
A Selective Histone Deacetylase Inhibitor Induces Autophagy and Cell Death via SCNN1A Downregulation in Glioblastoma Cells

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture, Chemicals, and Antibodies
2.2. Cell Proliferation Assay
2.3. Colony Formation Assay
2.4. Immunoblotting
2.5. Annexin V/PI Staining
2.6. RNA Extraction and Quantitative Analysis of mRNA
2.7. RNA Sequencing
2.8. Lentivirus Infection
2.9. Transient Transfection
2.10. Statistical Analyses
3. Results
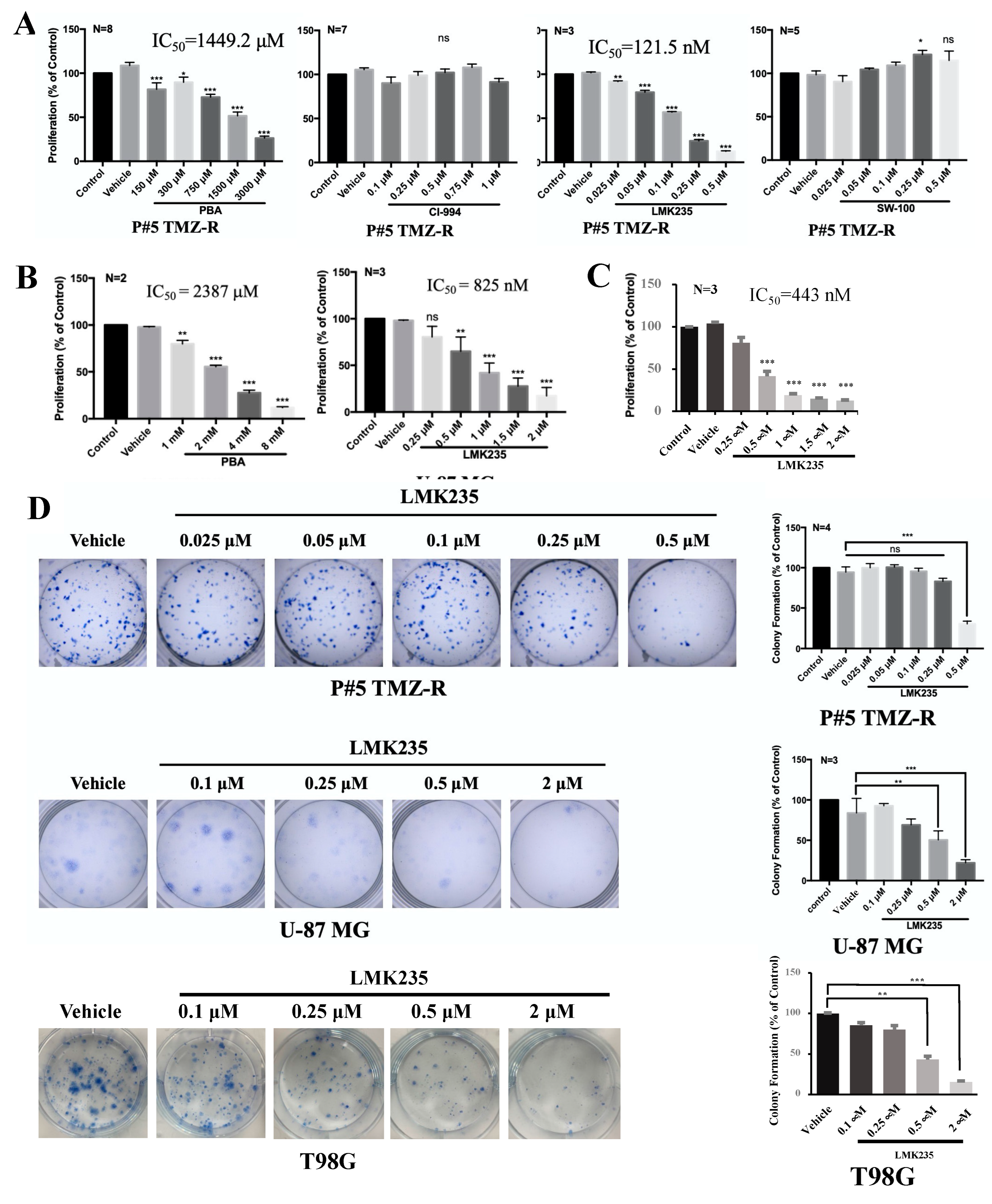
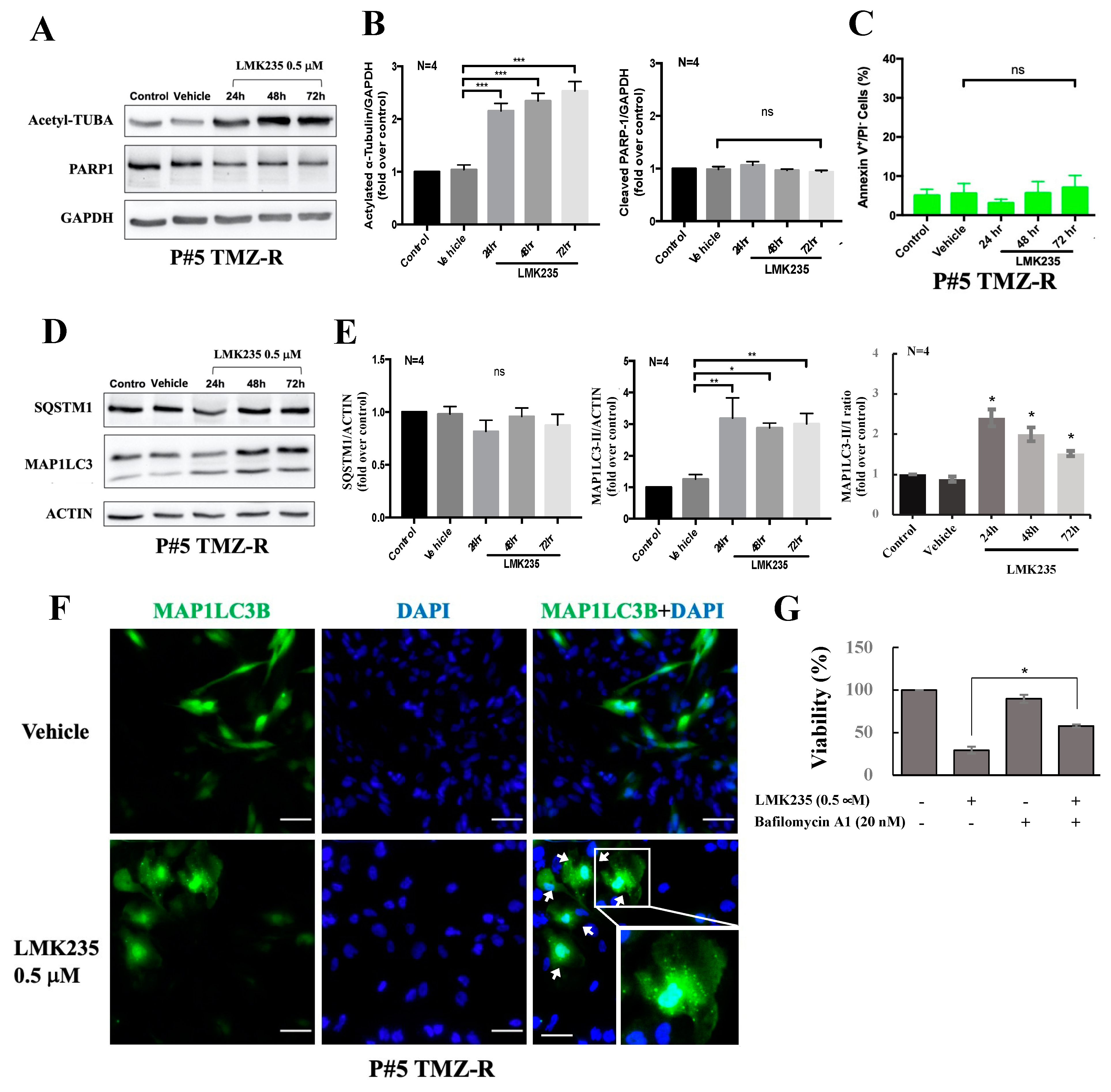
3.1. The HDAC4/HDAC5 Inhibitor LMK235 Reduces Viability and Colony Formation and Induces Autophagy in GBM Cells
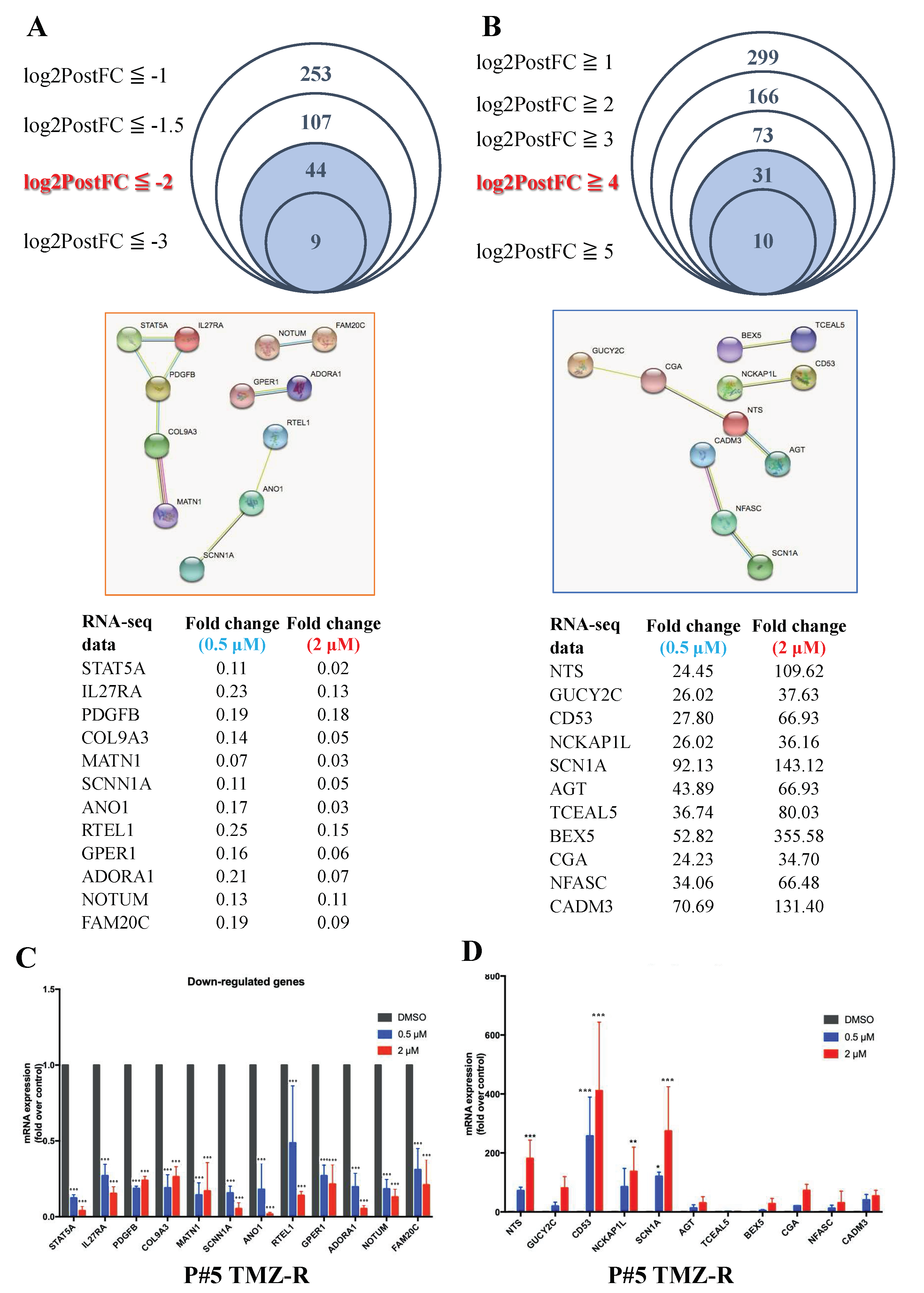
3.2. LMK235 Led to 597 Differentially Expressed Genes in Both 0.5 µM and 2 µM LMK235-Treated GBM Cells
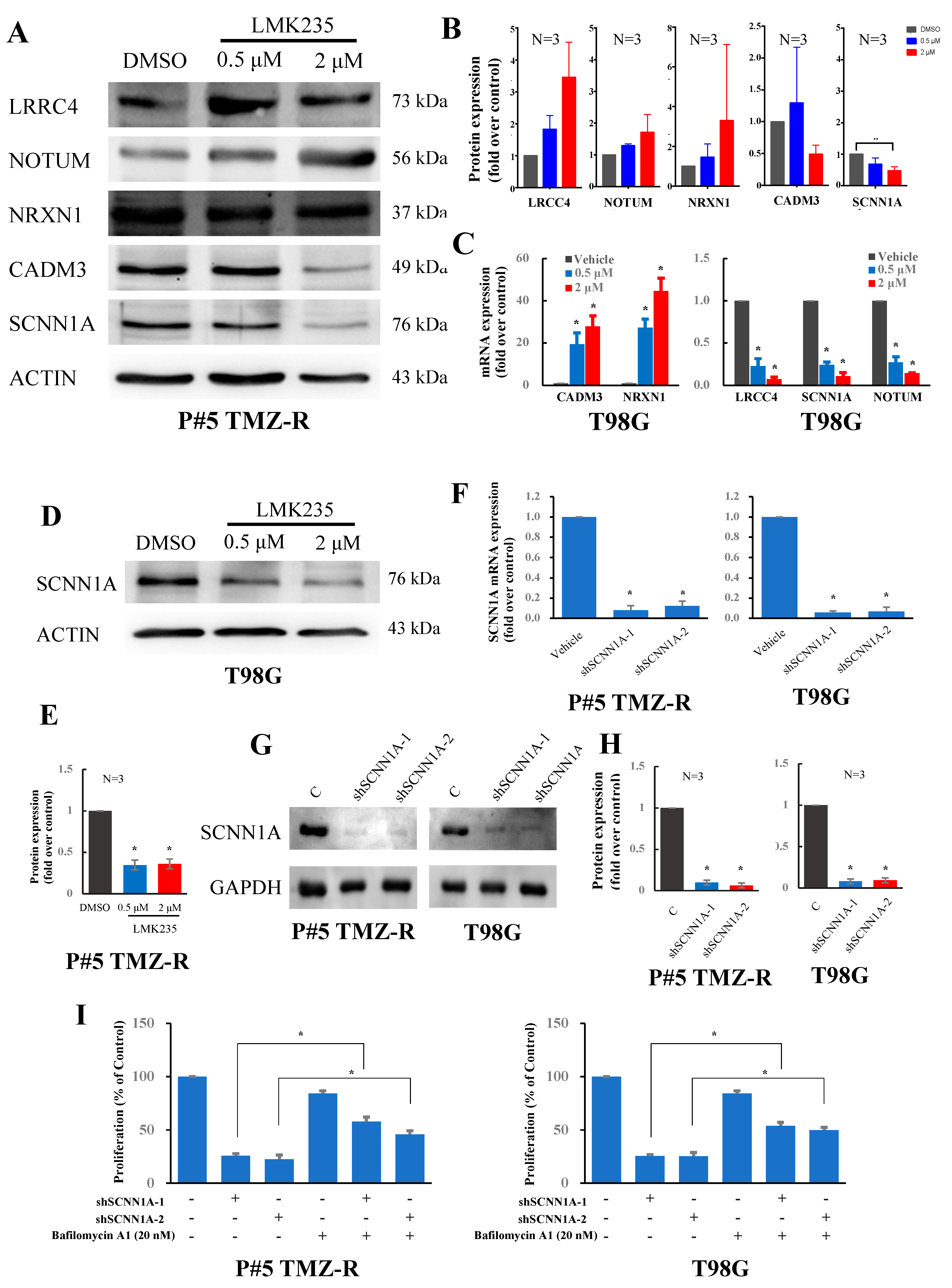
3.3. LMK235 Reduces SCNN1A Expression in GBM Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Davis, F.G.; McCarthy, B.J. Current epidemiological trends and surveillance issues in brain tumors. Expert Rev. Anticancer Ther. 2001, 1, 395–401. [Google Scholar] [CrossRef]
- Jansen, M.; Yip, S.; Louis, D.N. Molecular pathology in adult gliomas: Diagnostic, prognostic, and predictive markers. Lancet Neurol. 2010, 9, 717–726. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Soderberg-Naucler, C.; Rahbar, A.; Stragliotto, G. Survival in patients with glioblastoma receiving valganciclovir. N. Engl. J. Med. 2013, 369, 985–986. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N.; Brat, D.J.; Verhaak, R.G.; Aldape, K.D.; Yung, W.K.; Salama, S.R.; Cooper, L.A.; Rheinbay, E.; Miller, C.R.; Vitucci, M.; et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e821. [Google Scholar] [CrossRef]
- Chiang, K.; Zielinska, A.E.; Shaaban, A.M.; Sanchez-Bailon, M.P.; Jarrold, J.; Clarke, T.L.; Zhang, J.; Francis, A.; Jones, L.J.; Smith, S.; et al. PRMT5 Is a Critical Regulator of Breast Cancer Stem Cell Function via Histone Methylation and FOXP1 Expression. Cell Rep. 2017, 21, 3498–3513. [Google Scholar] [CrossRef]
- Swaroop, A.; Oyer, J.A.; Will, C.M.; Huang, X.; Yu, W.; Troche, C.; Bulic, M.; Durham, B.H.; Wen, Q.J.; Crispino, J.D.; et al. An activating mutation of the NSD2 histone methyltransferase drives oncogenic reprogramming in acute lymphocytic leukemia. Oncogene 2019, 38, 671–686. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, X.; Li, Y.; Quan, C.; Zheng, L.; Huang, K. Lung Cancer Therapy Targeting Histone Methylation: Opportunities and Challenges. Comput. Struct. Biotechnol. J. 2018, 16, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Martinez, R.; Schackert, G. Epigenetic aberrations in malignant gliomas: An open door leading to better understanding and treatment. Epigenetics 2007, 2, 147–150. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dubuc, A.M.; Mack, S.; Unterberger, A.; Northcott, P.A.; Taylor, M.D. The epigenetics of brain tumors. Methods Mol. Biol. 2012, 863, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chen, X.; Mao, L.; Zahid, K.R.; Wen, J.; Zhang, L.; Zhang, M.; Duan, J.; Duan, J.; Yin, X.; et al. Histone deacetylase 1 promotes glioblastoma cell proliferation and invasion via activation of PI3K/AKT and MEK/ERK signaling pathways. Brain Res. 2018, 1692, 154–162. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, Y.; Chen, J.; Tan, Q.; Xie, C.; Li, C.; Zhan, W.; Wang, M. Silencing of histone deacetylase 2 suppresses malignancy for proliferation, migration, and invasion of glioblastoma cells and enhances temozolomide sensitivity. Cancer Chemother. Pharmacol. 2016, 78, 1289–1296. [Google Scholar] [CrossRef]
- Zhong, S.; Fan, Y.; Wu, B.; Wang, Y.; Jiang, S.; Ge, J.; Hua, C.; Zhao, G.; Chen, Y.; Xu, H. HDAC3 Expression Correlates with the Prognosis and Grade of Patients with Glioma: A Diversification Analysis Based on Transcriptome and Clinical Evidence. World Neurosurg. 2018, 119, e145–e158. [Google Scholar] [CrossRef]
- Santos-Barriopedro, I.; Li, Y.; Bahl, S.; Seto, E. HDAC8 affects MGMT levels in glioblastoma cell lines via interaction with the proteasome receptor ADRM1. Genes Cancer 2019, 10, 119–133. [Google Scholar] [CrossRef]
- Cai, J.Y.; Xu, T.T.; Wang, Y.; Chang, J.J.; Li, J.; Chen, X.Y.; Chen, X.; Yin, Y.F.; Ni, X.J. Histone deacetylase HDAC4 promotes the proliferation and invasion of glioma cells. Int. J. Oncol. 2018, 53, 2758–2768. [Google Scholar] [CrossRef]
- Auzmendi-Iriarte, J.; Saenz-Antonanzas, A.; Mikelez-Alonso, I.; Carrasco-Garcia, E.; Tellaetxe-Abete, M.; Lawrie, C.H.; Sampron, N.; Cortajarena, A.L.; Matheu, A. Characterization of a new small-molecule inhibitor of HDAC6 in glioblastoma. Cell Death Dis. 2020, 11, 417. [Google Scholar] [CrossRef]
- Yang, R.; Wu, Y.; Wang, M.; Sun, Z.; Zou, J.; Zhang, Y.; Cui, H. HDAC9 promotes glioblastoma growth via TAZ-mediated EGFR pathway activation. Oncotarget 2015, 6, 7644–7656. [Google Scholar] [CrossRef]
- Lucio-Eterovic, A.K.; Cortez, M.A.; Valera, E.T.; Motta, F.J.; Queiroz, R.G.; Machado, H.R.; Carlotti, C.G., Jr.; Neder, L.; Scrideli, C.A.; Tone, L.G. Differential expression of 12 histone deacetylase (HDAC) genes in astrocytomas and normal brain tissue: Class II and IV are hypoexpressed in glioblastomas. BMC Cancer 2008, 8, 243. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Hu, P.; Tang, F.; Lian, H.; Chen, X.; Zhang, Y.; He, X.; Liu, W.; Xie, C. HDAC6 promotes cell proliferation and confers resistance to temozolomide in glioblastoma. Cancer Lett. 2016, 379, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lin, R.; Zhang, Z.; Wei, Q.; Zhong, Z.; Huang, J.; Xu, Y. Sirtuin 1 knockdown inhibits glioma cell proliferation and potentiates temozolomide toxicity via facilitation of reactive oxygen species generation. Oncol. Lett. 2019, 17, 5343–5350. [Google Scholar] [CrossRef] [PubMed]
- Mu, P.; Liu, K.; Lin, Q.; Yang, W.; Liu, D.; Lin, Z.; Shao, W.; Ji, T. Sirtuin 7 promotes glioma proliferation and invasion through activation of the ERK/STAT3 signaling pathway. Oncol. Lett. 2019, 17, 1445–1452. [Google Scholar] [CrossRef]
- Li, Y.; Dai, D.; Lu, Q.; Fei, M.; Li, M.; Wu, X. Sirt2 suppresses glioma cell growth through targeting NF-kappaB-miR-21 axis. Biochem. Biophys. Res. Commun. 2013, 441, 661–667. [Google Scholar] [CrossRef]
- Luo, K.; Huang, W.; Tang, S. Sirt3 enhances glioma cell viability by stabilizing Ku70-BAX interaction. Onco. Targets Ther. 2018, 11, 7559–7567. [Google Scholar] [CrossRef]
- Galanis, E.; Anderson, S.K.; Lafky, J.M.; Uhm, J.H.; Giannini, C.; Kumar, S.K.; Kimlinger, T.K.; Northfelt, D.W.; Flynn, P.J.; Jaeckle, K.A.; et al. Phase II study of bevacizumab in combination with sorafenib in recurrent glioblastoma (N0776): A north central cancer treatment group trial. Clin. Cancer Res. 2013, 19, 4816–4823. [Google Scholar] [CrossRef]
- Ghiaseddin, A.; Reardon, D.; Massey, W.; Mannerino, A.; Lipp, E.S.; Herndon, J.E., 2nd; McSherry, F.; Desjardins, A.; Randazzo, D.; Friedman, H.S.; et al. Phase II Study of Bevacizumab and Vorinostat for Patients with Recurrent World Health Organization Grade 4 Malignant Glioma. Oncologist 2018, 23, e121–e157. [Google Scholar] [CrossRef]
- Peters, K.B.; Lipp, E.S.; Miller, E.; Herndon, J.E., 2nd; McSherry, F.; Desjardins, A.; Reardon, D.A.; Friedman, H.S. Phase I/II trial of vorinostat, bevacizumab, and daily temozolomide for recurrent malignant gliomas. J. Neurooncol. 2018, 137, 349–356. [Google Scholar] [CrossRef]
- Chen, J.C.; Lee, I.N.; Huang, C.; Wu, Y.P.; Chung, C.Y.; Lee, M.H.; Lin, M.H.; Yang, J.T. Valproic acid-induced amphiregulin secretion confers resistance to temozolomide treatment in human glioma cells. BMC Cancer 2019, 19, 756. [Google Scholar] [CrossRef]
- Asklund, T.; Kvarnbrink, S.; Holmlund, C.; Wibom, C.; Bergenheim, T.; Henriksson, R.; Hedman, H. Synergistic killing of glioblastoma stem-like cells by bortezomib and HDAC inhibitors. Anticancer Res. 2012, 32, 2407–2413. [Google Scholar] [PubMed]
- Liu, J.R.; Yu, C.W.; Hung, P.Y.; Hsin, L.W.; Chern, J.W. High-selective HDAC6 inhibitor promotes HDAC6 degradation following autophagy modulation and enhanced antitumor immunity in glioblastoma. Biochem. Pharmacol. 2019, 163, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Li, P. Growth Suppression of Glioma Cells Using HDAC6 Inhibitor, Tubacin. Open Med. 2018, 13, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Xia, Y.; Hu, K.; Zeng, S.; Wu, L.; Liu, S.; Zhi, C.; Lai, M.; Chen, D.; Xie, L.; et al. Histone deacetylase 6 promotes growth of glioblastoma through the MKK7/JNK/c-Jun signaling pathway. J. Neurochem. 2020, 152, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.T.; Chen, P.F.; Chuang, J.Y.; Gean, P.W.; Hsueh, Y.S. A heat shock protein 90 inhibitor reduces oncoprotein expression and induces cell death in heterogeneous glioblastoma cells with EGFR, PDGFRA, CDK4, and NF1 aberrations. Life Sci. 2021, 288, 120176. [Google Scholar] [CrossRef]
- Chang, K.Y.; Hsu, T.I.; Hsu, C.C.; Tsai, S.Y.; Liu, J.J.; Chou, S.W.; Liu, M.S.; Liou, J.P.; Ko, C.Y.; Chen, K.Y.; et al. Specificity protein 1-modulated superoxide dismutase 2 enhances temozolomide resistance in glioblastoma, which is independent of O(6)-methylguanine-DNA methyltransferase. Redox Biol. 2017, 13, 655–664. [Google Scholar] [CrossRef]
- Kim, M.S.; Akhtar, M.W.; Adachi, M.; Mahgoub, M.; Bassel-Duby, R.; Kavalali, E.T.; Olson, E.N.; Monteggia, L.M. An essential role for histone deacetylase 4 in synaptic plasticity and memory formation. J. Neurosci. 2012, 32, 10879–10886. [Google Scholar] [CrossRef]
- de Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef]
- Mottet, D.; Pirotte, S.; Lamour, V.; Hagedorn, M.; Javerzat, S.; Bikfalvi, A.; Bellahcene, A.; Verdin, E.; Castronovo, V. HDAC4 represses p21(WAF1/Cip1) expression in human cancer cells through a Sp1-dependent, p53-independent mechanism. Oncogene 2009, 28, 243–256. [Google Scholar] [CrossRef]
- Sferra, R.; Pompili, S.; Festuccia, C.; Marampon, F.; Gravina, G.L.; Ventura, L.; Di Cesare, E.; Cicchinelli, S.; Gaudio, E.; Vetuschi, A. The possible prognostic role of histone deacetylase and transforming growth factor beta/Smad signaling in high grade gliomas treated by radio-chemotherapy: A preliminary immunohistochemical study. Eur. J. Histochem. 2017, 61, 2732. [Google Scholar] [CrossRef]
- Marampon, F.; Megiorni, F.; Camero, S.; Crescioli, C.; McDowell, H.P.; Sferra, R.; Vetuschi, A.; Pompili, S.; Ventura, L.; De Felice, F.; et al. HDAC4 and HDAC6 sustain DNA double strand break repair and stem-like phenotype by promoting radioresistance in glioblastoma cells. Cancer Lett. 2017, 397, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.V.; Borges, K.S.; Moreno, D.A.; Queiroz, R.G.; Machado, H.R.; Carlotti, C.G., Jr.; Scrideli, C.A.; Tone, L.G. Abnormal methylation of histone deacetylase genes: Implications on etiology and epigenetic therapy of astrocytomas. Anticancer Res. 2011, 31, 1337–1343. [Google Scholar] [PubMed]
- Dali-Youcef, N.; Froelich, S.; Moussallieh, F.M.; Chibbaro, S.; Noel, G.; Namer, I.J.; Heikkinen, S.; Auwerx, J. Gene expression mapping of histone deacetylases and co-factors, and correlation with survival time and 1H-HRMAS metabolomic profile in human gliomas. Sci. Rep. 2015, 5, 9087. [Google Scholar] [CrossRef] [PubMed]
- Marek, L.; Hamacher, A.; Hansen, F.K.; Kuna, K.; Gohlke, H.; Kassack, M.U.; Kurz, T. Histone deacetylase (HDAC) inhibitors with a novel connecting unit linker region reveal a selectivity profile for HDAC4 and HDAC5 with improved activity against chemoresistant cancer cells. J. Med. Chem. 2013, 56, 427–436. [Google Scholar] [CrossRef]
- Li, A.; Liu, Z.; Li, M.; Zhou, S.; Xu, Y.; Xiao, Y.; Yang, W. HDAC5, a potential therapeutic target and prognostic biomarker, promotes proliferation, invasion and migration in human breast cancer. Oncotarget 2016, 7, 37966–37978. [Google Scholar] [CrossRef]
- Li, X.; He, Z.; Cheng, B.; Fang, Q.; Ma, D.; Lu, T.; Wei, D.; Kuang, X.; Tang, S.; Xiong, J.; et al. Effect of BCLAF1 on HDAC inhibitor LMK-235-mediated apoptosis of diffuse large B cell lymphoma cells and its mechanism. Cancer Biol. Ther. 2018, 19, 825–834. [Google Scholar] [CrossRef]
- Wanek, J.; Gaisberger, M.; Beyreis, M.; Mayr, C.; Helm, K.; Primavesi, F.; Jager, T.; Di Fazio, P.; Jakab, M.; Wagner, A.; et al. Pharmacological inhibition of class IIA HDACs by LMK-235 in pancreatic neuroendocrine tumor cells. Int. J. Mol. Sci. 2018, 19, 3128. [Google Scholar] [CrossRef]
- Li, X.; Guo, Y.; Kuang, X.; Zhao, L.; Li, H.; Cheng, B.; Wang, W.; Zhang, Z.; Liu, P.; Wang, J. Histone deacetylase inhibitor LMK-235-mediated HO-1 expression induces apoptosis in multiple myeloma cells via the JNK/AP-1 signaling pathway. Life Sci. 2019, 223, 146–157. [Google Scholar] [CrossRef]
- Barrera, L.N.; Rushworth, S.A.; Bowles, K.M.; MacEwan, D.J. Bortezomib induces heme oxygenase-1 expression in multiple myeloma. Cell Cycle 2012, 11, 2248–2252. [Google Scholar] [CrossRef]
- Trazzi, S.; Fuchs, C.; Viggiano, R.; De Franceschi, M.; Valli, E.; Jedynak, P.; Hansen, F.K.; Perini, G.; Rimondini, R.; Kurz, T.; et al. HDAC4: A key factor underlying brain developmental alterations in CDKL5 disorder. Hum. Mol. Genet. 2016, 25, 3887–3907. [Google Scholar] [CrossRef]
- Gao, F.; Wang, D.; Liu, X.; Wu, Y.H.; Wang, H.T.; Sun, S.L. Sodium channel 1 subunit alpha SCNN1A exerts oncogenic function in pancreatic cancer via accelerating cellular growth and metastasis. Arch. Biochem. Biophys. 2022, 727, 109323. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Hu, X.; Nan, J.; Zhang, X.; Jin, X. HOXD9induced SCNN1A upregulation promotes pancreatic cancer cell proliferation, migration and predicts prognosis by regulating epithelialmesenchymal transformation. Mol. Med. Rep. 2021, 24, 819. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Zhang, Y.; Peng, X. Knocking down Sterol regulatory element binding protein 2 (SREBF2) inhibits the Serine Protease 8 (PRSS8) /sodium channel epithelial 1alpha subunit (SCNN1A) axis to reduce the cell proliferation, migration and epithelial-mesenchymal transformation of ovarian cancer. Bioengineered 2021, 12, 9390–9400. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Ling, Z.H.; Wang, H.; Wang, X.Y.; Gui, J. Upregulation of SCNN1A promotes cell proliferation, migration, and predicts poor prognosis in ovarian cancer through regulating epithelial-mesenchymal transformation. Cancer Biother. Radiopharm. 2019, 34, 642–649. [Google Scholar] [CrossRef]
- He, M.; Liu, S.; Gallolu Kankanamalage, S.; Borromeo, M.D.; Girard, L.; Gazdar, A.F.; Minna, J.D.; Johnson, J.E.; Cobb, M.H. The epithelial sodium channel (alphaENaC) is a downstream therapeutic target of ASCL1 in pulmonary neuroendocrine tumors. Transl. Oncol. 2018, 11, 292–299. [Google Scholar] [CrossRef]
- Varley, K.E.; Gertz, J.; Roberts, B.S.; Davis, N.S.; Bowling, K.M.; Kirby, M.K.; Nesmith, A.S.; Oliver, P.G.; Grizzle, W.E.; Forero, A.; et al. Recurrent read-through fusion transcripts in breast cancer. Breast Cancer Res. Treat. 2014, 146, 287–297. [Google Scholar] [CrossRef]
- Caren, H.; Djos, A.; Nethander, M.; Sjoberg, R.M.; Kogner, P.; Enstrom, C.; Nilsson, S.; Martinsson, T. Identification of epigenetically regulated genes that predict patient outcome in neuroblastoma. BMC Cancer 2011, 11, 66. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, T.; Han, Q.; Chen, M.; You, J.; Fang, F.; Peng, L.; Wu, B. HDAC inhibitor LMK235 promotes the odontoblast differentiation of dental pulp cells. Mol. Med. Rep. 2018, 17, 1445–1452. [Google Scholar] [CrossRef]
- Mazzocchi, M.; Goulding, S.R.; Wyatt, S.L.; Collins, L.M.; Sullivan, A.M.; O’Keeffe, G.W. LMK235, a small molecule inhibitor of HDAC4/5, protects dopaminergic neurons against neurotoxin- and alpha-synuclein-induced degeneration in cellular models of Parkinson’s disease. Mol. Cell Neurosci. 2021, 115, 103642. [Google Scholar] [CrossRef]
- Chen, Z.; Zhou, B.; Wang, X.; Zhou, G.; Zhang, W.; Yi, B.; Wang, W.; Liu, W. Synergistic effects of mechanical stimulation and crimped topography to stimulate natural collagen development for tendon engineering. Acta Biomater. 2022, 145, 297–315. [Google Scholar] [CrossRef]
- Schluter, A.; Aksan, B.; Fioravanti, R.; Valente, S.; Mai, A.; Mauceri, D. Histone Deacetylases Contribute to Excitotoxicity-Triggered Degeneration of Retinal Ganglion Cells In Vivo. Mol. Neurobiol. 2019, 56, 8018–8034. [Google Scholar] [CrossRef] [PubMed]
- Graule, J.; Uth, K.; Fischer, E.; Centeno, I.; Galvan, J.A.; Eichmann, M.; Rau, T.T.; Langer, R.; Dawson, H.; Nitsche, U.; et al. CDX2 in colorectal cancer is an independent prognostic factor and regulated by promoter methylation and histone deacetylation in tumors of the serrated pathway. Clin. Epigenetics 2018, 10, 120. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Fang, Q.; Ma, D.; Yu, K.; Cheng, B.; Tang, S.; Lu, T.; Wang, W.; Wang, J. Up-regulation of HO-1 promotes resistance of B-cell acute lymphocytic leukemia cells to HDAC4/5 inhibitor LMK-235 via the Smad7 pathway. Life Sci. 2018, 207, 386–394. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, H.H.; Chang, Y.-Y.; Tsai, B.-C.; Chen, L.-J.; Chang, A.-C.; Chuang, J.-Y.; Gean, P.-W.; Hsueh, Y.-S. A Selective Histone Deacetylase Inhibitor Induces Autophagy and Cell Death via SCNN1A Downregulation in Glioblastoma Cells. Cancers 2022, 14, 4537. https://doi.org/10.3390/cancers14184537
Chang HH, Chang Y-Y, Tsai B-C, Chen L-J, Chang A-C, Chuang J-Y, Gean P-W, Hsueh Y-S. A Selective Histone Deacetylase Inhibitor Induces Autophagy and Cell Death via SCNN1A Downregulation in Glioblastoma Cells. Cancers. 2022; 14(18):4537. https://doi.org/10.3390/cancers14184537
Chicago/Turabian StyleChang, Hui Hua, Yao-Yuan Chang, Bing-Chen Tsai, Li-Jyun Chen, An-Chi Chang, Jian-Ying Chuang, Po-Wu Gean, and Yuan-Shuo Hsueh. 2022. "A Selective Histone Deacetylase Inhibitor Induces Autophagy and Cell Death via SCNN1A Downregulation in Glioblastoma Cells" Cancers 14, no. 18: 4537. https://doi.org/10.3390/cancers14184537
APA StyleChang, H. H., Chang, Y.-Y., Tsai, B.-C., Chen, L.-J., Chang, A.-C., Chuang, J.-Y., Gean, P.-W., & Hsueh, Y.-S. (2022). A Selective Histone Deacetylase Inhibitor Induces Autophagy and Cell Death via SCNN1A Downregulation in Glioblastoma Cells. Cancers, 14(18), 4537. https://doi.org/10.3390/cancers14184537