
Bis(chloroacetamidino)-Derived Heteroarene-Fused Anthraquinones Bind to and Cause Proteasomal Degradation of tNOX, Leading to c-Flip Downregulation and Apoptosis in Oral Cancer Cells

 , , , and
, , , and 
Abstract
Simple Summary
Abstract
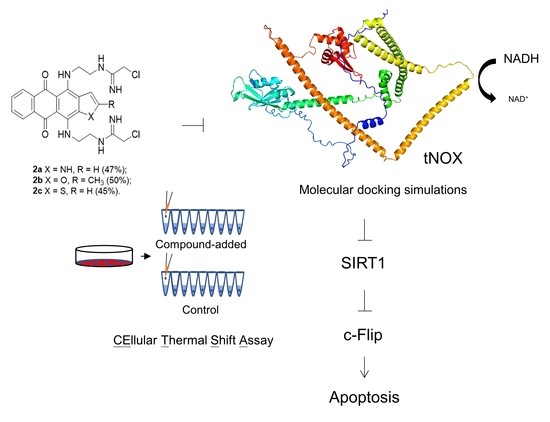
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemistry
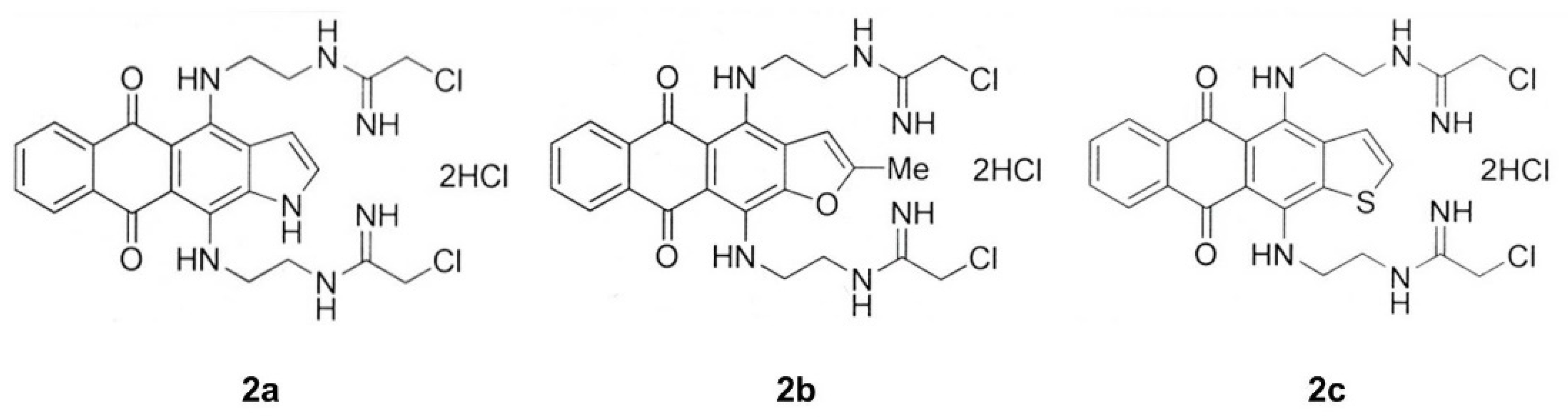
2.2. 4,11-bis(2-(2-chloroacetamidine)ethylamino)naphtho[2,3-f]indole-5,10-dione dihydrochloride (2a)
2.3. 4,11-bis(2-(2-chloroacetamidine)ethylamino)-2-methylanthra[2,3-b]furan-5,10-dione dihydrochloride (2b)
2.4. 4,11-bis(2-(2-chloroacetamidine)ethylamino)anthra[2,3-b]thiophene-5,10-dione dihydrochloride (2c)
2.5. Cell Culture and Reagents
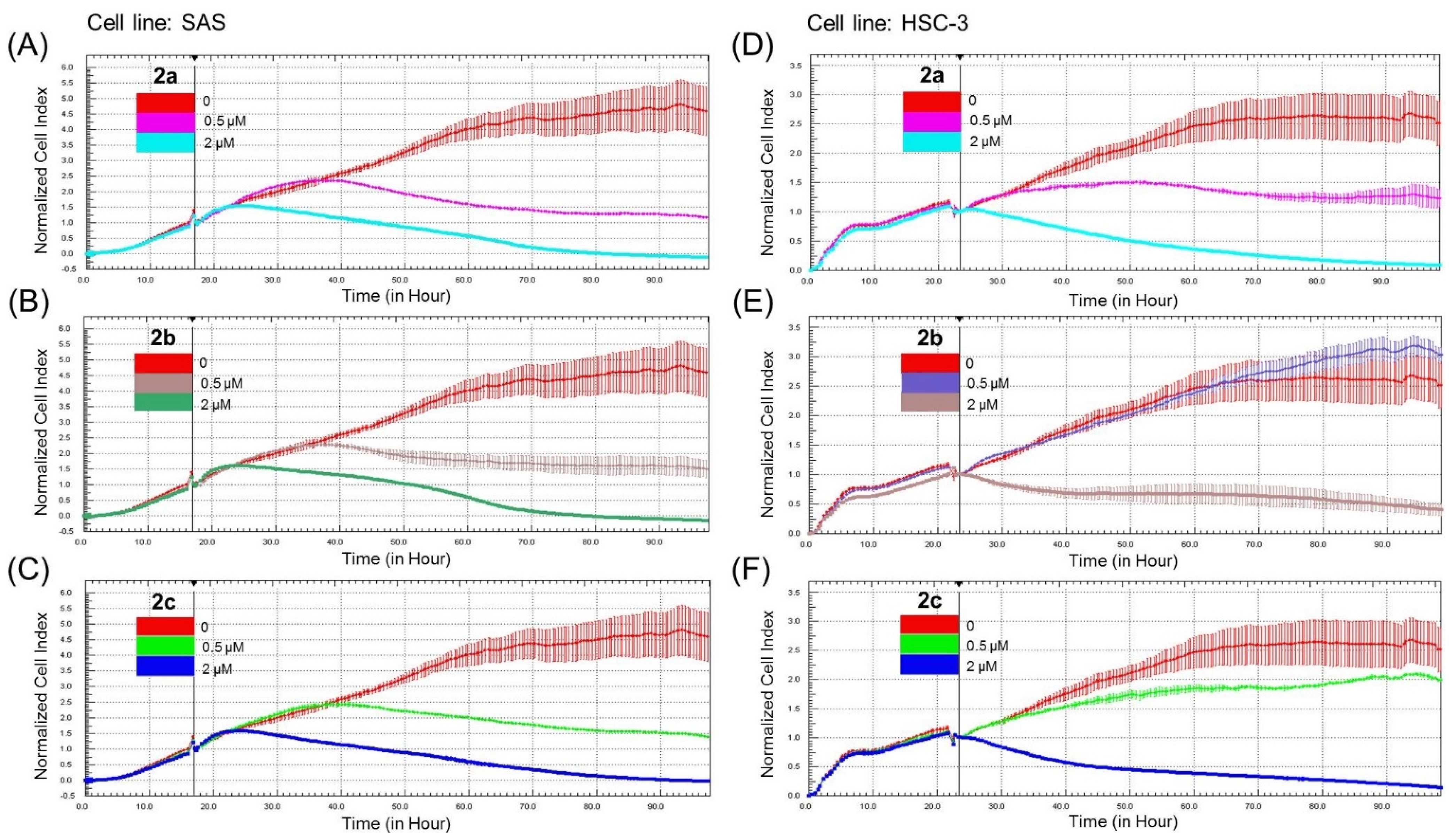
2.6. Continuous Monitoring of Cell Growth by Cell Impedance Measurements
2.7. Apoptosis Determination
2.8. Reverse Transcriptase–Polymerase Chain Reaction (RT-PCR)
2.9. Identification of tNOX as a Cellular Target by Cellular Thermal Shift Assays (CETSA)
2.10. Immunoblotting and Immunoprecipitation
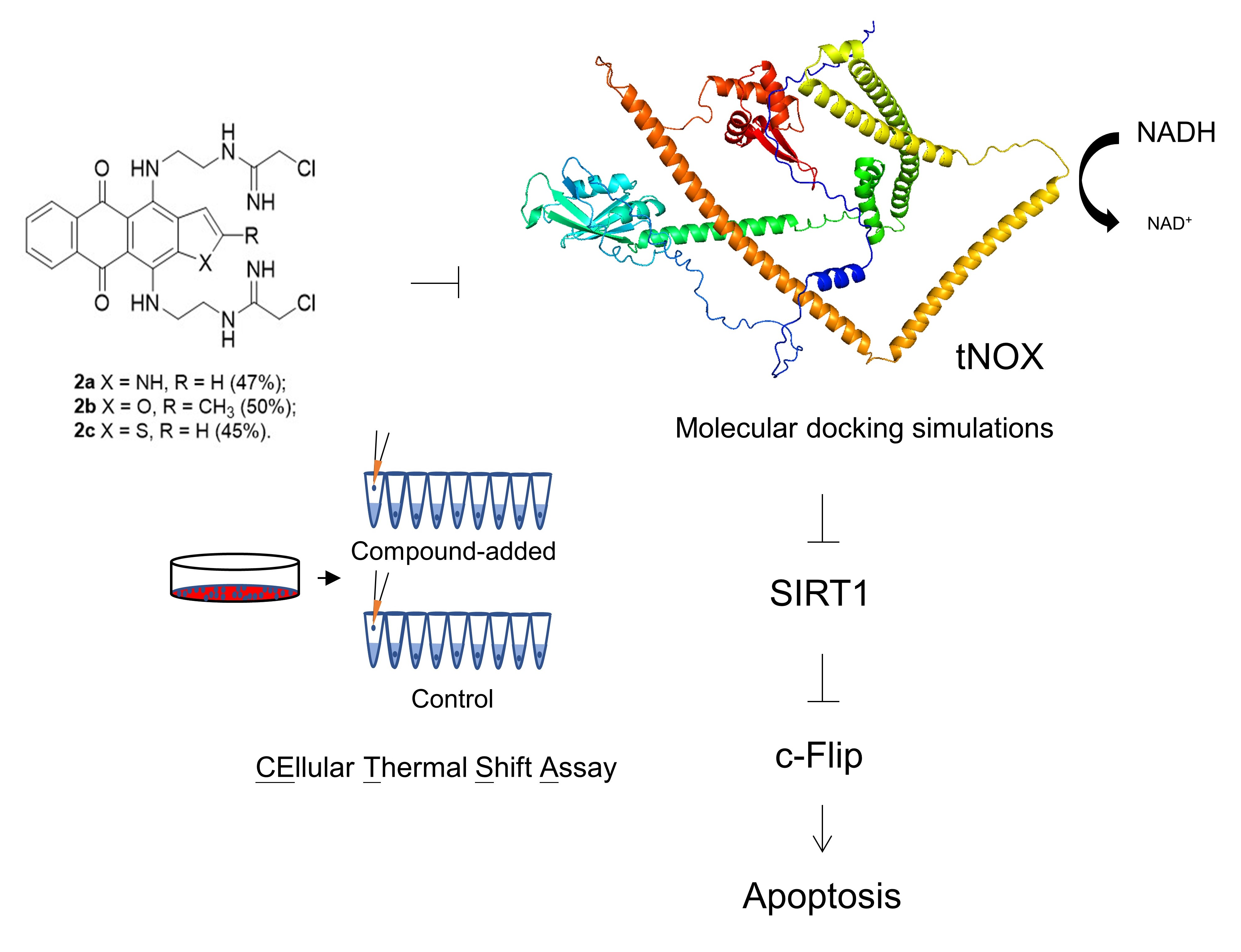
2.11. Molecular Docking Simulation
2.12. Statistics
3. Results
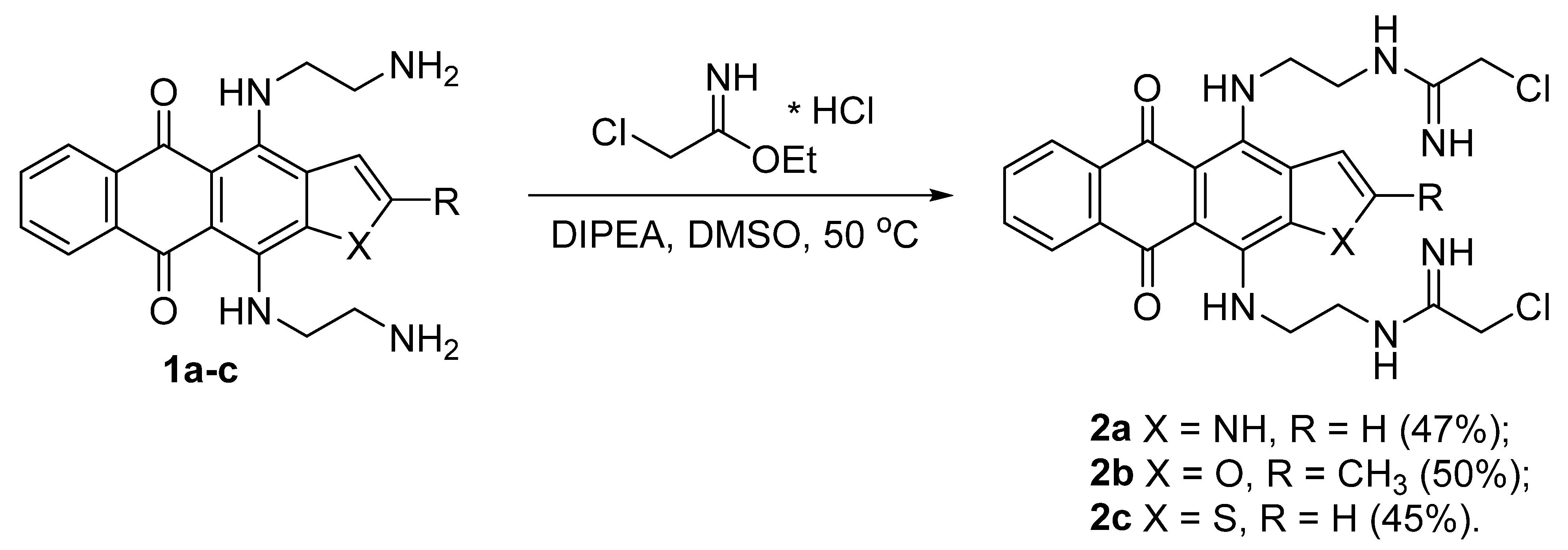
3.1. Synthesis of Heteroarene-Fused Anthraquinones
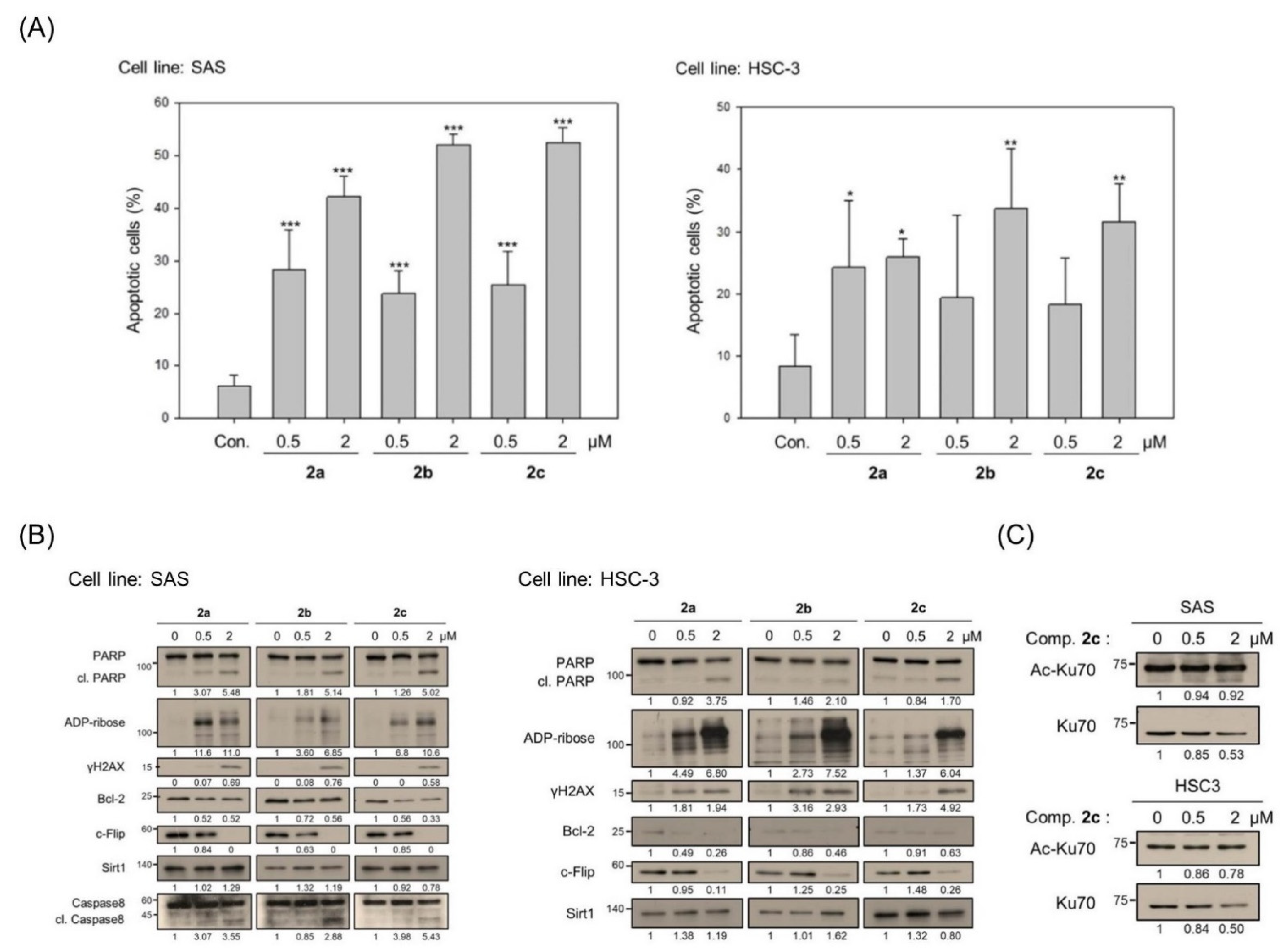
3.2. Bis(chloroacetamidino)heteroarene-Fused Anthraquinones Attenuate the Proliferation of Oral Cancer Cells through Induction of Apoptosis
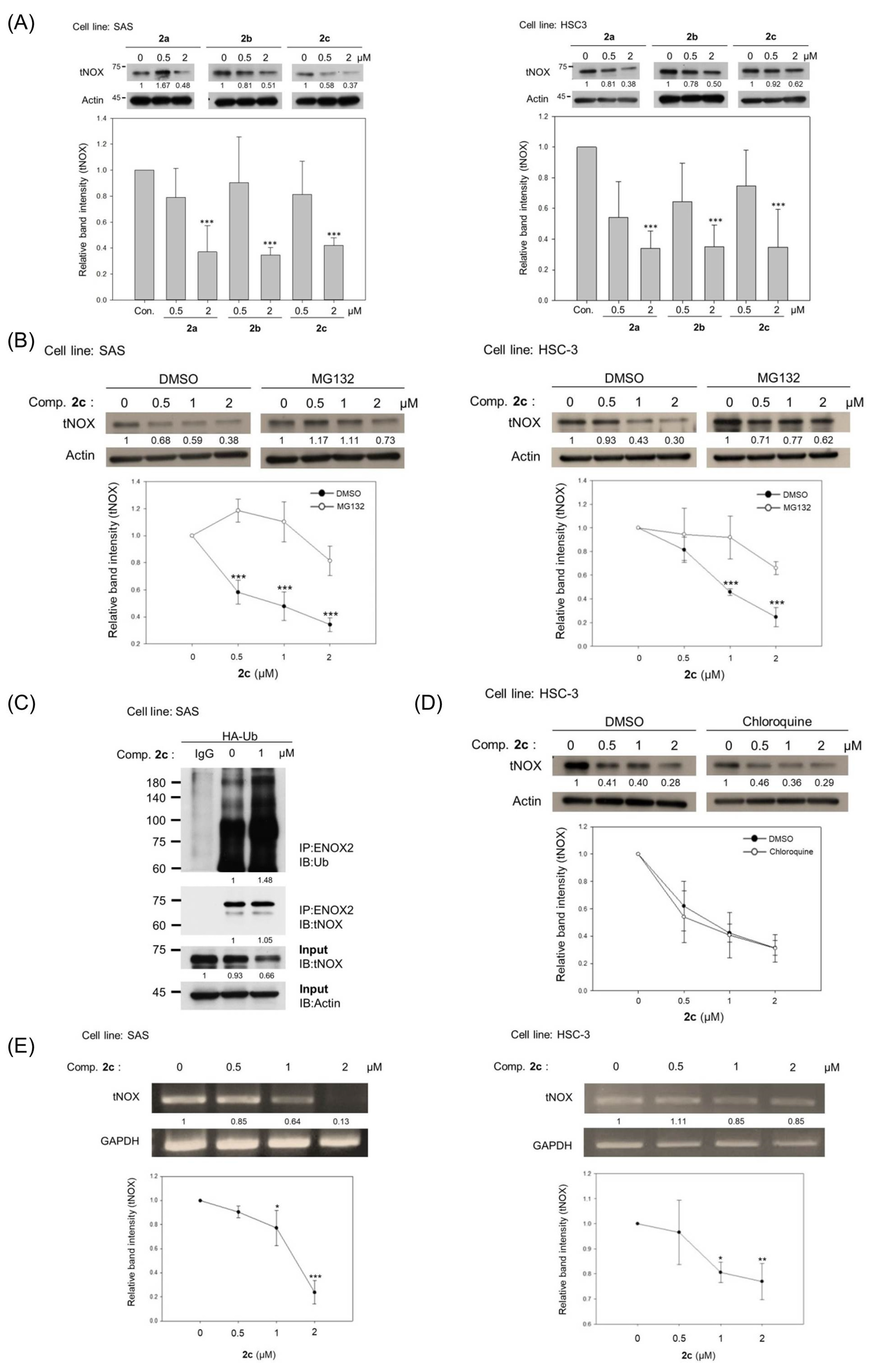
3.3. Bis(chloroacetamidino)heteroarene-Fused Anthraquinones Downregulate tNOX Expression at the Transcriptional and Protein Levels
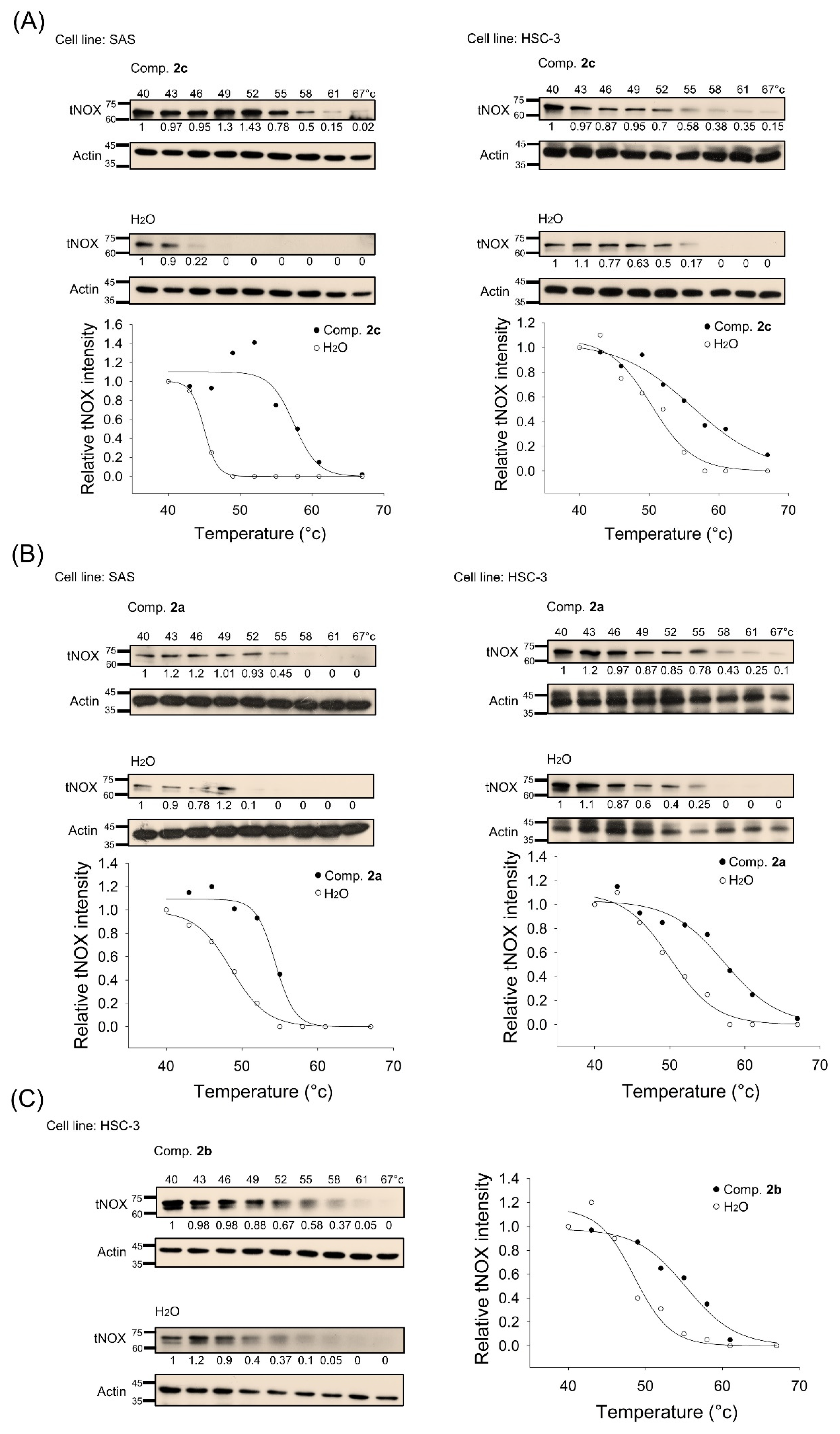
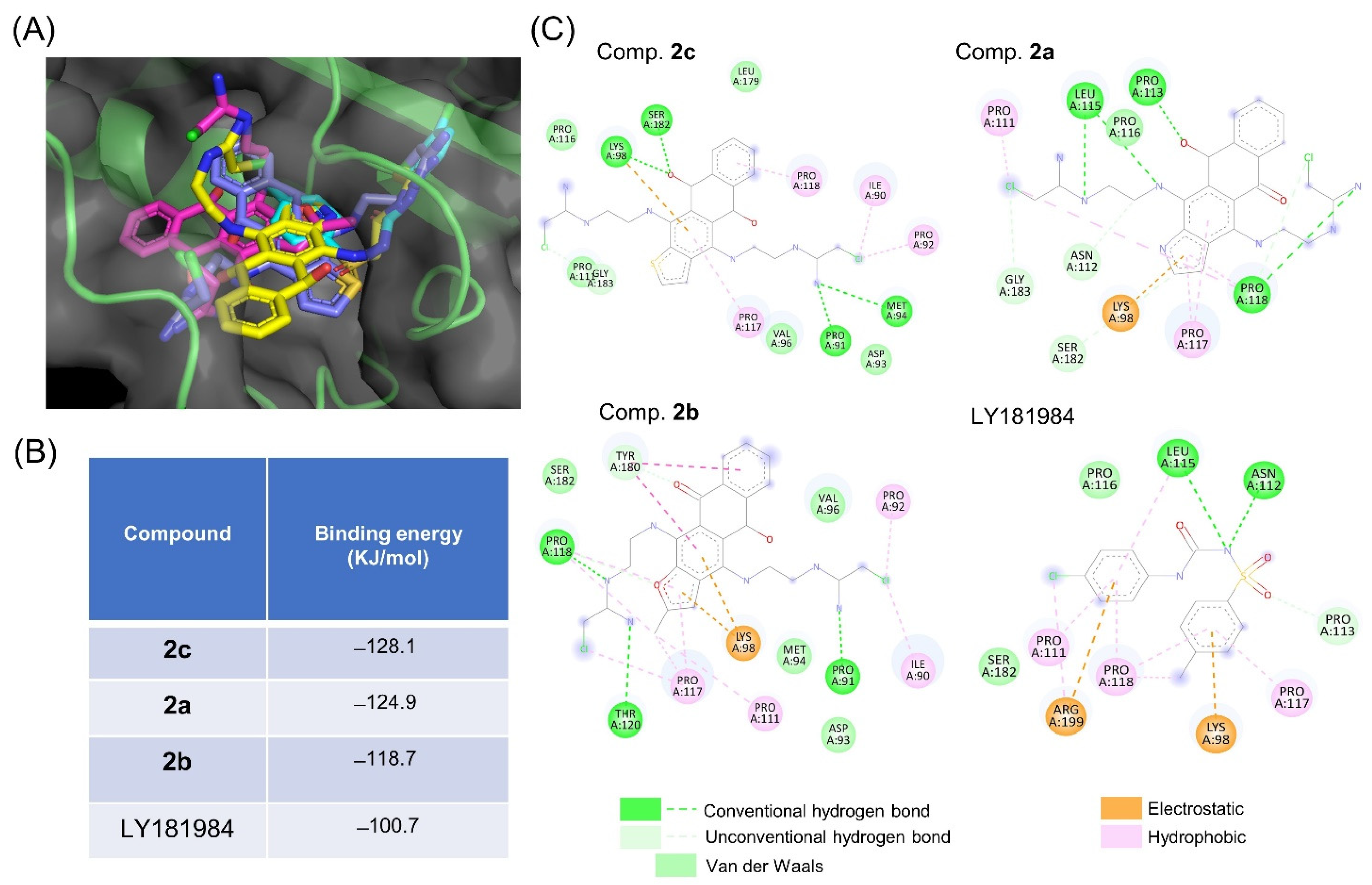
3.4. tNOX Acts as a Cellular Target of bis(chloroacetamidino)heteroarene-Fused Anthraquinones, as Shown by Cellular Thermal Shift Assays (CETSA) and Molecular Docking Simulations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CETSA | cellular thermal shift assay |
| c-Flip | cellular FLICE-like inhibitory protein |
| DIPEA | diisopropylethylamine |
| ESI | electron spray 118 ionization |
| HDAC | histone deacetylase |
| SIRT1 | silent mating-type information regulation 1 (Sirtuin 1) |
| TM | melting temperature |
| tNOX | tumor-associated NADH oxidase |
| Top1 | topoisomerase 1 |
References
- Faulds, D.; Balfour, J.A.; Chrisp, P.; Langtry, H.D. Mitoxantrone—A Review of Its Pharmacodynamic and Pharmacokinetic Properties, and Therapeutic Potential in the Chemotherapy of Cancer. Drugs 1991, 41, 400–449. [Google Scholar] [CrossRef] [PubMed]
- Stuart-Harris, R.C.; Bozek, T.; Pavlidis, N.A.; Smith, I.E. Mitoxantrone: An active new agent in the treatment of advanced breast cancer. Cancer Chemother. Pharmacol. 1984, 12, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Fox, K.R.; Waring, M.J.; Brown, J.R.; Neidle, S. DNA sequence preferences for the anti-cancer drug mitoxanthrone and related anthraquinones revealed by DNase I footprinting. FEBS Lett. 1986, 202, 289–294. [Google Scholar] [CrossRef]
- Tikhomirov, A.S.; Shtil, A.A.; Shchekotikhin, A.E. Advances in the Discovery of Anthraquinone-Based Anticancer Agents. Recent Pat. Anti-Cancer Drug Discov. 2018, 13, 159–183. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Wang, C.M.; Li, D.R.; Hou, H.X. Novel anthraquinone compounds as anticancer agents and their potential mechanism. Future Med. Chem. 2020, 12, 627–644. [Google Scholar] [CrossRef]
- Wright, E.P.; Day, H.A.; Ibrahim, A.M.; Kumar, J.; Boswell, L.J.; Huguin, C.; Stevenson, C.E.; Pors, K.; Waller, Z.A. Mitoxantrone and Analogues Bind and Stabilize i-Motif Forming DNA Sequences. Sci. Rep. 2016, 6, 39456. [Google Scholar] [CrossRef]
- Al-Otaibi, J.S.; Spittle, P.T.; El Gogary, T.M. Interaction of anthraquinone anti-cancer drugs with DNA:Experimental and computational quantum chemical study. J. Mol. Struct. 2017, 1127, 751–760. [Google Scholar] [CrossRef]
- Al-Otaibi, J.S.; El Gogary, T.M. Synthesis of novel anthraquinones: Molecular structure, molecular chemical reactivity descriptors and interactions with DNA as antibiotic and anti-cancer drugs. J. Mol. Struct. 2017, 1130, 799–809. [Google Scholar] [CrossRef]
- Ongaro, A.; Ribaudo, G.; Braud, E.; Etheve-Quelquejeu, M.; De Franco, M.; Garbay, C.; Demange, L.; Gresh, N.; Zagotto, G. Design and synthesis of a peptide derivative of ametantrone targeting the major groove of the d(GGCGCC)(2) palindromic sequence. New J. Chem. 2020, 44, 3624–3631. [Google Scholar] [CrossRef]
- Xie, X.W.; Liu, Z.P.; Li, X. Design, synthesis, bioevaluation of LFC- and PA-tethered anthraquinone analogues of mitoxantrone. Bioorg. Chem. 2020, 101, 104005. [Google Scholar] [CrossRef]
- Chen, C.L.; Liu, F.L.; Lee, C.C.; Chen, T.C.; Chang, W.W.; Guh, J.H.; Ahmed Ali, A.A.; Chang, D.M.; Huang, H.S. Ring fusion strategy for the synthesis of anthra[2,3-d]oxazole-2-thione-5,10-dione homologues as DNA topoisomerase inhibitors and as antitumor agents. Eur. J. Med. Chem. 2014, 87, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Cao, Z.; Wang, J.; Chen, X.; Chen, Y.Q.; Li, Y.; Liu, J.; Zhao, Y.; Wang, A.; He, B. A Series of Novel HDAC Inhibitors with Anthraquinone as a Cap Group. Chem. Pharm. Bull. (Tokyo) 2020, 68, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.C.; Guh, J.H.; Hsu, H.W.; Chen, C.L.; Lee, C.C.; Wu, C.L.; Lee, Y.R.; Lin, J.J.; Yu, D.S.; Huang, H.S. Synthesis and biological evaluation of anthra [1,9-cd]pyrazol-6(2H)-one scaffold derivatives as potential anticancer agents. Arab. J. Chem. 2019, 12, 2864–2881. [Google Scholar] [CrossRef]
- Wang, Q.; Ma, C.; Li, X.; Wang, X.; Rong, R.; Wei, C.; Zhang, P.; Li, X. Synthesis of novel sugar or azasugar modified anthra[1,2-d] imidazole-6,11-dione derivatives and biological evaluation. Carbohydr. Res. 2018, 460, 29–33. [Google Scholar] [CrossRef]
- Fann, L.Y.; Chen, Y.; Chu, D.C.; Weng, S.J.; Chu, H.C.; Wu, A.T.H.; Lee, J.F.; Ali, A.A.A.; Chen, T.C.; Huang, H.S.; et al. Identification and preclinical evaluation of the small molecule, NSC745887, for treating glioblastomas via suppressing DcR3-associated signaling pathways. Oncotarget 2018, 9, 11922–11937. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shchekotikhin, A.E.; Glazunova, V.A.; Dezhenkova, L.G.; Shevtsova, E.K.; Traven, V.F.; Balzarini, J.; Huang, H.S.; Shtil, A.A.; Preobrazhenskaya, M.N. The first series of 4,11-bis[(2-aminoethyl)amino]anthra[2,3-b]furan-5,10-diones: Synthesis and anti-proliferative characteristics. Eur. J. Med. Chem. 2011, 46, 423–428. [Google Scholar] [CrossRef]
- Shchekotikhin, A.E.; Dezhenkova, L.G.; Tsvetkov, V.B.; Luzikov, Y.N.; Volodina, Y.L.; Tatarskiy, V.V., Jr.; Kalinina, A.A.; Treshalin, M.I.; Treshalina, H.M.; Romanenko, V.I.; et al. Discovery of antitumor anthra[2,3-b]furan-3-carboxamides: Optimization of synthesis and evaluation of antitumor properties. Eur. J. Med. Chem. 2016, 112, 114–129. [Google Scholar] [CrossRef]
- Volodina, Y.L.; Dezhenkova, L.G.; Tikhomirov, A.S.; Tatarskiy, V.V.; Kaluzhny, D.N.; Moisenovich, A.M.; Moisenovich, M.M.; Isagulieva, A.K.; Shtil, A.A.; Tsvetkov, V.B.; et al. New anthra[2,3-b]furancarboxamides: A role of positioning of the carboxamide moiety in antitumor properties. Eur. J. Med. Chem. 2019, 165, 31–45. [Google Scholar] [CrossRef]
- Tikhomirov, A.S.; Lin, C.Y.; Volodina, Y.L.; Dezhenkova, L.G.; Tatarskiy, V.V.; Schols, D.; Shtil, A.A.; Kaur, P.; Chueh, P.J.; Shchekotikhin, A.E. New antitumor anthra[2,3-b]furan-3-carboxamides: Synthesis and structure-activity relationship. Eur. J. Med. Chem. 2018, 148, 128–139. [Google Scholar] [CrossRef]
- Tikhomirov, A.S.; Litvinova, V.A.; Andreeva, D.V.; Tsvetkov, V.B.; Dezhenkova, L.G.; Volodina, Y.L.; Kaluzhny, D.N.; Treshalin, I.D.; Schols, D.; Ramonova, A.A.; et al. Amides of pyrrole- and thiophene-fused anthraquinone derivatives: A role of the heterocyclic core in antitumor properties. Eur. J. Med. Chem. 2020, 199, 112294. [Google Scholar] [CrossRef]
- Tikhomirov, A.S.; Shchekotikhin, A.E.; Lee, Y.H.; Chen, Y.A.; Yeh, C.A.; Tatarskiy, V.V., Jr.; Dezhenkova, L.G.; Glazunova, V.A.; Balzarini, J.; Shtil, A.A.; et al. Synthesis and Characterization of 4,11-Diaminoanthra[2,3-b]furan-5,10-diones: Tumor Cell Apoptosis through tNOX-Modulated NAD(+)/NADH Ratio and SIRT1. J. Med. Chem. 2015, 58, 9522–9534. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Islam, A.; Su, C.J.; Tikhomirov, A.S.; Shchekotikhin, A.E.; Chuang, S.M.; Chueh, P.J.; Chen, Y.L. Engagement with tNOX (ENOX2) to Inhibit SIRT1 and Activate p53-Dependent and -Independent Apoptotic Pathways by Novel 4,11-Diaminoanthra[2,3-b]furan-5,10-diones in Hepatocellular Carcinoma Cells. Cancers 2019, 11, 420. [Google Scholar] [CrossRef] [PubMed]
- Morre, D.J.; Chueh, P.J.; Morre, D.M. Capsaicin inhibits preferentially the NADH oxidase and growth of transformed cells in culture. Proc. Natl. Acad. Sci. USA 1995, 92, 1831–1835. [Google Scholar] [CrossRef]
- Bruno, M.; Brightman, A.O.; Lawrence, J.; Werderitsh, D.; Morré, D.M.; Morré, D.J. Stimulation of NADH oxidase activity from rat liver plasma membranes by growth factors and hormones is decreased or absent with hepatoma plasma membranes. Biochem. J. 1992, 284 Pt 3, 625–628. [Google Scholar] [CrossRef]
- Chueh, P.J. Cell membrane redox systems and transformation. Antioxid. Redox Signal. 2000, 2, 177–187. [Google Scholar] [CrossRef]
- Chueh, P.J.; Wu, L.Y.; Morre, D.M.; Morre, D.J. tNOX is both necessary and sufficient as a cellular target for the anticancer actions of capsaicin and the green tea catechin (-)-epigallocatechin-3-gallate. Biofactors 2004, 20, 235–249. [Google Scholar]
- Liu, S.C.; Yang, J.J.; Shao, K.N.; Chueh, P.J. RNA interference targeting tNOX attenuates cell migration via a mechanism that involves membrane association of Rac. Biochem. Biophys. Res. Commun. 2008, 365, 672–677. [Google Scholar] [CrossRef]
- Lin, M.H.; Lee, Y.H.; Cheng, H.L.; Chen, H.Y.; Jhuang, F.H.; Chueh, P.J. Capsaicin Inhibits Multiple Bladder Cancer Cell Phenotypes by Inhibiting Tumor-Associated NADH Oxidase (tNOX) and Sirtuin1 (SIRT1). Molecules 2016, 21, 849. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.L.; Lee, Y.H.; Yuan, T.M.; Chen, S.W.; Chueh, P.J. Update on a tumor-associated NADH oxidase in gastric cancer cell growth. World J. Gastroenterol. 2016, 22, 2900–2905. [Google Scholar] [CrossRef]
- Mao, L.C.; Wang, H.M.; Lin, Y.Y.; Chang, T.K.; Hsin, Y.H.; Chueh, P.J. Stress-induced down-regulation of tumor-associated NADH oxidase during apoptosis in transformed cells. FEBS Lett. 2008, 582, 3445–3450. [Google Scholar] [CrossRef]
- Liu, N.C.; Hsieh, P.F.; Hsieh, M.K.; Zeng, Z.M.; Cheng, H.L.; Liao, J.W.; Chueh, P.J. Capsaicin-mediated tNOX (ENOX2) up-regulation enhances cell proliferation and migration in vitro and in vivo. J. Agric. Food Chem. 2012, 60, 2758–2765. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Chen, H.Y.; Su, L.J.; Chueh, P.J. Sirtuin 1 (SIRT1) Deacetylase Activity and NAD(+)/NADH Ratio Are Imperative for Capsaicin-Mediated Programmed Cell Death. J. Agric. Food Chem. 2015, 63, 7361–7370. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Islam, A.; Yuan, T.M.; Chen, S.W.; Liu, P.F.; Chueh, P.J. Regulation of tNOX expression through the ROS-p53-POU3F2 axis contributes to cellular responses against oxaliplatin in human colon cancer cells. J. Exp. Clin. Cancer Res. 2018, 37, 161. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Cheng, H.L.; Lee, Y.H.; Yuan, T.M.; Chen, S.W.; Lin, Y.Y.; Chueh, P.J. Tumor-associated NADH oxidase (tNOX)-NAD+-sirtuin 1 axis contributes to oxaliplatin-induced apoptosis of gastric cancer cells. Oncotarget 2017, 8, 15338–15348. [Google Scholar] [CrossRef] [PubMed]
- Morre, D.J.; Hostetler, B.; Taggart, D.J.; Morre, D.M.; Musk, A.W.; Robinson, B.W.; Creaney, J. ENOX2-based early detection (ONCOblot) of asbestos-induced malignant mesothelioma 4–10 years in advance of clinical symptoms. Clin. Proteom. 2016, 13, 2. [Google Scholar] [CrossRef][Green Version]
- Chang, C.F.; Islam, A.; Liu, P.F.; Zhan, J.H.; Chueh, P.J. Capsaicin acts through tNOX (ENOX2) to induce autophagic apoptosis in p53-mutated HSC-3 cells but autophagy in p53-functional SAS oral cancer cells. Am. J. Cancer Res. 2020, 10, 3230–3247. [Google Scholar]
- Ronconi, G.; Lessiani, G.; Spinas, E.; Kritas, S.K.; Caraffa, A.; Saggini, A.; Antinolfi, P.; Pizzicannella, J.; Toniato, E.; Conti, P. ENOX2 (or tNOX): A new and old molecule with cancer activity involved in tumor prevention and therapy. J. Biol. Regul. Homeost. Agents 2016, 30, 649–653. [Google Scholar]
- Davies, S.L.; Bozzo, J. Spotlight on tNOX: A tumor-selective target for cancer therapies. Drug News Perspect. 2006, 19, 223–225. [Google Scholar] [CrossRef]
- Morre, D.J.; Kim, C.; Paulik, M.; Morre, D.M.; Faulk, W.P. Is the drug-responsive NADH oxidase of the cancer cell plasma membrane a molecular target for adriamycin? J. Bioenerg. Biomembr. 1997, 29, 269–280. [Google Scholar] [CrossRef]
- Cogoi, S.; Zorzet, S.; Shchekotikhin, A.E.; Xodo, L.E. Potent Apoptotic Response Induced by Chloroacetamidine Anthrathiophenediones in Bladder Cancer Cells. J. Med. Chem. 2015, 58, 5476–5485. [Google Scholar] [CrossRef]
- Shchekotikhin, A.E.; Glazunova, V.A.; Luzikov, Y.N.; Buyanov, V.N.; Susova, O.Y.; Shtil, A.A.; Preobrazhenskaya, M.N. Synthesis and structure-activity relationship studies of 4,11-diaminonaphtho[2,3-f]indole-5,10-diones. Bioorg. Med. Chem. 2006, 14, 5241–5251. [Google Scholar] [CrossRef] [PubMed]
- Shchekotikhin, A.E.; Glazunova, V.A.; Dezhenkova, L.G.; Luzikov, Y.N.; Sinkevich, Y.B.; Kovalenko, L.V.; Buyanov, V.N.; Balzarini, J.; Huang, F.C.; Lin, J.J.; et al. Synthesis and cytotoxic properties of 4,11-bis[(aminoethyl)amino]anthra[2,3-b]thiophene-5,10-diones, novel analogues of antitumor anthracene-9,10-diones. Bioorg. Med. Chem. 2009, 17, 1861–1869. [Google Scholar] [CrossRef]
- Chen, C.F.; Huang, S.; Liu, S.C.; Chueh, P.J. Effect of polyclonal antisera to recombinant tNOX protein on the growth of transformed cells. Biofactors 2006, 28, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.J. MolView: A program for analyzing and displaying atomic structures on the Macintosh personal computer. J. Mol. Graph. 1995, 13, 122–125, 115. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Hsu, K.C.; Chen, Y.F.; Lin, S.R.; Yang, J.M. iGEMDOCK: A graphical environment of enhancing GEMDOCK using pharmacological interactions and post-screening analysis. BMC Bioinform. 2011, 12, 1–11. [Google Scholar] [CrossRef]
- Aly, A.A.; Brase, S.; Gomaa, M.A.M. Amidines: Their synthesis, reactivity, and applications in heterocycle synthesis. Arkivoc 2018, 6, 85–138. [Google Scholar] [CrossRef]
- Ichwan, S.J.; Yamada, S.; Sumrejkanchanakij, P.; Ibrahim-Auerkari, E.; Eto, K.; Ikeda, M.A. Defect in serine 46 phosphorylation of p53 contributes to acquisition of p53 resistance in oral squamous cell carcinoma cells. Oncogene 2006, 25, 1216–1224. [Google Scholar] [CrossRef]
- Sakai, E.; Tsuchida, N. Most human squamous cell carcinomas in the oral cavity contain mutated p53 tumor-suppressor genes. Oncogene 1992, 7, 927–933. [Google Scholar]
- Kim, M.J.; Hong, K.S.; Kim, H.B.; Lee, S.H.; Bae, J.H.; Kim, D.W.; Dao, T.T.; Oh, W.K.; Kang, C.D.; Kim, S.H. Ku70 acetylation and modulation of c-Myc/ATF4/CHOP signaling axis by SIRT1 inhibition lead to sensitization of HepG2 cells to TRAIL through induction of DR5 and down-regulation of c-FLIP. Int. J. Biochem. Cell B 2013, 45, 711–723. [Google Scholar] [CrossRef]
- Kerr, E.; Holohan, C.; McLaughlin, K.M.; Majkut, J.; Dolan, S.; Redmond, K.; Riley, J.; McLaughlin, K.; Stasik, I.; Crudden, M.; et al. Identification of an acetylation-dependant Ku70/FLIP complex that regulates FLIP expression and HDAC inhibitor-induced apoptosis. Cell Death Differ. 2012, 19, 1317–1327. [Google Scholar] [CrossRef]
- Martinez Molina, D.; Jafari, R.; Ignatushchenko, M.; Seki, T.; Larsson, E.A.; Dan, C.; Sreekumar, L.; Cao, Y.; Nordlund, P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 2013, 341, 84–87. [Google Scholar] [CrossRef]
- Martinez Molina, D.; Nordlund, P. The Cellular Thermal Shift Assay: A Novel Biophysical Assay for In Situ Drug Target Engagement and Mechanistic Biomarker Studies. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 141–161. [Google Scholar] [CrossRef]
- Morre, D.J.; Wu, L.Y.; Morre, D.M. The antitumor sulfonylurea N-(4-methylphenylsulfonyl)-N′-(4-chlorophenyl) urea (LY181984) inhibits NADH oxidase activity of HeLa plasma membranes. Biochim. Biophys. Acta 1995, 1240, 11–17. [Google Scholar] [CrossRef]
- Yu, J.; Zhang, L.; Peng, J.; Ward, R.; Hao, P.; Wang, J.; Zhang, N.; Yang, Y.; Guo, X.; Xiang, C.; et al. Dictamnine, a novel c-Met inhibitor, suppresses the proliferation of lung cancer cells by downregulating the PI3K/AKT/mTOR and MAPK signaling pathways. Biochem. Pharmacol. 2022, 195, 114864. [Google Scholar] [CrossRef]
- Karvonen, H.; Raivola, J.; Ungureanu, D. Cellular thermal shift assay (CETSA) for determining the drug binding affinity using Ba/F3 clones stably expressing receptor pseudokinases. Methods Enzymol. 2022, 667, 339–363. [Google Scholar] [CrossRef]
- Fang, Z.; Mu, B.; Liu, Y.; Guo, N.; Xiong, L.; Guo, Y.; Xia, A.; Zhang, R.; Zhang, H.; Yao, R.; et al. Discovery of a potent, selective and cell active inhibitor of m(6)A demethylase ALKBH5. Eur. J. Med. Chem. 2022, 238, 114446. [Google Scholar] [CrossRef]
- Sumiyoshi, A.; Shibata, S.; Zhelev, Z.; Miller, T.; Lazarova, D.; Aoki, I.; Obata, T.; Higashi, T.; Bakalova, R. Targeting Glioblastoma via Selective Alteration of Mitochondrial Redox State. Cancers 2022, 14, 485. [Google Scholar] [CrossRef]
- Su, Y.C.; Lin, Y.H.; Zeng, Z.M.; Shao, K.N.; Chueh, P.J. Chemotherapeutic agents enhance cell migration and epithelial-to-mesenchymal transition through transient up-regulation of tNOX (ENOX2) protein. Biochim. Biophys. Acta 2012, 1820, 1744–1752. [Google Scholar] [CrossRef]
- Islam, A.; Hsieh, P.F.; Liu, P.F.; Chou, J.C.; Liao, J.W.; Hsieh, M.K.; Chueh, P.J. Capsaicin exerts therapeutic effects by targeting tNOX-SIRT1 axis and augmenting ROS-dependent autophagy in melanoma cancer cells. Am. J. Cancer Res. 2021, 11, 4199–4219. [Google Scholar]
- Chueh, P.J.; Morré, D.M.; Morré, D.J. A site-directed mutagenesis analysis of tNOX functional domains. Biochim. Biophys. Acta 2002, 1594, 74–83. [Google Scholar] [CrossRef]
- Horowitz, S.; Trievel, R.C. Carbon-oxygen hydrogen bonding in biological structure and function. J. Biol. Chem. 2012, 287, 41576–41582. [Google Scholar] [CrossRef]
- Fulda, S. Tumor resistance to apoptosis. Int. J. Cancer 2009, 124, 511–515. [Google Scholar] [CrossRef]
- Ivanisenko, N.V.; Seyrek, K.; Hillert-Richter, L.K.; Konig, C.; Espe, J.; Bose, K.; Lavrik, I.N. Regulation of extrinsic apoptotic signaling by c-FLIP: Towards targeting cancer networks. Trends Cancer 2022, 8, 190–209. [Google Scholar] [CrossRef]
- Schleich, K.; Buchbinder, J.H.; Pietkiewicz, S.; Kahne, T.; Warnken, U.; Ozturk, S.; Schnolzer, M.; Naumann, M.; Krammer, P.H.; Lavrik, I.N. Molecular architecture of the DED chains at the DISC: Regulation of procaspase-8 activation by short DED proteins c-FLIP and procaspase-8 prodomain. Cell Death Differ. 2016, 23, 681–694. [Google Scholar] [CrossRef]
- Day, T.W.; Huang, S.; Safa, A.R. c-FLIP knockdown induces ligand-independent DR5-, FADD-, caspase-8-, and caspase-9-dependent apoptosis in breast cancer cells. Biochem. Pharmacol. 2008, 76, 1694–1704. [Google Scholar] [CrossRef]
- Riley, J.S.; Hutchinson, R.; McArt, D.G.; Crawford, N.; Holohan, C.; Paul, I.; Van Schaeybroeck, S.; Salto-Tellez, M.; Johnston, P.G.; Fennell, D.A.; et al. Prognostic and therapeutic relevance of FLIP and procaspase-8 overexpression in non-small cell lung cancer. Cell Death Dis. 2013, 4, e951. [Google Scholar] [CrossRef]
- Micheau, O.; Lens, S.; Gaide, O.; Alevizopoulos, K.; Tschopp, J. NF-kappaB signals induce the expression of c-FLIP. Mol. Cell. Biol. 2001, 21, 5299–5305. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Q.; Zhu, N.; Yu, M.; Shen, B.; Xiang, J.; Lin, A. Cyclic AMP inhibits JNK activation by CREB-mediated induction of c-FLIPL and MKP-1, thereby antagonizing UV-induced apoptosis. Cell Death Differ. 2008, 15, 1654–1662. [Google Scholar] [CrossRef]
- Mahalingam, D.; Natoni, A.; Keane, M.; Samali, A.; Szegezdi, E. Early growth response-1 is a regulator of DR5-induced apoptosis in colon cancer cells. Br. J. Cancer 2010, 102, 754–764. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, L.; Yang, H.; Huang, X.; Otu, H.; Libermann, T.A.; DeWolf, W.C.; Khosravi-Far, R.; Olumi, A.F. c-Fos as a proapoptotic agent in TRAIL-induced apoptosis in prostate cancer cells. Cancer Res. 2007, 67, 9425–9434. [Google Scholar] [CrossRef]
- Skurk, C.; Maatz, H.; Kim, H.S.; Yang, J.; Abid, M.R.; Aird, W.C.; Walsh, K. The Akt-regulated forkhead transcription factor FOXO3a controls endothelial cell viability through modulation of the caspase-8 inhibitor FLIP. J. Biol. Chem. 2004, 279, 1513–1525. [Google Scholar] [CrossRef]
- Ricci, M.S.; Jin, Z.; Dews, M.; Yu, D.; Thomas-Tikhonenko, A.; Dicker, D.T.; El-Deiry, W.S. Direct repression of FLIP expression by c-myc is a major determinant of TRAIL sensitivity. Mol. Cell. Biol. 2004, 24, 8541–8555. [Google Scholar] [CrossRef]
- Humphreys, L.; Espona-Fiedler, M.; Longley, D.B. FLIP as a therapeutic target in cancer. FEBS J. 2018, 285, 4104–4123. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Wang, Y.; Jing, Y. Apoptosis Induction byHistone Deacetylase Inhibitors in Cancer Cells: Role of Ku70. Int. J. Mol. Sci. 2019, 20, 1601. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.B.; Kim, M.J.; Lee, S.H.; Lee, J.W.; Bae, J.H.; Kim, D.W.; Dao, T.T.; Oh, W.K.; Kang, C.D.; Kim, S.H. Amurensin G, a novel SIRT1 inhibitor, sensitizes TRAIL-resistant human leukemic K562 cells to TRAIL-induced apoptosis. Biochem. Pharmacol. 2012, 84, 402–410. [Google Scholar] [CrossRef]
- Kruyt, F.A.E. TRAIL and cancer therapy. Cancer Lett. 2008, 263, 14–25. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, J.S.; Chen, C.-Y.; Tikhomirov, A.S.; Islam, A.; Liang, R.-H.; Weng, C.-W.; Wu, W.-H.; Shchekotikhin, A.E.; Chueh, P.J. Bis(chloroacetamidino)-Derived Heteroarene-Fused Anthraquinones Bind to and Cause Proteasomal Degradation of tNOX, Leading to c-Flip Downregulation and Apoptosis in Oral Cancer Cells. Cancers 2022, 14, 4719. https://doi.org/10.3390/cancers14194719
Chang JS, Chen C-Y, Tikhomirov AS, Islam A, Liang R-H, Weng C-W, Wu W-H, Shchekotikhin AE, Chueh PJ. Bis(chloroacetamidino)-Derived Heteroarene-Fused Anthraquinones Bind to and Cause Proteasomal Degradation of tNOX, Leading to c-Flip Downregulation and Apoptosis in Oral Cancer Cells. Cancers. 2022; 14(19):4719. https://doi.org/10.3390/cancers14194719
Chicago/Turabian StyleChang, Jeng Shiun, Chien-Yu Chen, Alexander S. Tikhomirov, Atikul Islam, Ru-Hao Liang, Chia-Wei Weng, Wei-Hou Wu, Andrey E. Shchekotikhin, and Pin Ju Chueh. 2022. "Bis(chloroacetamidino)-Derived Heteroarene-Fused Anthraquinones Bind to and Cause Proteasomal Degradation of tNOX, Leading to c-Flip Downregulation and Apoptosis in Oral Cancer Cells" Cancers 14, no. 19: 4719. https://doi.org/10.3390/cancers14194719
APA StyleChang, J. S., Chen, C.-Y., Tikhomirov, A. S., Islam, A., Liang, R.-H., Weng, C.-W., Wu, W.-H., Shchekotikhin, A. E., & Chueh, P. J. (2022). Bis(chloroacetamidino)-Derived Heteroarene-Fused Anthraquinones Bind to and Cause Proteasomal Degradation of tNOX, Leading to c-Flip Downregulation and Apoptosis in Oral Cancer Cells. Cancers, 14(19), 4719. https://doi.org/10.3390/cancers14194719