.; Cox, J.; Schmidt-Supprian, M.; Mann, M. Super-SILAC allows classification of diffuse large B-cell lymphoma subtypes by their protein expression profiles. Mol. Cell. Proteom. 2012, 11, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Hart, M.; Rheinheimer, S.; Leidinger, P.; Backes, C.; Menegatti, J.; Fehlmann, T.; Grasser, F.; Keller, A.; Meese, E. Identification of miR-34a-target interactions by a combined network based and experimental approach. Oncotarget 2016, 7, 34288–34299. [Google Scholar] [CrossRef]
- Fruhwald, J.; Camacho Londono, J.; Dembla, S.; Mannebach, S.; Lis, A.; Drews, A.; Wissenbach, U.; Oberwinkler, J.; Philipp, S.E. Alternative splicing of a protein domain indispensable for function of transient receptor potential melastatin 3 (TRPM3) ion channels. J. Biol. Chem. 2012, 287, 36663–36672. [Google Scholar] [CrossRef] [PubMed]
- Hart, M.; Wach, S.; Nolte, E.; Szczyrba, J.; Menon, R.; Taubert, H.; Hartmann, A.; Stoehr, R.; Wieland, W.; Grasser, F.A.; et al. The proto-oncogene ERG is a target of microRNA miR-145 in prostate cancer. FEBS J. 2013, 280, 2105–2116. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef] [PubMed]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Diener, C.; Hart, M.; Kehl, T.; Rheinheimer, S.; Ludwig, N.; Krammes, L.; Pawusch, S.; Lenhof, K.; Tanzer, T.; Schub, D.; et al. Quantitative and time-resolved miRNA pattern of early human T cell activation. Nucleic Acids Res. 2020, 48, 10164–10183. [Google Scholar] [CrossRef]
- Beitzinger, M.; Meister, G. Experimental identification of microRNA targets by immunoprecipitation of Argonaute protein complexes. Methods Mol. Biol. 2011, 732, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Alles, J.; Menegatti, J.; Motsch, N.; Hart, M.; Eichner, N.; Reinhardt, R.; Meister, G.; Grasser, F.A. miRNA expression profiling of Epstein-Barr virus-associated NKTL cell lines by Illumina deep sequencing. FEBS Open Bio 2016, 6, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Alles, J.; Hasler, D.; Kazmi, S.M.A.; Tesson, M.; Hamilton, A.; Schlegel, L.; Marx, S.; Eichner, N.; Reinhardt, R.; Meister, G.; et al. Epstein-Barr Virus EBER Transcripts Affect miRNA-Mediated Regulation of Specific Targets and Are Processed to Small RNA Species. Non-Coding RNA 2015, 1, 170–191. [Google Scholar] [CrossRef] [PubMed]
- Schlierf, A.; Altmann, E.; Quancard, J.; Jefferson, A.B.; Assenberg, R.; Renatus, M.; Jones, M.; Hassiepen, U.; Schaefer, M.; Kiffe, M.; et al. Targeted inhibition of the COP9 signalosome for treatment of cancer. Nat. Commun. 2016, 7, 13166. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, P.M.; Bansal, N.; Kerrigan, J.E.; Abali, E.E.; Scotto, K.W.; Bertino, J.R. NAD+ Kinase as a Therapeutic Target in Cancer. Clin. Cancer Res. 2016, 22, 5189–5195. [Google Scholar] [CrossRef] [PubMed]
- Ezell, S.A.; Wang, S.; Bihani, T.; Lai, Z.; Grosskurth, S.E.; Tepsuporn, S.; Davies, B.R.; Huszar, D.; Byth, K.F. Differential regulation of mTOR signaling determines sensitivity to AKT inhibition in diffuse large B cell lymphoma. Oncotarget 2016, 7, 9163–9174. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cheng, Y.; Zhu, Y.; Xu, J.; Yang, M.; Chen, P.; Xu, W.; Zhao, J.; Geng, L.; Gong, S. PKN2 in colon cancer cells inhibits M2 phenotype polarization of tumor-associated macrophages via regulating DUSP6-Erk1/2 pathway. Mol. Cancer 2018, 17, 13. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Kong, J.; Lin, Z.; Yang, Y.; Jin, T.; Xu, M.; Sun, J.; Chen, L. Ezrin promotes breast cancer progression by modulating AKT signals. Br. J. Cancer 2019, 120, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Berrien-Elliott, M.M.; Sun, Y.; Neal, C.; Ireland, A.; Trissal, M.C.; Sullivan, R.P.; Wagner, J.A.; Leong, J.W.; Wong, P.; Mah-Som, A.Y.; et al. MicroRNA-142 Is Critical for the Homeostasis and Function of Type 1 Innate Lymphoid Cells. Immunity 2019, 51, 479–490.e6. [Google Scholar] [CrossRef]
- Ramsingh, G.; Koboldt, D.C.; Trissal, M.; Chiappinelli, K.B.; Wylie, T.; Koul, S.; Chang, L.W.; Nagarajan, R.; Fehniger, T.A.; Goodfellow, P.; et al. Complete characterization of the microRNAome in a patient with acute myeloid leukemia. Blood 2010, 116, 5316–5326. [Google Scholar] [CrossRef]
- Pascual, M.; Mena-Varas, M.; Robles, E.F.; Garcia-Barchino, M.J.; Panizo, C.; Hervas-Stubbs, S.; Alignani, D.; Sagardoy, A.; Martinez-Ferrandis, J.I.; Bunting, K.L.; et al. PD-1/PD-L1 immune checkpoint and p53 loss facilitate tumor progression in activated B-cell diffuse large B-cell lymphomas. Blood 2019, 133, 2401–2412. [Google Scholar] [CrossRef]
- Macario, A.J.L.; Conway de Macario, E. Chaperonins in cancer: Expression, function, and migration in extracellular vesicles. Semin. Cancer Biol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.X.; Song, W.; Jiang, M.P.; Yang, S.J.; Zhang, J.; Wang, D.D.; Tang, J.H. Systematic Characterization of Expression Profiles and Prognostic Values of the Eight Subunits of the Chaperonin TRiC in Breast Cancer. Front. Genet. 2021, 12, 637887. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Yang, L.; Feng, H.; Zheng, L.; Meng, H.; Li, X. CCT6A may act as a potential biomarker reflecting tumor size, lymphatic metastasis, FIGO stage, and prognosis in cervical cancer patients. J. Clin. Lab. Anal. 2021, 35, e23793. [Google Scholar] [CrossRef] [PubMed]
- Delforoush, M.; Berglund, M.; Edqvist, P.H.; Sundstrom, C.; Gullbo, J.; Enblad, G. Expression of possible targets for new proteasome inhibitors in diffuse large B-cell lymphoma. Eur. J. Haematol. 2017, 98, 52–56. [Google Scholar] [CrossRef]
- Beitzinger, M.; Peters, L.; Zhu, J.Y.; Kremmer, E.; Meister, G. Identification of human microRNA targets from isolated argonaute protein complexes. RNA Biol. 2007, 4, 76–84. [Google Scholar] [CrossRef]
- Tanaka, K.; Takeda, S.; Mitsuoka, K.; Oda, T.; Kimura-Sakiyama, C.; Maeda, Y.; Narita, A. Structural basis for cofilin binding and actin filament disassembly. Nat. Commun. 2018, 9, 1860. [Google Scholar] [CrossRef]
- Hebert, M.; Potin, S.; Sebbagh, M.; Bertoglio, J.; Breard, J.; Hamelin, J. Rho-ROCK-dependent ezrin-radixin-moesin phosphorylation regulates Fas-mediated apoptosis in Jurkat cells. J. Immunol. 2008, 181, 5963–5973. [Google Scholar] [CrossRef]
- Schmidt, A.; Durgan, J.; Magalhaes, A.; Hall, A. Rho GTPases regulate PRK2/PKN2 to control entry into mitosis and exit from cytokinesis. EMBO J. 2007, 26, 1624–1636. [Google Scholar] [CrossRef]
- Murray, E.R.; Menezes, S.; Henry, J.C.; Williams, J.L.; Alba-Castellon, L.; Baskaran, P.; Quetier, I.; Desai, A.; Marshall, J.J.T.; Rosewell, I.; et al. Disruption of pancreatic stellate cell myofibroblast phenotype promotes pancreatic tumor invasion. Cell Rep. 2022, 38, 110227. [Google Scholar] [CrossRef]
- Wallroth, A.; Koch, P.A.; Marat, A.L.; Krause, E.; Haucke, V. Protein kinase N controls a lysosomal lipid switch to facilitate nutrient signalling via mTORC1. Nat. Cell Biol. 2019, 21, 1093–1101. [Google Scholar] [CrossRef]
- de la Cruz Lopez, K.G.; Toledo Guzman, M.E.; Sanchez, E.O.; Garcia Carranca, A. mTORC1 as a Regulator of Mitochondrial Functions and a Therapeutic Target in Cancer. Front. Oncol. 2019, 9, 1373. [Google Scholar] [CrossRef] [PubMed]
- Yusufova, N.; Kloetgen, A.; Teater, M.; Osunsade, A.; Camarillo, J.M.; Chin, C.R.; Doane, A.S.; Venters, B.J.; Portillo-Ledesma, S.; Conway, J.; et al. Histone H1 loss drives lymphoma by disrupting 3D chromatin architecture. Nature 2021, 589, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Khachaturov, V.; Xiao, G.Q.; Kinoshita, Y.; Unger, P.D.; Burstein, D.E. Histone H1.5, a novel prostatic cancer marker: An immunohistochemical study. Hum. Pathol. 2014, 45, 2115–2119. [Google Scholar] [CrossRef] [PubMed]
- Ohishi, T.; Yoshida, H.; Katori, M.; Migita, T.; Muramatsu, Y.; Miyake, M.; Ishikawa, Y.; Saiura, A.; Iemura, S.I.; Natsume, T.; et al. Tankyrase-Binding Protein TNKS1BP1 Regulates Actin Cytoskeleton Rearrangement and Cancer Cell Invasion. Cancer Res. 2017, 77, 2328–2338. [Google Scholar] [CrossRef]
- Ye, Z.; Wang, D.; Lu, Y.; He, Y.; Yu, J.; Wei, W.; Chen, C.; Wang, R.; Zhang, L.; Zhang, L.; et al. Vacuolin-1 inhibits endosomal trafficking and metastasis via CapZbeta. Oncogene 2021, 40, 1775–1791. [Google Scholar] [CrossRef]
- Lee, Y.J.; Jeong, S.H.; Hong, S.C.; Cho, B.I.; Ha, W.S.; Park, S.T.; Choi, S.K.; Jung, E.J.; Ju, Y.T.; Jeong, C.Y.; et al. Prognostic value of CAPZA1 overexpression in gastric cancer. Int. J. Oncol. 2013, 42, 1569–1577. [Google Scholar] [CrossRef]
- Kwon, M.J.; Kim, R.N.; Song, K.; Jeon, S.; Jeong, H.M.; Kim, J.S.; Han, J.; Hong, S.; Oh, E.; Choi, J.S.; et al. Genes co-amplified with ERBB2 or MET as novel potential cancer-promoting genes in gastric cancer. Oncotarget 2017, 8, 92209–92226. [Google Scholar] [CrossRef]
- Huang, G.; Li, H.; Zhang, H. Abnormal Expression of Mitochondrial Ribosomal Proteins and Their Encoding Genes with Cell Apoptosis and Diseases. Int. J. Mol. Sci. 2020, 21, 8879. [Google Scholar] [CrossRef]
- Richardson, K.S.; Zundel, W. The emerging role of the COP9 signalosome in cancer. Mol. Cancer Res. 2005, 3, 645–653. [Google Scholar] [CrossRef]
- Wang, W.X.; Prajapati, P.; Nelson, P.T.; Springer, J.E. The Mitochondria-Associated ER Membranes Are Novel Subcellular Locations Enriched for Inflammatory-Responsive MicroRNAs. Mol. Neurobiol. 2020, 57, 2996–3013. [Google Scholar] [CrossRef]
- Kasahara, M.; Flajnik, M.F. Origin and evolution of the specialized forms of proteasomes involved in antigen presentation. Immunogenetics 2019, 71, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.; Du, M.Q.; McAllister-Lucas, L.M.; Lucas, P.C.; Bailey, N.G.; Hogaboam, C.M.; Lim, M.S.; Elenitoba-Johnson, K.S. Conversion of the LIMA1 tumour suppressor into an oncogenic LMO-like protein by API2-MALT1 in MALT lymphoma. Nat. Commun. 2015, 6, 5908. [Google Scholar] [CrossRef] [PubMed]
- Damacharla, D.; Thamilselvan, V.; Zhang, X.; Mestareehi, A.; Yi, Z.; Kowluru, A. Quantitative proteomics reveals novel interaction partners of Rac1 in pancreatic beta-cells: Evidence for increased interaction with Rac1 under hyperglycemic conditions. Mol. Cell. Endocrinol. 2019, 494, 110489. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Igarashi, M.; Fukuda, H.; Nakagama, H.; Katoh, M. Cancer genetics and genomics of human FOX family genes. Cancer Lett. 2013, 328, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Senyilmaz, D.; Virtue, S.; Xu, X.; Tan, C.Y.; Griffin, J.L.; Miller, A.K.; Vidal-Puig, A.; Teleman, A.A. Regulation of mitochondrial morphology and function by stearoylation of TFR1. Nature 2015, 525, 124–128. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, K.A.; Yu, D.; Zeller, K.I.; Kim, J.W.; Racke, F.; Thomas-Tikhonenko, A.; Dang, C.V. Activation of transferrin receptor 1 by c-Myc enhances cellular proliferation and tumorigenesis. Mol. Cell. Biol. 2006, 26, 2373–2386. [Google Scholar] [CrossRef]
- Wu, J.; Minikes, A.M.; Gao, M.; Bian, H.; Li, Y.; Stockwell, B.R.; Chen, Z.N.; Jiang, X. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 2019, 572, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Yang, Z.; Xia, Q.; Gao, S.; Sun, S.; Luo, X.; Li, Z.; Zhang, X.; Li, X. ACADSB regulates ferroptosis and affects the migration, invasion, and proliferation of colorectal cancer cells. Cell Biol. Int. 2020, 44, 2334–2343. [Google Scholar] [CrossRef] [PubMed]
- Sukma Dewi, I.; Celik, S.; Karlsson, A.; Hollander, Z.; Lam, K.; McManus, J.W.; Tebbutt, S.; Ng, R.; Keown, P.; McMaster, R.; et al. Exosomal miR-142-3p is increased during cardiac allograft rejection and augments vascular permeability through down-regulation of endothelial RAB11FIP2 expression. Cardiovasc. Res. 2017, 113, 440–452. [Google Scholar] [CrossRef]
- Guay, C.; Kruit, J.K.; Rome, S.; Menoud, V.; Mulder, N.L.; Jurdzinski, A.; Mancarella, F.; Sebastiani, G.; Donda, A.; Gonzalez, B.J.; et al. Lymphocyte-Derived Exosomal MicroRNAs Promote Pancreatic beta Cell Death and May Contribute to Type 1 Diabetes Development. Cell Metab. 2019, 29, 348–361.e6. [Google Scholar] [CrossRef]
- Cortes-Troncoso, J.; Jang, S.I.; Perez, P.; Hidalgo, J.; Ikeuchi, T.; Greenwell-Wild, T.; Warner, B.M.; Moutsopoulos, N.M.; Alevizos, I. T cell exosome-derived miR-142-3p impairs glandular cell function in Sjogren’s syndrome. JCI Insight 2020, 5, e133497. [Google Scholar] [CrossRef] [PubMed]
- Guiot, J.; Cambier, M.; Boeckx, A.; Henket, M.; Nivelles, O.; Gester, F.; Louis, E.; Malaise, M.; Dequiedt, F.; Louis, R.; et al. Macrophage-derived exosomes attenuate fibrosis in airway epithelial cells through delivery of antifibrotic miR-142-3p. Thorax 2020, 75, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Chao, G.; Li, W.; Zhu, J.; Li, F.; Qi, B.; Wei, Y.; Chen, S.; Zhou, G.; Lu, X.; et al. Activated CD4(+) T cells-derived exosomal miR-142-3p boosts post-ischemic ventricular remodeling by activating myofibroblast. Aging (Albany NY) 2020, 12, 7380–7396. [Google Scholar] [CrossRef]
- Zhou, C.; Zhang, Y.; Yan, R.; Huang, L.; Mellor, A.L.; Yang, Y.; Chen, X.; Wei, W.; Wu, X.; Yu, L.; et al. Exosome-derived miR-142-5p remodels lymphatic vessels and induces IDO to promote immune privilege in the tumour microenvironment. Cell Death Differ. 2021, 28, 715–729. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Garcia-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BJAB | SUDHL4 | SUDHL4 vs. BJAB | |||
|---|---|---|---|---|---|
| Gene Symbol | FC KO vs. WT | Gene Symbol | FC KO vs. WT | Gene Symbol | FC Difference |
| CASP1 | 0.002 | TUBB8 | 0.004 | CAMK2N2 | −90.240 |
| HEBP2 | 0.002 | GPATCH1 | 0.005 | APCDD1 | −43.254 |
| CARD16 | 0.003 | PSCA | 0.007 | DAB2IP | −41.563 |
| DCN | 0.004 | TTN | 0.011 | MAP3K6 | −34.677 |
| THEMIS | 0.006 | LMX1B | 0.011 | BCL2L15 | −33.949 |
| LRFN3 | 0.007 | NRIP1 | 0.012 | FBXL7 | −32.339 |
| SLC38A4 | 0.009 | SLC6A2 | 0.013 | ART5 | −27.928 |
| CD274 | 0.009 | ENTPD8 | 0.014 | GPR171 | −24.575 |
| ADCYAP1 | 0.009 | CD200R1 | 0.016 | H6PD | −23.742 |
| OR51S1 | 0.011 | CCDC120 | 0.016 | CD3G | −23.354 |
| CBS | 0.011 | NAV2 | 0.017 | MORN4 | −21.185 |
| FAM183A | 0.012 | PCDH15 | 0.017 | CELSR1 | −19.788 |
| PSG5 | 0.012 | CCDC144A | 0.017 | GPRASP2 | −19.247 |
| SMOC2 | 0.012 | FOXR1 | 0.017 | C12orf40 | −18.125 |
| TRAPPC3L | 0.013 | SMPDL3A | 0.019 | FOS | −15.809 |
| XAF1 | 0.016 | ST8SIA1 | 0.019 | TGM7 | −14.890 |
| CLIP4 | 0.016 | RXFP1 | 0.020 | AS3MT | −14.816 |
| CARD17 | 0.016 | PLXNB3 | 0.020 | PYGL | −13.197 |
| NT5E | 0.020 | DNASE2B | 0.020 | GUCA1A | −12.201 |
| RIC3 | 0.020 | OIT3 | 0.021 | C16orf62 | −11.145 |
| PDCD1LG2 | 20.835 | FOXL1 | 55.254 | BDNF | 37.896 |
| PLEKHH2 | 20.971 | FBXW2 | 57.711 | TMPRSS15 | 39.814 |
| PPP4R4 | 22.315 | MAGEB16 | 58.134 | WISP1 | 40.863 |
| IGF2BP2 | 22.854 | KIF5A | 58.160 | SEMA5A | 43.383 |
| COL4A4 | 23.466 | GPR85 | 66.176 | GJB1 | 46.221 |
| FOXL1 | 25.999 | FOXC2 | 68.900 | PLAU | 46.481 |
| CHRD | 27.345 | TDRD6 | 70.648 | SULF2 | 47.733 |
| NPHP1 | 28.583 | GAL3ST2 | 71.346 | CNPY1 | 50.162 |
| FBXL7 | 29.758 | PAGE4 | 73.636 | RSG1 | 51.428 |
| IRS1 | 35.501 | OR2H1 | 75.353 | IFI44L | 58.035 |
| OPLAH | 47.738 | KITLG | 78.372 | AKAP14 | 63.312 |
| DHRS9 | 52.841 | CACNA1B | 83.562 | CLIP4 | 64.945 |
| HOXB6 | 64.707 | SDC4 | 98.925 | MAGEB16 | 65.662 |
| AIM2 | 66.156 | SKI | 102.922 | FOXC2 | 71.354 |
| TCEAL8 | 67.141 | LOC100505841 | 103.228 | OR2H1 | 80.166 |
| CAMK2N2 | 84.172 | VAMP3 | 112.579 | NT5E | 81.031 |
| RIMKLA | 96.113 | ZNF680 | 262.647 | OR51S1 | 94.888 |
| ACAP2 | 127.043 | LRRC1 | 2774.524 | XAF1 | 95.632 |
| ARMCX5 | 144.324 | ZNF521 | 2876.359 | CACNA1B | 105.876 |
| TCEAL8 | 158.819 | KCNC2 | 3110.916 | SLC38A4 | 120.136 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menegatti, J.; Nakel, J.; Stepanov, Y.K.; Caban, K.M.; Ludwig, N.; Nord, R.; Pfitzner, T.; Yazdani, M.; Vilimova, M.; Kehl, T.; et al. Changes of Protein Expression after CRISPR/Cas9 Knockout of miRNA-142 in Cell Lines Derived from Diffuse Large B-Cell Lymphoma. Cancers 2022, 14, 5031. https://doi.org/10.3390/cancers14205031
Menegatti J, Nakel J, Stepanov YK, Caban KM, Ludwig N, Nord R, Pfitzner T, Yazdani M, Vilimova M, Kehl T, et al. Changes of Protein Expression after CRISPR/Cas9 Knockout of miRNA-142 in Cell Lines Derived from Diffuse Large B-Cell Lymphoma. Cancers. 2022; 14(20):5031. https://doi.org/10.3390/cancers14205031
Chicago/Turabian StyleMenegatti, Jennifer, Jacqueline Nakel, Youli K. Stepanov, Karolina M. Caban, Nicole Ludwig, Ruth Nord, Thomas Pfitzner, Maryam Yazdani, Monika Vilimova, Tim Kehl, and et al. 2022. "Changes of Protein Expression after CRISPR/Cas9 Knockout of miRNA-142 in Cell Lines Derived from Diffuse Large B-Cell Lymphoma" Cancers 14, no. 20: 5031. https://doi.org/10.3390/cancers14205031
APA StyleMenegatti, J., Nakel, J., Stepanov, Y. K., Caban, K. M., Ludwig, N., Nord, R., Pfitzner, T., Yazdani, M., Vilimova, M., Kehl, T., Lenhof, H.-P., Philipp, S. E., Meese, E., Fröhlich, T., Grässer, F. A., & Hart, M. (2022). Changes of Protein Expression after CRISPR/Cas9 Knockout of miRNA-142 in Cell Lines Derived from Diffuse Large B-Cell Lymphoma. Cancers, 14(20), 5031. https://doi.org/10.3390/cancers14205031