
Mutation Patterns in Portuguese Families with Hereditary Breast and Ovarian Cancer Syndrome

 ,
,  , ,
, , 
Abstract
Simple Summary
Abstract
1. Introduction
2. Genetic and Clinical Features of Hereditary Breast and Ovarian Cancer
2.1. The Founder Effect
2.2. Portuguese Founder Mutations
3. BRCA Gene Testing and Screening
4. Future Perspectives
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Breast Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/breast-cancer (accessed on 15 June 2021).
- Cabasag, C.J.; Fagan, P.J.; Ferlay, J.; Vignat, J.; Laversanne, M.; Liu, L.; van der Aa, A.M.; Bray, F.; Soerjomataram, I. Ovarian cancer today and tomorrow: A global assessment by world region and Human Development Index using GLOBOCAN 2020. Int. J. Cancer 2022. [Google Scholar] [CrossRef] [PubMed]
- Menon, U.; Gentry-Maharaj, A.; Burnell, M.; Singh, N.; Ryan, A.; Karpinskyj, C.; Carlino, G.; Taylor, J.; Massingham, S.K.; Raikou, M.; et al. Ovarian cancer population screening and mortality after long-term follow-up in the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS): A randomised controlled trial. Lancet 2021, 397, 2182–2193. [Google Scholar] [CrossRef]
- Yoshida, R. Hereditary breast and ovarian cancer (HBOC): Review of its molecular characteristics, screening, treatment, and prognosis. Breast Cancer 2020, 28, 1167–1180. [Google Scholar] [CrossRef] [PubMed]
- Felix, G.E.S.; Zheng, Y.; Olopade, O.I. Mutations in context: Implications of BRCA testing in diverse populations. Fam. Cancer 2017, 17, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Teugels, E.; De Brakeleer, S.; Goelen, G.; Lissens, W.; Sermijn, E.; De Grève, J. De novo Alu element insertions targeted to a sequence common to the BRCA1 and BRCA2 genes. Hum. Mutat. 2005, 26, 284. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, H.; Takei, J. Management of hereditary breast and ovarian cancer. Int. J. Clin. Oncol. 2017, 23, 45–51. [Google Scholar] [CrossRef]
- Toss, A.; Tomasello, C.; Razzaboni, E.; Contu, G.; Grandi, G.; Cagnacci, A.; Schilder, R.J.; Cortesi, L. Hereditary Ovarian Cancer: Not OnlyBRCA1 and 2 Genes. BioMed Res. Int. 2015, 2015, 341723. [Google Scholar] [CrossRef]
- Mavaddat, N.; Barrowdale, D.; Andrulis, I.L.; Domchek, S.M.; Eccles, D.; Nevanlinna, H.; Ramus, S.J.; Spurdle, A.; Robson, M.; Sherman, M.; et al. Pathology of Breast and Ovarian Cancers among BRCA1 and BRCA2 Mutation Carriers: Results from the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA). Cancer Epidemiol. Biomark. Prev. 2012, 21, 134–147. [Google Scholar] [CrossRef]
- Ibrahim, M.; Yadav, S.; Ogunleye, F.; Zakalik, D. Male BRCA mutation carriers: Clinical characteristics and cancer spectrum. BMC Cancer 2018, 18, 179. [Google Scholar] [CrossRef]
- Vietri, M.T.; Caliendo, G.; D’Elia, G.; Resse, M.; Casamassimi, A.; Minucci, P.B.; Cioffi, M.; Molinari, A.M. BRCA and PALB2 mutations in a cohort of male breast cancer with one bilateral case. Eur. J. Med. Genet. 2020, 63, 103883. [Google Scholar] [CrossRef]
- Bordeleau, L.; Panchal, S.; Goodwin, P. Prognosis of BRCA-associated breast cancer: A summary of evidence. Breast Cancer Res. Treat. 2010, 119, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Verhoog, L.C.; Berns, E.M.; Brekelmans, C.T.; Seynaeve, C.; Meijers-Heijboer, E.J.; Klijn, J.G. Prognostic significance of germline BRCA2 mutations in hereditary breast cancer patients. J. Clin. Oncol. 2000, 18, 119S–124S. [Google Scholar] [PubMed]
- van den Broek, A.J.; Schmidt, M.K.; van ‘t Veer, L.J.; Tollenaar, R.A.; van Leeuwen, F.E. Worse Breast Cancer Prognosis of BRCA1/BRCA2 Mutation Carriers: What’s the Evidence? A Systematic Review with Meta-Analysis. PLoS ONE 2015, 10, e0120189. [Google Scholar] [CrossRef]
- Zhong, Q.; Peng, H.-L.; Zhao, X.; Zhang, L.; Hwang, W.-T. Effects of BRCA1- and BRCA2-related mutations on ovarian and breast cancer survival: A meta-analysis. Clin. Cancer Res. 2015, 21, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yang, D.; Sun, Y.; Shmulevich, I.; Xue, F.; Sood, A.K.; Zhang, W. Differing clinical impact of BRCA1 and BRCA2 mutations in serous ovarian cancer. Pharmacogenomics 2012, 13, 1523–1535. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, V.; Illuzzi, G.; Héberlé, E.; Dantzer, F. De la découverte du poly(ADP-ribose) aux inhibiteurs PARP en thérapie du cancer. Bull. Cancer 2015, 102, 863–873. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Tutt, A.N.; Garber, J.E.; Kaufman, B.; Viale, G.; Fumagalli, D.; Rastogi, P.; Gelber, R.D.; de Azambuja, E.; Fielding, A.; Balmaña, J.; et al. Adjuvant Olaparib for Patients with BRCA1- or BRCA2-Mutated Breast Cancer. New Engl. J. Med. 2021, 384, 2394–2405. [Google Scholar] [CrossRef] [PubMed]
- Tung, N.M.; Garber, J.E. BRCA1/2 testing: Therapeutic implications for breast cancer management. Br. J. Cancer 2018, 119, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Neamatzadeh, H.; Shiryazdi, S.M.; Kalantar, S.M. BRCA1 and BRCA2 mutations in Iranian breast cancer patients: A systematic review. J. Res. Med. Sci. 2015, 20, 284–293. [Google Scholar] [PubMed]
- Ferla, R.; Calò, V.; Cascio, S.; Rinaldi, G.; Badalamenti, G.; Carreca, I.; Surmacz, E.; Colucci, G.; Bazan, V.; Russo, A. Founder mutations in BRCA1 and BRCA2 genes. Ann. Oncol. 2007, 18, vi93–vi98. [Google Scholar] [CrossRef]
- Petrucelli, N.; Daly, M.B.; Pal, T. BRCA1- and BRCA2-Associated Hereditary Breast and Ovarian Cancer. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Machado, P.M.; Brandão, R.D.; Cavaco, B.M.; Eugénio, J.; Bento, S.; Nave, M.; Rodrigues, P.; Fernandes, A.; Vaz, F. Screening for a BRCA2 Rearrangement in High-Risk Breast/Ovarian Cancer Families: Evidence for a Founder Effect and Analysis of the Associated Phenotypes. J. Clin. Oncol. 2007, 25, 2027–2034. [Google Scholar] [CrossRef]
- Peixoto, A.; Santos, C.; Rocha, P.; Pinheiro, M.; Príncipe, S.; Pereira, D.; Rodrigues, H.; Castro, F.; Abreu, J.; Gusmão, L.; et al. The c.156_157insAlu BRCA2 rearrangement accounts for more than one-fourth of deleterious BRCA mutations in northern/central Portugal. Breast Cancer Res. Treat. 2009, 114, 31–38. [Google Scholar] [CrossRef]
- Peixoto, A.; Santos, C.; Pinheiro, M.; Pinto, P.; Soares, M.J.; Rocha, P.; Gusmão, L.; Amorim, A.; Van Der Hout, A.; Gerdes, A.-M.; et al. International distribution and age estimation of the Portuguese BRCA2 c.156_157insAlu founder mutation. Breast Cancer Res. Treat. 2011, 127, 671–679. [Google Scholar] [CrossRef]
- Peixoto, A.; Santos, C.; Pinto, P.; Pinheiro, M.; Rocha, P.; Pinto, C.; Bizarro, S.; Veiga, I.; Principe, A.; Maia, S.; et al. The role of targetedBRCA1/BRCA2mutation analysis in hereditary breast/ovarian cancer families of Portuguese ancestry. Clin. Genet. 2015, 88, 41–48. [Google Scholar] [CrossRef]
- Miguel, I.; Rodrigues, F.; Fragoso, S.; Freixo, J.; Clara, A.; Luis, A.; Bento, S.; Fernandes, M.; Bacelar, F.; Câmara, S.; et al. Hereditary breast cancer and ancestry in the Madeira archipelago: An exploratory study. Ecancermedicalscience 2021, 15, 1261. [Google Scholar] [CrossRef]
- Macedo, F.; Soares, R.F.; Pereira, T.C.; Monteiro, A.; Bonito, N.; Broco, S.; Carvalho, T.; Mariano, M.; Sousa, G. Founder mutations BRCA2 c.156_157insAlu in Portuguese population. Ann. Oncol. 2019, 30, iii20. [Google Scholar] [CrossRef]
- Tuazon, A.M.D.A.; Lott, P.; Bohórquez, M.; Benavides, J.; Ramirez, C.; Criollo, A.; Estrada-Florez, A.; Mateus, G.; Velez, A.; Carmona, J.; et al. Haplotype analysis of the internationally distributed BRCA1 c.3331_3334delCAAG founder mutation reveals a common ancestral origin in Iberia. Breast Cancer Res. 2020, 22, 108. [Google Scholar] [CrossRef] [PubMed]
- King, M.-C.; Marks, J.H.; Mandell, J.B. Breast and Ovarian Cancer Risks Due to Inherited Mutations in BRCA1 and BRCA2. Science 2003, 302, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Domchek, S.M. Association of Risk-Reducing Surgery in BRCA1 or BRCA2 Mutation Carriers with Cancer Risk and Mortality. JAMA 2010, 304, 967–975. [Google Scholar] [CrossRef]
- Ludwig, K.K.; Neuner, J.; Butler, A.; Geurts, J.L.; Kong, A.L. Risk reduction and survival benefit of prophylactic surgery in BRCA mutation carriers, a systematic review. Am. J. Surg. 2016, 212, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Paluch-Shimon, S.; Cardoso, F.; Sessa, C.; Balmana, J.; Cardoso, M.J.; Gilbert, F.; Senkus, E. Prevention and screening in BRCA mutation carriers and other breast/ovarian hereditary cancer syndromes: ESMO Clinical Practice Guidelines for cancer prevention and screening. Ann. Oncol. 2016, 27, v103–v110. [Google Scholar] [CrossRef] [PubMed]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 2010, 376, 235–244. [Google Scholar] [CrossRef]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of Poly(ADP-Ribose) Polymerase in Tumors fromBRCAMutation Carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef]
- Abacan, M.; Alsubaie, L.; Barlow-Stewart, K.; Caanen, B.; Cordier, C.; Courtney, E.; Davoine, E.; Edwards, J.; Elackatt, N.J.; Gardiner, K.; et al. The Global State of the Genetic Counseling Profession. Eur. J. Hum. Genet. 2019, 27, 183–197. [Google Scholar] [CrossRef]
- Gradishar, W.J.; Moran, M.S.; Abraham, J.; Aft, R.; Agnese, D.; Allison, K.H.; Anderson, B.; Burstein, H.J.; Chew, H.; Dang, C.; et al. Breast Cancer, Version 3.2022, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2022, 20, 691–722. [Google Scholar] [CrossRef]
- Robertson, L.; Hanson, H.; Seal, S.; Warren-Perry, M.; Hughes, D.; Howell, I.; Turnbull, C.; Houlston, R.; Shanley, S.; Butler, S.; et al. BRCA1 testing should be offered to individuals with triple-negative breast cancer diagnosed below 50 years. Br. J. Cancer 2012, 106, 1234–1238. [Google Scholar] [CrossRef]
- Forbes, C.; Fayter, D.; de Kock, S.; Quek, R.G. A systematic review of international guidelines and recommendations for the genetic screening, diagnosis, genetic counseling, and treatment of BRCA-mutated breast cancer. Cancer Manag. Res. 2019, 11, 2321–2337. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.; Kyriakides, S.; Ohno, S.; Penault-Llorca, F.; Poortmans, P.; Rubio, I.T.; Zackrisson, S.; Senkus, E.; ESMO Guidelines Committee. Early breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up†. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 1194–1220. [Google Scholar] [CrossRef]
- Boughey, J.C.; Hartmann, L.C.; Anderson, S.S.; Degnim, A.C.; Vierkant, R.A.; Reynolds, C.A.; Frost, M.H.; Pankratz, V.S. Evaluation of the Tyrer-Cuzick (International Breast Cancer Intervention Study) Model for Breast Cancer Risk Prediction in Women With Atypical Hyperplasia. J. Clin. Oncol. 2010, 28, 3591–3596. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.A.; Iversen, S.I., Jr.; Gudbjartsson, D.F.; Hiller, E.H.; Garber, J.E.; Peshkin, B.N.; Lerman, C.; Watson, P.; Lynch, H.T.; Hilsenbeck, S.G.; et al. BRCAPRO Validation, Sensitivity of Genetic Testing of BRCA1/BRCA2, and Prevalence of Other Breast Cancer Susceptibility Genes. J. Clin. Oncol. 2002, 20, 2701–2712. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Mavaddat, N.; Wilcox, A.N.; Msc, A.P.C.; Carver, T.; Hartley, S.; de Villiers, C.B.; Izquierdo, A.; Simard, J.; Schmidt, M.K.; et al. BOADICEA: A comprehensive breast cancer risk prediction model incorporating genetic and nongenetic risk factors. Genet. Med. 2019, 21, 1708–1718. [Google Scholar] [CrossRef] [PubMed]
- Beitsch, P.D.; Whitworth, P.W.; Hughes, K.; Patel, R.; Rosen, B.; Compagnoni, G.; Baron, P.; Simmons, R.; Smith, L.A.; Grady, I.; et al. Underdiagnosis of Hereditary Breast Cancer: Are Genetic Testing Guidelines a Tool or an Obstacle? J. Clin. Oncol. 2019, 37, 453–460. [Google Scholar] [CrossRef]
- Manchanda, R.; Legood, R.; Burnell, M.; McGuire, A.; Raikou, M.; Loggenberg, K.; Wardle, J.; Sanderson, S.; Gessler, S.; Side, L.; et al. Cost-effectiveness of Population Screening for BRCA Mutations in Ashkenazi Jewish Women Compared with Family History–Based Testing. JNCI J. Natl. Cancer Inst. 2015, 107, 380. [Google Scholar] [CrossRef]
- Palmero, E.I.; Carraro, D.M.; Alemar, B.; Moreira, M.A.M.; Ribeiro-Dos-Santos, Â.; Abe-Sandes, K.; Galvão, H.C.R.; Reis, R.M.; Souza, C.D.P.; Campacci, N.; et al. The germline mutational landscape of BRCA1 and BRCA2 in Brazil. Sci. Rep. 2018, 8, 9188. [Google Scholar] [CrossRef]
- Silva, T.P.; Pereira, C.A.; Raposo, A.C.; Oliveira, A.R.; Arez, M.; Cabral, J.M.; Milagre, I.; Carmo-Fonseca, M.; da Rocha, S.T. Generation and characterization of induced pluripotent stem cells heterozygous for the Portuguese BRCA2 founder mutation. Stem Cell Res. 2021, 53, 102364. [Google Scholar] [CrossRef]
- Rauch, A. Proceedings of the 22nd Annual Meeting of the Portuguese Society of Human Genetics. Medicine 2019, 98, e15772. [Google Scholar] [CrossRef]
- Santos, R. Proceedings of the 23rd Annual Meeting of the Portuguese Society of Human Genetics. Medicine 2020, 99, e19291. [Google Scholar] [CrossRef]
- Vietri, M.T.; Caliendo, G.; D’Elia, G.; Resse, M.; Casamassimi, A.; Minucci, P.B.; Ioio, C.D.; Cioffi, M.; Molinari, A.M. Five Italian Families with Two Mutations in BRCA Genes. Genes 2020, 11, 1451. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Studies | Aim | Number of Patients/Families | Founder Mutation Investigated | Conclusions |
|---|---|---|---|---|
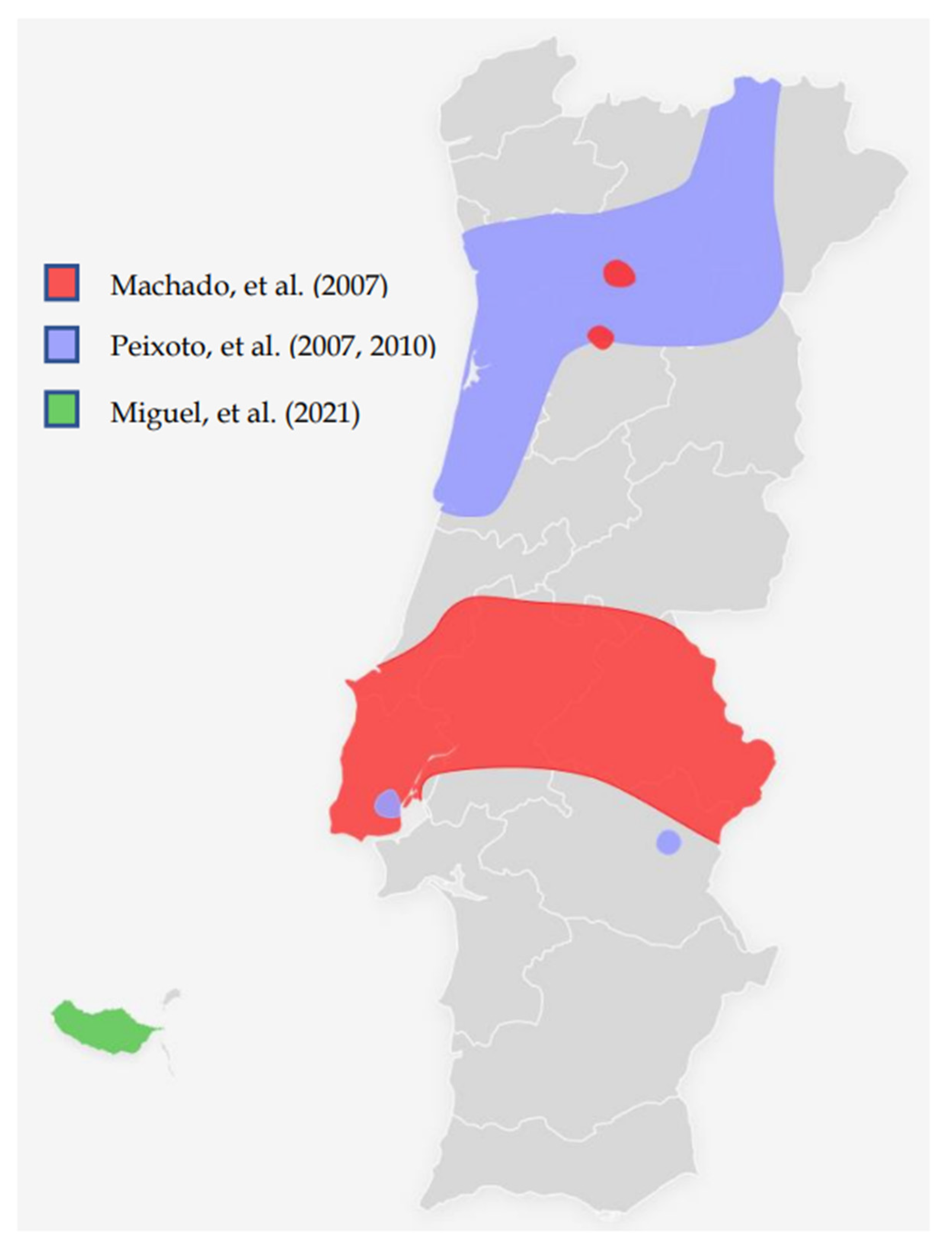
| Machado, et al. (2007) [27] | Molecular and phenotypic characterisation of a large insertion in exon 3 of BRCA2. | 210 patients from Central/southern Portugal | c.156_157insAlu | c.156_157insAlu is a founder mutation of Portuguese origin and is the most frequent BRCA2 rearrangement |
| Peixoto, et al. (2008) [28] | To evaluate the contribution of the c.156_157insAlu BRCA2 mutation to inherited predisposition to BC and OC in families originating mostly from northern/central Portugal. | 210 families from Northern/central Portugal | c.156_157insAlu | This rearrangement is responsible for more than half of all pathogenic BRCA2 mutations and about one-fourth of all pathogenic variants in HBOC families |
| Peixoto, et al. (2010) [29] | To gain insight into the ancestral origin and population spread of the c.156_157insAlu BRCA2 mutation. | 5443 families (149 from Portugal and 5294 from other countries than Portugal) | c.156_157insAlu | c.156_157insAlu BRCA2 rearrangement is a Portuguese founder mutation that originated about 558 ± 215 years ago, accounting for the majority of the BRCA2 mutations and about one-third of all pathogenic germline mutations in Portuguese HBOC families. |
| Peixoto, et al. (2015) [30] | To describe the mutational spectrum and evaluate the impact of founder mutations in the genetic testing criteria and strategy for molecular testing of HBOC families of Portuguese ancestry. | 1050 families (524 fully screened for BRCA1/BRCA2 mutations) | c.156_157insAlu Other possible founder mutations pointed out: c.3331_3334del c.2037delinsCC | Of the 119 families with pathogenic mutations, 40 (33.6%) had the BRCA2 c.156_157insAlu rearrangement, 15 (12.6%) the BRCA1 c.3331_3334del mutation and 7 (5.9%) the BRCA1 c.2037delinsCC mutation. The c.2037delinsCC mutation has not been described in other populations. |
| Miguel, et al. (2021) [31] | To evaluate the Hereditary Breast/Ovarian Cancer (HBOC) families with Madeira ancestry enrolled in the HBOC programme occurring in Instituto Português de Oncologia de Lisboa | 3566 patients (19 from Madeira Island, 3547 from other parts of Portugal) | c.156_157insAlu | BRCA1/2 detection rates were 27.9% and 10.5% for Madeira and the whole group, respectively. In all patients detected with BRCA1/2 mutations, 22.8 % had the c.156_157insAlu BRCA2 rearrangement. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vicente, R.; Alpuim Costa, D.; Vitorino, M.; Mendes, A.D.; Santos, C.; Fontes-Sousa, M. Mutation Patterns in Portuguese Families with Hereditary Breast and Ovarian Cancer Syndrome. Cancers 2022, 14, 4717. https://doi.org/10.3390/cancers14194717
Vicente R, Alpuim Costa D, Vitorino M, Mendes AD, Santos C, Fontes-Sousa M. Mutation Patterns in Portuguese Families with Hereditary Breast and Ovarian Cancer Syndrome. Cancers. 2022; 14(19):4717. https://doi.org/10.3390/cancers14194717
Chicago/Turabian StyleVicente, Rodrigo, Diogo Alpuim Costa, Marina Vitorino, Ana Duarte Mendes, Catarina Santos, and Mário Fontes-Sousa. 2022. "Mutation Patterns in Portuguese Families with Hereditary Breast and Ovarian Cancer Syndrome" Cancers 14, no. 19: 4717. https://doi.org/10.3390/cancers14194717
APA StyleVicente, R., Alpuim Costa, D., Vitorino, M., Mendes, A. D., Santos, C., & Fontes-Sousa, M. (2022). Mutation Patterns in Portuguese Families with Hereditary Breast and Ovarian Cancer Syndrome. Cancers, 14(19), 4717. https://doi.org/10.3390/cancers14194717