
Utilizing an Endogenous Progesterone Receptor Reporter Gene for Drug Screening and Mechanistic Study in Endometrial Cancer


Abstract
Simple Summary
Abstract

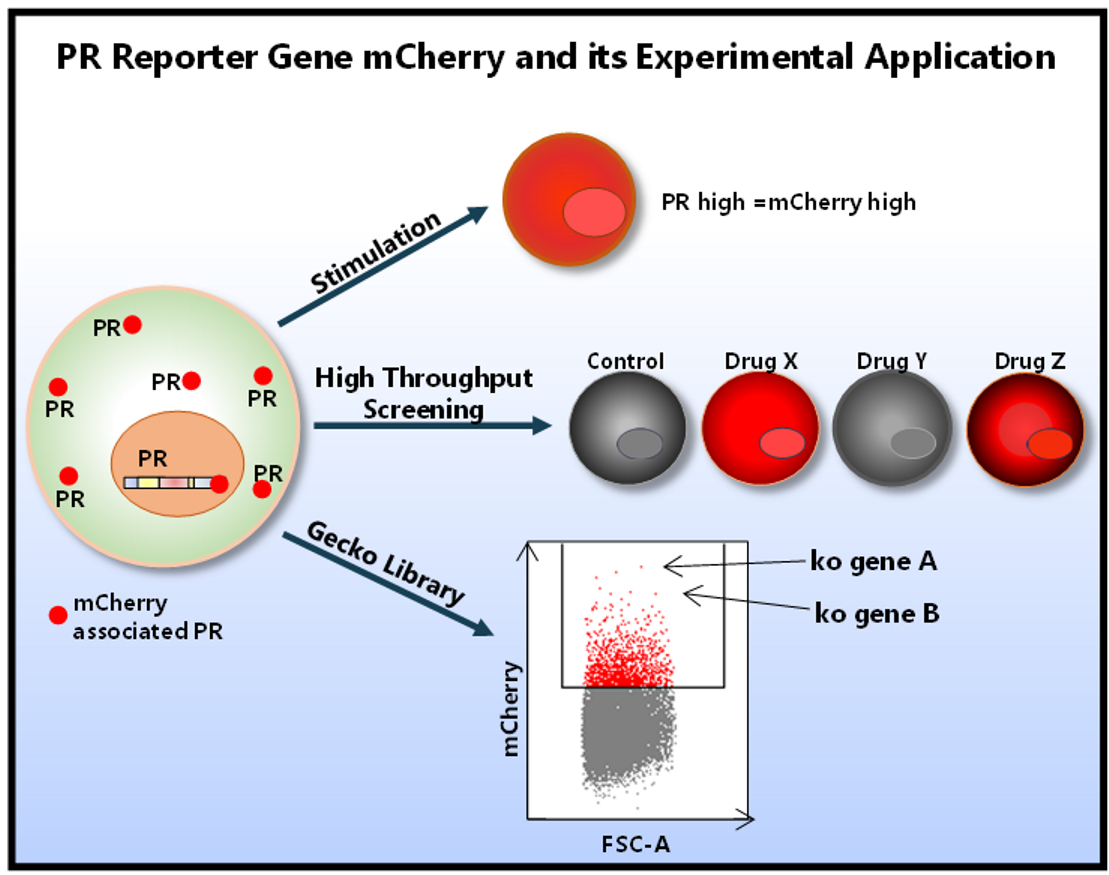{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Antibodies and Reagents
2.3. Generation of the Double Strand Break at Exon 8 of PGR
2.4. Alt-R Genome Editing Detection Assay
2.5. Construction of Hygromycin-mCherry Homology Donor Vector
2.6. Co-Transfections and Validation of the Correct Clones
2.7. Monitoring PR Expression Using High Content Imaging System
2.8. Screen Potential PR Repressors Using GeCKO Library
2.9. Real-Time PCR
2.10. Western Blotting
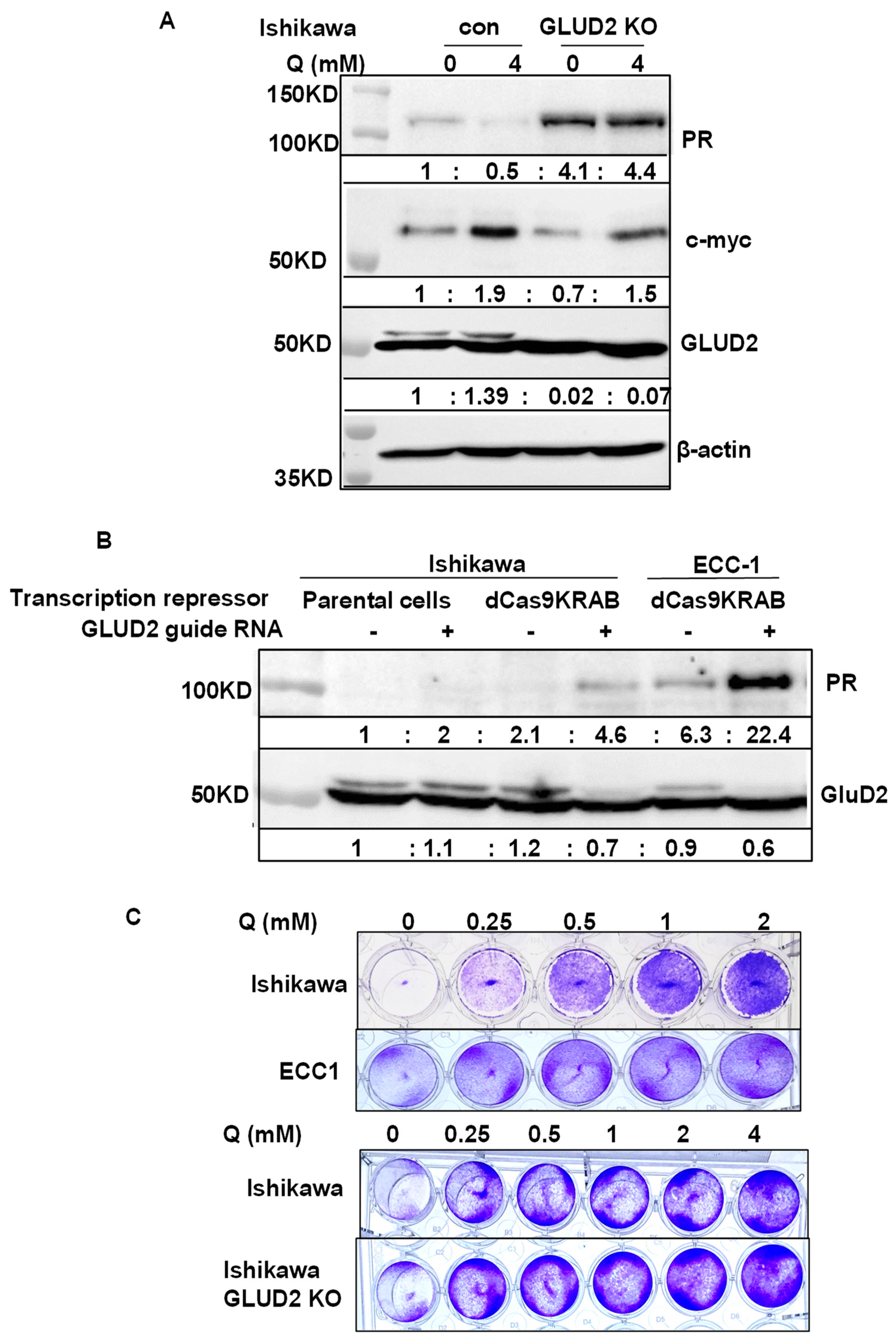
2.11. Knockout APH1A, SGPP2, SETDB1, and SOX9 in Ishikawa Cells
2.12. CRISPR-Mediated Promoter Repression of GLUD2
2.13. Quantification and Statistical Analysis
3. Results
3.1. Construction of an Endogenous PR Reporter Gene
3.2. Validating the Correlation between mCherry, Hygromycin, and PR Expression
3.3. Screen for and Validate Potential PR Inducers
3.4. Discovery of Potential PR Repressors Using the Genome-Wide CRISPR Knockout (GeCKO) Library
4. Discussion
4.1. Overview of Reporter Genes
4.2. PR Inducers
4.3. PR Downstream Genes
4.4. Reported PR Repressors
4.5. Caution in Choosing sgRNA Backbones
4.6. Unbiased Method to Screen GeCKO Library
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Proietti, C.J.; Cenciarini, M.E.; Elizalde, P.V. Revisiting progesterone receptor (PR) actions in breast cancer: Insights into PR repressive functions. Steroids 2018, 133, 75–81. [Google Scholar] [CrossRef]
- Diep, C.H.; Daniel, A.R.; Mauro, L.J.; Knutson, T.P.; Lange, C.A. Progesterone action in breast, uterine, and ovarian cancers. J. Mol. Endocrinol. 2015, 54, R31–R53. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Zaino, R.J.; Filiaci, V.J.; Leslie, K.K. Relationship of estrogen and progesterone receptors to clinical outcome in metastatic endometrial carcinoma: A Gynecologic Oncology Group Study. Gynecol. Oncol. 2007, 106, 325–333. [Google Scholar] [CrossRef]
- Yang, S.; Thiel, K.W.; De Geest, K.; Leslie, K.K. Endometrial cancer: Reviving progesterone therapy in the molecular age. Discov. Med. 2011, 12, 205–212. [Google Scholar]
- Ishibashi, H.; Suzuki, T.; Suzuki, S.; Niikawa, H.; Lu, L.; Miki, Y.; Moriya, T.; Hayashi, S.; Handa, M.; Kondo, T.; et al. Progesterone receptor in non-small cell lung cancer–A potent prognostic factor and possible target for endocrine therapy. Cancer Res. 2005, 65, 6450–6458. [Google Scholar] [CrossRef]
- Sieh, W.; Kobel, M.; Longacre, T.A.; Bowtell, D.D.; deFazio, A.; Goodman, M.T.; Hogdall, E.; Deen, S.; Wentzensen, N.; Moysich, K.B.; et al. Hormone-receptor expression and ovarian cancer survival: An Ovarian Tumor Tissue Analysis consortium study. Lancet Oncol. 2013, 14, 853–862. [Google Scholar] [CrossRef]
- Guan, J.; Xie, L.; Luo, X.; Yang, B.; Zhang, H.; Zhu, Q.; Chen, X. The prognostic significance of estrogen and progesterone receptors in grade I and II endometrioid endometrial adenocarcinoma: Hormone receptors in risk stratification. J. Gynecol. Oncol. 2019, 30, e13. [Google Scholar] [CrossRef] [PubMed]
- Raglan, O.; Kalliala, I.; Markozannes, G.; Cividini, S.; Gunter, M.J.; Nautiyal, J.; Gabra, H.; Paraskevaidis, E.; Martin-Hirsch, P.; Tsilidis, K.K.; et al. Risk factors for endometrial cancer: An umbrella review of the literature. Int. J. Cancer 2018, 145, 1719–1730. [Google Scholar] [CrossRef]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. CA Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Vereide, A.B.; Kaino, T.; Sager, G.; Arnes, M.; Orbo, A. Effect of levonorgestrel IUD and oral medroxyprogesterone acetate on glandular and stromal progesterone receptors (PRA and PRB), and estrogen receptors (ER-alpha and ER-beta) in human endometrial hyperplasia. Gynecol. Oncol. 2006, 101, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Orbo, A.; Arnes, M.; Pettersen, I.; Larsen, K.; Hanssen, K.; Moe, B. Down-regulated progesterone receptor A and B coinciding with successful treatment of endometrial hyperplasia by the levonorgestrel impregnated intrauterine system. Acta Obstet. Gynecol. Scand. 2010, 89, 1438–1446. [Google Scholar] [CrossRef] [PubMed]
- Khan, J.A.; Amazit, L.; Bellance, C.; Guiochon-Mantel, A.; Lombes, M.; Loosfelt, H. p38 and p42/44 MAPKs differentially regulate progesterone receptor A and B isoform stabilization. Mol. Endocrinol. 2011, 25, 1710–1724. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.J.; Zhao, J.; Li, H.Y.; Man, J.H.; He, K.; Zhou, T.; Pan, X.; Li, A.L.; Gong, W.L.; Jin, B.F.; et al. CUE domain containing 2 regulates degradation of progesterone receptor by ubiquitin-proteasome. EMBO J. 2007, 26, 1831–1842. [Google Scholar] [CrossRef]
- Cochrane, D.R.; Spoelstra, N.S.; Richer, J.K. The role of miRNAs in progesterone action. Mol. Cell Endocrinol. 2012, 357, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Tessel, M.A.; Krett, N.L.; Rosen, S.T. Steroid receptor and microRNA regulation in cancer. Curr. Opin. Oncol. 2010, 22, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.L.; Liang, X.H.; Su, R.W.; Lei, W.; Jia, B.; Feng, X.H.; Li, Z.X.; Yang, Z.M. Combined analysis of microRNome and 3′-UTRome reveals a species-specific regulation of progesterone receptor expression in the endometrium of rhesus monkey. J. Biol. Chem. 2012, 287, 13899–13910. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Li, Q.; Feng, L.; Ding, W. MiR-126-3p regulates progesterone receptors and involves development and lactation of mouse mammary gland. Mol. Cell. Biochem. 2011, 355, 17–25. [Google Scholar] [CrossRef]
- Lee, I.I.; Kim, J.J. Influence of AKT on progesterone action in endometrial diseases. Biol. Reprod. 2014, 91, 63. [Google Scholar] [CrossRef]
- Lee, I.I.; Maniar, K.; Lydon, J.P.; Kim, J.J. Akt regulates progesterone receptor B-dependent transcription and angiogenesis in endometrial cancer cells. Oncogene 2016, 35, 5191–5201. [Google Scholar] [CrossRef]
- Pant, A.; Lee, I.I.; Lu, Z.; Rueda, B.R.; Schink, J.; Kim, J.J. Inhibition of AKT with the orally active allosteric AKT inhibitor, MK-2206, sensitizes endometrial cancer cells to progestin. PLoS ONE 2012, 7, e41593. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Liu, X.; Ma, D.; Feng, Y.; Zhong, N. Down-regulation of the progesterone receptor by the methylation of progesterone receptor gene in endometrial cancer cells. Cancer Genet. Cytogenet. 2007, 175, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Dharia, A.; Oh, B.R.; Tanaka, Y.; Fujimoto, S.; Dahiya, R. Progesterone receptor B gene inactivation and CpG hypermethylation in human uterine endometrial cancer. Cancer Res. 2001, 61, 97–102. [Google Scholar] [PubMed]
- Xiong, Y.; Dowdy, S.C.; Gonzalez Bosquet, J.; Zhao, Y.; Eberhardt, N.L.; Podratz, K.C.; Jiang, S.-W. Epigenetic-mediated upregulation of progesterone receptor B gene in endometrial cancer cell lines. Gynecol. Oncol. 2005, 99, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Jia, Y.; Liu, X.; Winters, C.; Wang, X.; Zhang, Y.; Devor, E.J.; Hovey, A.M.; Reyes, H.D.; Xiao, X.; et al. Systematic dissection of the mechanisms underlying progesterone receptor downregulation in endometrial cancer. Oncotarget 2014, 5, 9783–9797. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Xiao, X.; Jia, Y.; Liu, X.; Zhang, Y.; Devor, E.J.; Meng, X.; Thiel, K.W.; Leslie, K.K. Epigenetic Modification Restores Functional PR Expression in Endometrial Cancer Cells. Curr. Pharm. Des. 2014, 20, 1874–1880. [Google Scholar] [CrossRef]
- Hagan, C.R.; Regan, T.M.; Dressing, G.E.; Lange, C.A. ck2-dependent phosphorylation of progesterone receptors (PR) on Ser81 regulates PR-B isoform-specific target gene expression in breast cancer cells. Mol. Cell. Biol. 2011, 31, 2439–2452. [Google Scholar] [CrossRef]
- Saito-Kanatani, M.; Urano, T.; Hiroi, H.; Momoeda, M.; Ito, M.; Fujii, T.; Inoue, S. Identification of TRIM22 as a progesterone-responsive gene in Ishikawa endometrial cancer cells. J. Steroid Biochem. Mol. Biol. 2015, 154, 217–225. [Google Scholar] [CrossRef]
- Jacobsen, B.M.; Jambal, P.; Schittone, S.A.; Horwitz, K.B. ALU repeats in promoters are position-dependent co-response elements (coRE) that enhance or repress transcription by dimeric and monomeric progesterone receptors. Mol. Endocrinol. 2009, 23, 989–1000. [Google Scholar] [CrossRef]
- Korch, C.; Spillman, M.A.; Jackson, T.A.; Jacobsen, B.M.; Murphy, S.K.; Lessey, B.A.; Jordan, V.C.; Bradford, A.P. DNA profiling analysis of endometrial and ovarian cell lines reveals misidentification, redundancy and contamination. Gynecol. Oncol. 2012, 127, 241–248. [Google Scholar] [CrossRef]
- Devor, E.J.; Gonzalez-Bosquet, J.; Thiel, K.W.; Leslie, K.K. Genomic characterization of five commonly used endometrial cancer cell lines. Int. J. Oncol. 2020, 57, 1348–1357. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Kavlashvili, T.; Jia, Y.; Dai, D.; Meng, X.; Thiel, K.W.; Leslie, K.K.; Yang, S. Inverse Relationship between Progesterone Receptor and Myc in Endometrial Cancer. PLoS ONE 2016, 11, e0148912. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Joung, J.; Konermann, S.; Gootenberg, J.S.; Abudayyeh, O.O.; Platt, R.J.; Brigham, M.D.; Sanjana, N.E.; Zhang, F. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc. 2017, 12, 828–863. [Google Scholar] [CrossRef]
- Li, M.; Wang, Y.; Liu, M.; Lan, X. Multimodality reporter gene imaging: Construction strategies and application. Theranostics 2018, 8, 2954–2973. [Google Scholar] [CrossRef]
- Liu, Y.; Hermes, J.; Li, J.; Tudor, M. Endogenous Locus Reporter Assays. Methods Mol. Biol. 2018, 1755, 163–177. [Google Scholar] [CrossRef]
- Greco, R.; Oliveira, G.; Stanghellini, M.T.; Vago, L.; Bondanza, A.; Peccatori, J.; Cieri, N.; Marktel, S.; Mastaglio, S.; Bordignon, C.; et al. Improving the safety of cell therapy with the TK-suicide gene. Front. Pharmacol. 2015, 6, 95. [Google Scholar] [CrossRef]
- Ando, H.; Miyamoto, T.; Kashima, H.; Higuchi, S.; Ida, K.; Mvunta, D.H.; Shiozawa, T. Panobinostat Enhances Growth Suppressive Effects of Progestin on Endometrial Carcinoma by Increasing Progesterone Receptor and Mitogen-Inducible Gene-6. Horm. Cancer 2017, 8, 257–267. [Google Scholar] [CrossRef]
- Winder, A.; Unno, K.; Yu, Y.; Lurain, J.; Kim, J.J. The allosteric AKT inhibitor, MK2206, decreases tumor growth and invasion in patient derived xenografts of endometrial cancer. Cancer Biol. Ther. 2017, 18, 958–964. [Google Scholar] [CrossRef]
- Delcuve, G.P.; Khan, D.H.; Davie, J.R. Roles of histone deacetylases in epigenetic regulation: Emerging paradigms from studies with inhibitors. Clin. Epigenetics 2012, 4, 5. [Google Scholar] [CrossRef]
- Delcuve, G.P.; Khan, D.H.; Davie, J.R. Targeting class I histone deacetylases in cancer therapy. Expert Opin. Ther. Targets 2013, 17, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Bantscheff, M.; Hopf, C.; Savitski, M.M.; Dittmann, A.; Grandi, P.; Michon, A.M.; Schlegl, J.; Abraham, Y.; Becher, I.; Bergamini, G.; et al. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol. 2011, 29, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Bates, S.E.; Eisch, R.; Ling, A.; Rosing, D.; Turner, M.; Pittaluga, S.; Prince, H.M.; Kirschbaum, M.H.; Allen, S.L.; Zain, J.; et al. Romidepsin in peripheral and cutaneous T-cell lymphoma: Mechanistic implications from clinical and correlative data. Br. J. Haematol. 2015, 170, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Berdeja, J.G.; Patel, M.R.; Flinn, I.; Gerecitano, J.F.; Neelapu, S.S.; Kelly, K.R.; Copeland, A.R.; Akins, A.; Clancy, M.S.; et al. Safety, tolerability, and preliminary activity of CUDC-907, a first-in-class, oral, dual inhibitor of HDAC and PI3K, in patients with relapsed or refractory lymphoma or multiple myeloma: An open-label, dose-escalation, phase 1 trial. Lancet Oncol. 2016, 17, 622–631. [Google Scholar] [CrossRef]
- Oki, Y.; Kelly, K.R.; Flinn, I.; Patel, M.R.; Gharavi, R.; Ma, A.; Parker, J.; Hafeez, A.; Tuck, D.; Younes, A. CUDC-907 in relapsed/refractory diffuse large B-cell lymphoma, including patients with MYC-alterations: Results from an expanded phase I trial. Haematologica 2017, 102, 1923–1930. [Google Scholar] [CrossRef]
- Sborov, D.W.; Canella, A.; Hade, E.M.; Mo, X.; Khountham, S.; Wang, J.; Ni, W.; Poi, M.; Coss, C.; Liu, Z.; et al. A phase 1 trial of the HDAC inhibitor AR-42 in patients with multiple myeloma and T- and B-cell lymphomas. Leuk. Lymphoma 2017, 58, 2310–2318. [Google Scholar] [CrossRef] [PubMed]
- Wartman, L.D.; Fiala, M.A.; Fletcher, T.; Hawkins, E.R.; Cashen, A.; DiPersio, J.F.; Jacoby, M.A.; Stockerl-Goldstein, K.E.; Pusic, I.; Uy, G.L.; et al. A phase I study of carfilzomib for relapsed or refractory acute myeloid and acute lymphoblastic leukemia. Leuk. Lymphoma 2016, 57, 728–730. [Google Scholar] [CrossRef]
- Avet-Loiseau, H.; Fonseca, R.; Siegel, D.; Dimopoulos, M.A.; Spicka, I.; Masszi, T.; Hajek, R.; Rosinol, L.; Goranova-Marinova, V.; Mihaylov, G.; et al. Carfilzomib significantly improves the progression-free survival of high-risk patients in multiple myeloma. Blood 2016, 128, 1174–1180. [Google Scholar] [CrossRef]
- Murphy, T.; Yee, K.W.L. Cytarabine and daunorubicin for the treatment of acute myeloid leukemia. Expert Opin. Pharmacother. 2017, 18, 1765–1780. [Google Scholar] [CrossRef]
- Yang, S.; Thiel, K.W.; Leslie, K.K. Progesterone: The ultimate endometrial tumor suppressor. Trends Endocrinol. Metab. 2011, 22, 145–152. [Google Scholar] [CrossRef]
- Diep, C.H.; Knutson, T.P.; Lange, C.A. Active FOXO1 Is a Key Determinant of Isoform-Specific Progesterone Receptor Transactivation and Senescence Programming. Mol. Cancer Res. 2016, 14, 141–162. [Google Scholar] [CrossRef] [PubMed]
- Takano, M.; Lu, Z.; Goto, T.; Fusi, L.; Higham, J.; Francis, J.; Withey, A.; Hardt, J.; Cloke, B.; Stavropoulou, A.V.; et al. Transcriptional cross talk between the forkhead transcription factor forkhead box O1A and the progesterone receptor coordinates cell cycle regulation and differentiation in human endometrial stromal cells. Mol. Endocrinol. 2007, 21, 2334–2349. [Google Scholar] [CrossRef] [PubMed]
- Ward, E.C.; Hoekstra, A.V.; Blok, L.J.; Hanifi-Moghaddam, P.; Lurain, J.R.; Singh, D.K.; Buttin, B.M.; Schink, J.C.; Kim, J.J. The Regulation and Function of the Forkhead Transcription Factor, Forkhead Box O1, Is Dependent on the Progesterone Receptor in Endometrial Carcinoma. Endocrinology 2008, 149, 1942–1950. [Google Scholar] [CrossRef] [PubMed]
- Stratmann, A.; Haendler, B. The histone demethylase JARID1A regulates progesterone receptor expression. FEBS J. 2011, 278, 1458–1469. [Google Scholar] [CrossRef] [PubMed]
- Purcell, D.J.; Jeong, K.W.; Bittencourt, D.; Gerke, D.S.; Stallcup, M.R. A distinct mechanism for coactivator versus corepressor function by histone methyltransferase G9a in transcriptional regulation. J. Biol. Chem. 2011, 286, 41963–41971. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Zhang, P.; Deng, W.; Oesterreich, S.; Lu, Y.; Mills, G.B.; Lee, A.V. Insulin-like growth factor-I inhibits progesterone receptor expression in breast cancer cells via the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin pathway: Progesterone receptor as a potential indicator of growth factor activity in breast cancer. Mol. Endocrinol. 2003, 17, 575–588. [Google Scholar] [PubMed]
- Zhao, G.; Liu, Z.; Ilagan, M.X.; Kopan, R. Gamma-secretase composed of PS1/Pen2/Aph1a can cleave notch and amyloid precursor protein in the absence of nicastrin. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 1648–1656. [Google Scholar] [CrossRef]
- Ruoming, W.; Zhen, Y.; Tengteng, Z.; Jisheng, H. Tumor suppressor microRNA-31 inhibits gastric carcinogenesis by targeting Smad4 and SGPP2. Cancer Gene Ther. 2015, 22, 564–572. [Google Scholar] [CrossRef]
- Karanth, A.V.; Maniswami, R.R.; Prashanth, S.; Govindaraj, H.; Padmavathy, R.; Jegatheesan, S.K.; Mullangi, R.; Rajagopal, S. Emerging role of SETDB1 as a therapeutic target. Expert Opin. Ther. Targets 2017, 21, 319–331. [Google Scholar] [CrossRef]
- Rodriguez-Paredes, M.; Martinez de Paz, A.; Simo-Riudalbas, L.; Sayols, S.; Moutinho, C.; Moran, S.; Villanueva, A.; Vazquez-Cedeira, M.; Lazo, P.A.; Carneiro, F.; et al. Gene amplification of the histone methyltransferase SETDB1 contributes to human lung tumorigenesis. Oncogene 2014, 33, 2807–2813. [Google Scholar] [CrossRef]
- Xiao, J.F.; Sun, Q.Y.; Ding, L.W.; Chien, W.; Liu, X.Y.; Mayakonda, A.; Jiang, Y.Y.; Loh, X.Y.; Ran, X.B.; Doan, N.B.; et al. The c-MYC-BMI1 axis is essential for SETDB1-mediated breast tumourigenesis. J. Pathol. 2018, 246, 89–102. [Google Scholar] [CrossRef]
- Cho, S.; Park, J.S.; Kang, Y.K. AGO2 and SETDB1 cooperate in promoter-targeted transcriptional silencing of the androgen receptor gene. Nucleic Acids Res. 2014, 42, 13545–13556. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.; Hu, S.; Zhang, H.; Du, G.; Li, X.; Li, X.; Li, X. Upregulated SOX9 expression indicates worse prognosis in solid tumors: A systematic review and meta-analysis. Oncotarget 2017, 8, 113163–113173. [Google Scholar] [CrossRef]
- Gonzalez, G.; Mehra, S.; Wang, Y.; Akiyama, H.; Behringer, R.R. Sox9 overexpression in uterine epithelia induces endometrial gland hyperplasia. Differ. Res. Biol. Divers. 2016, 92, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Saegusa, M.; Hashimura, M.; Suzuki, E.; Yoshida, T.; Kuwata, T. Transcriptional up-regulation of Sox9 by NF-kappaB in endometrial carcinoma cells, modulating cell proliferation through alteration in the p14(ARF)/p53/p21(WAF1) pathway. Am. J. Pathol. 2012, 181, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Fazilaty, H.; Gardaneh, M.; Akbari, P.; Zekri, A.; Behnam, B. SLUG and SOX9 Cooperatively Regulate Tumor Initiating Niche Factors in Breast Cancer. Cancer Microenviron. Off. J. Int. Cancer Microenviron. Soc. 2016, 9, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; McKnight, N.C.; Zhang, T.; Lu, M.L.; Balk, S.P.; Yuan, X. SOX9 is expressed in normal prostate basal cells and regulates androgen receptor expression in prostate cancer cells. Cancer Res. 2007, 67, 528–536. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Yoon, H.; Ringel, A.E.; Jeanfavre, S.; Clish, C.B.; Haigis, M.C. Metabolic recycling of ammonia via glutamate dehydrogenase supports breast cancer biomass. Science 2017, 358, 941–946. [Google Scholar] [CrossRef]
- Chen, R.; Nishimura, M.C.; Kharbanda, S.; Peale, F.; Deng, Y.; Daemen, A.; Forrest, W.F.; Kwong, M.; Hedehus, M.; Hatzivassiliou, G.; et al. Hominoid-specific enzyme GLUD2 promotes growth of IDH1R132H glioma. Proc. Natl. Acad. Sci. USA 2014, 111, 14217–14222. [Google Scholar] [CrossRef]
- Borompokas, N.; Papachatzaki, M.M.; Kanavouras, K.; Mastorodemos, V.; Zaganas, I.; Spanaki, C.; Plaitakis, A. Estrogen modification of human glutamate dehydrogenases is linked to enzyme activation state. J. Biol. Chem. 2010, 285, 31380–31387. [Google Scholar] [CrossRef] [PubMed]
- Spanaki, C.; Kotzamani, D.; Petraki, Z.; Drakos, E.; Plaitakis, A. Expression of human GLUD1 and GLUD2 glutamate dehydrogenases in steroid producing tissues. Mol. Cell Endocrinol. 2015, 415, 1–11. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Zhou, W.; Meng, X.; Murray, S.D.; Li, L.; Fronk, A.; Lazaro-Camp, V.J.; Wen, K.-k.; Wu, M.; Dupuy, A.; et al. Utilizing an Endogenous Progesterone Receptor Reporter Gene for Drug Screening and Mechanistic Study in Endometrial Cancer. Cancers 2022, 14, 4883. https://doi.org/10.3390/cancers14194883
Li Y, Zhou W, Meng X, Murray SD, Li L, Fronk A, Lazaro-Camp VJ, Wen K-k, Wu M, Dupuy A, et al. Utilizing an Endogenous Progesterone Receptor Reporter Gene for Drug Screening and Mechanistic Study in Endometrial Cancer. Cancers. 2022; 14(19):4883. https://doi.org/10.3390/cancers14194883
Chicago/Turabian StyleLi, Yiyang, Wei Zhou, Xiangbing Meng, Sarina D. Murray, Long Li, Abby Fronk, Vanessa J. Lazaro-Camp, Kuo-kuang Wen, Meng Wu, Adam Dupuy, and et al. 2022. "Utilizing an Endogenous Progesterone Receptor Reporter Gene for Drug Screening and Mechanistic Study in Endometrial Cancer" Cancers 14, no. 19: 4883. https://doi.org/10.3390/cancers14194883
APA StyleLi, Y., Zhou, W., Meng, X., Murray, S. D., Li, L., Fronk, A., Lazaro-Camp, V. J., Wen, K.-k., Wu, M., Dupuy, A., Leslie, K. K., & Yang, S. (2022). Utilizing an Endogenous Progesterone Receptor Reporter Gene for Drug Screening and Mechanistic Study in Endometrial Cancer. Cancers, 14(19), 4883. https://doi.org/10.3390/cancers14194883