
Comprehensive Analysis of Co-Mutations Identifies Cooperating Mechanisms of Tumorigenesis




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
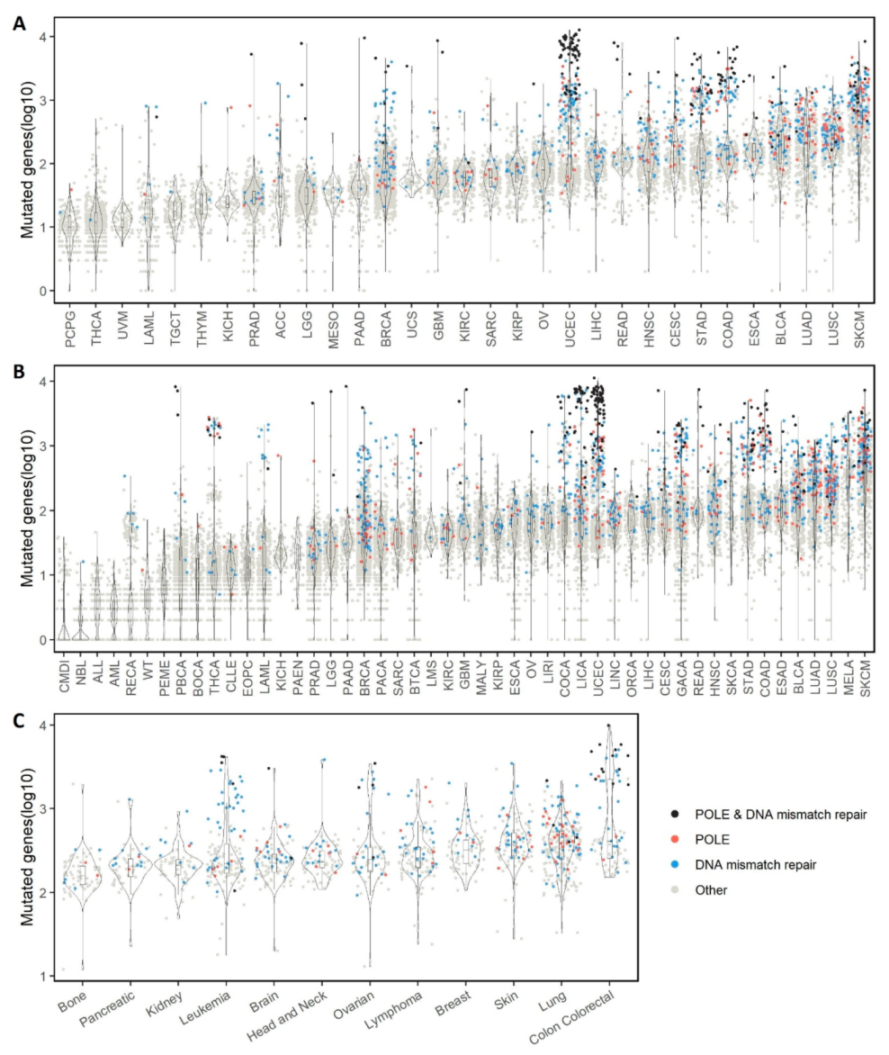
2.1. Overall Single Mutation Description
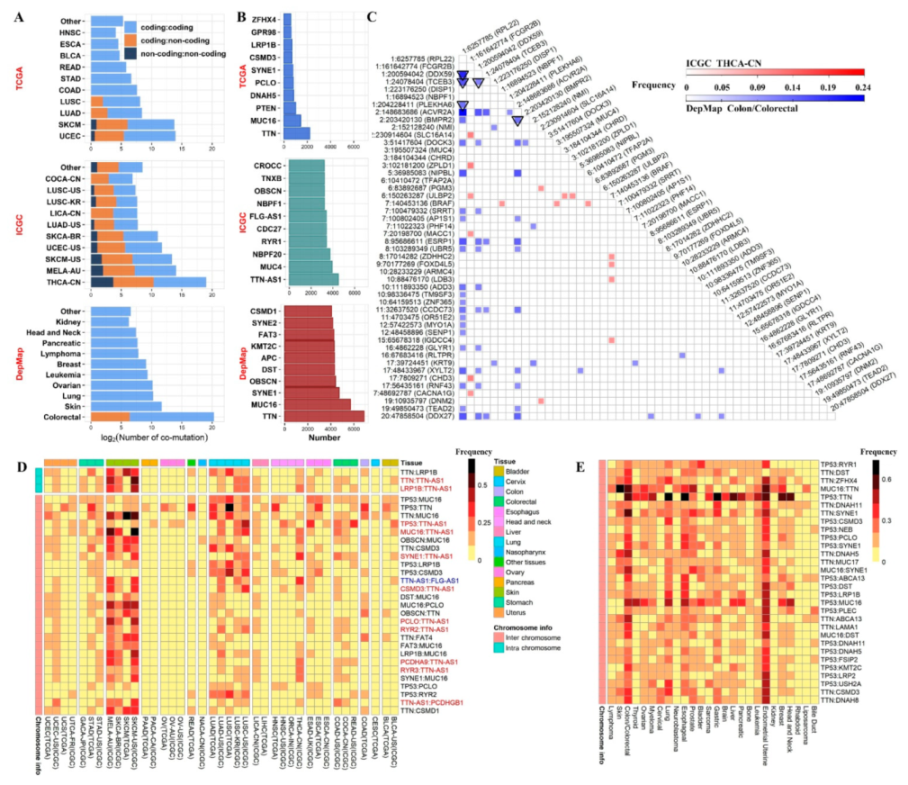
2.2. Overall Co-Mutation Description
2.3. Co-Mutation Disparity with Age, Sex, and Race
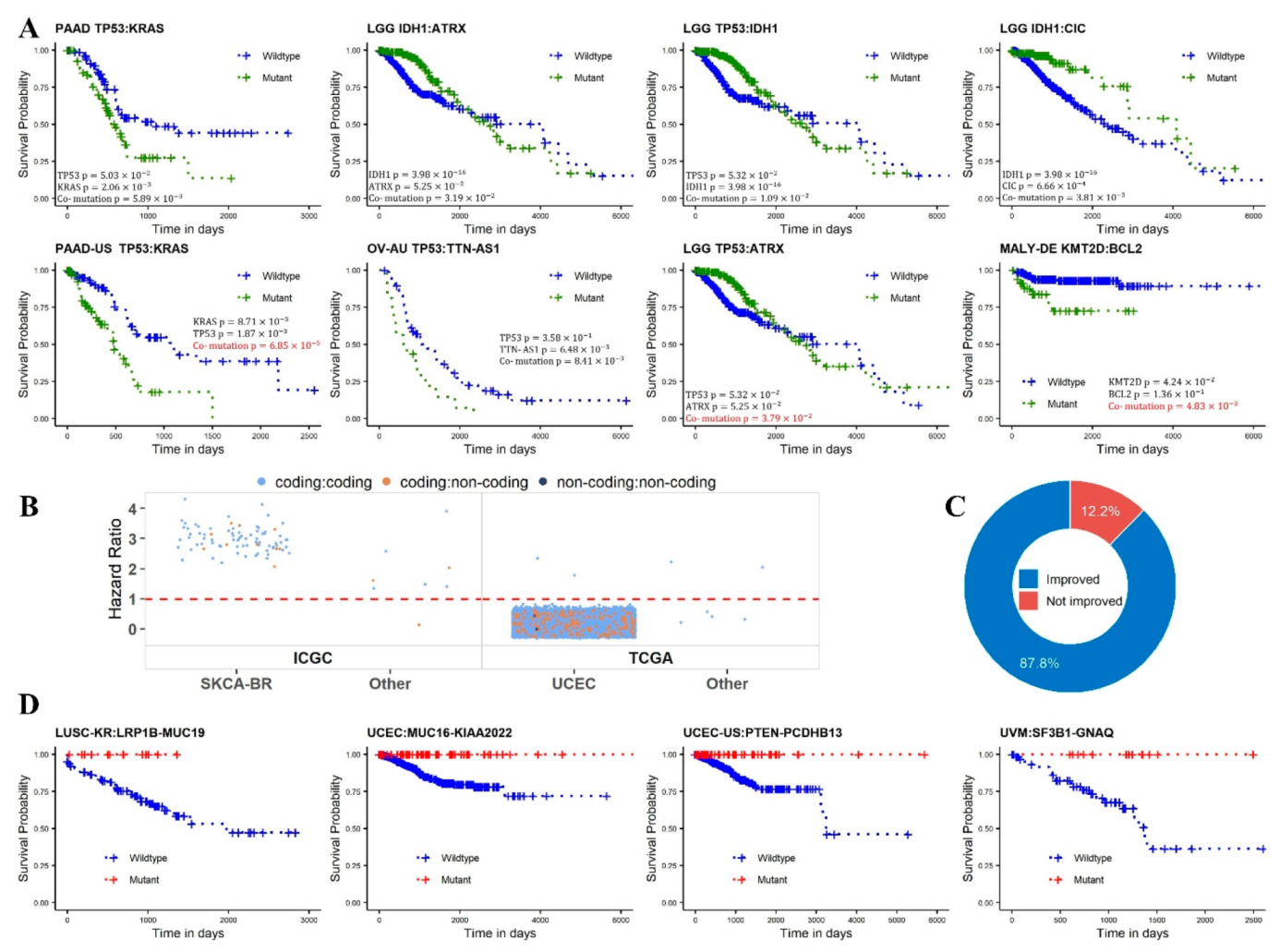
2.4. Survival Analysis
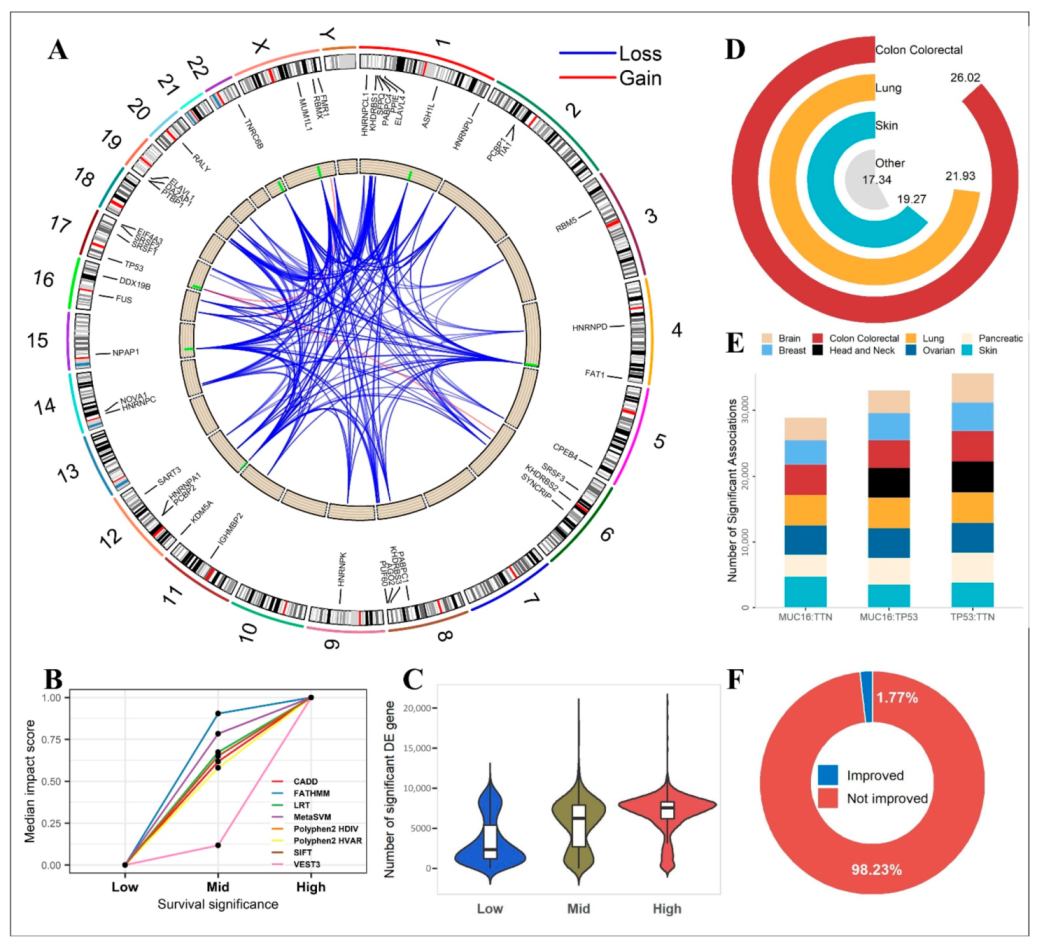
2.5. Functional Analysis
2.6. Comparisons with Clinical Cancer Gene Panels
3. Discussion
4. Materials and Methods
4.1. Data Acquisition
4.2. Mutation Annotation
4.3. Co-Mutation Definition
4.4. Phenotypic Variable Association Analysis
4.5. Survival Analysis
4.6. Regulatory Element Analysis
4.7. Mutation Impact Analysis
4.8. Clinical Cancer Gene Panels
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Skoulidis, F.; Heymach, J.V. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat. Rev. Cancer 2019, 19, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Offin, M.; Chan, J.M.; Tenet, M.; Rizvi, H.A.; Shen, R.; Riely, G.J.; Rekhtman, N.; Daneshbod, Y.; Quintanal-Villalonga, A.; Penson, A.; et al. Concurrent RB1 and TP53 Alterations Define a Subset of EGFR-Mutant Lung Cancers at risk for Histologic Transformation and Inferior Clinical Outcomes. J. Thorac. Oncol. 2019, 14, 1784–1793. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Byers, L.A.; Diao, L.; Papadimitrakopoulou, V.A.; Tong, P.; Izzo, J.; Behrens, C.; Kadara, H.; Parra, E.R.; Canales, J.R.; et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015, 5, 860–877. [Google Scholar] [CrossRef] [Green Version]
- Todisco, G.; Creignou, M.; Gallì, A.; Guglielmelli, P.; Rumi, E.; Roncador, M.; Rizzo, E.; Nannya, Y.; Pietra, D.; Elena, C.; et al. Co-mutation pattern, clonal hierarchy, and clone size concur to determine disease phenotype of SRSF2P95-mutated neoplasms. Leukemia 2021, 35, 2371–2381. [Google Scholar] [CrossRef]
- Wilson, M.R.; Reske, J.J.; Holladay, J.; Wilber, G.E.; Rhodes, M.; Koeman, J.; Adams, M.; Johnson, B.; Su, R.-W.; Joshi, N.R.; et al. ARID1A and PI3-kinase pathway mutations in the endometrium drive epithelial transdifferentiation and collective invasion. Nat. Commun. 2019, 10, 3554. [Google Scholar] [CrossRef]
- Wang, S.; Jiang, M.; Yang, Z.; Huang, X.; Li, N. The role of distinct co-mutation patterns with TP53 mutation in immunotherapy for NSCLC. Genes Dis. 2020, 9, 245–251. [Google Scholar] [CrossRef]
- Xian, R.R.; Xie, Y.; Haley, L.M.; Yonescu, R.; Pallavajjala, A.; Pittaluga, S.; Jaffe, E.S.; Duffield, A.S.; McCall, C.M.; Gheith, S.M.F.; et al. CREBBP and STAT6 co-mutation and 16p13 and 1p36 loss define the t(14;18)-negative diffuse variant of follicular lymphoma. Blood Cancer J. 2020, 10, 69. [Google Scholar] [CrossRef]
- Rosell, R.; Karachaliou, N. Co-mutations in EGFR driven non-small cell lung cancer. EBioMedicine 2019, 42, 18–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tessier-Cloutier, B.; Pors, J.; Thompson, E.; Ho, J.; Prentice, L.; McConechy, M.; Aguirre-Hernandez, R.; Miller, R.; Leung, S.; Proctor, L.; et al. Molecular characterization of invasive and in situ squamous neoplasia of the vulva and implications for morphologic diagnosis and outcome. Mod. Pathol. 2021, 34, 508–518. [Google Scholar] [CrossRef]
- Arbour, K.C.; Jordan, E.; Kim, H.R.; Dienstag, J.; Yu, H.A.; Sanchez-Vega, F.; Lito, P.; Berger, M.; Solit, D.B.; Hellmann, M.; et al. Effects of Co-occurring Genomic Alterations on Outcomes in Patients with KRAS-Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 334. [Google Scholar] [CrossRef] [Green Version]
- Wakita, S.; Yamaguchi, H.; Ueki, T.; Usuki, K.; Kurosawa, S.; Kobayashi, Y.; Kawata, E.; Tajika, K.; Gomi, S.; Koizumi, M.; et al. Complex molecular genetic abnormalities involving three or more genetic mutations are important prognostic factors for acute myeloid leukemia. Leukemia 2016, 30, 545–554. [Google Scholar] [CrossRef]
- Wang, F.; Zhao, N.; Gao, G.; Deng, H.-B.; Wang, Z.-H.; Deng, L.-L.; Yang, Y.; Lu, C. Prognostic value of TP53 co-mutation status combined with EGFR mutation in patients with lung adenocarcinoma. J. Cancer Res. Clin. Oncol. 2020, 146, 2851–2859. [Google Scholar] [CrossRef]
- Campbell, B.B.; Light, N.; Fabrizio, D.; Zatzman, M.; Fuligni, F.; de Borja, R.; Davidson, S.; Edwards, M.; Elvin, J.A.; Hodel, K.P.; et al. Comprehensive Analysis of Hypermutation in Human Cancer. Cell 2017, 171, 1042–1056. [Google Scholar] [CrossRef] [Green Version]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef] [Green Version]
- Risques, R.A.; Kennedy, S.R. Aging and the rise of somatic cancer-associated mutations in normal tissues. PLoS Genet. 2018, 14, >e1007108. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Artomov, M.; Goggins, W.; Daly, M.; Tsao, H. Gender Disparity and Mutation Burden in Metastatic Melanoma. JNCI-J. Natl. Cancer I. 2015, 107, djv221. [Google Scholar] [CrossRef] [PubMed]
- Wesa, A.K.; Mandic, M.; Taylor, J.L.; Moschos, S.; Kirkwood, J.M.; Kwok, W.W.; Finke, J.H.; Storkus, W.J. Circulating Type-1 Anti-Tumor CD4(+) T Cells are Preferentially Pro-Apoptotic in Cancer Patients. Front Oncol. 2014, 4, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.L.; Luo, C.L.; Shen, L.; Liu, Y.G.; Wang, Q.Q.; Zhang, C.; Guo, R.C.; Zhang, Y.N.; Xie, Z.Q.; Wei, N.; et al. SRSF1 Prevents DNA Damage and Promotes Tumorigenesis through Regulation of DBF4B Pre-mRNA Splicing. Cell Rep. 2017, 21, 3406–3413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Tebaldi, T.; Rejeski, K.; Joshi, P.; Stefani, G.; Taylor, A.; Song, Y.B.; Vasic, R.; Maziarz, J.; Balasubramanian, K.; et al. SRSF2 mutations drive oncogenesis by activating a global program of aberrant alternative splicing in hematopoietic cells. Leukemia 2018, 32, 2659–2671. [Google Scholar] [CrossRef]
- Song, X.; Wan, X.C.; Huang, T.Z.; Zeng, C.; Sastry, N.; Wu, B.L.; James, C.D.; Horbinski, C.; Nakano, I.; Zhang, W.; et al. SRSF3-Regulated RNA Alternative Splicing Promotes Glioblastoma Tumorigenicity by Affecting Multiple Cellular Processes. Cancer Res. 2019, 79, 5288–5301. [Google Scholar] [CrossRef] [Green Version]
- Yan, Q.X.; Zeng, P.; Zhou, X.Q.; Zhao, X.Y.; Chen, R.Q.; Qiao, J.; Feng, L.; Zhu, Z.J.; Zhang, G.Z.; Chen, C.R. RBMX suppresses tumorigenicity and progression of bladder cancer by interacting with the hnRNP A1 protein to regulate PKM alternative splicing. Oncogene 2021, 40, 2635–2650. [Google Scholar] [CrossRef] [PubMed]
- Argani, P.; Zhang, L.; Sung, Y.S.; White, M.J.; Miller, K.; Hopkins, M.; Small, D.; Pratilas, C.A.; Swanson, D.; Dickson, B.; et al. A novel RBMX-TFE3 gene fusion in a highly aggressive pediatric renal perivascular epithelioid cell tumor. Gene Chromosome Cancer 2020, 59, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.X.; Xu, X.N.; Pan, B.; Zeng, K.X.; Xu, M.; Liu, X.X.; He, B.S.; Pan, Y.Q.; Sun, H.L.; Wang, S.K. miR-150-5p suppresses tumor progression by targeting VEGFA in colorectal cancer. Aging 2018, 10, 3421–3437. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, B.S.J.; Dardshaw, K.; Halvorson, L.; Schaffer, J.; Corton, M. Williams Gynecology. 2012. Available online: https://accessmedicine.mhmedical.com/content.aspx?bookid=1758§ionid=118165489 (accessed on 12 December 2021).
- Ye, B.; Shi, J.; Kang, H.; Oyebamiji, O.; Hill, D.; Yu, H.; Ness, S.; Ye, F.; Ping, J.; He, J.; et al. Advancing Pan-cancer Gene Expression Survial Analysis by Inclusion of Non-coding RNA. RNA Biol. 2020, 17, 1666–1673. [Google Scholar] [CrossRef]
- Liu, J.F.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Jiang, L.; Duan, M.; Guo, F.; Tang, J.; Oybamiji, O.; Yu, H.; Ness, S.; Zhao, Y.-Y.; Mao, P.; Guo, Y. SMDB: Pivotal somatic sequence alterations reprogramming regulatory cascades. NAR Cancer 2020, 2, zcaa030. [Google Scholar] [CrossRef]
- Jiang, L.; Guo, F.; Tang, J.; Yu, H.; Ness, S.; Duan, M.; Mao, P.; Zhao, Y.-Y.; Guo, Y. SBSA: An online service for somatic binding sequence annotation. Nucleic Acids Res. 2022, 50, e4. [Google Scholar] [CrossRef]
- Giudice, G.; Sanchez-Cabo, F.; Torroja, C.; Lara-Pezzi, E. ATtRACT-a database of RNA-binding proteins and associated motifs. Database 2016, 2016, baw035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benoit Bouvrette, L.P.; Bovaird, S.; Blanchette, M.; Lecuyer, E. oRNAment: A database of putative RNA binding protein target sites in the transcriptomes of model species. Nucleic Acids Res. 2020, 48, D166–D173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berglund, A.C.; Sjolund, E.; Ostlund, G.; Sonnhammer, E.L. InParanoid 6: Eukaryotic ortholog clusters with inparalogs. Nucleic Acids Res. 2008, 36, D263–D266. [Google Scholar] [CrossRef] [PubMed]
- Paz, I.; Kosti, I.; Ares, M.; Jr Cline, M.; Mandel-Gutfreund, Y. RBPmap: A web server for mapping binding sites of RNA-binding proteins. Nucleic Acids Res. 2014, 42, W361–W367. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [Green Version]
- Li, J.H.; Liu, S.; Zhou, H.; Qu, L.H.; Yang, J.H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Meltzer, P.; Davis, S. RCircos: An R package for Circos 2D track plots. BMC Bioinform. 2013, 14, 244. [Google Scholar] [CrossRef] [Green Version]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Chun, S.; Fay, J.C. Identification of deleterious mutations within three human genomes. Genome Res. 2009, 19, 1553–1561. [Google Scholar] [CrossRef] [Green Version]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.A.; Edwards, K.J.; Day, I.N.M.; Gaunt, T.R. Predicting the Functional, Molecular, and Phenotypic Consequences of Amino Acid Substitutions using Hidden Markov Models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Carter, H.; Douville, C.; Stenson, P.D.; Cooper, D.N.; Karchin, R. Identifying Mendelian disease genes with the Variant Effect Scoring Tool. BMC Genom. 2013, 14, S3. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.L.; Wei, P.; Jian, X.Q.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X.M. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ping, J.; Oyebamiji, O.; Yu, H.; Ness, S.; Chien, J.; Ye, F.; Kang, H.; Samuels, D.; Ivanov, S.; Chen, D.; et al. MutEx: A multifaceted gateway for exploring integrative pan-cancer genomic data. Brief Bioinform. 2019, 21, 1479–1486. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, L.; Yu, H.; Ness, S.; Mao, P.; Guo, F.; Tang, J.; Guo, Y. Comprehensive Analysis of Co-Mutations Identifies Cooperating Mechanisms of Tumorigenesis. Cancers 2022, 14, 415. https://doi.org/10.3390/cancers14020415
Jiang L, Yu H, Ness S, Mao P, Guo F, Tang J, Guo Y. Comprehensive Analysis of Co-Mutations Identifies Cooperating Mechanisms of Tumorigenesis. Cancers. 2022; 14(2):415. https://doi.org/10.3390/cancers14020415
Chicago/Turabian StyleJiang, Limin, Hui Yu, Scott Ness, Peng Mao, Fei Guo, Jijun Tang, and Yan Guo. 2022. "Comprehensive Analysis of Co-Mutations Identifies Cooperating Mechanisms of Tumorigenesis" Cancers 14, no. 2: 415. https://doi.org/10.3390/cancers14020415
APA StyleJiang, L., Yu, H., Ness, S., Mao, P., Guo, F., Tang, J., & Guo, Y. (2022). Comprehensive Analysis of Co-Mutations Identifies Cooperating Mechanisms of Tumorigenesis. Cancers, 14(2), 415. https://doi.org/10.3390/cancers14020415