
Role of the Hedgehog Pathway and CAXII in Controlling Melanoma Cell Migration and Invasion in Hypoxia
 , , and
, , and
Abstract
Simple Summary
Abstract
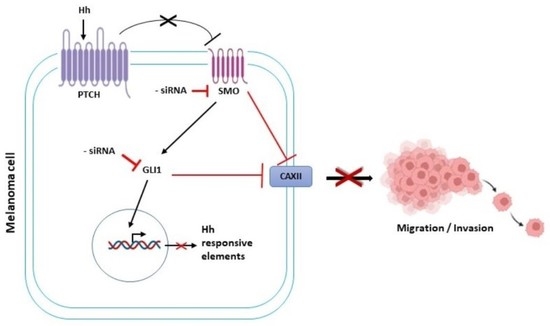
1. Introduction
2. Results
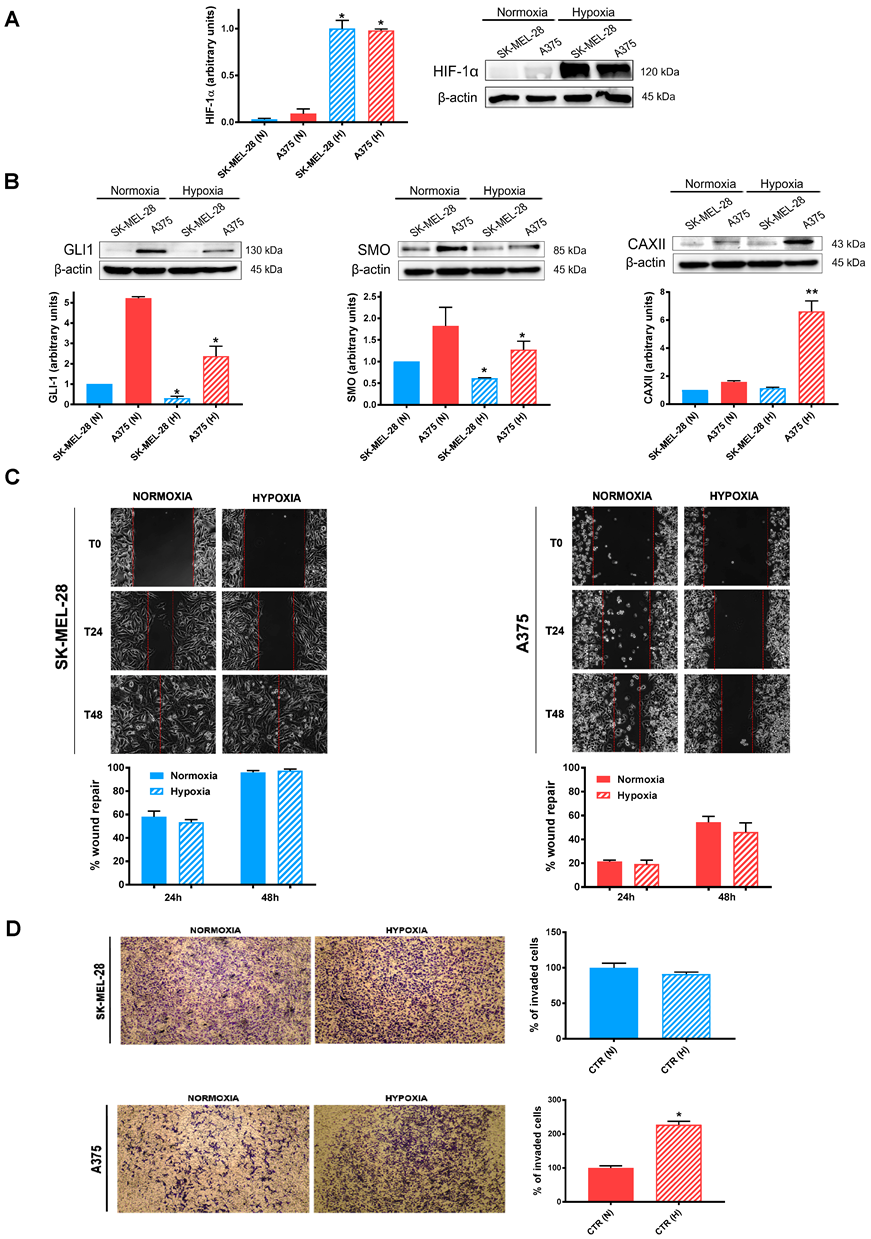
2.1. Effects of Hypoxia on Untreated SK-MEL-28 and A375
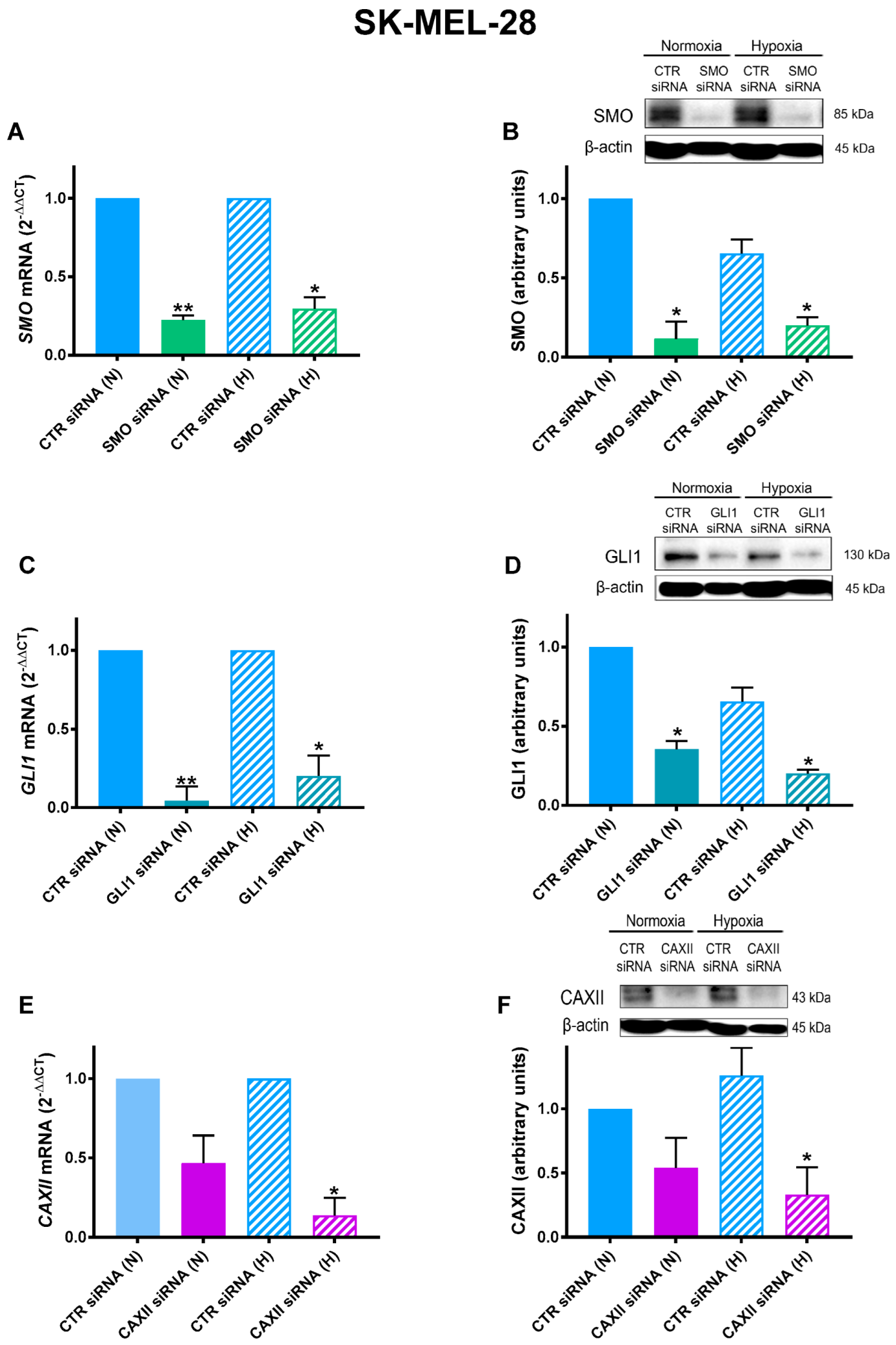
2.2. SMO, GLI1, and CAXII Transient Knockdown in SK-MEL-28
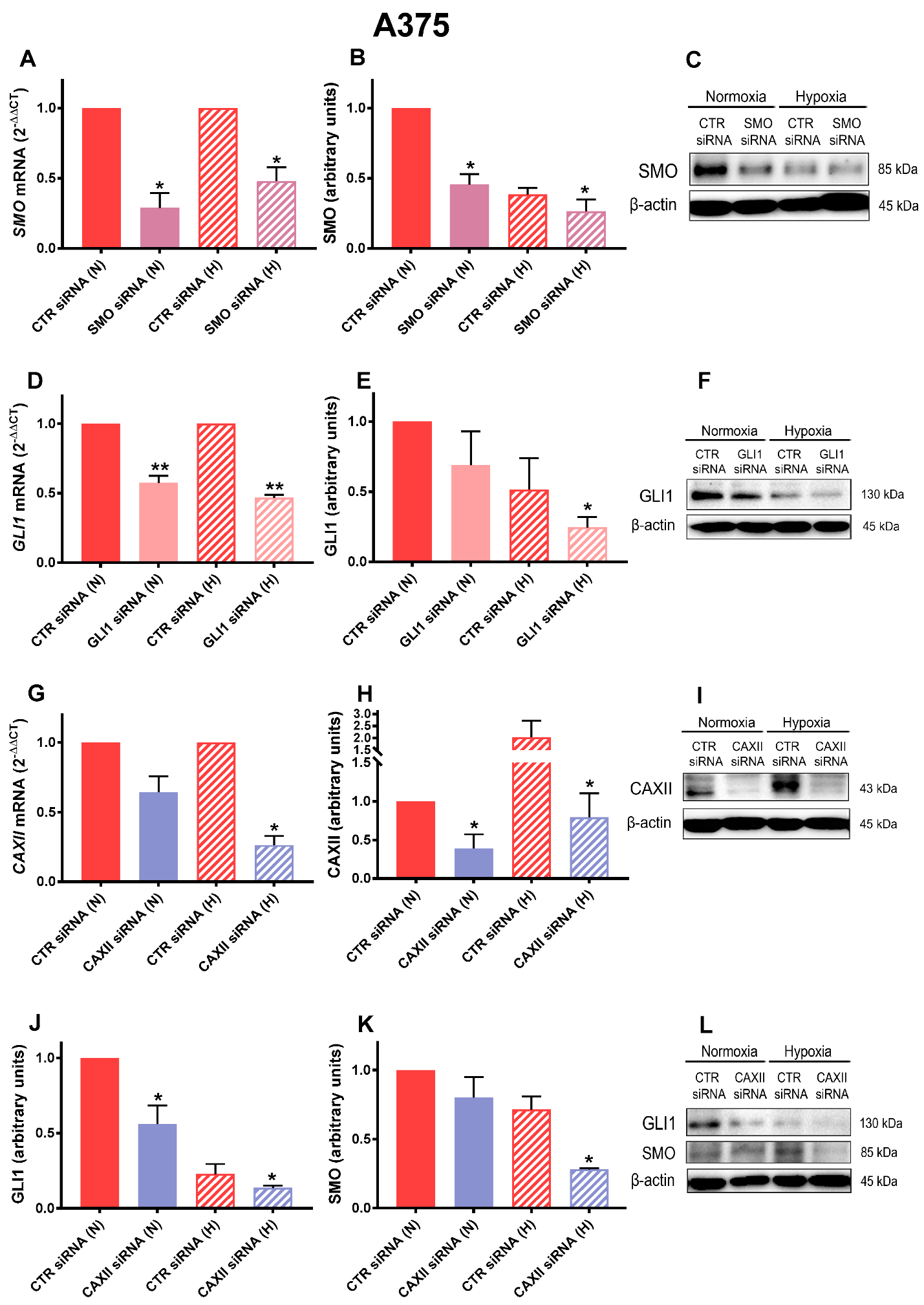
2.3. SMO, GLI1, and CAXII Transient Knockdown in A375
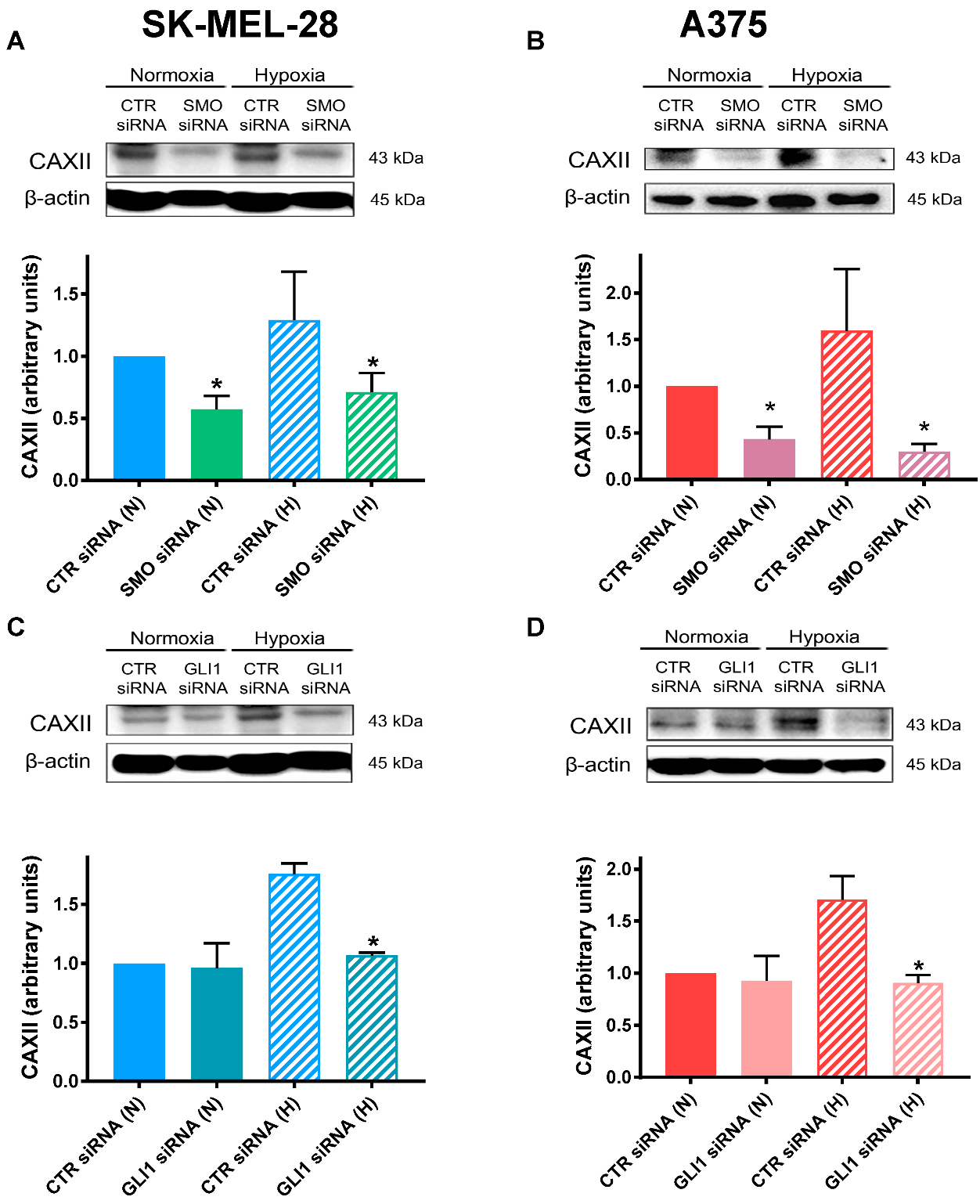
2.4. SMO and GLI1 Transient Knockdown Reduced CAXII Protein Levels
2.5. SK-MEL-28 Cell Migration Is Impaired by CAXII, SMO, and GLI1 Transient Knockdown
2.6. A375 Cell Migration Is Inhibited by CAXII, SMO, and GLI1 siRNA
2.7. SMO and GLI1 Transient Knockdown Impaired Melanoma Cell Invasion
2.8. CAXII Transient Knockdown Resulted in Decreased Cell Invasion and MMP-2/ MMP-9 Activity
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Western Blot
4.3. RNA Extraction and RT-qPCR
4.4. Wound-Healing Assay
4.5. Modified Boyden Chamber
4.6. Zymography
4.7. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rebecca, V.W.; Sondak, V.K.; Smalley, K.S. A brief history of melanoma: From mummies to mutations. Melanoma Res. 2012, 22, 114. [Google Scholar] [CrossRef] [PubMed]
- Little, E.G.; Eide, M.J. Update on the current state of melanoma incidence. Dermatol. Clin. 2012, 30, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Faiao-Flores, F.; Alves-Fernandes, D.; Pennacchi, P.C.; Sandri, S.; Vicente, A.L.S.A.; Scapulatempo-Neto, C.; Vazquez, V.D.L.; Reis, R.M.; Chauhan, J.; Goding, C. Targeting the hedgehog transcription factors GLI1 and GLI2 restores sensitivity to vemurafenib-resistant human melanoma cells. Oncogene 2017, 36, 1849–1861. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.E.; Shalin, S.C.; Tackett, A.J. Current state of melanoma diagnosis and treatment. Cancer Biol. Ther. 2019, 20, 1366–1379. [Google Scholar] [CrossRef] [PubMed]
- Scolyer, R.A.; Long, G.V.; Thompson, J.F. Evolving concepts in melanoma classification and their relevance to multidisciplinary melanoma patient care. Mol. Oncol. 2011, 5, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef]
- Yang, L.; Xie, G.; Fan, Q.; Xie, J. Activation of the hedgehog-signaling pathway in human cancer and the clinical implications. Oncogene 2010, 29, 469–481. [Google Scholar] [CrossRef]
- Stecca, B.; Mas, C.; Clement, V.; Zbinden, M.; Correa, R.; Piguet, V.; Beermann, F.; i Altaba, A.R. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 5895–5900. [Google Scholar] [CrossRef]
- Pandolfi, S.; Montagnani, V.; Lapucci, A.; Stecca, B. HEDGEHOG/GLI-E2F1 axis modulates iASPP expression and function and regulates melanoma cell growth. Cell Death Differ. 2015, 22, 2006–2019. [Google Scholar] [CrossRef]
- O’Reilly, K.E.; De Miera, E.V.-S.; Segura, M.F.; Friedman, E.; Poliseno, L.; Han, S.W.; Zhong, J.; Zavadil, J.; Pavlick, A.; Hernando, E. Hedgehog pathway blockade inhibits melanoma cell growth in vitro and in vivo. Pharmaceuticals 2013, 6, 1429–1450. [Google Scholar] [CrossRef]
- Santini, R.; Vinci, M.C.; Pandolfi, S.; Penachioni, J.Y.; Montagnani, V.; Olivito, B.; Gattai, R.; Pimpinelli, N.; Gerlini, G.; Borgognoni, L. Hedgehog-GLI signaling drives self-renewal and tumorigenicity of human melanoma-initiating cells. Stem Cells 2012, 30, 1808–1818. [Google Scholar] [CrossRef] [PubMed]
- Pietrobono, S.; Gaudio, E.; Gagliardi, S.; Zitani, M.; Carrassa, L.; Migliorini, F.; Petricci, E.; Manetti, F.; Makukhin, N.; Bond, A.G. Targeting non-canonical activation of GLI1 by the SOX2-BRD4 transcriptional complex improves the efficacy of HEDGEHOG pathway inhibition in melanoma. Oncogene 2021, 40, 3799–3814. [Google Scholar] [CrossRef]
- Infante, P.; Mori, M.; Alfonsi, R.; Ghirga, F.; Aiello, F.; Toscano, S.; Ingallina, C.; Siler, M.; Cucchi, D.; Po, A.; et al. Gli1/DNA interaction is a druggable target for Hedgehog-dependent tumors. EMBO J. 2015, 34, 200–217. [Google Scholar] [CrossRef] [PubMed]
- Lospinoso Severini, L.; Quaglio, D.; Basili, I.; Ghirga, F.; Bufalieri, F.; Caimano, M.; Balducci, S.; Moretti, M.; Romeo, I.; Loricchio, E. A smo/gli multitarget hedgehog pathway inhibitor impairs tumor growth. Cancers 2019, 11, 1518. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.L.; Matsui, W. Hedgehog pathway as a drug target: Smoothened inhibitors in development. Oncotargets 2012, 5, 47. [Google Scholar] [CrossRef]
- Amakye, D.; Jagani, Z.; Dorsch, M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat. Med. 2013, 19, 1410–1422. [Google Scholar] [CrossRef]
- Pietrobono, S.; Santini, R.; Gagliardi, S.; Dapporto, F.; Colecchia, D.; Chiariello, M.; Leone, C.; Valoti, M.; Manetti, F.; Petricci, E. Targeted inhibition of Hedgehog-GLI signaling by novel acylguanidine derivatives inhibits melanoma cell growth by inducing replication stress and mitotic catastrophe. Cell Death Dis. 2018, 9, 1–16. [Google Scholar] [CrossRef]
- Turner, N.; Ware, O.; Bosenberg, M. Genetics of metastasis: Melanoma and other cancers. Clin. Exp. Metastas 2018, 35, 379–391. [Google Scholar] [CrossRef]
- Wang, S.; Fan, W.; Wan, B.; Tu, M.; Jin, F.; Liu, F.; Xu, H.; Han, P. Characterization of long noncoding RNA and messenger RNA signatures in melanoma tumorigenesis and metastasis. PLoS ONE 2017, 12, e0172498. [Google Scholar] [CrossRef]
- Flockhart, R.J.; Webster, D.E.; Qu, K.; Mascarenhas, N.; Kovalski, J.; Kretz, M.; Khavari, P.A. BRAFV600E remodels the melanocyte transcriptome and induces BANCR to regulate melanoma cell migration. Genome Res. 2012, 22, 1006–1014. [Google Scholar] [CrossRef]
- Radford, K.J.; Thorne, R.F.; Hersey, P. Regulation of tumor cell motility and migration by CD63 in a human melanoma cell line. J. Immunol. 1997, 158, 3353–3358. [Google Scholar] [PubMed]
- Katerinaki, E.; Evans, G.; Lorigan, P.; MacNeil, S. TNF-α increases human melanoma cell invasion and migration in vitro: The role of proteolytic enzymes. Br. J. Cancer 2003, 89, 1123–1129. [Google Scholar] [CrossRef] [PubMed]
- Voura, E.B.; Ramjeesingh, R.A.; Montgomery, A.M.; Siu, C.-H. Involvement of integrin αvβ3and cell adhesion molecule l1 in transendothelial migration of melanoma cells. Mol. Biol. Cell 2001, 12, 2699–2710. [Google Scholar] [CrossRef] [PubMed]
- Sadok, A.; McCarthy, A.; Caldwell, J.; Collins, I.; Garrett, M.D.; Yeo, M.; Hooper, S.; Sahai, E.; Kuemper, S.; Mardakheh, F.K. Rho kinase inhibitors block melanoma cell migration and inhibit metastasis. Cancer Res. 2015, 75, 2272–2284. [Google Scholar] [CrossRef]
- Stock, C.; Gassner, B.; Hauck, C.R.; Arnold, H.; Mally, S.; Eble, J.A.; Dieterich, P.; Schwab, A. Migration of human melanoma cells depends on extracellular pH and Na+/H+ exchange. J. Physiol. 2005, 567, 225–238. [Google Scholar] [CrossRef]
- Mazurkiewicz, J.; Simiczyjew, A.; Dratkiewicz, E.; Ziętek, M.; Matkowski, R.; Nowak, D. Stromal cells present in the melanoma niche affect tumor invasiveness and its resistance to therapy. Int. J. Mol. Sci. 2021, 22, 529. [Google Scholar] [CrossRef]
- Gurzu, S.; Beleaua, M.A.; Jung, I. The role of tumor microenvironment in development and progression of malignant melanomas—A systematic review. Rom. J. Morphol. Embryol. 2018, 59, 23–28. [Google Scholar]
- Semenza, G.L. Hypoxia, clonal selection, and the role of HIF-1 in tumor progression. Crit. Rev. Biochem. Mol. Biol. 2000, 35, 71–103. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83. [Google Scholar] [CrossRef]
- Lartigau, E.; Randrianarivelo, H.; Avril, M.; Margulis, A.; Spatz, A.; Eschwege, F.; Guichard, M. Intratumoral oxygen tension in metastatic melanoma. Melanoma Res. 1997, 7, 400–406. [Google Scholar] [CrossRef]
- Vaupel, P.; Mayer, A. Hypoxia in cancer: Significance and impact on clinical outcome. Cancer Metastasis Rev. 2007, 26, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat Rev Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrases-an overview. Curr. Pharm. Des. 2008, 14, 603–614. [Google Scholar] [CrossRef]
- Zamanova, S.; Shabana, A.M.; Mondal, U.K.; Ilies, M.A. Carbonic anhydrases as disease markers. Expert Opin. Ther. Pat. 2019, 29, 509–533. [Google Scholar] [CrossRef]
- Carta, F.; Dumy, P.; Supuran, C.T.; Winum, J.-Y. Multivalent carbonic anhydrases inhibitors. Int. J. Mol. Sci. 2019, 20, 5352. [Google Scholar] [CrossRef]
- Maren, T.H. Carbonic anhydrase: Chemistry, physiology, and inhibition. Physiol. Rev. 1967, 47, 595–781. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic Anhydrases and Metabolism. Metabolites 2018, 9, 25. [Google Scholar]
- Chiche, J.; Ilc, K.; Laferriere, J.; Trottier, E.; Dayan, F.; Mazure, N.M.; Brahimi-Horn, M.C.; Pouysségur, J. Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular pH. Cancer Res. 2009, 69, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Tafreshi, N.K.; Lloyd, M.C.; Proemsey, J.B.; Bui, M.M.; Kim, J.; Gillies, R.J.; Morse, D.L. Evaluation of CAIX and CAXII expression in breast cancer at varied O 2 levels: CAIX is the superior surrogate imaging biomarker of tumor hypoxia. Mol. Imaging Biol. 2016, 18, 219–231. [Google Scholar] [CrossRef]
- McDonald, P.C.; Dedhar, S. Carbonic anhydrase IX (CAIX) as a mediator of hypoxia-induced stress response in cancer cells. Carbon. Anhydrase Mech. Regul. Links Dis. Ind. Appl. 2014, 75, 255–269. [Google Scholar]
- Chiche, J.; Brahimi-Horn, M.C.; Pouysségur, J. Tumour hypoxia induces a metabolic shift causing acidosis: A common feature in cancer. J. Cell. Mol. Med. 2010, 14, 771–794. [Google Scholar] [CrossRef]
- Kuchuk, O.; Tuccitto, A.; Citterio, D.; Huber, V.; Camisaschi, C.; Milione, M.; Vergani, B.; Villa, A.; Alison, M.R.; Carradori, S. pH regulators to target the tumor immune microenvironment in human hepatocellular carcinoma. Oncoimmunology 2018, 7, e1445452. [Google Scholar] [CrossRef] [PubMed]
- Potter, C.; Harris, A. Diagnostic, prognostic and therapeutic implications of carbonic anhydrases in cancer. Br. J. Cancer 2003, 89, 2–7. [Google Scholar] [CrossRef]
- Guerrini, G.; Criscuoli, M.; Filippi, I.; Naldini, A.; Carraro, F. Inhibition of smoothened in breast cancer cells reduces CAXII expression and cell migration. J. Cell. Physiol. 2018, 233, 9799–9811. [Google Scholar] [CrossRef] [PubMed]
- Giuntini, G.; Monaci, S.; Cau, Y.; Mori, M.; Naldini, A.; Carraro, F. Inhibition of Melanoma Cell Migration and Invasion Targeting the Hypoxic Tumor Associated CAXII. Cancers 2020, 12, 3018. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, G.; Durivault, J.; Filippi, I.; Criscuoli, M.; Monaci, S.; Pouyssegur, J.; Naldini, A.; Carraro, F.; Parks, S.K. Carbonic anhydrase XII expression is linked to suppression of Sonic hedgehog ligand expression in triple negative breast cancer cells. Biochem. Biophys. Res. Commun. 2019, 516, 408–413. [Google Scholar] [CrossRef]
- Hofmann, U.B.; Houben, R.; Brocker, E.B.; Becker, J.C. Role of matrix metalloproteinases in melanoma cell invasion. Biochimie 2005, 87, 307–314. [Google Scholar] [CrossRef]
- Napoli, S.; Scuderi, C.; Gattuso, G.; Bella, V.D.; Candido, S.; Basile, M.S.; Libra, M.; Falzone, L. Functional Roles of Matrix Metalloproteinases and Their Inhibitors in Melanoma. Cells 2020, 9, 1151. [Google Scholar] [CrossRef]
- Inaguma, S.; Kasai, K.; Ikeda, H. GLI1 facilitates the migration and invasion of pancreatic cancer cells through MUC5AC-mediated attenuation of E-cadherin. Oncogene 2011, 30, 714–723. [Google Scholar] [CrossRef]
- Feldmann, G.; Dhara, S.; Fendrich, V.; Bedja, D.; Beaty, R.; Mullendore, M.; Karikari, C.; Alvarez, H.; Iacobuzio-Donahue, C.; Jimeno, A. Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: A new paradigm for combination therapy in solid cancers. Cancer Res. 2007, 67, 2187–2196. [Google Scholar] [CrossRef]
- Cheng, W.-T.; Xu, K.; Tian, D.-Y.; Zhang, Z.-G.; Liu, L.-J.; Chen, Y. Role of Hedgehog signaling pathway in proliferation and invasiveness of hepatocellular carcinoma cells. Int. J. Oncol. 2009, 34, 829–836. [Google Scholar]
- Liao, X.; Siu, M.K.; Au, C.W.; Wong, E.S.; Chan, H.Y.; Ip, P.P.; Ngan, H.Y.; Cheung, A.N. Aberrant activation of hedgehog signaling pathway in ovarian cancers: Effect on prognosis, cell invasion and differentiation. Carcinogenesis 2009, 30, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Dratkiewicz, E.; Simiczyjew, A.; Mazurkiewicz, J.; Ziętek, M.; Matkowski, R.; Nowak, D. Hypoxia and Extracellular Acidification as Drivers of Melanoma Progression and Drug Resistance. Cells 2021, 10, 862. [Google Scholar] [CrossRef]
- Rossi, S.; Cordella, M.; Tabolacci, C.; Nassa, G.; D’Arcangelo, D.; Senatore, C.; Pagnotto, P.; Magliozzi, R.; Salvati, A.; Weisz, A.; et al. TNF-alpha and metalloproteases as key players in melanoma cells aggressiveness. J. Exp. Clin. Cancer Res. 2018, 37, 326. [Google Scholar] [CrossRef] [PubMed]
- Michaylira, C.Z.; Nakagawa, H. Hypoxic microenvironment as a cradle for melanoma development and progression. Cancer Biol. 2006, 5, 476–479. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.K.; Taipale, J.; Cooper, M.K.; Beachy, P.A. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002, 16, 2743–2748. [Google Scholar] [CrossRef]
- Buart, S.; Terry, S.; Noman, M.Z.; Lanoy, E.; Boutros, C.; Fogel, P.; Dessen, P.; Meurice, G.; Gaston-Mathe, Y.; Vielh, P.; et al. Transcriptional response to hypoxic stress in melanoma and prognostic potential of GBE1 and BNIP3. Oncotarget 2017, 8, 108786–108801. [Google Scholar] [CrossRef]
- Spivak-Kroizman, T.R.; Hostetter, G.; Posner, R.; Aziz, M.; Hu, C.; Demeure, M.J.; Von Hoff, D.; Hingorani, S.R.; Palculict, T.B.; Izzo, J.; et al. Hypoxia triggers hedgehog-mediated tumor-stromal interactions in pancreatic cancer. Cancer Res. 2013, 73, 3235–3247. [Google Scholar] [CrossRef]
- Monaci, S.; Aldinucci, C.; Rossi, D.; Giuntini, G.; Filippi, I.; Ulivieri, C.; Marotta, G.; Sozzani, S.; Carraro, F.; Naldini, A. Hypoxia Shapes Autophagy in LPS-Activated Dendritic Cells. Front. Immunol. 2020, 11, 573646. [Google Scholar] [CrossRef]
- Criscuoli, M.; Filippi, I.; Osti, D.; Aldinucci, C.; Guerrini, G.; Pelicci, G.; Carraro, F.; Naldini, A. The Shc protein RAI promotes an adaptive cell survival program in hypoxic neuroblastoma cells. J. Cell. Physiol. 2017, 233, 4282–4293. [Google Scholar] [CrossRef]
- Livak, K.J.; Marmaro, J.; Todd, J.A. Towards fully automated genome–wide polymorphism screening. Nat. Genet. 1995, 9, 341–342. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GENE | Sense | Antisense |
|---|---|---|
| GLI1 | GGAAAGCAGACUGACUGUGtt | CACAGUCAGUCUGCUUUCCcg |
| SMO | CUGUUAUUCUCUUCUACGUtt | ACGUAGAAGAGAAUAACAGcg |
| CAXII | CGGUUCCAAGUGGACUUAUtt | AUAAGUCCACUUGGAACCGcg |
| Scrambled | GGAUUUCUAUACGUUUAUUtt | AAUAAACGUAUAGAAAUCCcg |
| GENE | Fv | Rv |
|---|---|---|
| GLI1 | 5′ TTCCTACCAGAGTCCCAAGT | 5′ CCCTATGTCAAGCCCTATTT |
| SMO | 5′ CTTTGTCATCGTGTACTACGCC | 5′ CGAGAGAGGCTGGTAGGTC |
| CAXII | 5′ CTGCATCATGTATTTAGGGGC | 5′ GAGTTGCGCCTGTCAGAAAC |
| L32 | 5′ GCTGGAAGTGCTGCTGATGTG | 5′ CGATGGCTTTGCGGTTCTTGG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giuntini, G.; Coppola, F.; Falsini, A.; Filippi, I.; Monaci, S.; Naldini, A.; Carraro, F. Role of the Hedgehog Pathway and CAXII in Controlling Melanoma Cell Migration and Invasion in Hypoxia. Cancers 2022, 14, 4776. https://doi.org/10.3390/cancers14194776
Giuntini G, Coppola F, Falsini A, Filippi I, Monaci S, Naldini A, Carraro F. Role of the Hedgehog Pathway and CAXII in Controlling Melanoma Cell Migration and Invasion in Hypoxia. Cancers. 2022; 14(19):4776. https://doi.org/10.3390/cancers14194776
Chicago/Turabian StyleGiuntini, Gaia, Federica Coppola, Alessandro Falsini, Irene Filippi, Sara Monaci, Antonella Naldini, and Fabio Carraro. 2022. "Role of the Hedgehog Pathway and CAXII in Controlling Melanoma Cell Migration and Invasion in Hypoxia" Cancers 14, no. 19: 4776. https://doi.org/10.3390/cancers14194776
APA StyleGiuntini, G., Coppola, F., Falsini, A., Filippi, I., Monaci, S., Naldini, A., & Carraro, F. (2022). Role of the Hedgehog Pathway and CAXII in Controlling Melanoma Cell Migration and Invasion in Hypoxia. Cancers, 14(19), 4776. https://doi.org/10.3390/cancers14194776