
Identification and Functional Analysis of a Novel CTNNB1 Mutation in Pediatric Medulloblastoma

 ,
,  , , ,
, , ,  , ,
, ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Tumor Material
2.2. Histology and Immunohistochemistry
2.3. Fluorescence In Situ Hybridization
2.4. Tumor DNA Isolation and Mutational Analyses of the CTNNB1 Gene
2.5. Plasmids and Cell Culture
2.6. Protein Eextracts, Immunoblot Analysis, and Antibodies
2.7. Luciferase Reporter Assay
2.8. Statistical Analysis
3. Results
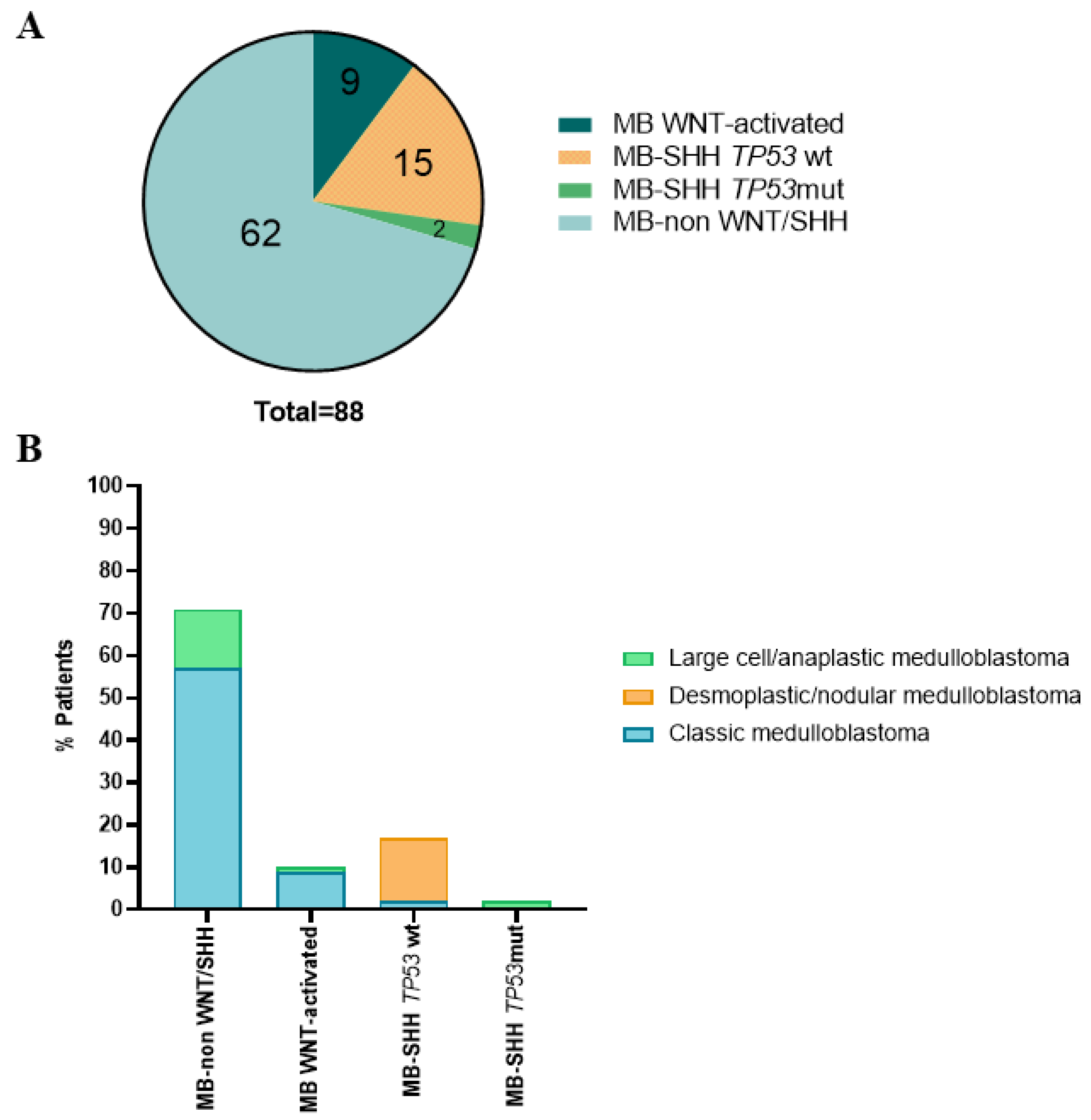
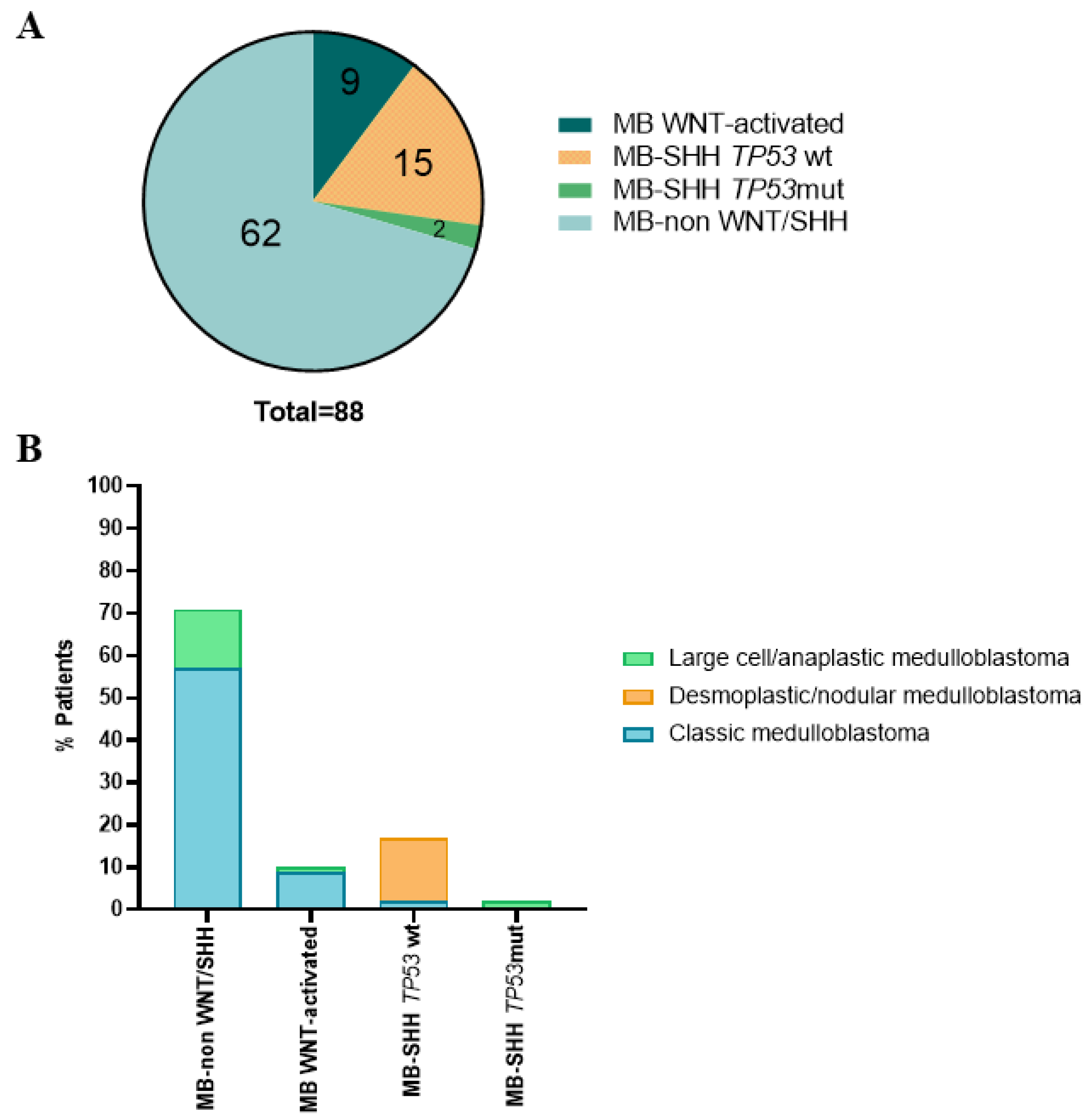
3.1. Histological and Molecular Subgroups of the Pediatric MB Cohort
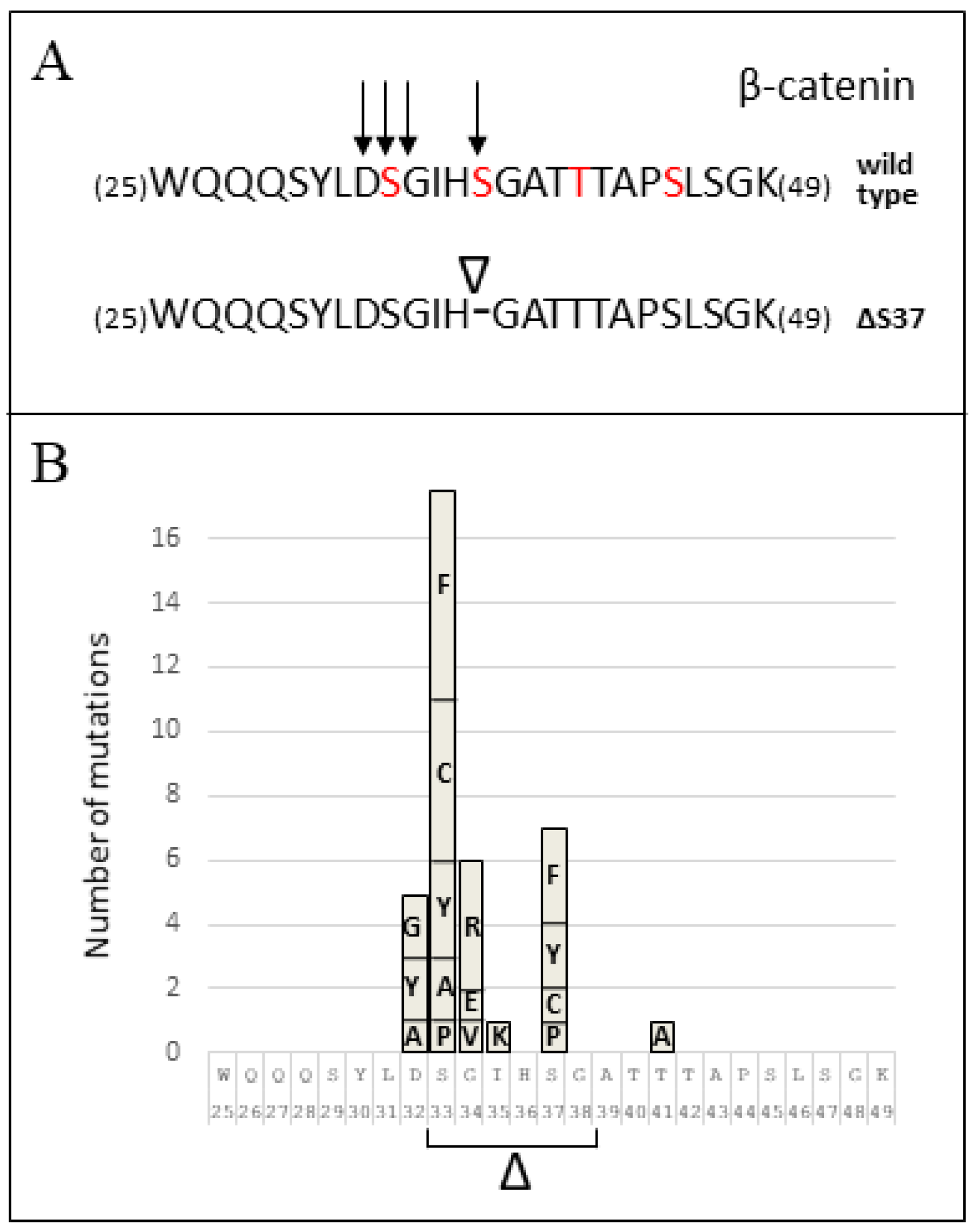
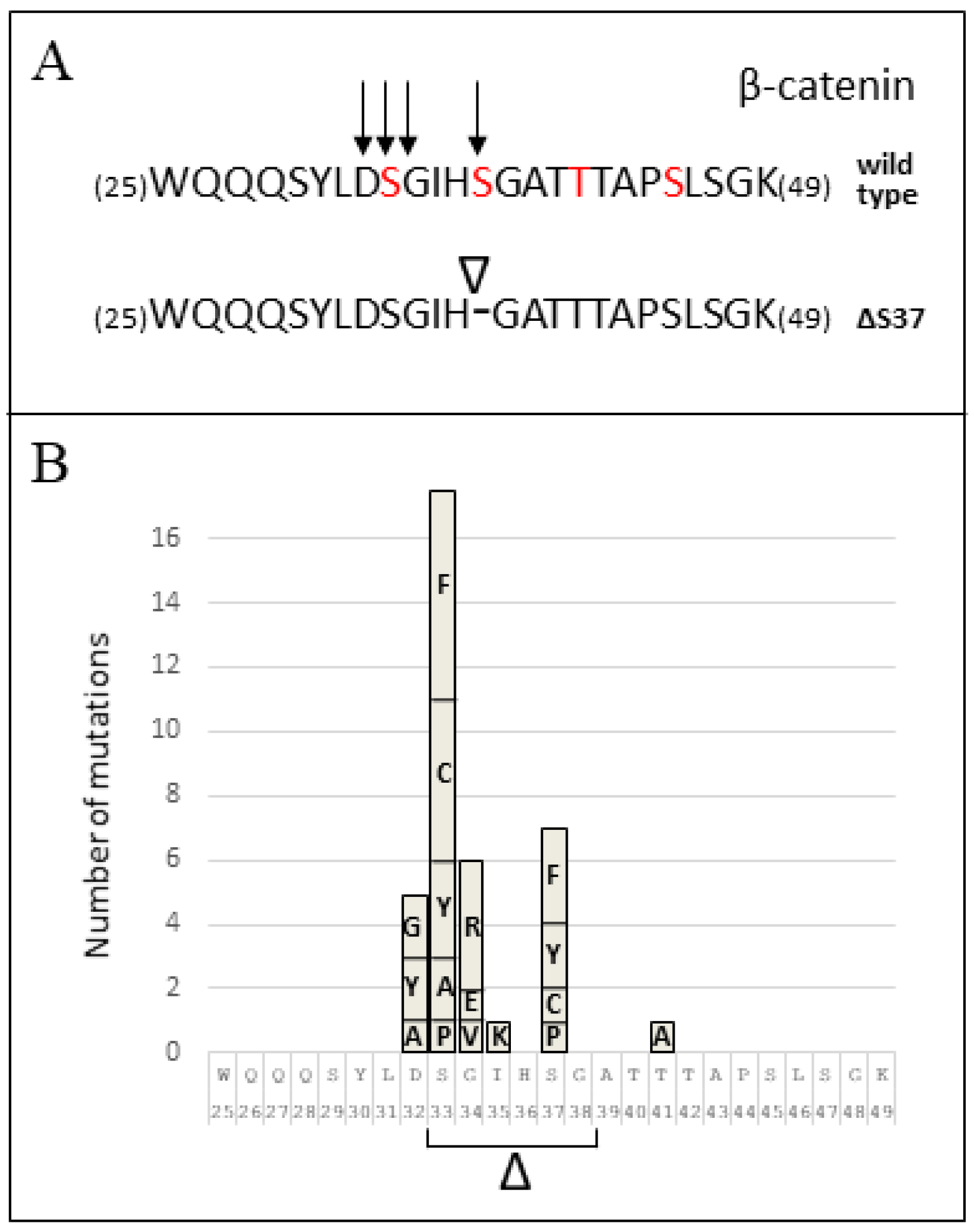
3.2. CTNNB1 Mutation Analysis
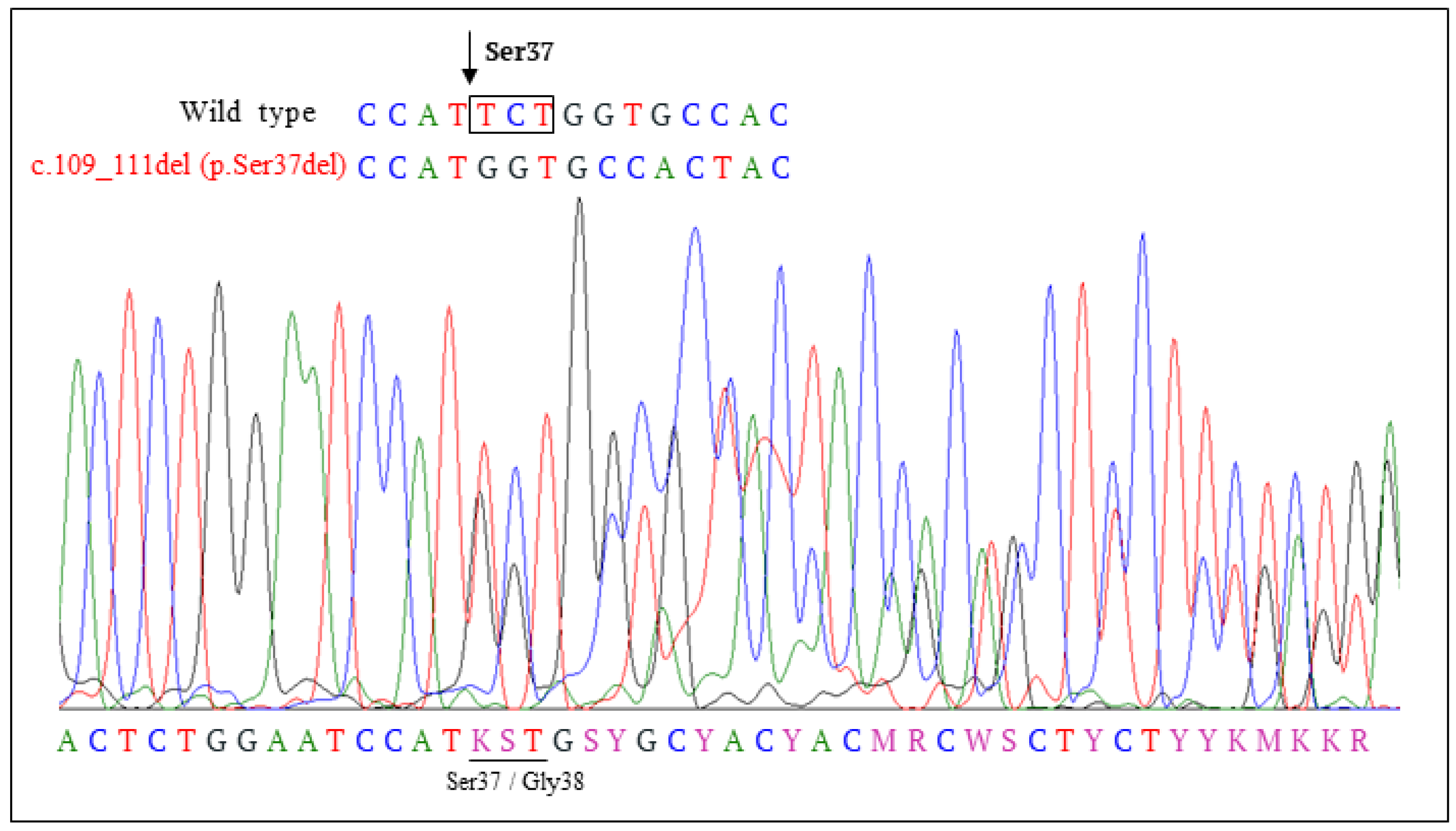
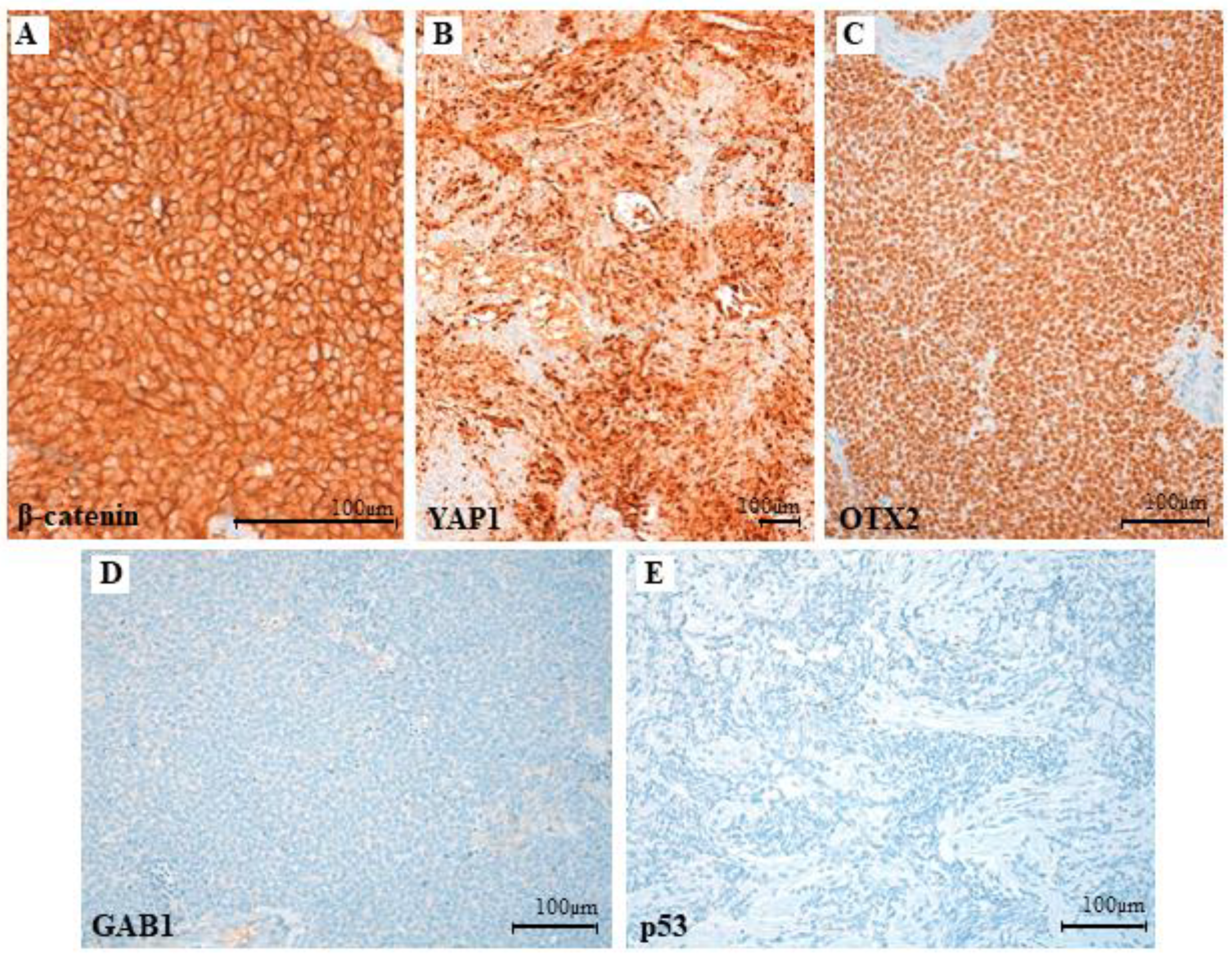
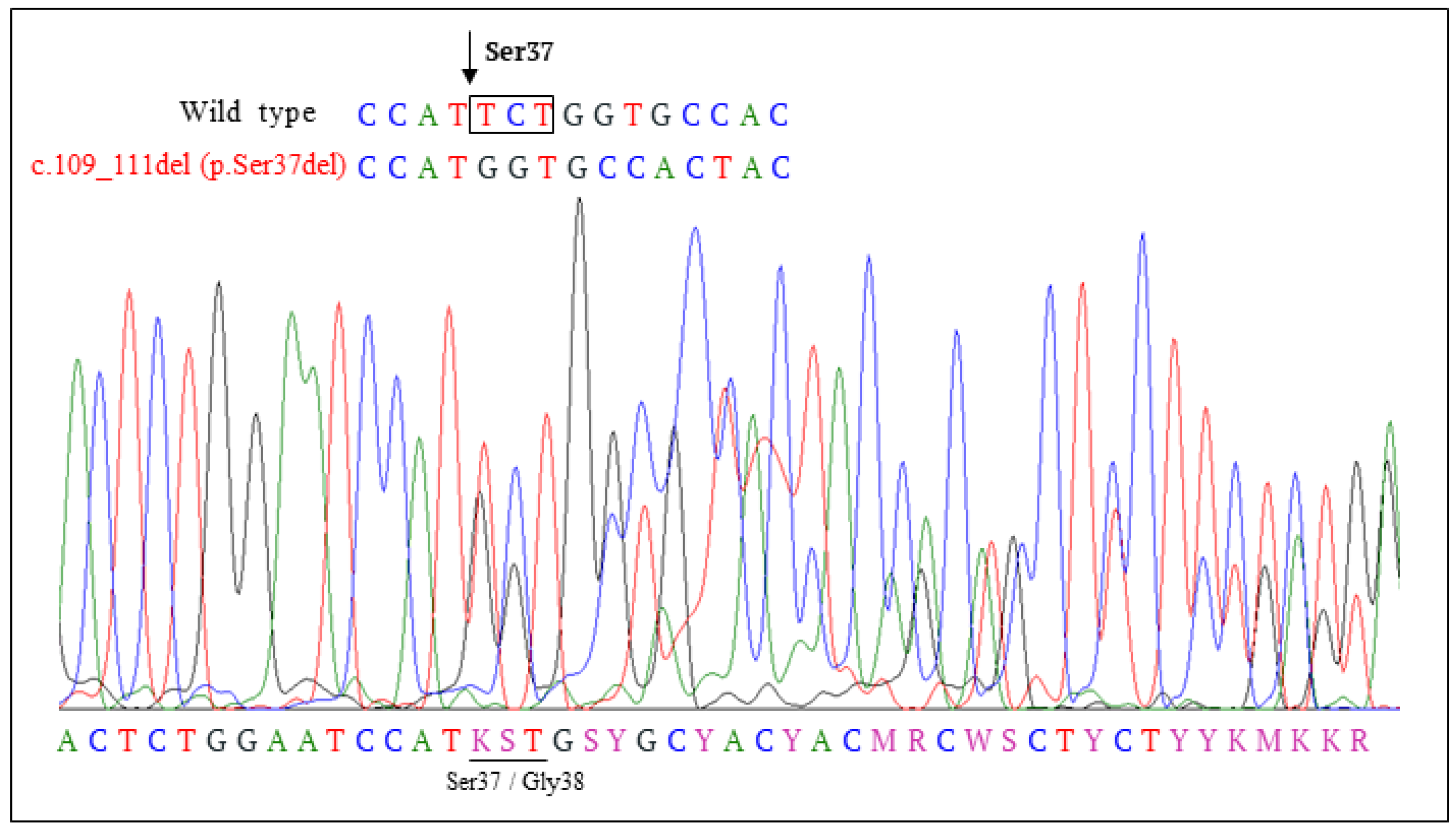
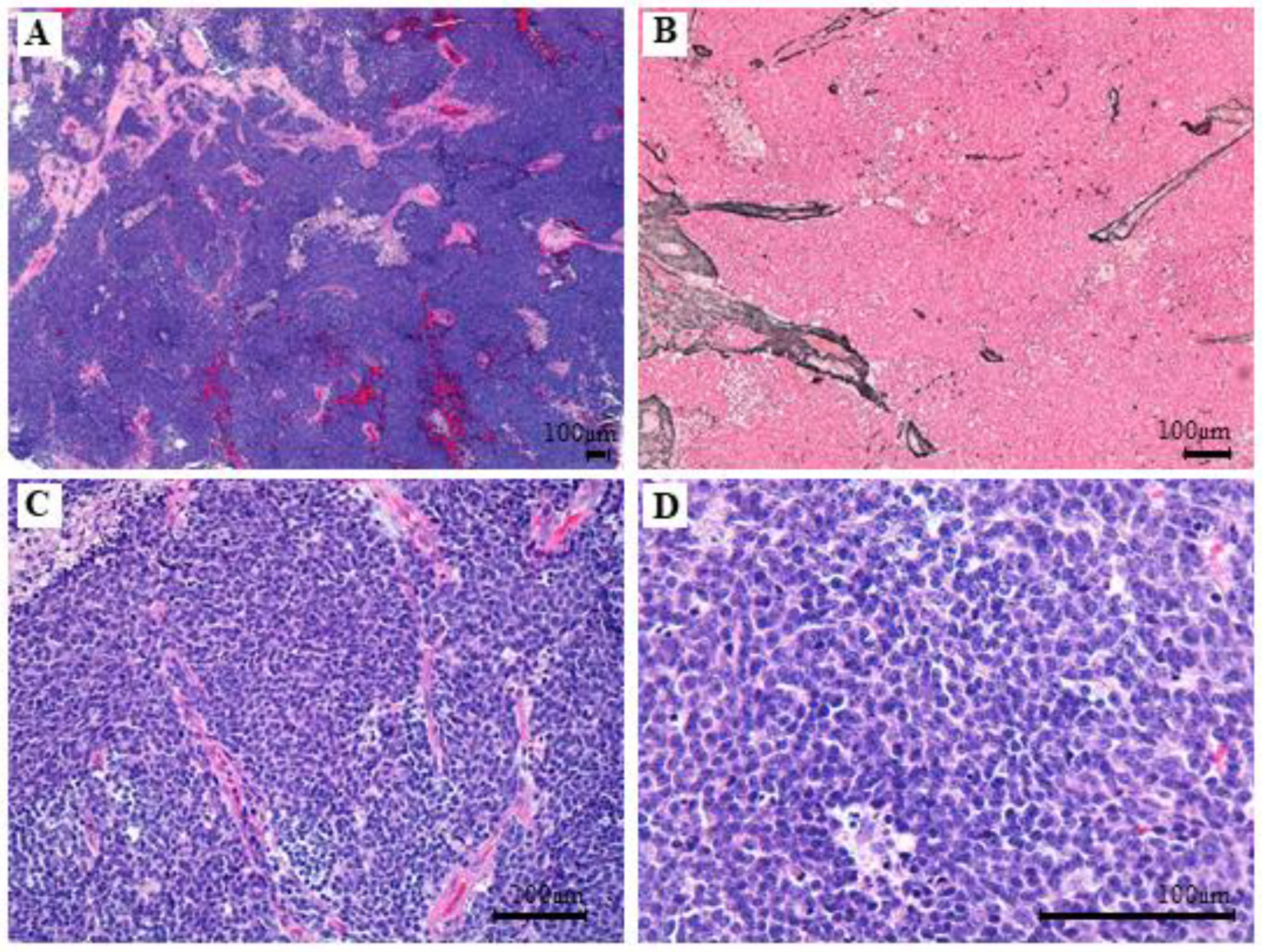
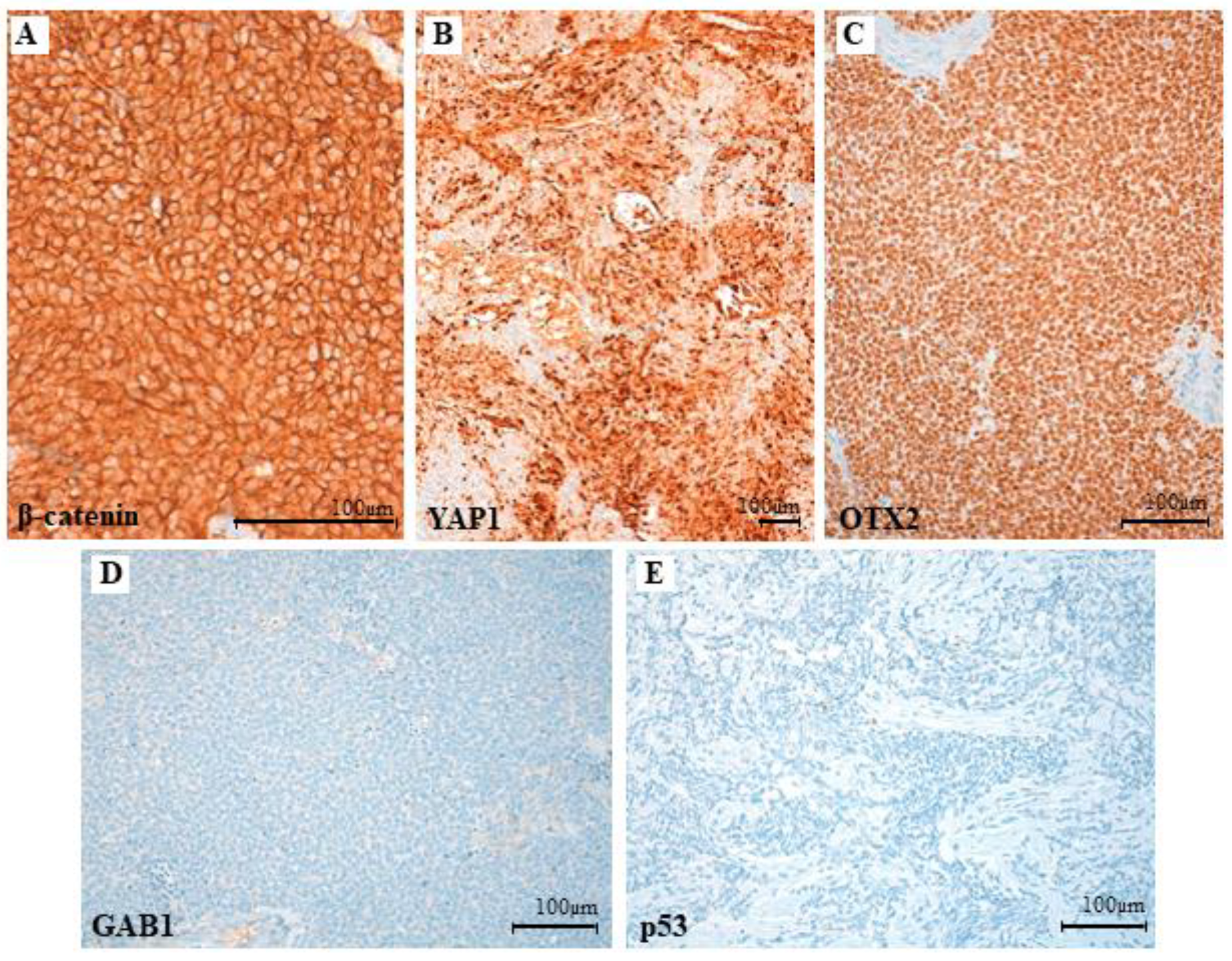
3.3. Clinical, Pathological and Molecular Characteristics of the Tumor with the Novel β-Catenin Variant c.109-111del (p.Ser37del)
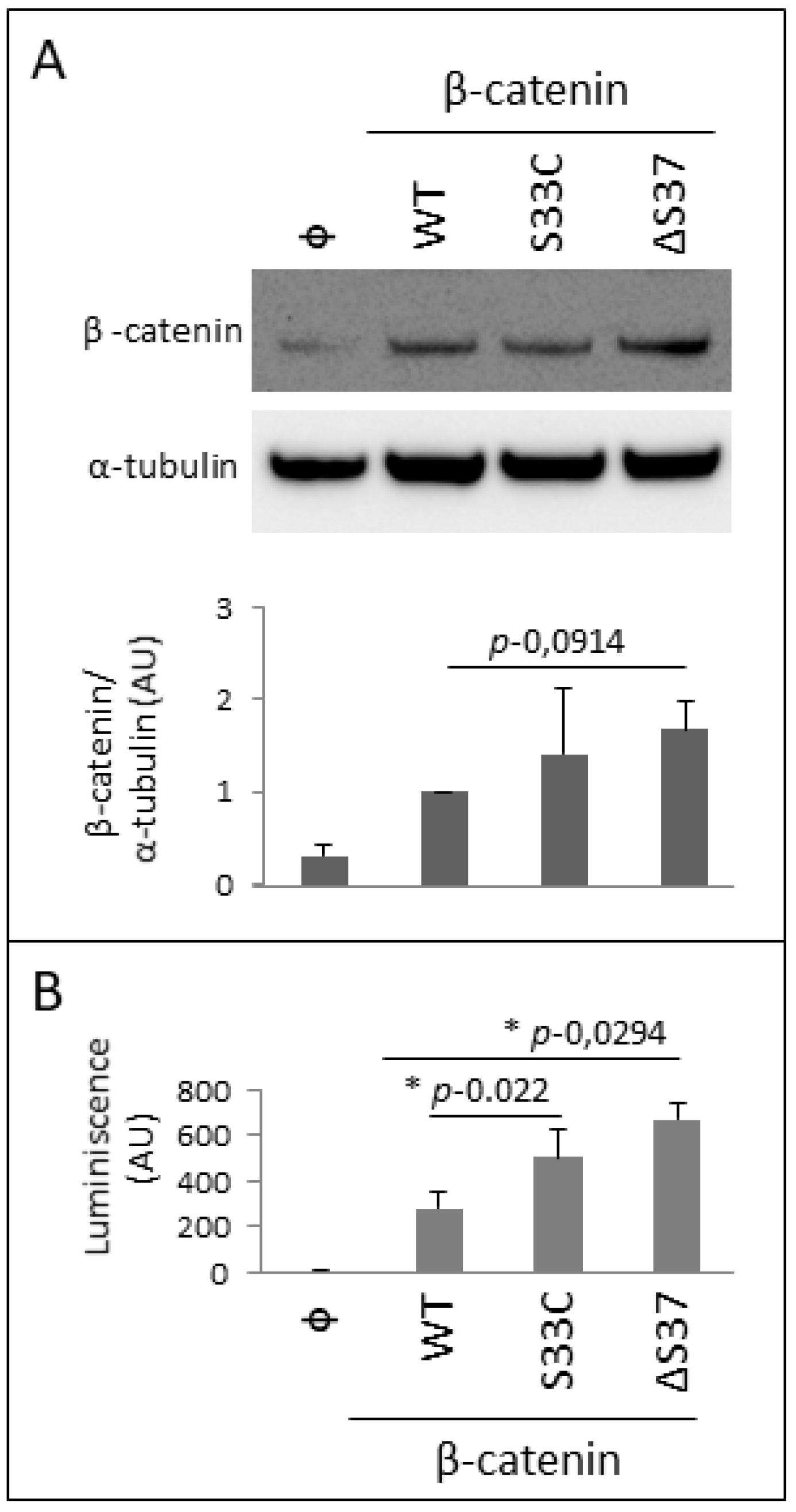
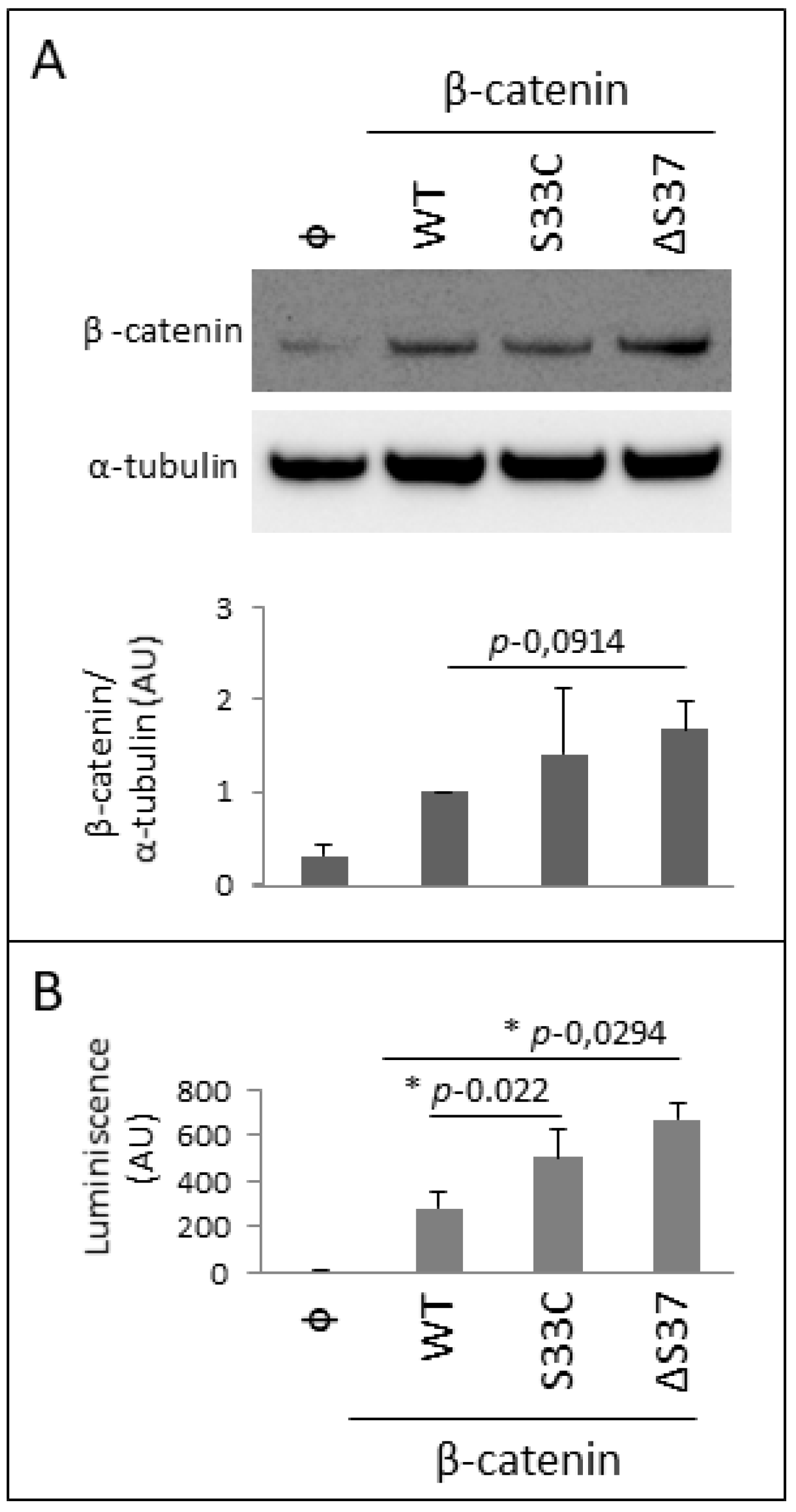
3.4. Functional Characterization of the Novel β-Catenin Variant c.109-111del (p.Ser37del)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Peris-Bonet, R.; Martínez-García, C.; Lacour, B.; Petrovich, S.; Giner-Ripoll, B.; Navajas, A.; Steliarova-Foucher, E. Childhood central nervous system tumours—Incidence and survival in Europe (1978–1997): Report from Automated Childhood Cancer Information System project. Eur. J. Cancer 2006, 42, 2064–2080. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Northcott, P.A.; Korshunov, A.; Witt, H.; Hielscher, T.; Eberhart, C.G.; Mack, S.C.; Bouffet, E.; Clifford, S.C.; Hawkins, C.E.; French, P.; et al. Medulloblastoma Comprises Four Distinct Molecular Variants. J. Clin. Oncol. 2011, 29, 1408–1414. [Google Scholar] [CrossRef]
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.-J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2011, 123, 465–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kool, M.; Korshunov, A.; Remke, M.; Jones, D.T.; Schlanstein, M.; Northcott, P.A.; Cho, Y.-J.; Koster, J.; Schouten-van Meeteren, A.; Van Vuurden, D.; et al. Molecular subgroups of medulloblastoma: An international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012, 123, 473–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, Y.-J.; Tsherniak, A.; Tamayo, P.; Santagata, S.; Ligon, A.; Greulich, H.; Berhoukim, R.; Amani, V.; Goumnerova, L.; Eberhart, C.G.; et al. Integrative Genomic Analysis of Medulloblastoma Identifies a Molecular Subgroup That Drives Poor Clinical Outcome. J. Clin. Oncol. 2011, 29, 1424–1430. [Google Scholar] [CrossRef] [PubMed]
- Schwalbe, E.; Williamson, D.; Lindsey, J.; Hamilton, D.; Ryan, S.L.; Megahed, H.; Garami, M.; Hauser, P.; Dembowska-Baginska, B.; Perek, D.; et al. DNA methylation profiling of medulloblastoma allows robust subclassification and improved outcome prediction using formalin-fixed biopsies. Acta Neuropathol. 2013, 125, 359–371. [Google Scholar] [CrossRef] [Green Version]
- Hovestadt, V.; Remke, M.; Kool, M.; Pietsch, T.; Northcott, P.A.; Fischer, R.; Cavalli, F.M.G.; Ramaswamy, V.; Zapatka, M.; Reifenberger, G.; et al. Robust molecular subgrouping and copy-number profiling of medulloblastoma from small amounts of archival tumour material using high-density DNA methylation arrays. Acta Neuropathol. 2013, 125, 913–916. [Google Scholar] [CrossRef] [Green Version]
- Pietsch, T.; Schmidt, R.; Remke, M.; Korshunov, A.; Hovestadt, V.; Jones, D.T.W.; Felsberg, J.; Kaulich, K.; Goschzik, T.; Kool, M.; et al. Prognostic significance of clinical, histopathological, and molecular characteristics of medulloblastomas in the prospective HIT2000 multicenter clinical trial cohort. Acta Neuropathol. 2014, 128, 137–149. [Google Scholar] [CrossRef] [Green Version]
- Pietsch, T.; Haberler, C. Update on the integrated histopathological and genetic classification of medulloblastoma—A practical diagnostic guideline. Clin. Neuropathol. 2016, 35, 344–352. [Google Scholar] [CrossRef] [Green Version]
- Juraschka, K.; Taylor, M.D. Medulloblastoma in the age of molecular subgroups: A review. J. Neurosurg. Pediatr. 2019, 24, 353–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Northcott, P.A.; Buchhalter, I.; Morrissy, A.S.; Hovestadt, V.; Weischenfeldt, J.; Ehrenberger, T.; Gröbner, S.; Segura-Wang, M.; Zichner, T.; Rudneva, V.A.; et al. The whole-genome landscape of medulloblastoma subtypes. Nature 2017, 547, 311–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; A Cree, I.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Ellison, D.W.; Onilude, O.E.; Lindsey, J.C.; Lusher, M.E.; Weston, C.L.; Taylor, R.E.; Pearson, A.D.; Clifford, S.C. β-Catenin Status Predicts a Favorable Outcome in Childhood Medulloblastoma: The United Kingdom Children’s Cancer Study Group Brain Tumour Committee. J. Clin. Oncol. 2005, 23, 7951–7957. [Google Scholar] [CrossRef]
- Koch, A.; Hrychyk, A.; Hartmann, W.; Waha, A.; Mikeska, T.; Waha, A.; Schüller, U.; Sörensen, N.; Berthold, F.; Goodyer, C.G.; et al. Mutations of the Wnt antagonistAXIN2(Conductin) result in TCF-dependent transcription in medulloblastomas. Int. J. Cancer 2007, 121, 284–291. [Google Scholar] [CrossRef]
- Koch, A.; Waha, A.; Berthold, F.; Wolter, M.; Reifenberger, J.; Hartmann, W.; Friedl, W.; Reifenberger, G.; Wiestler, O.D.; Pietsch, T. Somatic mutations ofWNT/wingless signaling pathway components in primitive neuroectodermal tumors. Int. J. Cancer 2001, 93, 445–449. [Google Scholar] [CrossRef]
- Surun, A.; Varlet, P.; Brugières, L.; Lacour, B.; Faure-Conter, C.; Leblond, P.; Bertozzi-Salomon, A.-I.; Berger, C.; André, N.; Sariban, E.; et al. Medulloblastomas associated with an APC germline pathogenic variant share the good prognosis of CTNNB1-mutated medulloblastomas. Neuro-Oncology 2019, 22, 128–138. [Google Scholar] [CrossRef]
- Anastas, J.N.; Moon, R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jeong, S. Mutation Hotspots in the β-Catenin Gene: Lessons from the Human Cancer Genome Databases. Mol. Cells 2019, 42, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Megy, S.; Bertho, G.; Gharbi-Benarous, J.; Baleux, F.; Benarous, R.; Girault, J.-P. Solution structure of a peptide derived from the oncogenic protein β-Catenin in its phosphorylated and nonphosphorylated states. Peptides 2005, 26, 227–241. [Google Scholar] [CrossRef]
- Al-Fageeh, M.; Li, Q.; Dashwood, W.M.; Myzak, M.C.; Dashwood, R.H. Phosphorylation and ubiquitination of oncogenic mutants of β-catenin containing substitutions at Asp32. Oncogene 2004, 23, 4839–4846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajjar, A.; Chintagumpala, M.; Ashley, D.; Kellie, S.; Kun, L.E.; Merchant, T.E.; Woo, S.; Wheeler, G.; Ahern, V.; Krasin, M.J.; et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): Long-term results from a prospective, multicentre trial. Lancet Oncol. 2006, 7, 813–820. [Google Scholar] [CrossRef]
- Clifford, S.C.; Lusher, M.E.; Lindsey, J.C.; Langdon, J.A.; Gilbertson, R.J.; Straughton, D.; Ellison, D.W. Wnt/Wingless Pathway Activation and Chromosome 6 Loss Characterise a Distinct Molecular Sub-Group of Medulloblastomas Associated with a Favourable Prognosis. Cell Cycle 2006, 5, 2666–2670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, M.C.; Fuller, C.; Hogg, T.L.; Dalton, J.; Finkelstein, D.; Lau, C.C.; Chintagumpala, M.; Adesina, A.; Ashley, D.M.; Kellie, S.J.; et al. Genomics Identifies Medulloblastoma Subgroups That Are Enriched for Specific Genetic Alterations. J. Clin. Oncol. 2006, 24, 1924–1931. [Google Scholar] [CrossRef]
- Giangaspero, F.; Perilongo, G.; Fondelli, M.P.; Brisigotti, M.; Carollo, C.; Burnelli, R.; Burger, P.C.; Garrè, M.L. Medulloblastoma with extensive nodularity: A variant with favorable prognosis. J. Neurosurg. 1999, 91, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.; Denkhaus, D.; Albrecht, S.; Leuschner, I.; von Schweinitz, D.; Pietsch, T. Child-hood Hepatoblastomas Frequently Carry a Mutated Degradation Targeting Box of the Be-ta-Catenin Gene. Cancer Res. 1999, 59, 269–273. [Google Scholar]
- Fuchs, M.; Müller, T.; Lerch, M.M.; Ullrich, A. Association of Human Protein-tyrosine Phosphatase κ with Members of the Armadillo Family. J. Biol. Chem. 1996, 271, 16712–16719. [Google Scholar] [CrossRef] [Green Version]
- Mingo, J.; Erramuzpe, A.; Luna, S.; Aurtenetxe, O.; Amo, L.; Diez, I.; Schepens, J.T.G.; Hendriks, W.; Cortes, J.M.; Pulido, R. One-Tube-Only Standardized Site-Directed Mutagenesis: An Alternative Approach to Generate Amino Acid Substitution Collections. PLoS ONE 2016, 11, e0160972. [Google Scholar] [CrossRef] [Green Version]
- Karlsen, K.F.; Tekle, C.; Andersson, Y.; Flatmark, K.; Fodstad, Ø.; Nunes-Xavier, C.E. Immunoregulatory protein B7-H3 promotes growth and decreases sensitivity to therapy in metastatic melanoma cells. Pigment. Cell Melanoma Res. 2017, 30, 467–476. [Google Scholar] [CrossRef] [Green Version]
- Ramaswamy, V.; Remke, M.; Bouffet, E.; Bailey, S.; Clifford, S.C.; Doz, F.; Kool, M.; Dufour, C.; Vassal, G.; Milde, T.; et al. Risk stratification of childhood medulloblastoma in the molecular era: The current consensus. Acta Neuropathol. 2016, 131, 821–831. [Google Scholar] [CrossRef] [Green Version]
- Stamos, J.L.; Weis, W.I. The β-Catenin Destruction Complex. Cold Spring Harb. Perspect. Biol. 2013, 5, a007898. [Google Scholar] [CrossRef]
- Arnold, A.; Tronser, M.; Sers, C.; Ahadova, A.; Endris, V.; Mamlouk, S.; Horst, D.; Möbs, M.; Bischoff, P.; Kloor, M.; et al. The majority of β-catenin mutations in colorectal cancer is homozygous. BMC Cancer 2020, 20, 1038. [Google Scholar] [CrossRef]
- Polakis, P. Wnt Signaling in Cancer. Cold Spring Harb. Perspect. Biol. 2012, 4, a008052. [Google Scholar] [CrossRef] [Green Version]
- Polakis, P. The many ways of Wnt in cancer. Curr. Opin. Genet. Dev. 2007, 17, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Giles, R.H.; van Es, J.H.; Clevers, H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim. Biophys. Acta 2003, 1653, 1–24. [Google Scholar] [CrossRef]
- Van Es, J.H.; Barker, N.; Clevers, H. You Wnt some, you lose some: Oncogenes in the Wnt signaling pathway. Curr. Opin. Genet. Dev. 2002, 13, 28–33. [Google Scholar] [CrossRef]
- Lee, H.-S.; Park, M.-H.; Yang, S.-J.; Park, K.C.; Kim, N.-S.; Kim, Y.-S.; Kim, D.I.; Yoo, H.-S.; Choi, E.-J.; Yeom, Y.I. Novel candidate targets of Wnt/β-catenin signaling in hepatoma cells. Life Sci. 2007, 80, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Shang, S.; Hua, F.; Hu, Z.-W. The regulation of β-catenin activity and function in cancer: Therapeutic opportunities. Oncotarget 2017, 8, 33972–33989. [Google Scholar] [CrossRef] [Green Version]
- Jamieson, C.; Sharma, M.; Henderson, B.R. Targeting the β-catenin nuclear transport pathway in cancer. Semin. Cancer Biol. 2014, 27, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Fodde, R.; Brabletz, T. Wnt/β-catenin signaling in cancer stemness and malignant behavior. Curr. Opin. Cell Biol. 2007, 19, 150–158. [Google Scholar] [CrossRef]
- Bienz, M. APC: The plot thickens. Curr. Opin. Genet. Dev. 1999, 9, 595–603. [Google Scholar] [CrossRef]
- Tillhon, M.; Cazzalini, O.; Nardo, T.; Necchi, D.; Sommatis, S.; Stivala, L.A.; Scovassi, A.I.; Prosperi, E. p300/CBP acetyl transferases interact with and acetylate the nucleotide excision repair factor XPG. DNA Repair 2012, 11, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Logan, C.Y.; Nusse, R. The Wnt Signaling Pathway in Development and Disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef] [Green Version]
- Eberhart, C.G.; Tihan, T.; Burger, P.C. Nuclear Localization and Mutation of β-Catenin in Medulloblastomas. J. Neuropathol. Exp. Neurol. 2000, 59, 333–337. [Google Scholar] [CrossRef]
- Fattet, S.; Haberler, C.; Legoix, P.; Varlet, P.; Lellouch-Tubiana, A.; Lair, S.; Manie, E.; Raquin, M.-A.; Bours, D.; Carpentier, S.; et al. Beta-catenin status in paediatric medulloblastomas: Correlation of immunohistochemical expression with mutational status, genetic profiles, and clinical characteristics. J. Pathol. 2009, 218, 86–94. [Google Scholar] [CrossRef]
- Lu, R.; Bosland, M.; Xia, Y.; Zhang, Y.-G.; Kato, I.; Sun, J. Presence of Salmonella AvrA in colorectal tumor and its precursor lesions in mouse intestine and human specimens. Oncotarget 2017, 8, 55104–55115. [Google Scholar] [CrossRef] [Green Version]
- Aberle, H.; Bauer, A.; Stappert, J.; Kispert, A.; Kemler, R. β-catenin is a target for the ubiquitin–proteasome pathway. EMBO J. 1997, 16, 3797–3804. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Kato, Y.; Zhang, Z.; Do, V.M.; Yankner, B.A.; He, X. -Trcp couples -catenin phosphorylation-degradation and regulates Xenopus axis formation. Proc. Natl. Acad. Sci. USA 1999, 96, 6273–6278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonetta, K.R.; Taygerly, J.; Boyle, K.; Basham, S.E.; Padovani, C.; Lou, Y.; Cummins, T.J.; Yung, S.L.; Von Soly, S.K.; Kayser, F.; et al. Prospective discovery of small molecule enhancers of an E3 ligase-substrate interaction. Nat. Commun. 2019, 10, 1402. [Google Scholar] [CrossRef]
- Rubinfeld, B.; Robbins, P.; El-Gamil, M.; Albert, I.; Porfiri, E.; Polakis, P. Stabilization of β-Catenin by Genetic Defects in Melanoma Cell Lines. Science 1997, 275, 1790–1792. [Google Scholar] [CrossRef]
- Manoranjan, B.; Venugopal, C.; Bakhshinyan, D.; Adile, A.A.; Richards, L.; Kameda-Smith, M.M.; Whitley, O.; Dvorkin-Gheva, A.; Subapanditha, M.; Savage, N.; et al. Wnt activation as a therapeutic strategy in medulloblastoma. Nat. Commun. 2020, 11, 4323. [Google Scholar] [CrossRef] [PubMed]
- Gajjar, A.J.; Robinson, G. Medulloblastoma—Translating discoveries from the bench to the bedside. Nat. Rev. Clin. Oncol. 2014, 11, 714–722. [Google Scholar] [CrossRef]
- Phoenix, T.N.; Patmore, D.M.; Boop, S.; Boulos, N.; Jacus, M.O.; Patel, Y.T.; Roussel, M.F.; Finkelstein, D.; Goumnerova, L.; Perreault, S.; et al. Medulloblastoma Genotype Dictates Blood Brain Barrier Phenotype. Cancer Cell 2016, 29, 508–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amino Acid Position 1 | Mutation 2 | This Study | Number of MB Cases 3 PeCan COSMIC BioPortal | Mutation Type | ||
|---|---|---|---|---|---|---|
| 32 | c.94G>A/p.(Asp32Asn)/D32N c.94G>T/p.(Asp32Tyr)/D32Y c.94G>C/p.(Asp32His)/D32H c.95A>C/p.(Asp32Ala)/D32A c.95A>G/p.(Asp32Gly)/D32G c.95A>T/p.(Asp32Val)/D32V | - 1 - - 1 - | - 2 - 1 2 - | 29 34 4 2 7 10 | 1 - - 1 2 - | Substitution-Missense Substitution-Missense Substitution-Missense Substitution-Missense Substitution-Missense Substitution-Missense |
| 33 | c.97_114del/p.(S33_G38del)/ΔS33-G38 c.97T>G/p.(Ser33Ala)/S33A c.97T>C/p.(Ser33Pro)/S33P c.98C>G/p.(Ser33Cys)/S33C c.98C>T/p.(Ser33Phe)/S33F c.98C>A/p.(Ser33Tyr)/S33Y | - - - 1 2 2 | 1 2 1 5 6 3 | - 3 5 45 40 22 | 1 - - 4 6 2 | Deletion-In frame Substitution-Missense Substitution-Missense Substitution-Missense Substitution-Missense Substitution-Missense |
| 34 | c.100G>A/p.(Gly34Arg)/G34R c.101G>A/p.(Gly34Glu)/G34E c.101G>T/p.(Gly34Val)/G34V | 1 - - | 4 2 1 | 25 17 6 | 5 1 1 | Substitution-Missense Substitution-Missense Substitution-Missense |
| 35 | ?/p.(Ile35Lys)/I35K c.104T>G/p.(Ile35Ser)/I35S | - - | 1 - | 1 1 | - - | Substitution-Missense Substitution-Missense |
| 37 | c.109_111del/p.(Ser37del)/ΔS37 c.109T>C/p.(Ser37Pro)/S37P c.110C>G/p.(Ser37Cys)/S37C c.110C>T/p.(Ser37Phe)/S37F c.110C>A/p.(Ser37Tyr)/S37Y | 1 - - - - | - 1 1 3 2 | - 6 10 17 10 | - 1 1 1 1 | Deletion-In frame Substitution-Missense Substitution-Missense Substitution-Missense Substitution-Missense |
| 40 | c.119C>G/p.(Thr40Ser)/T40S | - | - | 1 | - | Substitution-Missense |
| 41 | c.121A>G/p.(Thr41Ala)/T41A | - | 1 | 2 | 1 | Substitution-Missense |
| 45 | c.134C>T/p.(Ser45Phe)/S45F | - | - | 2 | - | Substitution-Missense |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alaña, L.; Nunes-Xavier, C.E.; Zaldumbide, L.; Martin-Guerrero, I.; Mosteiro, L.; Alba-Pavón, P.; Villate, O.; García-Obregón, S.; González-García, H.; Herraiz, R.; et al. Identification and Functional Analysis of a Novel CTNNB1 Mutation in Pediatric Medulloblastoma. Cancers 2022, 14, 421. https://doi.org/10.3390/cancers14020421
Alaña L, Nunes-Xavier CE, Zaldumbide L, Martin-Guerrero I, Mosteiro L, Alba-Pavón P, Villate O, García-Obregón S, González-García H, Herraiz R, et al. Identification and Functional Analysis of a Novel CTNNB1 Mutation in Pediatric Medulloblastoma. Cancers. 2022; 14(2):421. https://doi.org/10.3390/cancers14020421
Chicago/Turabian StyleAlaña, Lide, Caroline E. Nunes-Xavier, Laura Zaldumbide, Idoia Martin-Guerrero, Lorena Mosteiro, Piedad Alba-Pavón, Olatz Villate, Susana García-Obregón, Hermenegildo González-García, Raquel Herraiz, and et al. 2022. "Identification and Functional Analysis of a Novel CTNNB1 Mutation in Pediatric Medulloblastoma" Cancers 14, no. 2: 421. https://doi.org/10.3390/cancers14020421
APA StyleAlaña, L., Nunes-Xavier, C. E., Zaldumbide, L., Martin-Guerrero, I., Mosteiro, L., Alba-Pavón, P., Villate, O., García-Obregón, S., González-García, H., Herraiz, R., Astigarraga, I., Pulido, R., & García-Ariza, M. (2022). Identification and Functional Analysis of a Novel CTNNB1 Mutation in Pediatric Medulloblastoma. Cancers, 14(2), 421. https://doi.org/10.3390/cancers14020421