
Ethanol Ablation Therapy Drives Immune-Mediated Antitumor Effects in Murine Breast Cancer Models

 , , ,
, , , 
Abstract
Simple Summary
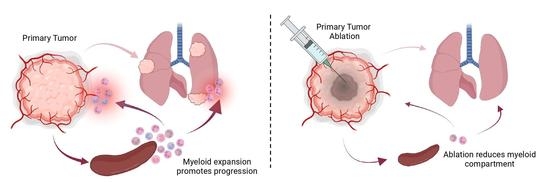
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. ECE Promotes CD8+ T Cell-Dependent Antitumor Effects in Poorly Invasive TNBC Models
2.2. ECE Modulates the Myeloid Compartment in Mice Bearing Highly Invasive 4T1 Tumors
2.3. ECE Therapy Suppresses Granulocytic Myeloid-Derived Suppressor Cells (MDSCs) in 4T1 Tumor-Bearing Mice
2.4. ECE Therapy Sensitizes 4T1 Tumors to Checkpoint Blockade Immunotherapy
3. Discussion
4. Materials and Methods
4.1. Tumor Cell Lines
4.2. Murine Mammary Tumor Models
4.3. Rat DMBA Induced Mammary Tumor Model
4.4. Murine Allograft Tumor Growth and Survival
4.5. Ethyl-Cellulose Ethanol (ECE) Ablations
4.6. Immunohistochemistry
4.7. Lung Metastases Quantification
4.8. Flow Cytometry
4.9. G-CSF ELISA
4.10. T-Cell Suppression Assay
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Thakur, K.K.; Bordoloi, D.; Kunnumakkara, A.B. Alarming Burden of Triple-Negative Breast Cancer in India. Clin. Breast Cancer 2018, 18, e393–e399. [Google Scholar] [CrossRef]
- McCarthy, A.M.; Friebel-Klingner, T.; Ehsan, S.; He, W.; Welch, M.; Chen, J.; Kontos, D.; Domchek, S.M.; Conant, E.F.; Semine, A.; et al. Relationship of established risk factors with breast cancer subtypes. Cancer Med. 2021, 10, 6456–6467. [Google Scholar] [CrossRef] [PubMed]
- Stead, L.A.; Lash, T.L.; Sobieraj, J.E.; Chi, D.D.; Westrup, J.L.; Charlot, M.; Blanchard, R.A.; Lee, J.C.; King, T.C.; Rosenberg, C.L. Triple-negative breast cancers are increased in black women regardless of age or body mass index. Breast Cancer Res. 2009, 11, R18. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.; Chandrashekar, D.S.; Shahin, S.; Agarwal, S.; Kim, H.-G.; Behring, M.; Shaikh, A.J.; Moloo, Z.; Eltoum, I.-E.A.; Yates, C.; et al. Comparative analysis of triple-negative breast cancer transcriptomics of Kenyan, African American and Caucasian Women. Transl. Oncol. 2021, 14, 101086. [Google Scholar] [CrossRef]
- Meara, J.G.; Leather, A.J.M.; Hagander, L.; Alkire, B.C.; Alonso, N.; Ameh, E.A.; Bickler, S.W.; Conteh, L.; Dare, A.J.; Davies, J.; et al. Global Surgery 2030: Evidence and solutions for achieving health, welfare, and economic development. Lancet 2015, 386, 569–624. [Google Scholar] [CrossRef]
- Nief, C.; Morhard, R.; Chelales, E.; Alvarez, D.A.; Bs, I.B.; Lam, C.T.; Sag, A.A.; Crouch, B.T.; Mueller, J.L.; Katz, D.; et al. Polymer-assisted intratumoral delivery of ethanol: Preclinical investigation of safety and efficacy in a murine breast cancer model. PLoS ONE 2021, 16, e0234535. [Google Scholar] [CrossRef]
- Morhard, R.; Mueller, J.L.; Tang, Q.; Nief, C.; Chelales, E.; Lam, C.T.; Alvarez, D.A.; Rubinstein, M.; Katz, D.F.; Ramanujam, N. Understanding Factors Governing Distribution Volume of Ethyl Cellulose-Ethanol to Optimize Ablative Therapy in the Liver. IEEE Trans. Biomed. Eng. 2020, 67, 2337–2348. [Google Scholar] [CrossRef]
- Chelales, E.; Morhard, R.; Nief, C.; Crouch, B.; Everitt, J.I.; Sag, A.A.; Ramanujam, N. Radiologic-pathologic analysis of increased ethanol localization and ablative extent achieved by ethyl cellulose. Sci. Rep. 2021, 11, 20700. [Google Scholar] [CrossRef]
- Morhard, R.; Nief, C.; Barrero Castedo, C.; Hu, F.; Madonna, M.; Mueller, J.L.; Dewhirst, M.W.; Katz, D.F.; Ramanujam, N. Development of enhanced ethanol ablation as an alternative to surgery in treatment of superficial solid tumors. Sci. Rep. 2017, 7, 8750. [Google Scholar] [CrossRef]
- Nief, C.A.; Chelales, E.; Morhard, R.; Chem, M.; Everitt, J.; Mueller, J.; Yao, J.; Dewhirst, M.W.; Ramanujam, N. Abstract 5243: Averting tumor growth in rodent breast cancer models with a liquid ablation approach. Cancer Res. 2020, 80, 5243. [Google Scholar] [CrossRef]
- Nief, C.A.; Gonzales, A.; Chelales, E.; Agudogo, J.S.; Crouch, B.T.; Nair, S.K.; Ramanujam, N. Targeting Tumor Acidosis and Regulatory T Cells Unmasks Anti-Metastatic Potential of Local Tumor Ablation in Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2022, 23, 8479. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, E.; Westerhuis, J.J.; Volk, M.; Hix, J.; Chakravarty, S.; Claucherty, E.; Zaluzec, E.; Ramsey, L.; Madaj, Z.; Hostetter, G.; et al. Ductal tree ablation by local delivery of ethanol prevents tumor formation in an aggressive mouse model of breast cancer. Breast Cancer Res. 2019, 21, 129. [Google Scholar] [CrossRef]
- Kenyon, E.; Zaluzec, E.K.; Powell, K.; Volk, M.; Chakravarty, S.; Hix, J.; Arora, R.; Westerhuis, J.J.; Kiupel, M.; Shapiro, E.M.; et al. Intraductal Delivery and X-ray Visualization of Ethanol-Based Ablative Solution for Prevention and Local Treatment of Breast Cancer in Mouse Models. J. Vis. Exp. 2022, 182, e63457. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.-H.E.; Morhard, R.; Ramanujam, N.; Nolan, M.W. Minimally invasive ethyl cellulose ethanol ablation in domesticated cats with naturally occurring head and neck cancers: Six cats. Vet. Comp. Oncol. 2021, 19, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Orlando, A.; D’Antoni, A.; Cammà, C.; Albanese, M.; Livraghi, T.; Torzilli, G.; Virdone, R.; Sciarrino, E.; Simonetti, R.G.; Maringhini, A.; et al. Treatment of small hepatocellular carcinoma with percutaneous ethanol injection: A validated prognostic model. Am. J. Gastroenterol. 2000, 95, 2921–2927. [Google Scholar] [CrossRef] [PubMed]
- Gamrekelashvili, J.; Ormandy, L.A.; Heimesaat, M.M.; Kirschning, C.J.; Manns, M.P.; Korangy, F.; Greten, T.F. Primary sterile necrotic cells fail to cross-prime CD8+ T cells. Oncoimmunology 2012, 1, 1017–1026. [Google Scholar] [CrossRef]
- Aaes, T.L.; Kaczmarek, A.; Delvaeye, T.; De Craene, B.; De Koker, S.; Heyndrickx, L.; Delrue, I.; Taminau, J.; Wiernicki, B.; De Groote, P.; et al. Vaccination with Necroptotic Cancer Cells Induces Efficient Anti-tumor Immunity. Cell Rep. 2016, 15, 274–287. [Google Scholar] [CrossRef]
- Costa, I.; Solanas, M.; Escrich, E. Histopathologic characterization of mammary neoplastic lesions induced with 7,12 dimethylbenz(alpha)anthracene in the rat: A comparative analysis with human breast tumors. Arch. Pathol. Lab. Med. 2002, 126, 915–927. [Google Scholar] [CrossRef]
- Demaria, S.; Ng, B.; Devitt, M.L.; Babb, J.S.; Kawashima, N.; Liebes, L.; Formenti, S.C. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 862–870. [Google Scholar] [CrossRef]
- Yang, L.; Yong, L.; Zhu, X.; Feng, Y.; Fu, Y.; Kong, D.; Lu, W.; Zhou, T.-Y. Disease progression model of 4T1 metastatic breast cancer. J. Pharmacokinet. Pharmacodyn. 2020, 47, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Huggins, C.; Grand, L.C.; Brillantes, F.P. Mammary cancer induced by a single feeding of polymucular hydrocarbons, and its suppression. Nature 1961, 189, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.-Y.; Chang, C.-J.; Cheng, J.-S. Survival, treatment regimens and medical costs of women newly diagnosed with metastatic triple-negative breast cancer. Sci. Rep. 2022, 12, 729. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Duan, J.-J.; Bian, X.-W.; Yu, S.-C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef] [PubMed]
- Kowanetz, M.; Wu, X.; Lee, J.; Tan, M.; Hagenbeek, T.; Qu, X.; Yu, L.; Ross, J.; Korsisaari, N.; Cao, T.; et al. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 21248–21255. [Google Scholar] [CrossRef]
- Bosiljcic, M.; Cederberg, R.A.; Hamilton, M.J.; LePard, N.E.; Harbourne, B.T.; Collier, J.L.; Halvorsen, E.C.; Shi, R.; Franks, S.E.; Kim, A.Y.; et al. Targeting myeloid-derived suppressor cells in combination with primary mammary tumor resection reduces metastatic growth in the lungs. Breast Cancer Res. 2019, 21, 103. [Google Scholar] [CrossRef]
- Ouzounova, M.; Lee, E.; Piranlioglu, R.; El Andaloussi, A.; Kolhe, R.; Demirci, M.F.; Marasco, D.; Asm, I.; Chadli, A.; Hassan, K.A.; et al. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat. Commun. 2017, 8, 14979. [Google Scholar] [CrossRef]
- Mouchemore, K.A.; Anderson, R.L.; Hamilton, J.A. Neutrophils, G-CSF and their contribution to breast cancer metastasis. FEBS J. 2018, 285, 665–679. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef]
- Ma, X.; Wang, M.; Yin, T.; Zhao, Y.; Wei, X. Myeloid-Derived Suppressor Cells Promote Metastasis in Breast Cancer After the Stress of Operative Removal of the Primary Cancer. Front. Oncol. 2019, 9, 855. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.S.; Gao, Y.; Welte, T.; Wang, H.; Liu, J.; Janghorban, M.; Sheng, K.; Niu, Y.; Goldstein, A.; Zhao, N.; et al. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat. Cell Biol. 2019, 21, 1113–1126. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Deguchi, K.; Zheng, R.; Tamai, H.; Wang, L.-X.; Cohen, P.A.; Shu, S. Tumor-induced CD11b+Gr-1+ myeloid cells suppress T cell sensitization in tumor-draining lymph nodes. J. Immunol. 2008, 181, 3291–3300. [Google Scholar] [CrossRef]
- Haile, L.A.; Greten, T.F.; Korangy, F. Immune suppression: The hallmark of myeloid derived suppressor cells. Immunol. Investig. 2012, 41, 581–594. [Google Scholar] [CrossRef]
- Kim, K.; Skora, A.D.; Li, Z.; Liu, Q.; Tam, A.J.; Blosser, R.L.; Diaz, L.A.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc. Natl. Acad. Sci. USA 2014, 111, 11774–11779. [Google Scholar] [CrossRef] [PubMed]
- Mall, C.; Sckisel, G.D.; Proia, D.A.; Mirsoian, A.; Grossenbacher, S.K.; Pai, C.-C.S.; Chen, M.; Monjazeb, A.M.; Kelly, K.; Blazar, B.R.; et al. Repeated PD-1/PD-L1 monoclonal antibody administration induces fatal xenogeneic hypersensitivity reactions in a murine model of breast cancer. Oncoimmunology 2016, 5, e1075114. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.; Montero, A.; Garret-Mayer, E.; Onicescu, G.; Vandenberg, T.; Hutchens, S.; Diaz-Montero, C. Elevated Circulating Myeloid Derived Suppressor Cells (MDSC) Are Associated with Inferior Overall Survival (OS) and Correlate with Circulating Tumor Cells (CTC) in Patients with Metastatic Breast Cancer. Cancer Res. 2009, 69, 4135. [Google Scholar] [CrossRef]
- Diaz-Montero, C.M.; Salem, M.L.; Nishimura, M.I.; Garrett-Mayer, E.; Cole, D.J.; Montero, A.J. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin–cyclophosphamide chemotherapy. Cancer Immunol. Immunother. 2009, 58, 49–59. [Google Scholar] [CrossRef]
- Adams, S.; Othus, M.; Patel, S.P.; Miller, K.D.; Chugh, R.; Schuetze, S.M.; Chamberlin, M.D.; Haley, B.J.; Storniolo, A.M.V.; Reddy, M.P.; et al. A Multicenter Phase II Trial of Ipilimumab and Nivolumab in Unresectable or Metastatic Metaplastic Breast Cancer: Cohort 36 of Dual Anti-CTLA-4 and Anti-PD-1 Blockade in Rare Tumors (DART, SWOG S1609). Clin. Cancer Res. 2022, 28, 271–278. [Google Scholar] [CrossRef]
- Majidpoor, J.; Mortezaee, K. The efficacy of PD-1/PD-L1 blockade in cold cancers and future perspectives. Clin. Immunol. 2021, 226, 108707. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-L1 Expression in Triple-Negative Breast Cancer. Cancer Immunol. Res. 2014, 2, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Aslakson, C.J.; Miller, F.R. Selective Events in the Metastatic Process Defined by Analysis of the Sequential Dissemination of Subpopulations of a Mouse Mammary Tumor1. Cancer Res. 1992, 52, 1399–1405. [Google Scholar] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nief, C.A.; Swartz, A.M.; Chelales, E.; Sheu, L.Y.; Crouch, B.T.; Ramanujam, N.; Nair, S.K. Ethanol Ablation Therapy Drives Immune-Mediated Antitumor Effects in Murine Breast Cancer Models. Cancers 2022, 14, 4669. https://doi.org/10.3390/cancers14194669
Nief CA, Swartz AM, Chelales E, Sheu LY, Crouch BT, Ramanujam N, Nair SK. Ethanol Ablation Therapy Drives Immune-Mediated Antitumor Effects in Murine Breast Cancer Models. Cancers. 2022; 14(19):4669. https://doi.org/10.3390/cancers14194669
Chicago/Turabian StyleNief, Corrine A., Adam M. Swartz, Erika Chelales, Lauren Y. Sheu, Brian T. Crouch, Nirmala Ramanujam, and Smita K. Nair. 2022. "Ethanol Ablation Therapy Drives Immune-Mediated Antitumor Effects in Murine Breast Cancer Models" Cancers 14, no. 19: 4669. https://doi.org/10.3390/cancers14194669
APA StyleNief, C. A., Swartz, A. M., Chelales, E., Sheu, L. Y., Crouch, B. T., Ramanujam, N., & Nair, S. K. (2022). Ethanol Ablation Therapy Drives Immune-Mediated Antitumor Effects in Murine Breast Cancer Models. Cancers, 14(19), 4669. https://doi.org/10.3390/cancers14194669