
CDK5RAP3, a New BRCA2 Partner That Regulates DNA Repair, Is Associated with Breast Cancer Survival

 , , , , , , ,
, , , , , , ,  and
and  add
Show full author list
add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell lines and Plasmids
2.3. Western Blot
2.4. siRNA Transfection Oligonucleotides
2.5. Analysis of BRCA2 Interactions
2.6. Laser Microirradiation Experiments
2.7. Survival and Clonogenic Assays
2.8. In Vitro Flow Cytometric Micronuclei (FCM) Assay
2.9. DNA Repair Assays
2.10. Analysis of Publicly Available Cancer Transcriptomic Datasets
3. Results
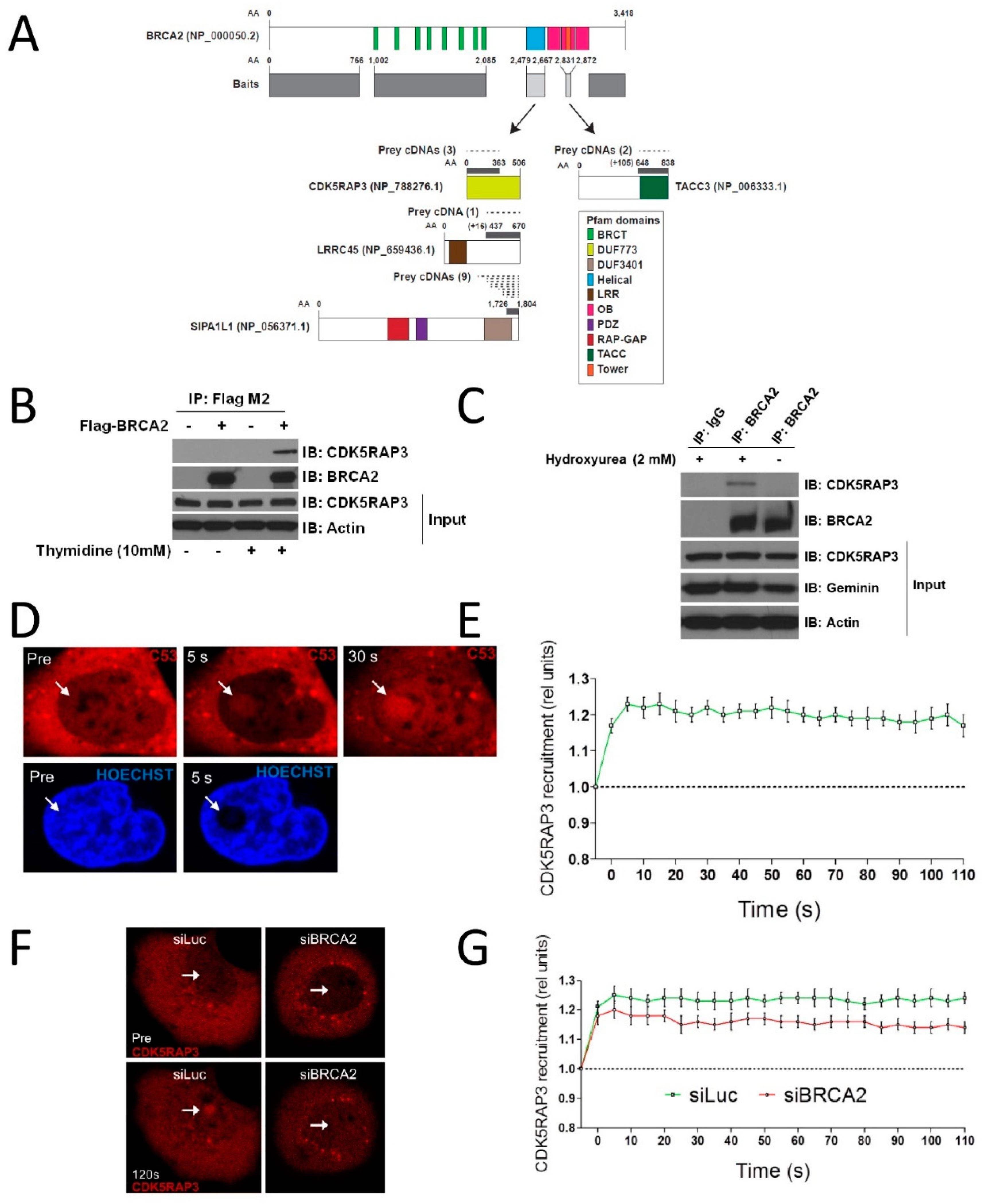
3.1. CDK5RAP3 Interacts with BRCA2 and Localizes to DNA Damage
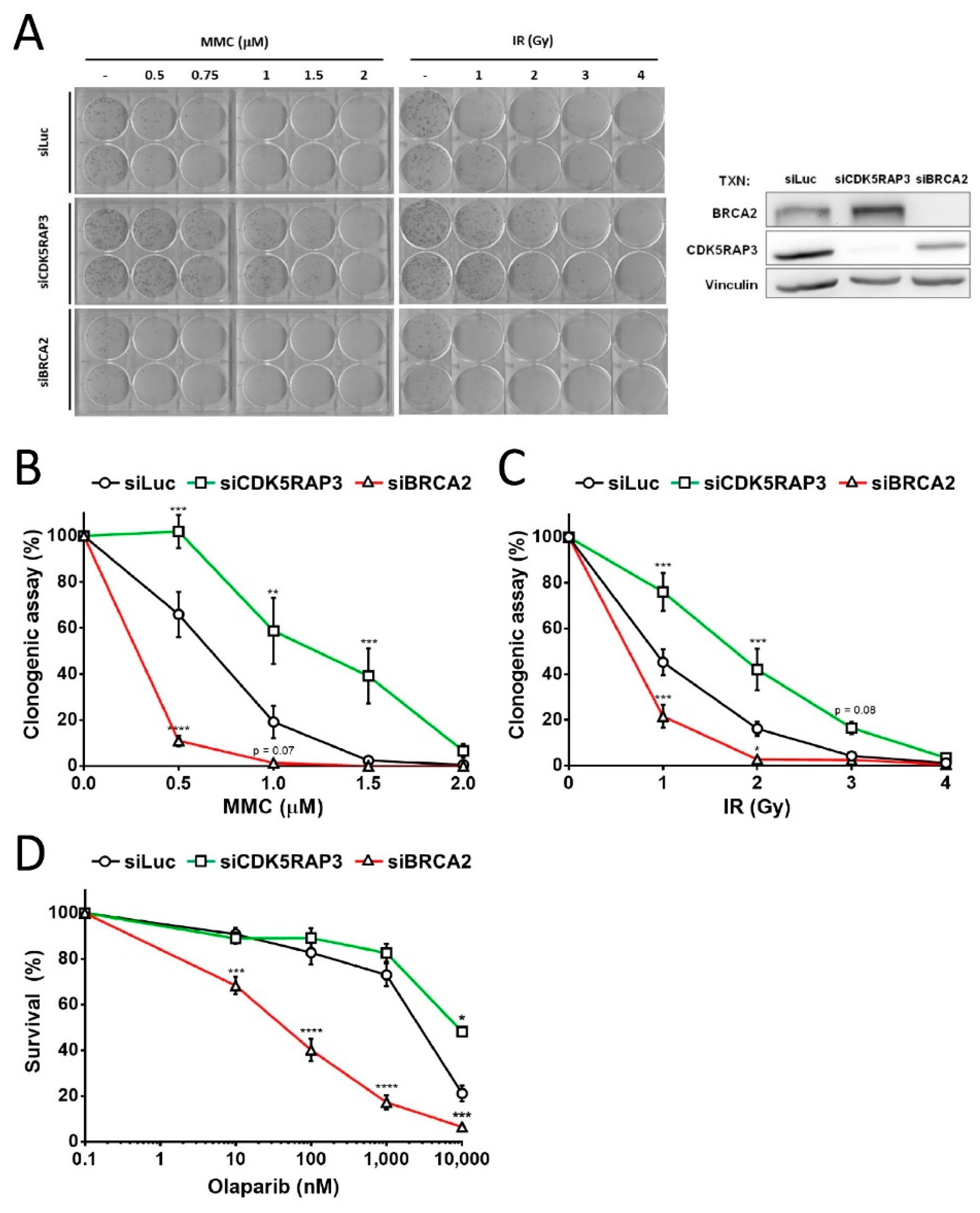
3.2. CDK5RAP3-Depleted Cells Are Resistant DNA Damage
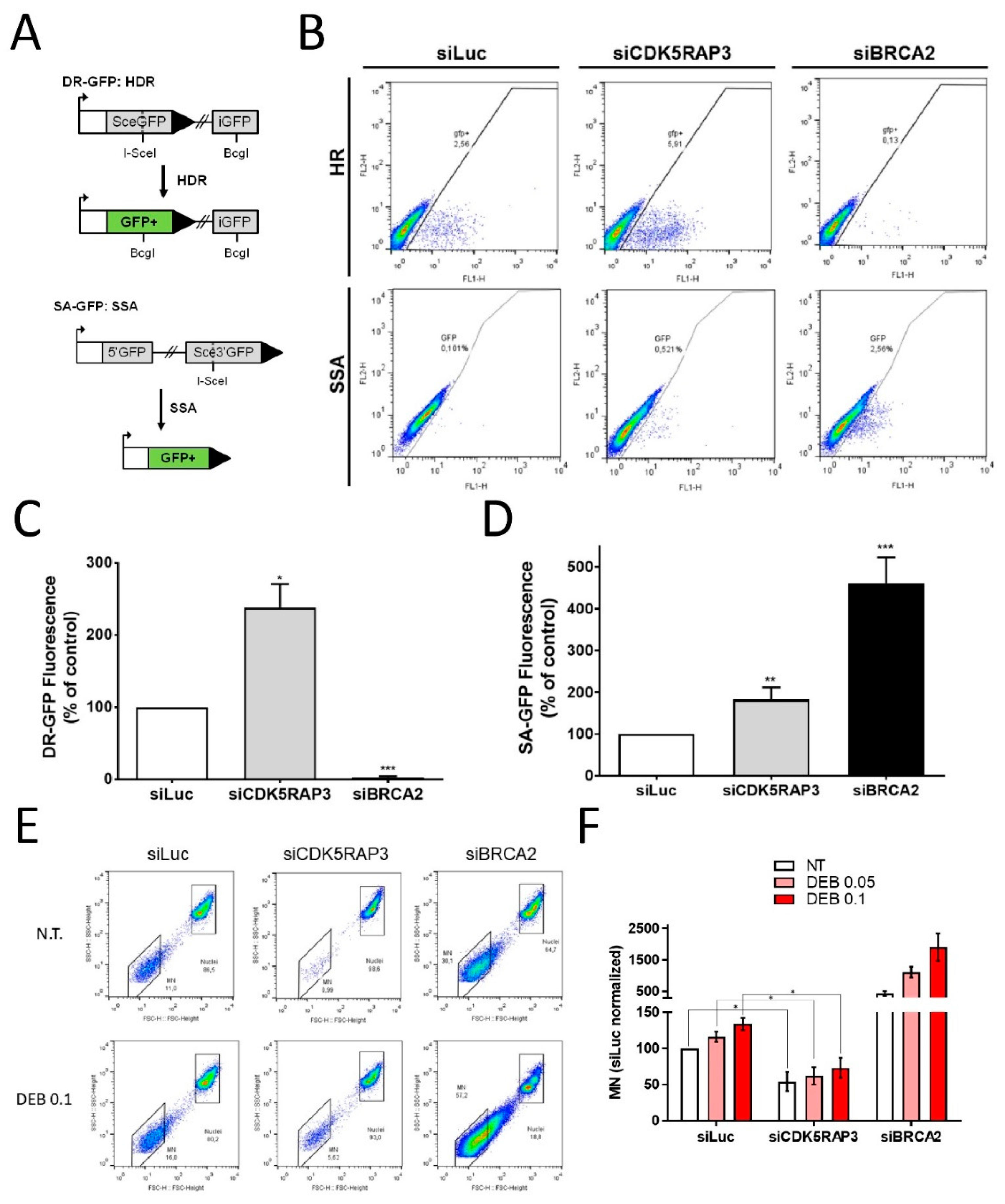
3.3. CDK5RAP3 Regulates DSB Repair and Genomic Instability
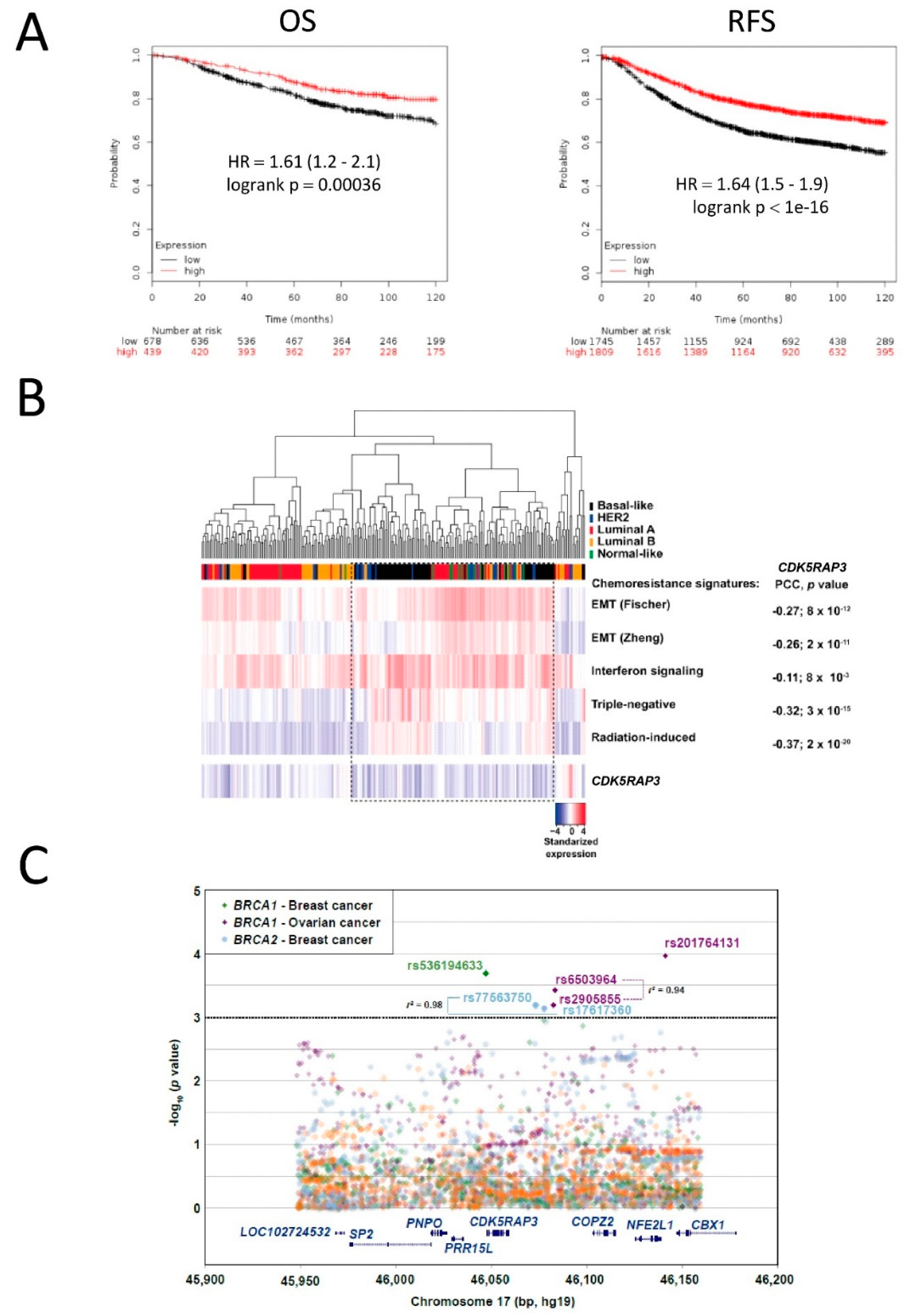
3.4. CDK5RAP3 Expression Is Associated with Poor Breast and Ovarian Cancer Survival
3.5. SNPs in CDK5RAP3 May Be Associated with Cancer Risk in BRCA1/2 Mutation Carriers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell. 2010, 40, 179–204. [Google Scholar]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2012, 12, 68–78. [Google Scholar]
- Lee, H. Cycling with BRCA2 from DNA repair to mitosis. Exp. Cell Res. 2014, 329, 78–84. [Google Scholar] [PubMed] [Green Version]
- Guidugli, L.; Carreira, A.; Caputo, S.M.; Ehlen, A.; Galli, A.; Monteiro, A.N.A.; Nwuhausen, S.L.; Hansen, T.V.O.; Couch, F.J.; Vreeswijk, M.P.G.; et al. Functional assays for analysis of variants of uncertain significance in BRCA2. Hum. Mutat. 2014, 35, 151–164. [Google Scholar] [PubMed] [Green Version]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous recombination and human health: The roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol. 2015, 7, a016600. [Google Scholar]
- Bogliolo, M.; Surrallés, J. Fanconi anemia: A model disease for studies on human genetics and advanced therapeutics. Curr. Opin. Genet. Dev. 2015, 33, 32–40. [Google Scholar] [PubMed] [Green Version]
- Easton, D.F.; Lesueur, F.; Decker, B.; Michailidou, K.; Li, J.; Allen, J.; Luccarini, C.; Pooley, K.A.; Shah, M.; Bolla, M.K.; et al. No evidence that protein truncating variants in BRIP1 are associated with breast cancer risk: Implications for gene panel testing. J. Med. Genet. 2016, 53, 298–309. [Google Scholar] [PubMed] [Green Version]
- Desmond, A.; Kurian, A.W.; Gabree, M.; Mills, M.A.; Anderson, M.J.; Kobayashi, Y.; Horick, N.; Yang, S.; Shannon, K.; Tung, N.; et al. Clinical Actionability of Multigene Panel Testing for Hereditary Breast and Ovarian Cancer Risk Assessment. JAMA Oncol. 2015, 1, 943–951. [Google Scholar]
- Knies, K.; Inano, S.; Ramírez, M.J.; Ishiai, M.; Surrallés, J.; Takata, M.; Schindler, D. Biallelic mutations in the ubiquitin ligase RFWD3 cause Fanconi anemia. J. Clin. Investig. 2017, 127, 3013–3027. [Google Scholar] [PubMed] [Green Version]
- Walsh, T.; King, M.-C. Ten genes for inherited breast cancer. Cancer Cell 2007, 11, 103–105. [Google Scholar]
- Rahman, N.; Seal, S.; Thompson, D.; Kelly, P.; Renwick, A.; Elliott, A.; Reid, S.; Spanova, K.; Barfoot, R.; Chagtai, T.; et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat. Genet. 2007, 39, 165–167. [Google Scholar] [PubMed] [Green Version]
- Southey, M.C.; Fab, K.C.; Teo, Z.L.; Dowty, J.G.; Odefrey, F.A.; Park, D.J.; Tischkowitz, M.; Sabbaghian, N.; Apicella, C.; Byrnes, G.B.; et al. A PALB2 mutation associated with high risk of breast cancer. Breast Cancer Res. 2010, 12, R109. [Google Scholar]
- Antoniou, A.C.; Casadei, S.; Heikkinen, T.; Barrowdale, D.; Pylkäs, K.; Roberts, J.; Lee, A.; Subramanian, D.; De Leeneer, K.; Fostira, F.; et al. Breast-cancer risk in families with mutations in PALB2. N. Engl. J. Med. 2014, 371, 497–506. [Google Scholar] [PubMed] [Green Version]
- Meindl, A.; Hellebrand, H.; Wiek, C.; Erven, V.; Wappenschmidt, B.; Niederacher, D.; Freund, M.; Lichtner, P.; Hartmann, L.; Schaal, H.; et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat. Genet. 2010, 42, 410–414. [Google Scholar]
- Peterlongo, P.; Catucci, I.; Colombo, M.; Caleca, L.; Mucaki, E.; Bogliolo, M.; Marin, M.; Damiola, F.; Bernard, L.; Pensotti, V.; et al. FANCM c.5791C>T nonsense mutation (rs144567652) induces exon skipping, affects DNA repair activity and is a familial breast cancer risk factor. Hum. Mol. Genet. 2015, 24, 5345–5355. [Google Scholar]
- Jiang, H.; Luo, S.; Li, H. Cdk5 activator-binding protein C53 regulates apoptosis induced by genotoxic stress via modulating the G2/M DNA damage checkpoint. J. Biol. Chem. 2005, 280, 20651–20659. [Google Scholar]
- Wang, J.; He, X.; Luo, Y.; Yarbrough, W.G. A novel ARF-binding protein (LZAP) alters ARF regulation of HDM2. Biochem. J. 2006, 393, 489–501. [Google Scholar]
- Wang, J.; An, H.; Mayo, M.W.; Baldwin, A.S.; Yarbrough, W.G. LZAP, a putative tumor suppressor, selectively inhibits NF-kappaB. Cancer Cell 2007, 12, 239–251. [Google Scholar] [PubMed] [Green Version]
- Jiang, H.; Wu, J.; He, C.; Yang, W.; Li, H. Tumor suppressor protein C53 antagonizes checkpoint kinases to promote cyclin-dependent kinase 1 activation. Cell Res. 2009, 19, 458–468. [Google Scholar]
- Zhao, J.-J.; Pan, K.; Li, J.-J.; Chen, Y.-B.; Chen, J.-G.; Lv, L.; Wang, D.-D.; Pan, Q.-Z.; Chen, M.-S.; Xia, J.-C. Identification of LZAP as a New Candidate Tumor Suppressor in Hepatocellular Carcinoma. PLoS ONE. 2011, 6, e26608. [Google Scholar]
- Montanuy, H.; Camps-Fajol, C.; Carreras-Puigvert, J.; Haggblad, M.; Lundgren, B.; Aza-Carmona, M.; Helleday, T.; Minguillon, J.; Surralles, J. High content drug screening for Fanconi anemia therapeutics. Orphanet. J. Rare Dis. 2020, 15, 170. [Google Scholar]
- Martrat, G.; Maxwell, C.M.; Tominaga, E.; Porta-de-la-Riva, M.; Bonifaci, N.; Gómez-Baldó, L.; Bogliolo, M.; Lazaro, C.; Blanco, I.; Brunet, J.; et al. Exploring the link between MORF4L1 and risk of breast cancer. Breast Cancer Res. 2011, 13, R40. [Google Scholar]
- Hernández, G.; Ramírez, M.J.; Minguillón, J.; Quiles, P.; de Garibay, G.R.; Aza-Carmona, M.; Bogliolo, M.; Pujol, R.; Prados, R.; Fernández, J.; et al. Decapping protein EDC4 regulates DNA repair and phenocopies BRCA1. Nat. Commun. 2018, 9, 967. [Google Scholar] [PubMed] [Green Version]
- Montanuy, H.; Martínez-Barriocanal, A.; Casado, J.A.; Rovirosa, L.; Ramírez, M.J.; Nieto, R.; Carrascoso-Rubio, C.; Riera, P.; González, A.; Lerma, E.; et al. Gefitinib and afatinib show potential efficacy for fanconi anemia-related head and neck cancer. Clin. Cancer Res. 2020, 26, 3044–3057. [Google Scholar]
- Pierce, A.J.; Johnson, R.D.; Thompson, L.H.; Jasin, M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999, 13, 2633–2638. [Google Scholar] [PubMed] [Green Version]
- Stark, J.M.; Pierce, A.J.; Oh, J.; Pastink, A.; Jasin, M. Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol. Cell Biol. 2004, 24, 9305–9316. [Google Scholar]
- Bennardo, N.; Cheng, A.; Huang, N.; Stark, J.M. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet 2008, 4, e1000110. [Google Scholar]
- Győrffy, B.; Surowiak, P.; Budczies, J.; Lánczky, A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS ONE 2013, 8, e82241. [Google Scholar]
- Ramírez, M.J.; Minguillón, J.; Loveless, S.; Lake, K.; Carrasco, E.; Stjepanovic, N.; Balmaña, J.; Català, A.; Mehta, P.A.; Surrallés, J. Chromosome fragility in the buccal epithelium in patients with Fanconi anemia. Cancer Lett. 2020, 472, 1–7. [Google Scholar]
- Avlasevich, S.L.; Bryce, S.M.; Cairns, S.E.; Dertinger, S.D. In vitro micronucleus scoring by flow cytometry: Differential staining of micronuclei versus apoptotic and necrotic chromatin enhances assay reliability. Environ. Mol. Mutagen. 2006, 47, 56–66. [Google Scholar] [PubMed]
- CGAN. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.C.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.S.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar]
- Weichselbaum, R.R.; Ishwaran, H.; Yoon, T.; Nuyten, D.S.A.; Baker, S.; Khodarev, N.; Su, A.W.; Shaikh, A.Y.; Roach, P.; Kreike, B.; et al. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 18490–18495. [Google Scholar] [PubMed] [Green Version]
- Oh, D.S.; Cheang, M.C.U.; Fan, C.; Perou, C.M. Radiation-induced gene signature predicts pathologic complete response to neoadjuvant chemotherapy in breast cancer patients. Radiat. Res. 2014, 181, 193–207. [Google Scholar]
- Liu, Y.-R.; Jiang, Y.-Z.; Xu, X.-E.; Hu, X.; Yu, K.-D.; Shao, Z.-M. Comprehensive Transcriptome Profiling Reveals Multigene Signatures in Triple-Negative Breast Cancer. Clin. Cancer Res. 2016, 22, 1653–1662. [Google Scholar]
- Rebbeck, T.R.; Mitra, N.; Domchek, S.M.; Wan, F.; Friebel, T.; Tran, T.V.; Singer, C.F.; Tea, M.-K.M.; Blum, J.L.; Tung, N.; et al. Modification of BRCA1-Associated Breast and Ovarian Cancer Risk by BRCA1-Interacting Genes. Cancer Res. 2011, 71, 5792–5805. [Google Scholar]
- Maxwell, C.A.; Benítez, J.; Gómez-Baldó, L.; Osorio, A.; Bonifaci, N.; Fernández-Ramires, R.; Costes, S.; Guinó, E.; Chen, H.; Evans, G.; et al. Interplay between BRCA1 and RHAMM Regulates Epithelial Apicobasal Polarization and May Influence Risk of Breast Cancer. PLoS Biol. 2011, 9, e1001199. [Google Scholar]
- Chenevix-Trench, G.; Milne, R.L.; Antoniou, A.C.; Couch, F.J.; Easton, D.F.; Goldgar, D.E.; CIMBA. An international initiative to identify genetic modifiers of cancer risk in BRCA1 and BRCA2 mutation carriers: The Consortium of Investigators of Modifiers of BRCA1 and BRCA2 (CIMBA). Breast Cancer Res. 2007, 9, 104. [Google Scholar]
- Tutt, A.; Bertwistle, D.; Valentine, J.; Gabriel, A.; Swift, S.; Ross, G.; Griffin, C.; Thacker, J.; Ashworth, A. Mutation in Brca2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences. EMBO J. 2001, 20, 4704–4716. [Google Scholar]
- Larminat, F.; Germanier, M.; Papouli, E.; Defais, M. Deficiency in BRCA2 leads to increase in non-conservative homologous recombination. Oncogene 2002, 21, 5188–5192. [Google Scholar]
- Guidugli, L.; Pankratz, V.S.; Singh, N.; Thompson, J.; Erding, C.A.; Engel, C.; Schmutzler, R.; Domchek, S.; Nathanson, K.; Radice, P.; et al. A classification model for BRCA2 DNA binding domain missense variants based on homology-directed repair activity. Cancer Res. 2013, 73, 265–275. [Google Scholar]
- Feng, Z.; Scott, S.P.; Bussen, W.; Sharma, G.G.; Guo, G.; Pandita, T.K.; Powell, S.N. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc. Natl. Acad. Sci. USA 2011, 108, 686–691. [Google Scholar] [PubMed] [Green Version]
- Lok, B.H.; Powell, S.N. Molecular pathways: Understanding the role of Rad52 in homologous recombination for therapeutic advancement. Clin. Cancer Res. 2012, 18, 6400–6406. [Google Scholar]
- Lok, B.H.; Carley, C.; Tchang, B.; Powell, S.N. RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene 2013, 32, 3552–3558. [Google Scholar]
- Ward, A.; Khanna, K.K.; Wiegmans, A.P. Targeting homologous recombination, new pre-clinical and clinical therapeutic combinations inhibiting RAD51. Cancer Treat Rev. 2015, 41, 35–45. [Google Scholar] [PubMed]
- Gavande, N.S.; VanderVere-Carozza, P.S.; Hinshaw, H.D.; Jalal, S.I.; Sears, C.R.; Pawelczak, K.S.; Turchi, J.J. DNA repair targeted therapy: The past or future of cancer treatment? Pharmacol. Ther. 2016, 160, 65–83. [Google Scholar] [PubMed]
- Chandramouly, G.; McDevitt, S.; Sullivan, K.; Kent, T.; Luz, A.; Glickman, J.F.; Andrake, M.; Skorski, T.; Pomerantz, R.T. Small-Molecule Disruption of RAD52 Rings as a Mechanism for Precision Medicine in BRCA-Deficient Cancers. Chem. Biol. 2015, 22, 1491–1504. [Google Scholar]
- Huang, F.; Goyal, N.; Sullivan, K.; Hanamshet, K.; Patel, M.; Mazina, O.M.; Wang, C.X.; An, W.F.; Spoonamore, J.; Metkar, S.; et al. Targeting BRCA1- and BRCA2-deficient cells with RAD52 small molecule inhibitors. Nucleic Acids Res. 2016, 44, 4189–4199. [Google Scholar]
- Sullivan, K.; Cramer-Morales, K.; McElroy, D.L.; Ostrov, D.A.; Haas, K.; Childers, W.; Hromas, R.; Skorski, T. Identification of a Small Molecule Inhibitor of RAD52 by Structure-Based Selection. PLoS ONE 2016, 11, e0147230. [Google Scholar]
- Jonkers, J.; Meuwissen, R.; van der Gulden, H.; Peterse, H.; van der Valk, M.; Berns, A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat. Genet. 2001, 29, 418–425. [Google Scholar]
- Jonkers, J.; Berns, A. Conditional mouse models of sporadic cancer. Nat. Rev. Cancer 2002, 2, 251–265. [Google Scholar] [PubMed]
- Holstege, H.; Joosse, S.A.; van Oostrom, C.T.M.; Nederlof, P.M.; de Vries, A.; Jonkers, J. High incidence of protein-truncating TP53 mutations in BRCA1-related breast cancer. Cancer Res. 2009, 69, 3625–3633. [Google Scholar] [PubMed] [Green Version]
- Bowman-Colin, C.; Xia, B.; Bunting, S.; Klijn, C.; Drost, R.; Bouwman, P.; Fineman, L.; Chen, X.; Culhane, A.; Cai, H.; et al. Palb2 synergizes with Trp53 to suppress mammary tumor formation in a model of inherited breast cancer. Proc. Natl. Acad Sci. USA 2013, 110, 8632–8637. [Google Scholar] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minguillón, J.; Ramírez, M.J.; Rovirosa, L.; Bustamante-Madrid, P.; Camps-Fajol, C.; Ruiz de Garibay, G.; Shimelis, H.; Montanuy, H.; Pujol, R.; Hernandez, G.; et al. CDK5RAP3, a New BRCA2 Partner That Regulates DNA Repair, Is Associated with Breast Cancer Survival. Cancers 2022, 14, 353. https://doi.org/10.3390/cancers14020353
Minguillón J, Ramírez MJ, Rovirosa L, Bustamante-Madrid P, Camps-Fajol C, Ruiz de Garibay G, Shimelis H, Montanuy H, Pujol R, Hernandez G, et al. CDK5RAP3, a New BRCA2 Partner That Regulates DNA Repair, Is Associated with Breast Cancer Survival. Cancers. 2022; 14(2):353. https://doi.org/10.3390/cancers14020353
Chicago/Turabian StyleMinguillón, Jordi, María José Ramírez, Llorenç Rovirosa, Pilar Bustamante-Madrid, Cristina Camps-Fajol, Gorka Ruiz de Garibay, Hermela Shimelis, Helena Montanuy, Roser Pujol, Gonzalo Hernandez, and et al. 2022. "CDK5RAP3, a New BRCA2 Partner That Regulates DNA Repair, Is Associated with Breast Cancer Survival" Cancers 14, no. 2: 353. https://doi.org/10.3390/cancers14020353
APA StyleMinguillón, J., Ramírez, M. J., Rovirosa, L., Bustamante-Madrid, P., Camps-Fajol, C., Ruiz de Garibay, G., Shimelis, H., Montanuy, H., Pujol, R., Hernandez, G., Bogliolo, M., Castillo, P., Soucy, P., Martrat, G., Gómez, A., Cuadras, D., García, M. J., Gayarre, J., CIMBA, ... Surrallés, J. (2022). CDK5RAP3, a New BRCA2 Partner That Regulates DNA Repair, Is Associated with Breast Cancer Survival. Cancers, 14(2), 353. https://doi.org/10.3390/cancers14020353