
Canine Oral Melanoma Genomic and Transcriptomic Study Defines Two Molecular Subgroups with Different Therapeutical Targets

 , , ,
, , ,  , and
, and 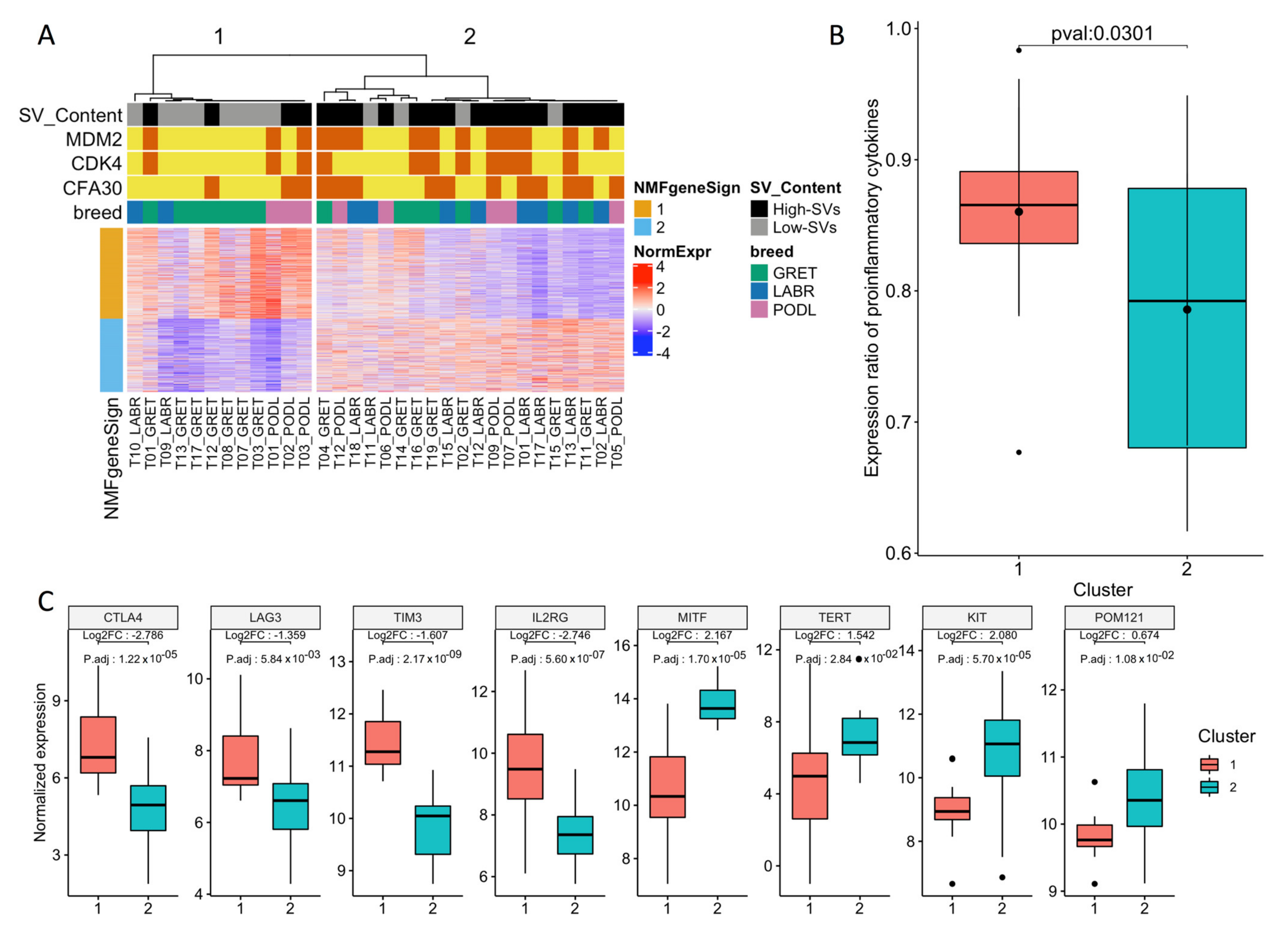{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Canine Cell Lines
2.3. Fluorescence In Situ Hybridization
2.4. RNAseq Clustering and Signature Analysis
2.5. Whole Genome Sequencing
2.6. Low Pass Sequencing
2.7. Mapping
2.8. Somatic Variant Calling
2.9. Somatic Copy Number Variant Calling
2.10. SV Calling
2.11. Modeling Chromothripsis Events
2.12. Classification of Dogs with “Low” versus “High” Content of Structural Variants (SV)
2.13. Targeted Copy Number and Expression Analysis
2.14. Western Blot
2.15. Transfection with Specific and Control siRNAs and Cell Proliferation Assays
3. Results
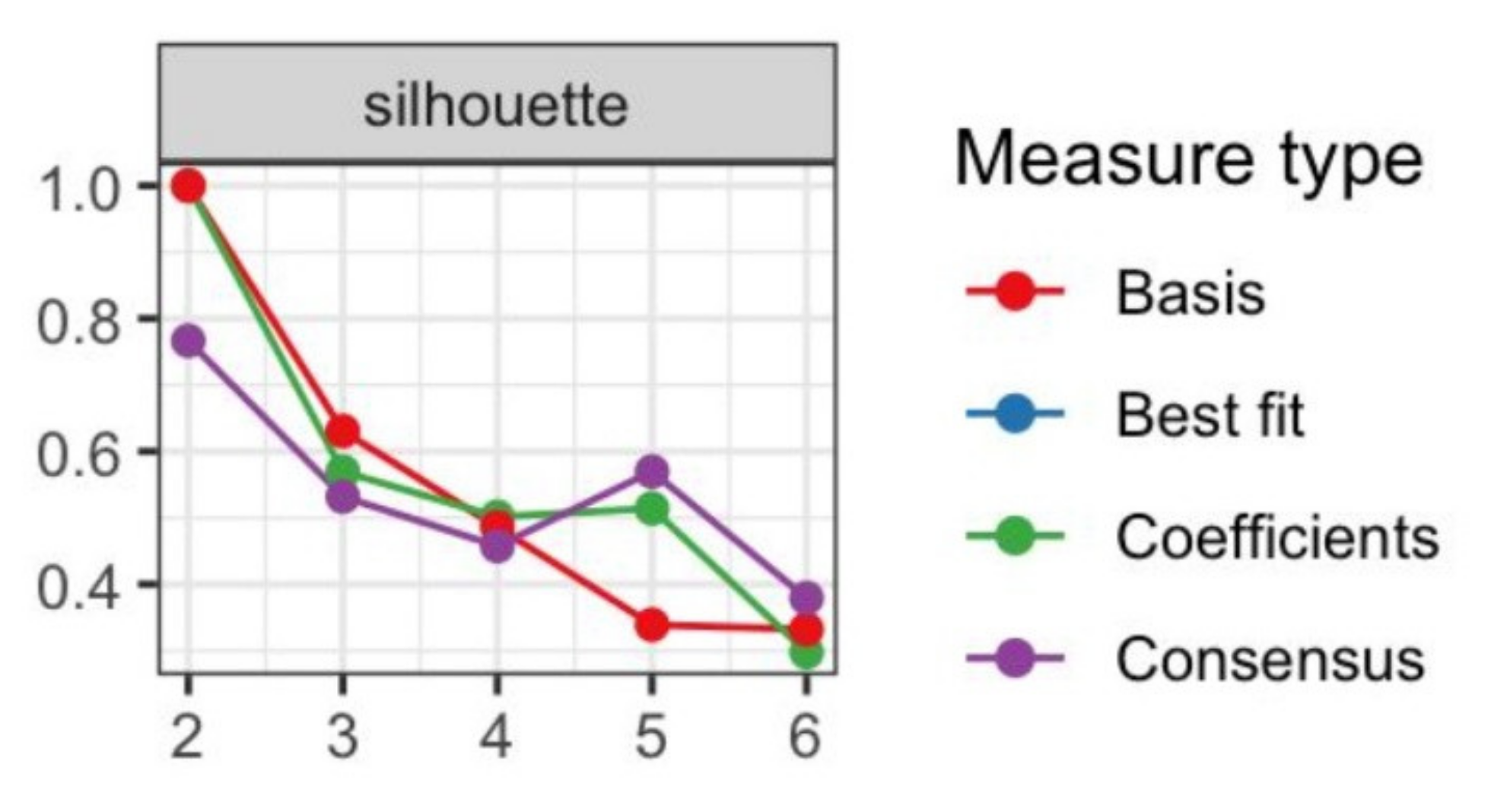
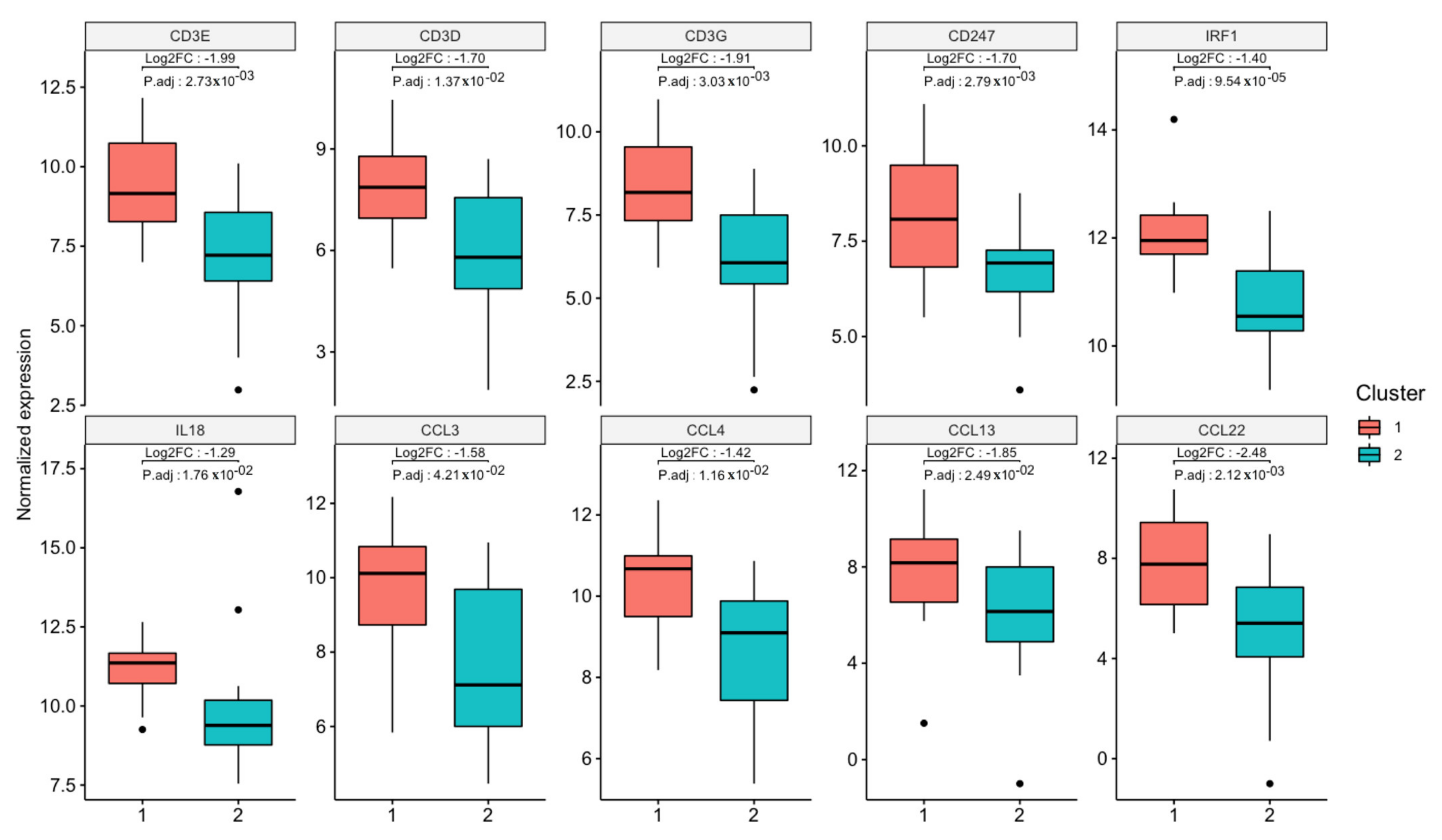
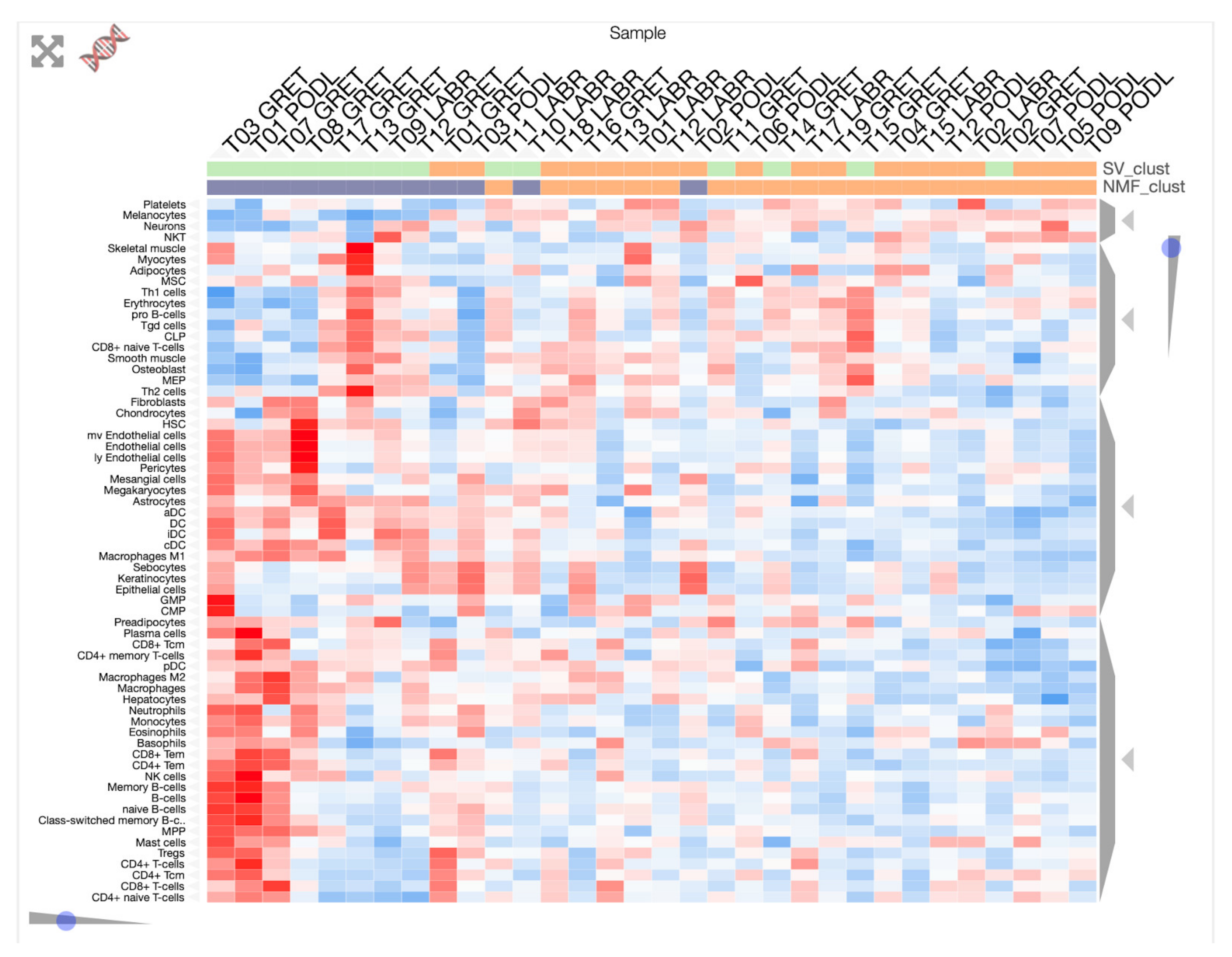
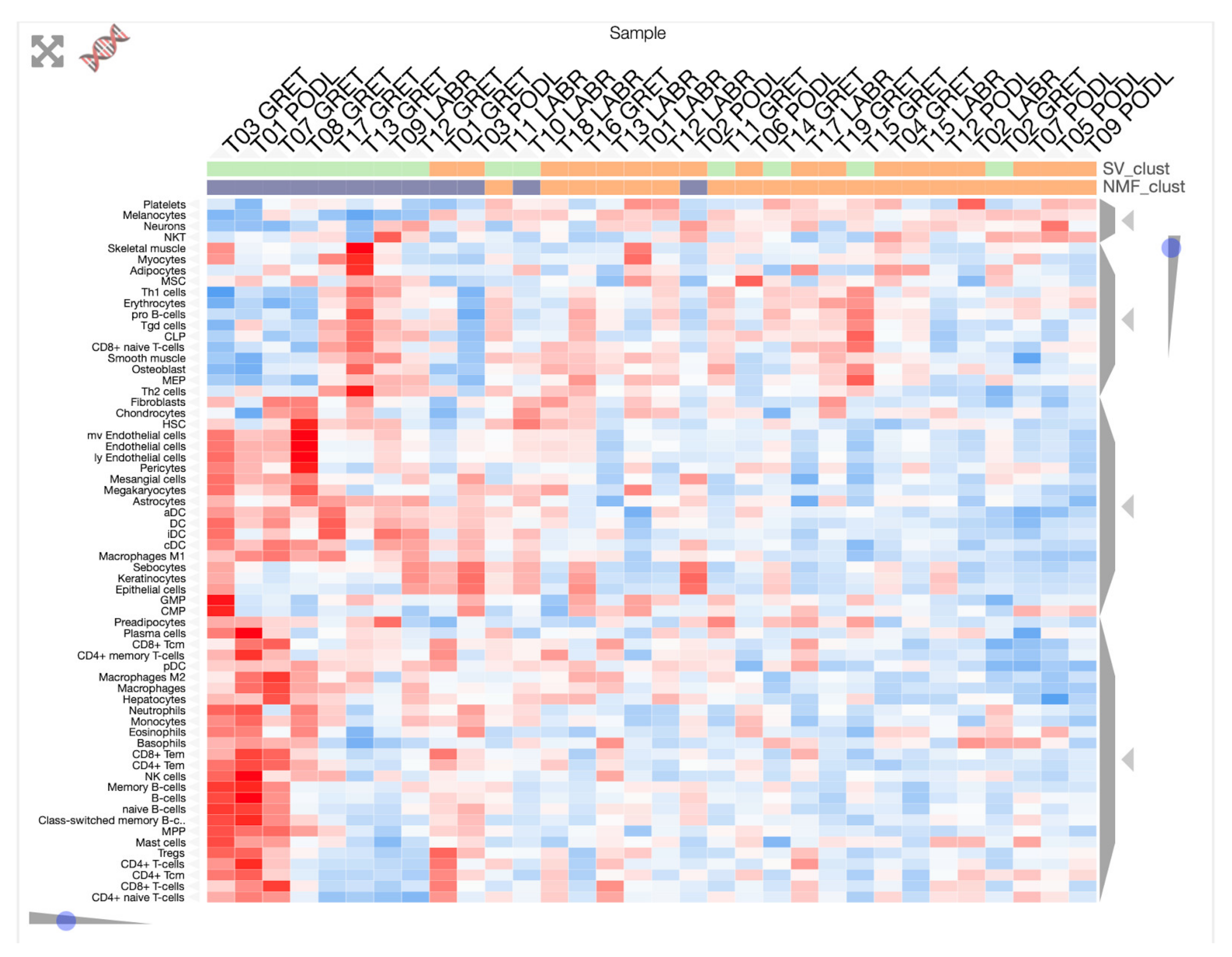
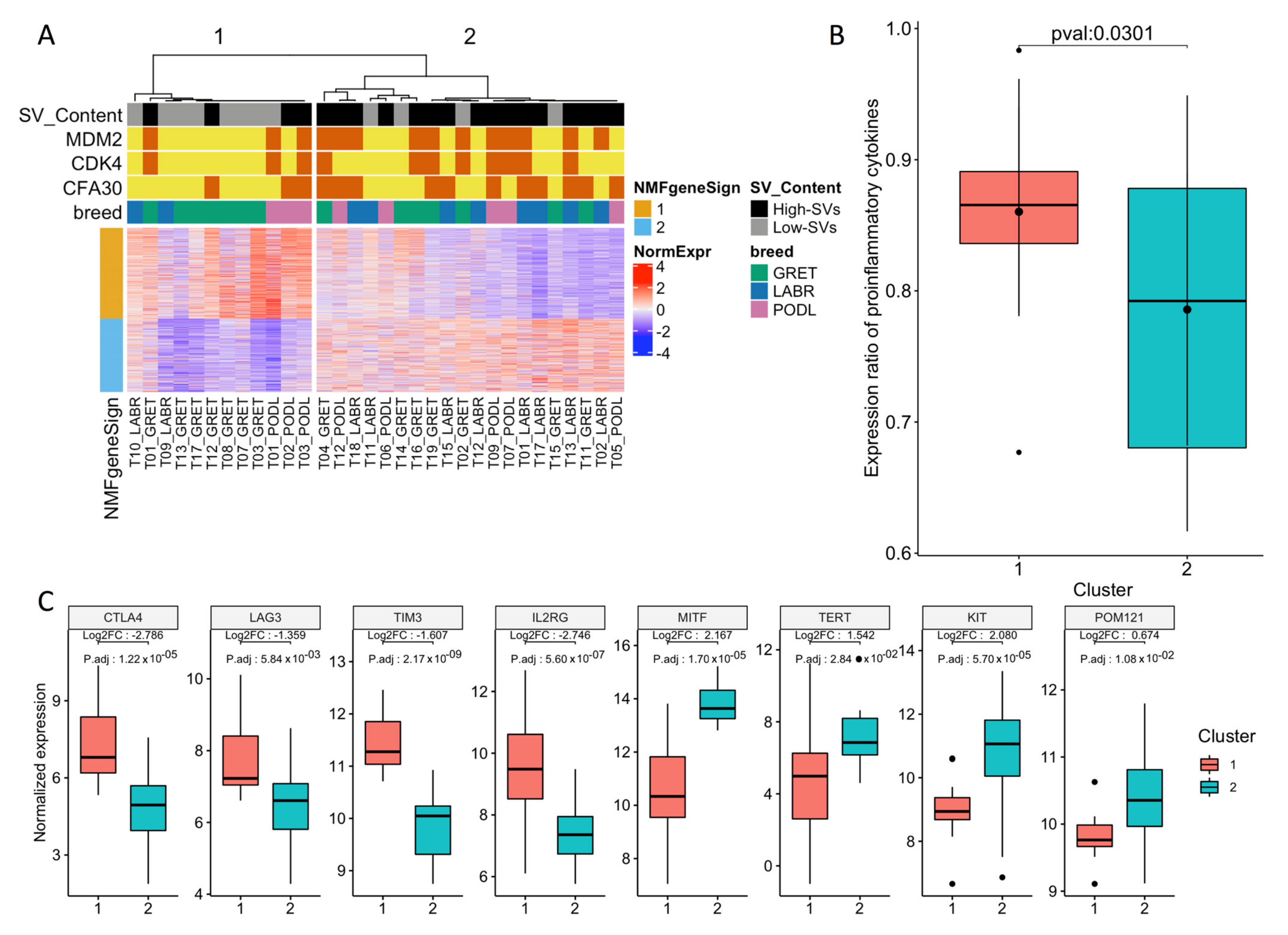
3.1. Transcriptome Landscape of Canine Oral Melanoma Highlights Two Distinct Subgroups
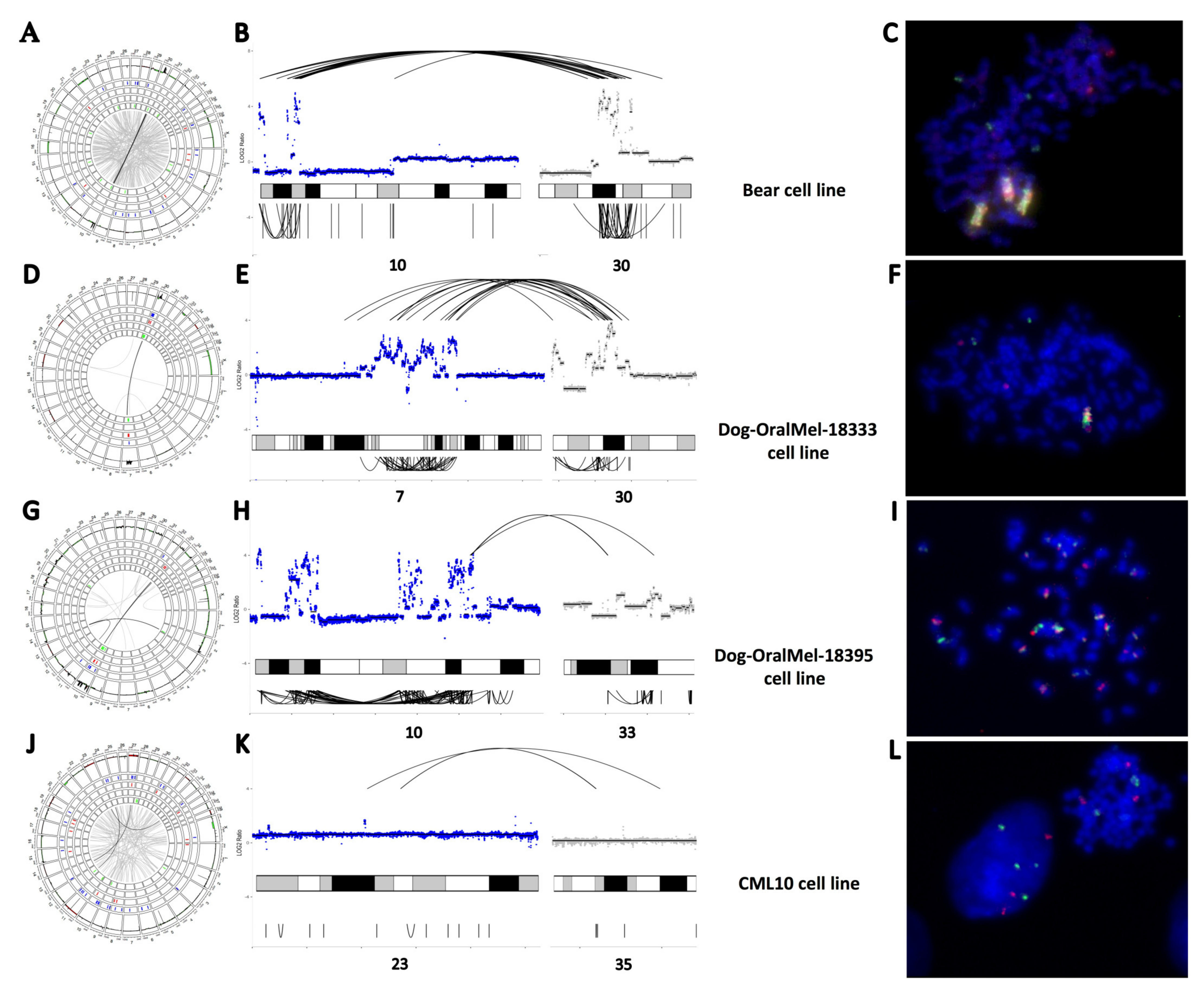
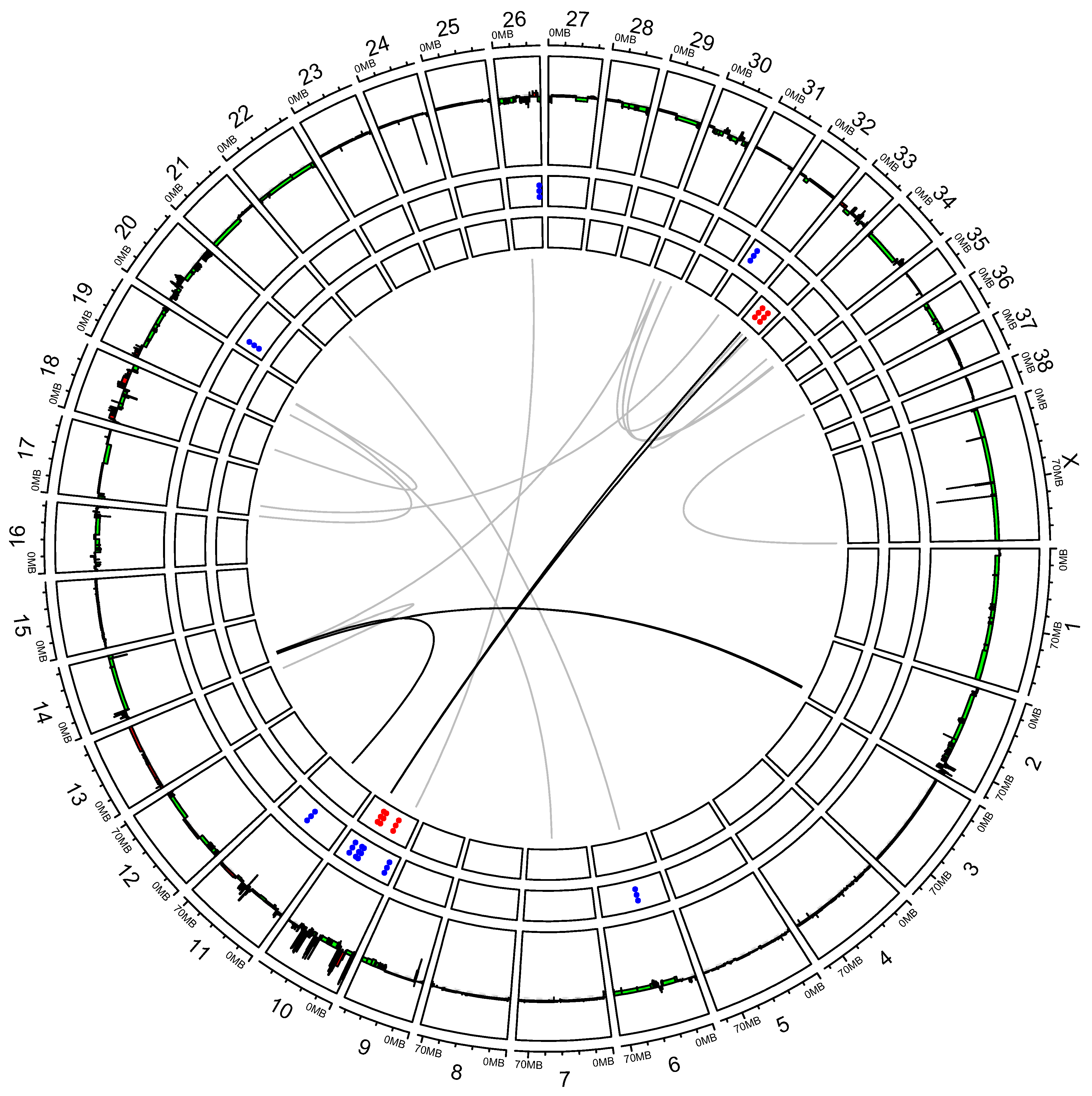
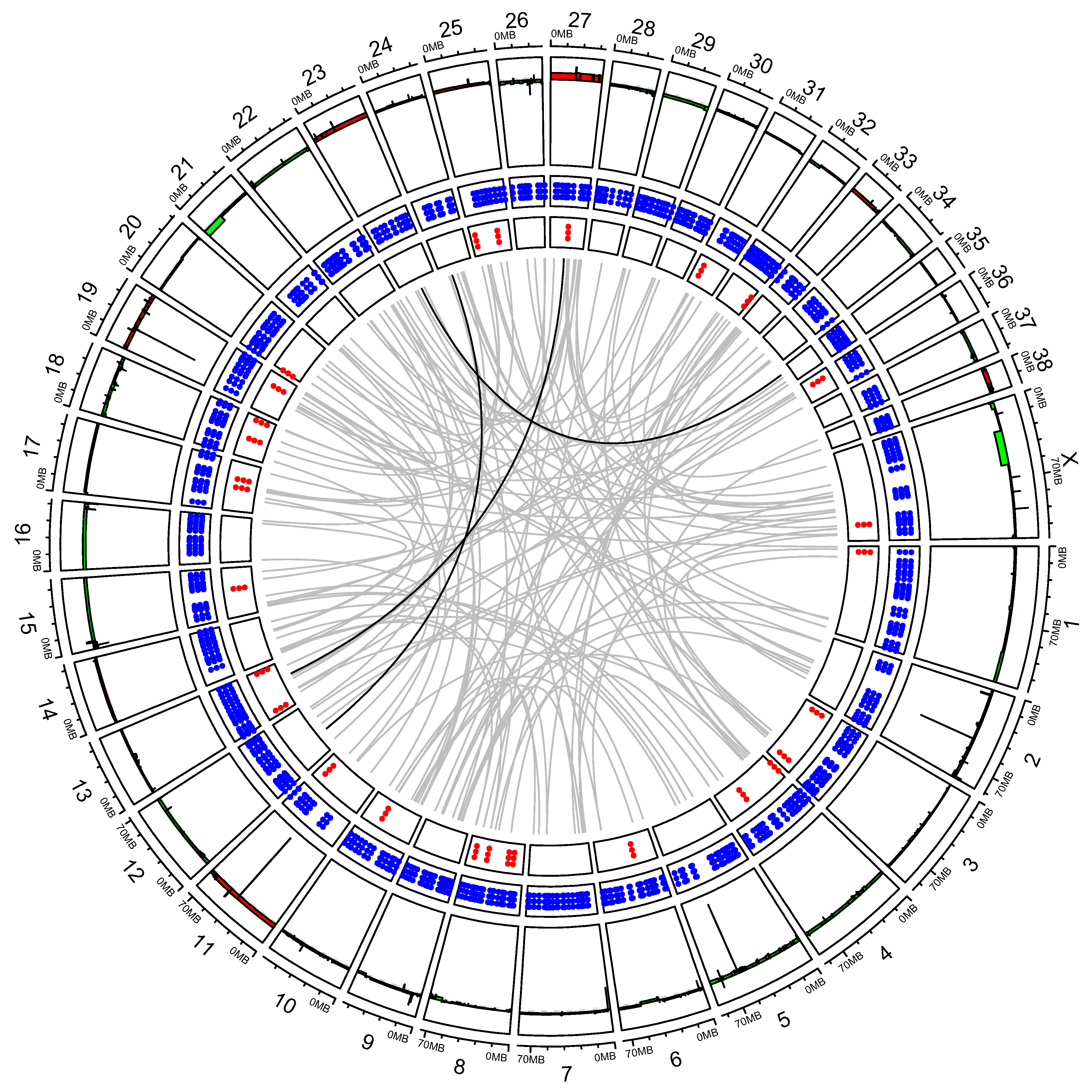
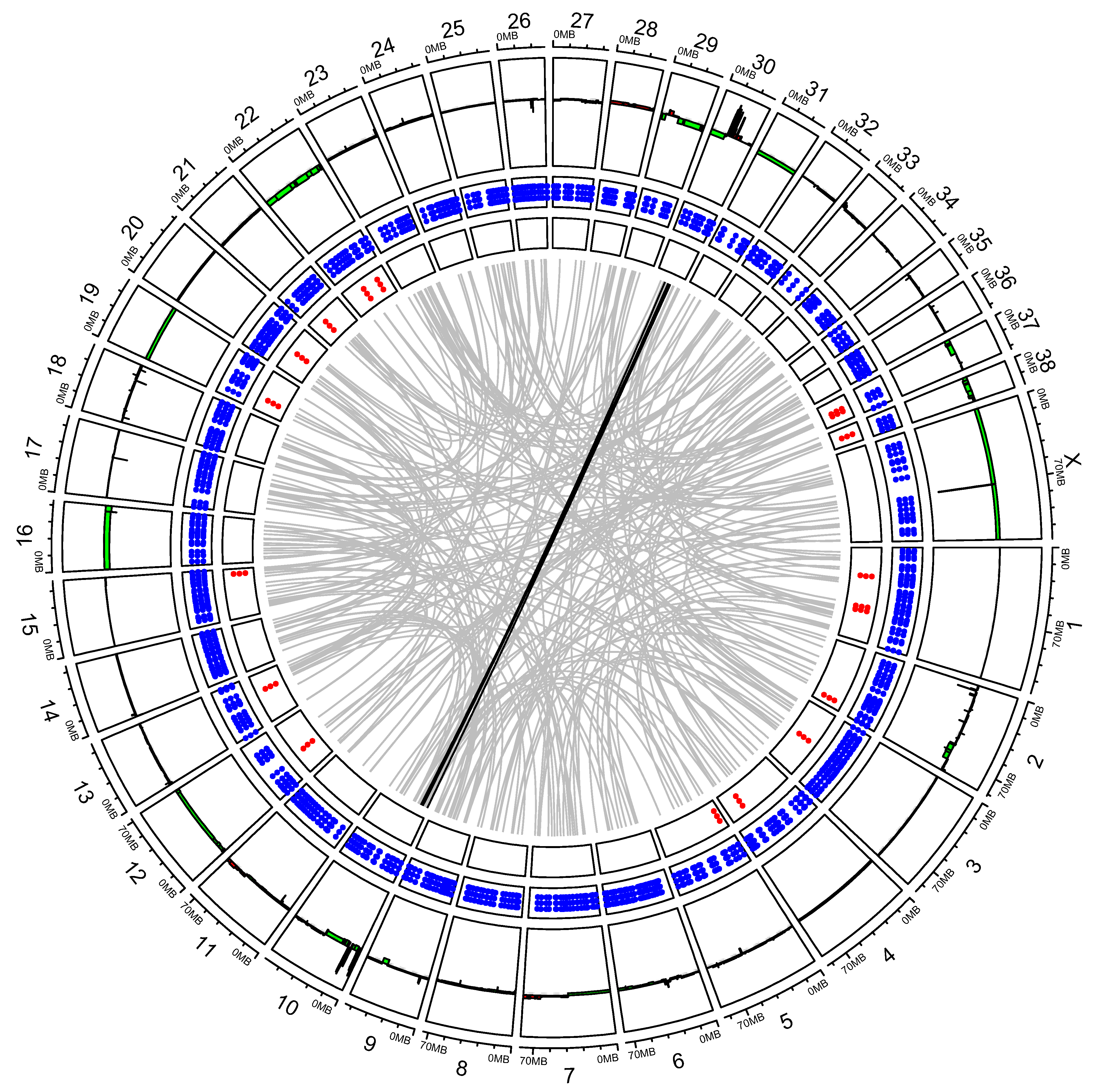
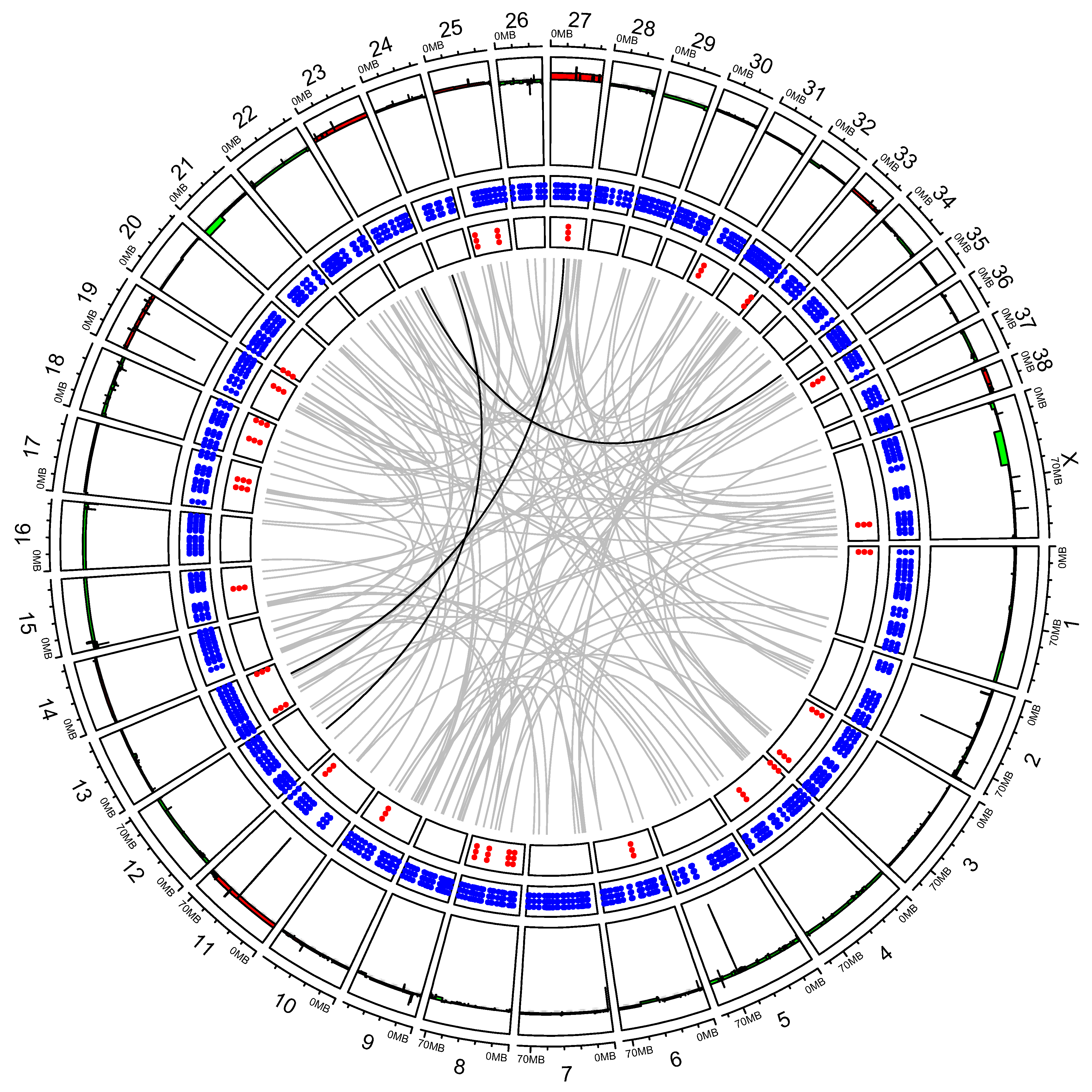
3.2. “High SV” MM Are Characterized by Focal Amplifications and Numerous Chromosomal Translocations
3.3. Exploring Candidate Oncogenes on CFA 30
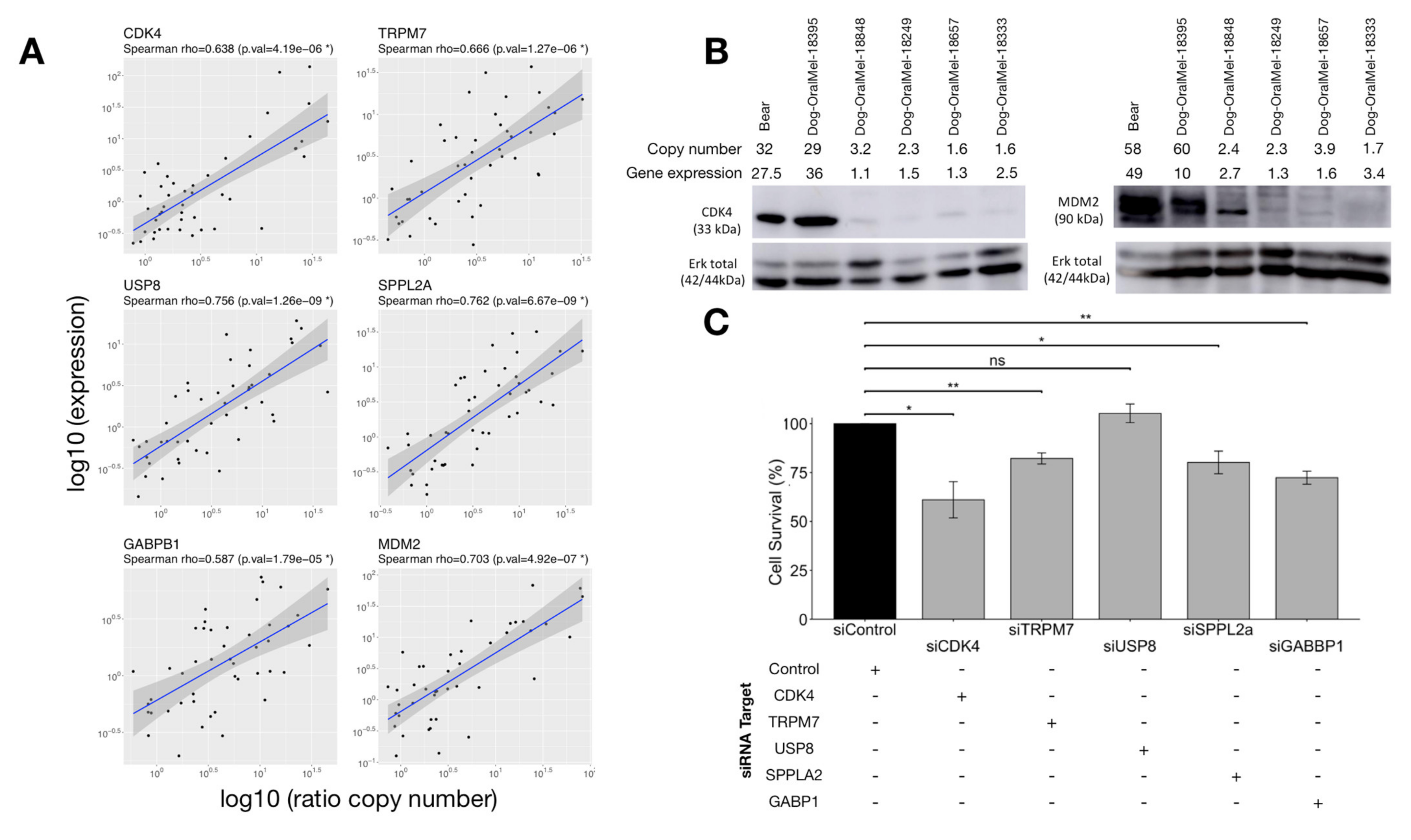
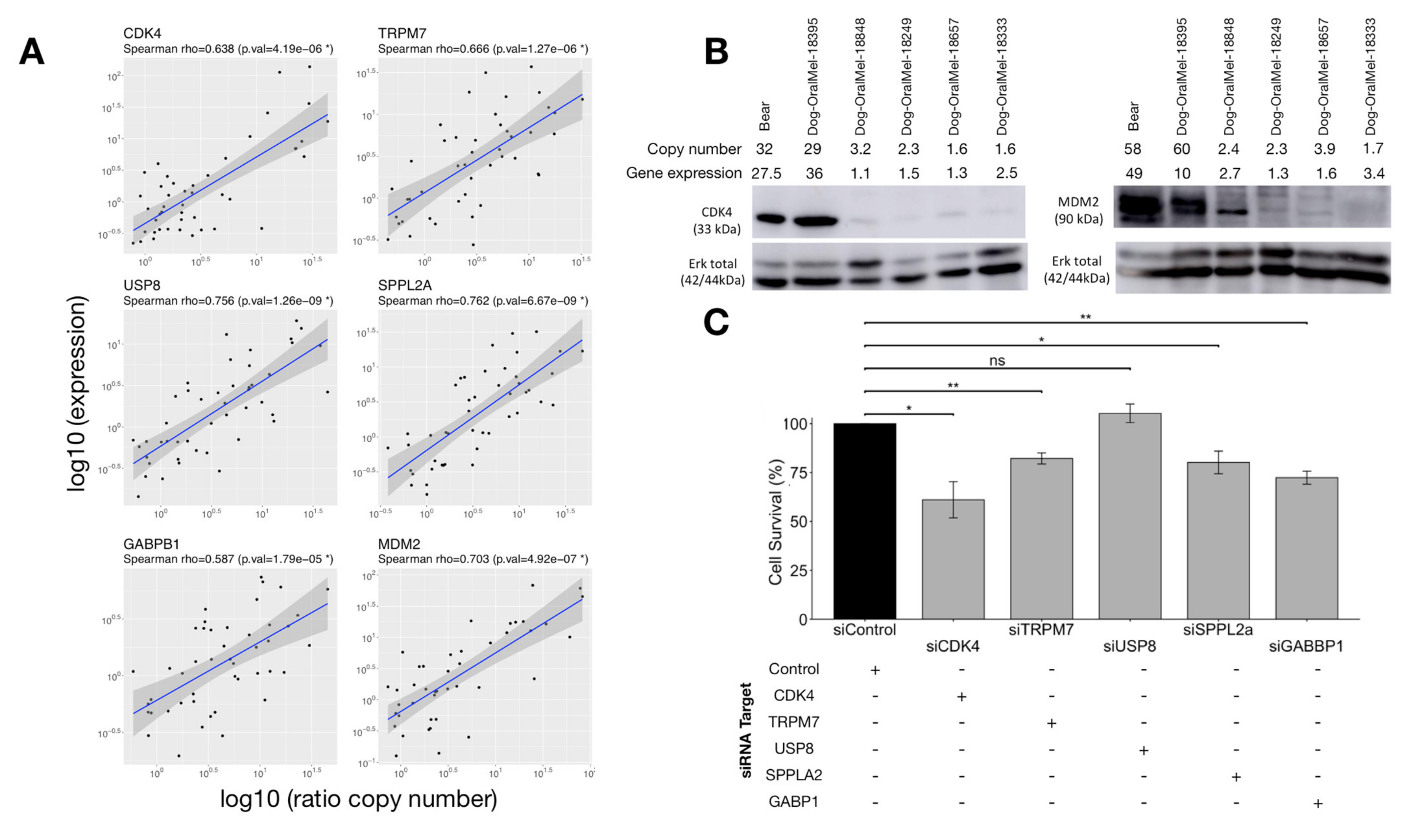
3.3.1. Correlation between Copy Numbers (CNA) and Expression of Candidate Oncogenes
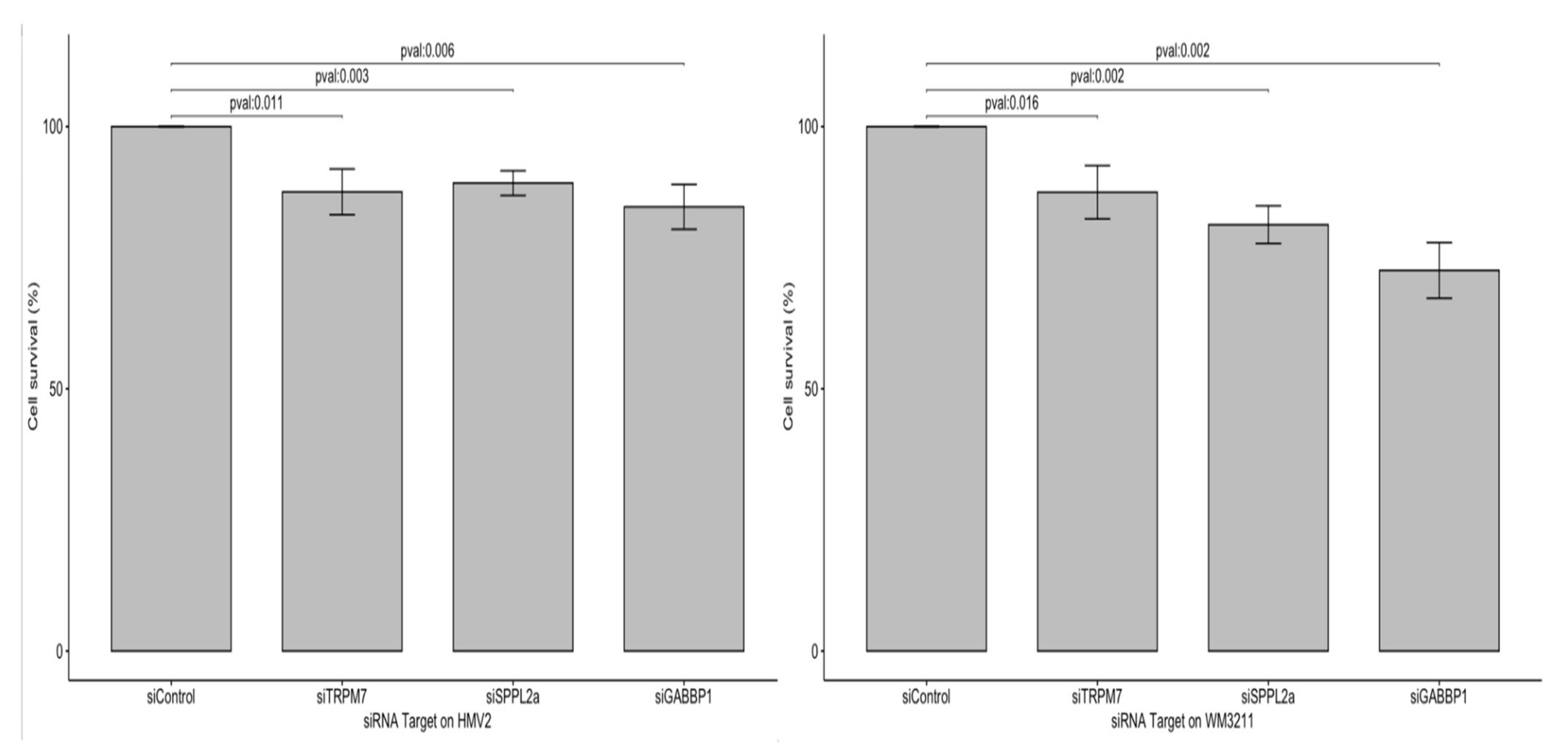
3.3.2. Effect of Candidate Oncogenes on MM Cell Proliferation
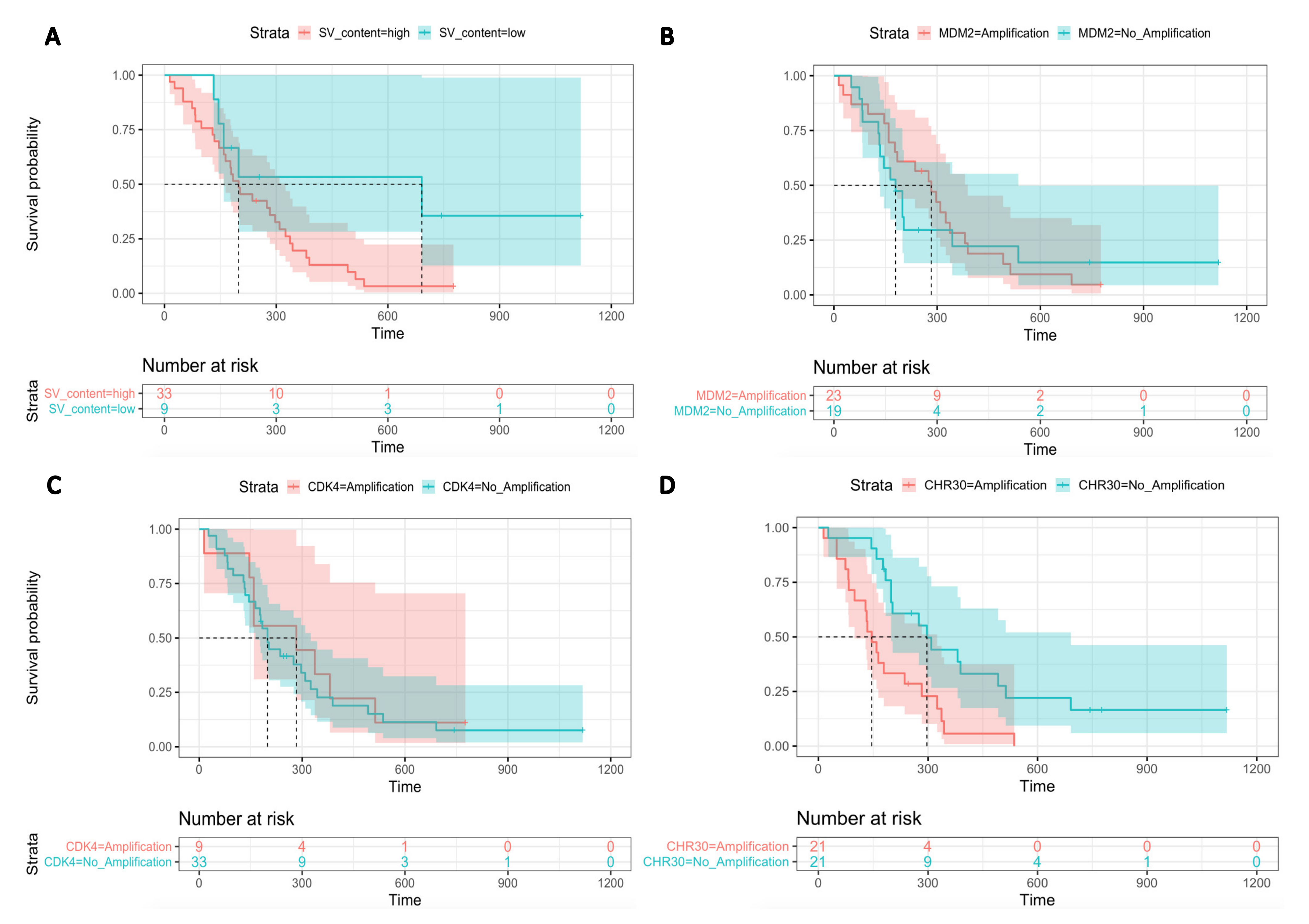
3.3.3. Effect of Candidate Oncogenes on Survival of Canine MM
3.4. Exploring the Value of CFA 30 Oncogenes in Human Melanomas
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A






References
- Williams, M.D. Update from the 4th Edition of the World Health Organization Classification of Head and Neck Tumours: Mucosal Melanomas. Head Neck Pathol. 2017, 11, 110–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yde, S.S.; Sjoegren, P.; Heje, M.; Stolle, L.B. Mucosal Melanoma: A Literature Review. Curr. Oncol. Rep. 2018, 20, 28. [Google Scholar] [CrossRef]
- Lerner, B.A.; Stewart, L.A.; Horowitz, D.P.; Carvajal, R.D. Mucosal Melanoma: New Insights and Therapeutic Options for a Unique and Aggressive Disease. Oncology 2017, 31, e23–e32. [Google Scholar] [PubMed]
- Chi, Z.; Li, S.; Sheng, X.; Si, L.; Cui, C.; Han, M.; Guo, J. Clinical presentation, histology, and prognoses of malignant melanoma in ethnic Chinese: A study of 522 consecutive cases. BMC Cancer 2011, 11, 85. [Google Scholar] [CrossRef] [Green Version]
- Kuk, D.; Shoushtari, A.N.; Barker, C.A.; Panageas, K.S.; Munhoz, R.R.; Momtaz, P.; Ariyan, C.E.; Brady, M.S.; Coit, D.G.; Bogatch, K.; et al. Prognosis of Mucosal, Uveal, Acral, Nonacral Cutaneous, and Unknown Primary Melanoma from the Time of First Metastasis. Oncologist 2016, 21, 848–854. [Google Scholar] [CrossRef] [Green Version]
- Bishop, K.D.; Olszewski, A.J. Epidemiology and survival outcomes of ocular and mucosal melanomas: A population-based analysis. Int. J. Cancer 2014, 134, 2961–2971. [Google Scholar] [CrossRef] [PubMed]
- Tacastacas, J.D.; Bray, J.; Cohen, Y.K.; Arbesman, J.; Kim, J.; Koon, H.B.; Honda, K.; Cooper, K.D.; Gerstenblith, M.R. Update on primary mucosal melanoma. J. Am. Acad. Dermatol. 2014, 71, 366–375. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Hamid, O.A.; Tarhini, A.; Schadendorf, D.; Chmielowski, B.; Collichio, F.A.; Pavlick, A.C.; Lewis, K.D.; Weil, S.C.; Heyburn, J.; et al. A phase 2 study of ontuxizumab, a monoclonal antibody targeting endosialin, in metastatic melanoma. Investig. New Drugs 2018, 36, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, A.; Sachpekidis, C.; Diella, F.; Doerks, A.; Kratz, A.-S.; Meisel, C.; Jackson, D.B.; Soldatos, T.G. Public Adverse Event Data Insights into the Safety of Pembrolizumab in Melanoma Patients. Cancers 2020, 12, 1008. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Gu, T.; Tian, X.; Li, W.; Zhao, R.; Yang, W.; Gao, Q.; Li, T.; Shim, J.-H.; Zhang, C.; et al. A Small Molecule Antagonist of PD-1/PD-L1 Interactions Acts as an Immune Checkpoint Inhibitor for NSCLC and Melanoma Immunotherapy. Front. Immunol. 2021, 12, 654463. [Google Scholar] [CrossRef]
- Furney, S.; Turajlic, S.; Stamp, G.; Thomas, J.M.; Hayes, A.; Strauss, D.; Gavrielides, M.; Xing, W.; Gore, M.; Larkin, J.; et al. The mutational burden of acral melanoma revealed by whole-genome sequencing and comparative analysis. Pigment. Cell Melanoma Res. 2014, 27, 835–838. [Google Scholar] [CrossRef]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.-M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.; van der Weyden, L.; Schott, C.R.; Foote, A.; Constantino-Casas, F.; Smith, S.; Dobson, J.M.; Murchison, E.P.; Wu, H.; Yeh, I.; et al. Cross-species genomic landscape comparison of human mucosal melanoma with canine oral and equine melanoma. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Newell, F.; Kong, Y.; Wilmott, J.S.; Johansson, P.A.; Ferguson, P.M.; Cui, C.; Li, Z.; Kazakoff, S.H.; Burke, H.; Dodds, T.J.; et al. Whole-genome landscape of mucosal melanoma reveals diverse drivers and therapeutic targets. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Shi, C.; Tao, W.; Li, J.; Wu, J.; Han, Y.; Yang, G.; Gu, Z.; Xu, S.; Wang, Y.; et al. Analysis of Mucosal Melanoma Whole-Genome Landscapes Reveals Clinically Relevant Genomic Aberrations. Clin. Cancer Res. 2019, 25, 3548–3560. [Google Scholar] [CrossRef] [Green Version]
- Lyu, J.; Song, Z.; Chen, J.; Shepard, M.J.; Song, H.; Ren, G.; Li, Z.; Guo, W.; Zhuang, Z.; Shi, Y. Whole-exome sequencing of oral mucosal melanoma reveals mutational profile and therapeutic targets. J. Pathol. 2018, 244, 358–366. [Google Scholar] [CrossRef]
- Hintzsche, J.D.; Gorden, N.T.; Amato, C.M.; Kim, J.; Wuensch, K.E.; Robinson, S.E.; Applegate, A.J.; Couts, K.L.; Medina, T.M.; Wells, K.R.; et al. Whole-exome sequencing identifies recurrent SF3B1 R625 mutation and comutation of NF1 and KIT in mucosal melanoma. Melanoma Res. 2017, 27, 189–199. [Google Scholar] [CrossRef]
- Broit, N.; Johansson, P.A.; Rodgers, C.B.; Walpole, S.T.; Newell, F.; Hayward, N.K.; Pritchard, A.L. Meta-Analysis and Systematic Review of the Genomics of Mucosal Melanoma. Mol. Cancer Res. 2021, 19, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, A.K.; Breen, M.; Choyke, P.; Dewhirst, M.; Fan, T.M.; Gustafson, D.L.; Helman, L.J.; Kastan, M.B.; Knapp, D.W.; Levin, W.J.; et al. Perspectives from man’s best friend: National Academy of Medicine’s Workshop on Comparative Oncology. Sci. Transl. Med. 2016, 8, 324ps5. [Google Scholar] [CrossRef] [Green Version]
- Dewhirst, M.W.; Page, R.L. Editorial: Emerging Translational Opportunities in Comparative Oncology with Companion Canine Cancers. Front. Oncol. 2020, 10, 270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulvé, R.; Rault, M.; Bahin, M.; Lagoutte, L.; Abadie, J.; De Brito, C.; Coindre, J.-M.; Botherel, N.; Rousseau, A.; Wucher, V.; et al. Discovery of Human-Similar Gene Fusions in Canine Cancers. Cancer Res. 2017, 77, 5721–5727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Weyden, L.; Patton, E.E.; Wood, G.; Foote, A.K.; Brenn, T.; Arends, M.J.; Adams, D.J. Cross-species models of human melanoma. J. Pathol. 2015, 238, 152–165. [Google Scholar] [CrossRef] [Green Version]
- Prouteau, A.; André, C. Canine Melanomas as Models for Human Melanomas: Clinical, Histological, and Genetic Comparison. Genes 2019, 10, 501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prouteau, A.; Chocteau, F.; De Brito, C.; Cadieu, E.; Primot, A.; Botherel, N.; Degorce, F.; Cornevin, L.; Lagadic, M.A.; Cabillic, F.; et al. Prognostic value of somatic focal amplifications on chromosome 30 in canine oral melanoma. Vet. Comp. Oncol. 2019, 18, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Verganti, S.; Berlato, D.; Blackwood, L.; Amores-Fuster, I.; Polton, G.A.; Elders, R.; Doyle, R.; Taylor, A.; Murphy, S. Use of Oncept melanoma vaccine in 69 canine oral malignant melanomas in the UK. J. Small Anim. Pract. 2017, 58, 10–16. [Google Scholar] [CrossRef]
- Igase, M.; Nemoto, Y.; Itamoto, K.; Tani, K.; Nakaichi, M.; Sakurai, M.; Sakai, Y.; Noguchi, S.; Kato, M.; Tsukui, T.; et al. A pilot clinical study of the therapeutic antibody against canine PD-1 for advanced spontaneous cancers in dogs. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef]
- Maekawa, N.; Konnai, S.; Takagi, S.; Kagawa, Y.; Okagawa, T.; Nishimori, A.; Ikebuchi, R.; Izumi, Y.; Deguchi, T.; Nakajima, C.; et al. A canine chimeric monoclonal antibody targeting PD-L1 and its clinical efficacy in canine oral malignant melanoma or undifferentiated sarcoma. Sci. Rep. 2017, 7, 8951. [Google Scholar] [CrossRef]
- Maekawa, N.; Konnai, S.; Nishimura, M.; Kagawa, Y.; Takagi, S.; Hosoya, K.; Ohta, H.; Kim, S.; Okagawa, T.; Izumi, Y.; et al. PD-L1 immunohistochemistry for canine cancers and clinical benefit of anti-PD-L1 antibody in dogs with pulmonary metastatic oral malignant melanoma. NPJ Precis. Oncol. 2021, 5, 10. [Google Scholar] [CrossRef]
- Hendricks, W.P.D.; Zismann, V.; Sivaprakasam, K.; Legendre, C.; Poorman, K.; Tembe, W.; Perdigones, N.; Kiefer, J.; Liang, W.; DeLuca, V.; et al. Somatic inactivating PTPRJ mutations and dysregulated pathways identified in canine malignant melanoma by integrated comparative genomic analysis. PLoS Genet. 2018, 14, e1007589. [Google Scholar] [CrossRef] [Green Version]
- Fowles, J.S.; Denton, C.L.; Gustafson, D.L. Comparative analysis of MAPK and PI3K/AKT pathway activation and inhibition in human and canine melanoma. Vet. Comp. Oncol. 2013, 13, 288–304. [Google Scholar] [CrossRef]
- Rahman, M.; Lai, Y.; Husna, A.A.; Chen, H.; Tanaka, Y.; Kawaguchi, H.; Hatai, H.; Miyoshi, N.; Nakagawa, T.; Fukushima, R.; et al. Transcriptome analysis of dog oral melanoma and its oncogenic analogy with human melanoma. Oncol. Rep. 2019, 43, 16–30. [Google Scholar] [CrossRef] [Green Version]
- Di Palma, S.; McConnell, A.; Verganti, S.; Starkey, M. Review on Canine Oral Melanoma: An Undervalued Authentic Genetic Model of Human Oral Melanoma? Vet. Pathol. 2021, 58, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Hitte, C.; Le Béguec, C.; Cadieu, E.; Wucher, V.; Primot, A.; Prouteau, A.; Botherel, N.; Hédan, B.; Lindblad-Toh, K.; André, C.; et al. Genome-Wide Analysis of Long Non-Coding RNA Profiles in Canine Oral Melanomas. Genes 2019, 10, 477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hédan, B.; Thomas, R.; Motsinger-Reif, A.; Abadie, J.; André, C.; Cullen, J.; Breen, M. Molecular cytogenetic characterization of canine histiocytic sarcoma: A spontaneous model for human histiocytic cancer identifies deletion of tumor suppressor genes and highlights influence of genetic background on tumor behavior. BMC Cancer 2011, 11, 201. [Google Scholar] [CrossRef] [Green Version]
- Wucher, V.; Legeai, F.; Hédan, B.; Rizk, G.; Lagoutte, L.; Leeb, T.; Jagannathan, V.; Cadieu, E.; David, A.; Lohi, H.; et al. FEELnc: A tool for long non-coding RNA annotation and its application to the dog transcriptome. Nucleic Acids Res. 2017, 45, e57. [Google Scholar] [CrossRef] [Green Version]
- Hoeppner, M.P.; Lundquist, A.; Pirun, M.; Meadows, J.; Zamani, N.; Johnson, J.; Sundström, G.; Cook, A.; Fitzgerald, M.G.; Swofford, R.; et al. An Improved Canine Genome and a Comprehensive Catalogue of Coding Genes and Non-Coding Transcripts. PLoS ONE 2014, 9, e91172. [Google Scholar] [CrossRef] [PubMed]
- Djebali, S.; Wucher, V.; Foissac, S.; Hitte, C.; Corre, E.; Derrien, T. Bioinformatics Pipeline for Transcriptome Sequencing Analysis. In Advanced Structural Safety Studies; Springer: Singapore, 2017; Volume 1468, pp. 201–219. [Google Scholar]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaujoux, R.; Seoighe, C. A flexible R package for nonnegative matrix factorization. BMC Bioinform. 2010, 11, 367. [Google Scholar] [CrossRef] [Green Version]
- Kolberg, L.; Raudvere, U.; Kuzmin, I.; Vilo, J.; Peterson, H. gprofiler2—An R package for gene list functional enrichment analysis and namespace conversion toolset g:Profiler. F1000Research 2020, 9, 709. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Patch, A.-M.; Nones, K.; Kazakoff, S.H.; Newell, F.; Wood, S.; Leonard, C.; Holmes, O.; Xu, Q.; Addala, V.; Creaney, J.; et al. Germline and somatic variant identification using BGISEQ-500 and HiSeq X Ten whole genome sequencing. PLoS ONE 2018, 13, e0190264. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Seshan, V.E.; Olshen, A. DNAcopy: DNA Copy Number Data Analysis, version 1.68.0; R Package: Madison, WI, USA, 2016. [Google Scholar]
- Rausch, T.; Zichner, T.; Schlattl, A.; Stütz, A.M.; Benes, V.; Korbel, J. DELLY: Structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012, 28, i333–i339. [Google Scholar] [CrossRef]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. circlize implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef] [Green Version]
- Korbel, J.O.; Campbell, P.J. Criteria for Inference of Chromothripsis in Cancer Genomes. Cell 2013, 152, 1226–1236. [Google Scholar] [CrossRef] [Green Version]
- Govind, S.K.; Zia, A.; Hennings-Yeomans, P.H.; Watson, J.D.; Fraser, M.; Anghel, C.; Wyatt, A.W.; van der Kwast, T.; Collins, C.C.; McPherson, J.D.; et al. ShatterProof: Operational detection and quantification of chromothripsis. BMC Bioinform. 2014, 15, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davoli, T.; Uno, H.; Wooten, E.C.; Elledge, S.J. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017, 355. [Google Scholar] [CrossRef] [Green Version]
- Ock, C.-Y.; Hwang, J.-E.; Keam, B.; Kim, S.-B.; Shim, J.-J.; Jang, H.-J.; Park, S.; Sohn, B.H.; Chan-Young, O.; Ajani, J.A.; et al. Genomic landscape associated with potential response to anti-CTLA-4 treatment in cancers. Nat. Commun. 2017, 8, 1050. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Kemper, K.; de Goeje, P.; Peeper, D.S.; Van Amerongen, R. Phenotype Switching: Tumor Cell Plasticity as a Resistance Mechanism and Target for Therapy. Cancer Res. 2014, 74, 5937–5941. [Google Scholar] [CrossRef] [Green Version]
- Hoek, K.S.; Goding, C.R. Cancer stem cells versus phenotype-switching in melanoma. Pigment Cell Melanoma Res. 2010, 23, 746–759. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, J.; Robert, L.; Paraiso, K.; Galvan, C.; Sheu, K.M.; Lay, J.; Wong, D.J.; Atefi, M.; Shirazi, R.; Wang, X.; et al. Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 2018, 33, 890–904. [Google Scholar] [CrossRef] [Green Version]
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018, 174, 843–855.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandamme, N.; Denecker, G.; Bruneel, K.; Blancke, G.; Akay, Ö.; Taminau, J.; De Coninck, J.; De Smedt, E.; Skrypek, N.; Van Loocke, W.; et al. The EMT Transcription Factor ZEB2 Promotes Proliferation of Primary and Metastatic Melanoma While Suppressing an Invasive, Mesenchymal-Like Phenotype. Cancer Res. 2020, 80, 2983–2995. [Google Scholar] [CrossRef]
- Corre, S.; Tardif, N.; Mouchet, N.; LeClair, H.M.; Boussemart, L.; Gautron, A.; Bachelot, L.; Perrot, A.; Soshilov, A.; Rogiers, A.; et al. Sustained activation of the Aryl hydrocarbon Receptor transcription factor promotes resistance to BRAF-inhibitors in melanoma. Nat. Commun. 2018, 9, 4775. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, A.; Imrichova, H.; Atak, Z.K.; Dewaele, M.; Rambow, F.; Hulselmans, G.; Christiaens, V.; Svetlichnyy, D.; Luciani, F.; Mooter, L.L.V.D.; et al. Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat. Commun. 2015, 6, 6683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keenan, T.E.; Burke, K.P.; Van Allen, E.M. Genomic correlates of response to immune checkpoint blockade. Nat. Med. 2019, 25, 389–402. [Google Scholar] [CrossRef]
- Poorman, K.; Borst, L.; Moroff, S.; Roy, S.; Labelle, P.; Motsinger-Reif, A.; Breen, M. Comparative cytogenetic characterization of primary canine melanocytic lesions using array CGH and fluorescence in situ hybridization. Chromosom. Res. 2014, 23, 171–186. [Google Scholar] [CrossRef] [Green Version]
- Brocca, G.; Ferraresso, S.; Zamboni, C.; Martinez-Merlo, E.M.; Ferro, S.; Goldschmidt, M.H.; Castagnaro, M. Array Comparative Genomic Hybridization Analysis Reveals Significantly Enriched Pathways in Canine Oral Melanoma. Front. Oncol. 2019, 9, 1397. [Google Scholar] [CrossRef] [Green Version]
- Rode, A.; Maass, K.K.; Willmund, K.V.; Ernst, A.; Lichter, P. Chromothripsis in cancer cells: An update. Int. J. Cancer 2015, 138, 2322–2333. [Google Scholar] [CrossRef] [PubMed]
- Furgason, J.M.; Koncar, R.F.; Michelhaugh, S.K.; Sarkar, F.H.; Mittal, S.; Sloan, A.E.; Barnholtz-Sloan, J.; Bahassi, E.M. Whole genome sequence analysis links chromothripsis to EGFR, MDM2, MDM4, and CDK4 amplification in glioblastoma. Oncoscience 2015, 2, 618–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Xu, S.-H.; Chen, Z.; Zeng, Q.-X.; Li, Z.-J.; Chen, Z.-M. TRPM7 overexpression enhances the cancer stem cell-like and metastatic phenotypes of lung cancer through modulation of the Hsp90α/uPA/MMP2 signaling pathway. BMC Cancer 2018, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wu, N.; Wang, Y.; Zhang, X.; Xia, B.; Tang, J.; Cai, J.; Zhao, Z.; Liao, Q.; Wang, J. TRPM7 promotes the epithelial–mesenchymal transition in ovarian cancer through the calcium-related PI3K/AKT oncogenic signaling. J. Exp. Clin. Cancer Res. 2019, 38, 1–15. [Google Scholar] [CrossRef]
- Yee, N.S. Role of TRPM7 in Cancer: Potential as Molecular Biomarker and Therapeutic Target. Pharmaceuticals 2017, 10, 39. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Cai, C.; Wu, J.; Cai, S.; Ye, C.; Chen, H.; Yang, Z.; Zeng, H.; Shen, Q.; Zou, F. TRPM7 mediates breast cancer cell migration and invasion through the MAPK pathway. Cancer Lett. 2013, 333, 96–102. [Google Scholar] [CrossRef]
- Chen, S.-C.; Yen, M.-C.; Chen, F.-W.; Wu, L.-Y.; Yang, S.-J.; Kuo, P.-L.; Hsu, Y.-L. Knockdown of GA-binding protein subunit β1 inhibits cell proliferation via p21 induction in renal cell carcinoma. Int. J. Oncol. 2018, 53, 886–894. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.; Kim, K.; Kim, H.-R.; Ylaya, K.; Do, S.-I.; Hewitt, S.M.; Park, H.-S.; Roe, J.-S.; Chung, J.-Y.; Song, J. Deubiquitylation and stabilization of Notch1 intracellular domain by ubiquitin-specific protease 8 enhance tumorigenesis in breast cancer. Cell Death Differ. 2020, 27, 1341–1354. [Google Scholar] [CrossRef]
- Yan, M.; Zhao, C.; Wei, N.; Wu, X.; Cui, J.; Xing, Y. High Expression of Ubiquitin-Specific Protease 8 (USP8) Is Associated with Poor Prognosis in Patients with Cervical Squamous Cell Carcinoma. Med Sci. Monit. 2018, 24, 4934–4943. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.-F.; Martinez-Barricarte, R.; Kennedy, J.; Mele, F.; Lazarov, T.; Deenick, E.K.; Ma, C.; Breton, G.; Lucero, K.B.; Langlais, D.; et al. Disruption of an antimycobacterial circuit between dendritic and helper T cells in human SPPL2a deficiency. Nat. Immunol. 2018, 19, 973–985. [Google Scholar] [CrossRef]
- Duan, B.; Wang, C.; Liu, Z.; Yang, X. USP8 is a Novel Therapeutic Target in Melanoma Through Regulating Receptor Tyrosine Kinase Levels. Cancer Manag. Res. 2021, 13, 4181–4189. [Google Scholar] [CrossRef]
- McNeill, M.S.; Paulsen, J.; Bonde, G.; Burnight, E.; Hsu, M.-Y.; Cornell, R.A. Cell Death of Melanophores in Zebrafish trpm7 Mutant Embryos Depends on Melanin Synthesis. J. Investig. Dermatol. 2007, 127, 2020–2030. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Carlson, J.A.; Slominski, A. Role of TRPM in melanocytes and melanoma. Exp. Dermatol. 2012, 21, 650–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, W.S.; Hendricks, W.; Kiefer, J.; Schmidt, J.; Sekar, S.; Carpten, J.; Craig, D.W.; Adkins, J.; Cuyugan, L.; Manojlovic, Z.; et al. Integrated genomic analyses reveal frequent TERT aberrations in acral melanoma. Genome Res. 2017, 27, 524–532. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, B.; Adissu, H.A.; Wei, B.-R.; Michael, H.T.; Merlino, G.; Simpson, R.M. Naturally Occurring Canine Melanoma as a Predictive Comparative Oncology Model for Human Mucosal and Other Triple Wild-Type Melanomas. Int. J. Mol. Sci. 2018, 19, 394. [Google Scholar] [CrossRef] [Green Version]
- Simpson, R.M.; Bastian, B.; Michael, H.; Webster, J.D.; Prasad, M.L.; Conway, C.M.; Prieto, V.M.; Gary, J.M.; Goldschmidt, M.; Esplin, D.G.; et al. Sporadic naturally occurring melanoma in dogs as a preclinical model for human melanoma. Pigment. Cell Melanoma Res. 2014, 27, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Gillard, M.; Cadieu, E.; De Brito, C.; Abadie, J.; Vergier, B.; Devauchelle, P.; Degorce, F.; Dréano, S.; Primot, A.; Dorso, L.; et al. Naturally occurring melanomas in dogs as models for non-UV pathways of human melanomas. Pigment. Cell Melanoma Res. 2014, 27, 90–102. [Google Scholar] [CrossRef]
- Sweis, R.; Spranger, S.; Bao, R.; Paner, G.P.; Stadler, W.M.; Steinberg, G.; Gajewski, T.F. Molecular Drivers of the Non–T-cell-Inflamed Tumor Microenvironment in Urothelial Bladder Cancer. Cancer Immunol. Res. 2016, 4, 563–568. [Google Scholar] [CrossRef] [Green Version]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [Green Version]
- Chan, T.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, D.; Kemmerling, R.; Davis, S.; Camps, J.; Meltzer, P.S.; Ried, T.; Gaiser, T. Chromothripsis and Focal Copy Number Alterations Determine Poor Outcome in Malignant Melanoma. Cancer Res. 2013, 73, 1454–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rücker, F.G.; Dolnik, A.; Blätte, T.J.; Teleanu, V.; Ernst, A.; Thol, F.; Heuser, M.; Ganser, A.; Döhner, H.; Döhner, K.; et al. Chromothripsis is linked to TP53 alteration, cell cycle impairment, and dismal outcome in acute myeloid leukemia with complex karyotype. Haematologica 2018, 103, e17–e20. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.Y.; Baptiste, C.; Mbbs, R.B.H.; Tergas, A.I.; Feldman, R.; Jones, N.L.; Chatterjee-Paer, S.; Bus-Kwolfski, A.; Wright, J.D.; Burke, W.M. Vulvar and vaginal melanoma: A unique subclass of mucosal melanoma based on a comprehensive molecular analysis of 51 cases compared with 2253 cases of nongynecologic melanoma. Cancer 2017, 123, 1333–1344. [Google Scholar] [CrossRef]
- Sheng, X.; Kong, Y.; Li, Y.; Zhang, Q.; Si, L.; Cui, C.; Chi, Z.; Tang, B.; Mao, L.; Lian, B.; et al. GNAQ and GNA11 mutations occur in 9.5% of mucosal melanoma and are associated with poor prognosis. Eur. J. Cancer 2016, 65, 156–163. [Google Scholar] [CrossRef]
- Yeh, I.; Jorgenson, E.; Shen, L.; Xu, M.; North, J.P.; Shain, A.H.; Reuss, D.; Wu, H.; Robinson, W.; Olshen, A.; et al. Targeted Genomic Profiling of Acral Melanoma. JNCI J. Natl. Cancer Inst. 2019, 111, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Forschner, A.; Hilke, F.-J.; Bonzheim, I.; Gschwind, A.; Demidov, G.; Amaral, T.; Ossowski, S.; Riess, O.; Schroeder, C.; Martus, P.; et al. MDM2, MDM4 and EGFR Amplifications and Hyperprogression in Metastatic Acral and Mucosal Melanoma. Cancers 2020, 12, 540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurmeister, P.; Wrede, N.; Hoffmann, I.; Vollbrecht, C.; Heim, D.; Hummel, M.; Wolkenstein, P.; Koch, I.; Heynol, V.; Schmitt, W.D.; et al. Mucosal melanomas of different anatomic sites share a common global DNA methylation profile with cutaneous melanoma but show location-dependent patterns of genetic and epigenetic alterations. J. Pathol. 2021. [Google Scholar] [CrossRef]
- Akincilar, S.C.; Unal, B.; Tergaonkar, V. Reactivation of telomerase in cancer. Cell. Mol. Life Sci. 2016, 73, 1659–1670. [Google Scholar] [CrossRef] [Green Version]
- Bell, R.J.A.; Rube, H.T.; Kreig, A.; Mancini, A.; Fouse, S.D.; Nagarajan, R.P.; Choi, S.; Hong, C.; He, D.; Pekmezci, M.; et al. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science 2015, 348, 1036–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancini, A.; Xavier-Magalhães, A.; Woods, W.S.; Nguyen, K.-T.; Amen, A.M.; Hayes, J.L.; Fellmann, C.; Gapinske, M.; McKinney, A.M.; Hong, C.; et al. Disruption of the β1L Isoform of GABP Reverses Glioblastoma Replicative Immortality in a TERT Promoter Mutation-Dependent Manner. Cancer Cell 2018, 34, 513–528.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barthel, F.P.; Wei, W.; Tang, M.; Martinez-Ledesma, E.; Hu, X.; Amin, S.B.; Akdemir, K.C.; Seth, S.; Song, X.; Wang, Q.; et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat. Genet. 2017, 49, 349–357. [Google Scholar] [CrossRef]
- Yuan, X.; Larsson, C.; Xu, D. Mechanisms underlying the activation of TERT transcription and telomerase activity in human cancer: Old actors and new players. Oncogene 2019, 38, 6172–6183. [Google Scholar] [CrossRef] [Green Version]
- Meng, S.; Alanazi, R.; Ji, D.; Bandura, J.; Luo, Z.-W.; Fleig, A.; Feng, Z.-P.; Sun, H.-S. Role of TRPM7 kinase in cancer. Cell Calcium 2021, 96, 102400. [Google Scholar] [CrossRef] [PubMed]
- Tawa, G.J.; Braisted, J.; Gerhold, D.; Grewal, G.; Mazcko, C.; Breen, M.; Sittampalam, G.; LeBlanc, A.K. Transcriptomic profiling in canines and humans reveals cancer specific gene modules and biological mechanisms common to both species. PLoS Comput. Biol. 2021, 17, e1009450. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, N.F.; Gundersen, G.W.; Rahman, A.; Grimes, M.L.; Rikova, K.; Hornbeck, P.; Ma’Ayan, A. Clustergrammer, a web-based heatmap visualization and analysis tool for high-dimensional biological data. Sci. Data 2017, 4, 170151. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prouteau, A.; Mottier, S.; Primot, A.; Cadieu, E.; Bachelot, L.; Botherel, N.; Cabillic, F.; Houel, A.; Cornevin, L.; Kergal, C.; et al. Canine Oral Melanoma Genomic and Transcriptomic Study Defines Two Molecular Subgroups with Different Therapeutical Targets. Cancers 2022, 14, 276. https://doi.org/10.3390/cancers14020276
Prouteau A, Mottier S, Primot A, Cadieu E, Bachelot L, Botherel N, Cabillic F, Houel A, Cornevin L, Kergal C, et al. Canine Oral Melanoma Genomic and Transcriptomic Study Defines Two Molecular Subgroups with Different Therapeutical Targets. Cancers. 2022; 14(2):276. https://doi.org/10.3390/cancers14020276
Chicago/Turabian StyleProuteau, Anais, Stephanie Mottier, Aline Primot, Edouard Cadieu, Laura Bachelot, Nadine Botherel, Florian Cabillic, Armel Houel, Laurence Cornevin, Camille Kergal, and et al. 2022. "Canine Oral Melanoma Genomic and Transcriptomic Study Defines Two Molecular Subgroups with Different Therapeutical Targets" Cancers 14, no. 2: 276. https://doi.org/10.3390/cancers14020276
APA StyleProuteau, A., Mottier, S., Primot, A., Cadieu, E., Bachelot, L., Botherel, N., Cabillic, F., Houel, A., Cornevin, L., Kergal, C., Corre, S., Abadie, J., Hitte, C., Gilot, D., Lindblad-Toh, K., André, C., Derrien, T., & Hedan, B. (2022). Canine Oral Melanoma Genomic and Transcriptomic Study Defines Two Molecular Subgroups with Different Therapeutical Targets. Cancers, 14(2), 276. https://doi.org/10.3390/cancers14020276