
Polyethylenimine, an Autophagy-Inducing Platinum-Carbene-Based Drug Carrier with Potent Toxicity towards Glioblastoma Cancer Stem Cells
 , , ,
, , ,  ,
,  ,
,  and
and
Abstract
Simple Summary
Abstract
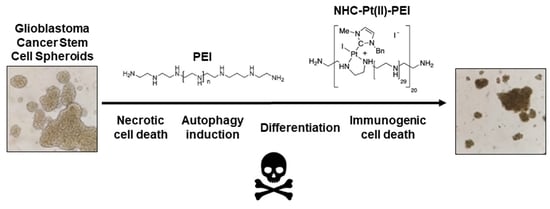
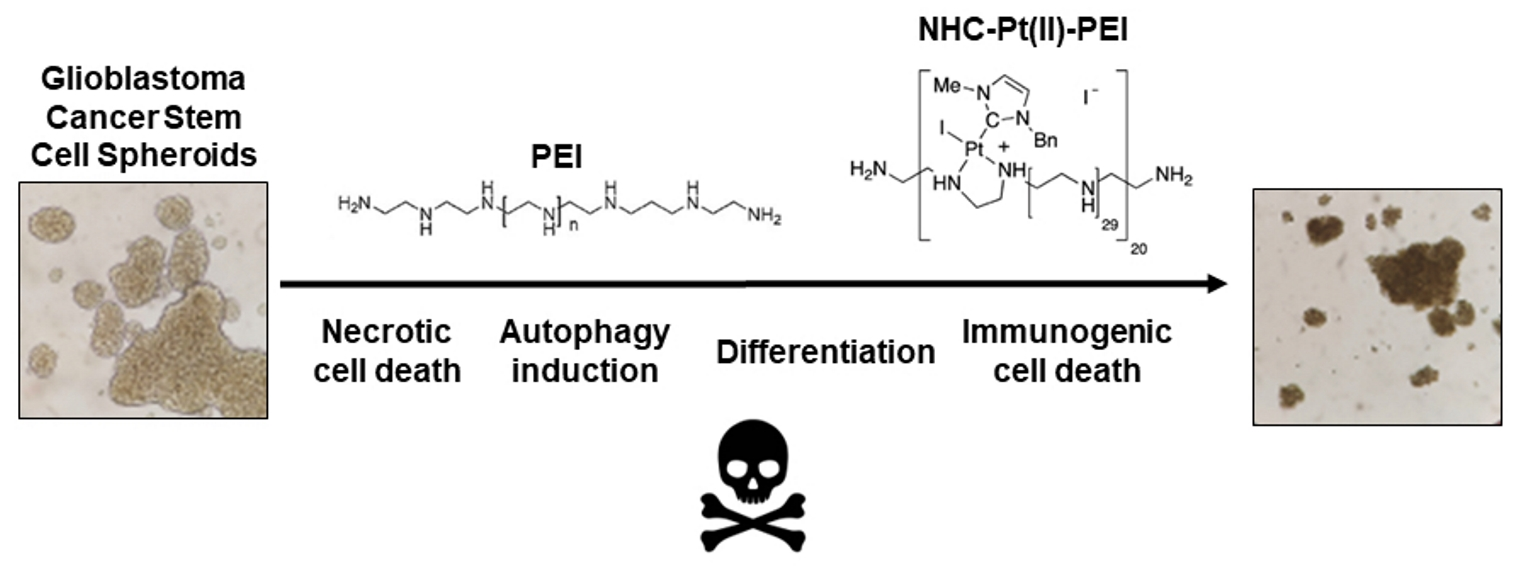
1. Introduction
2. Materials and Methods
2.1. Storage of Compounds
2.2. Cell Culture and Treatment Conditions
2.2.1. U87-MG Cells
2.2.2. Dental Pulp Stem Cells
2.2.3. Glioblastoma Stem Cells
2.2.4. RAW Macrophages
2.3. Cell Viability Assay
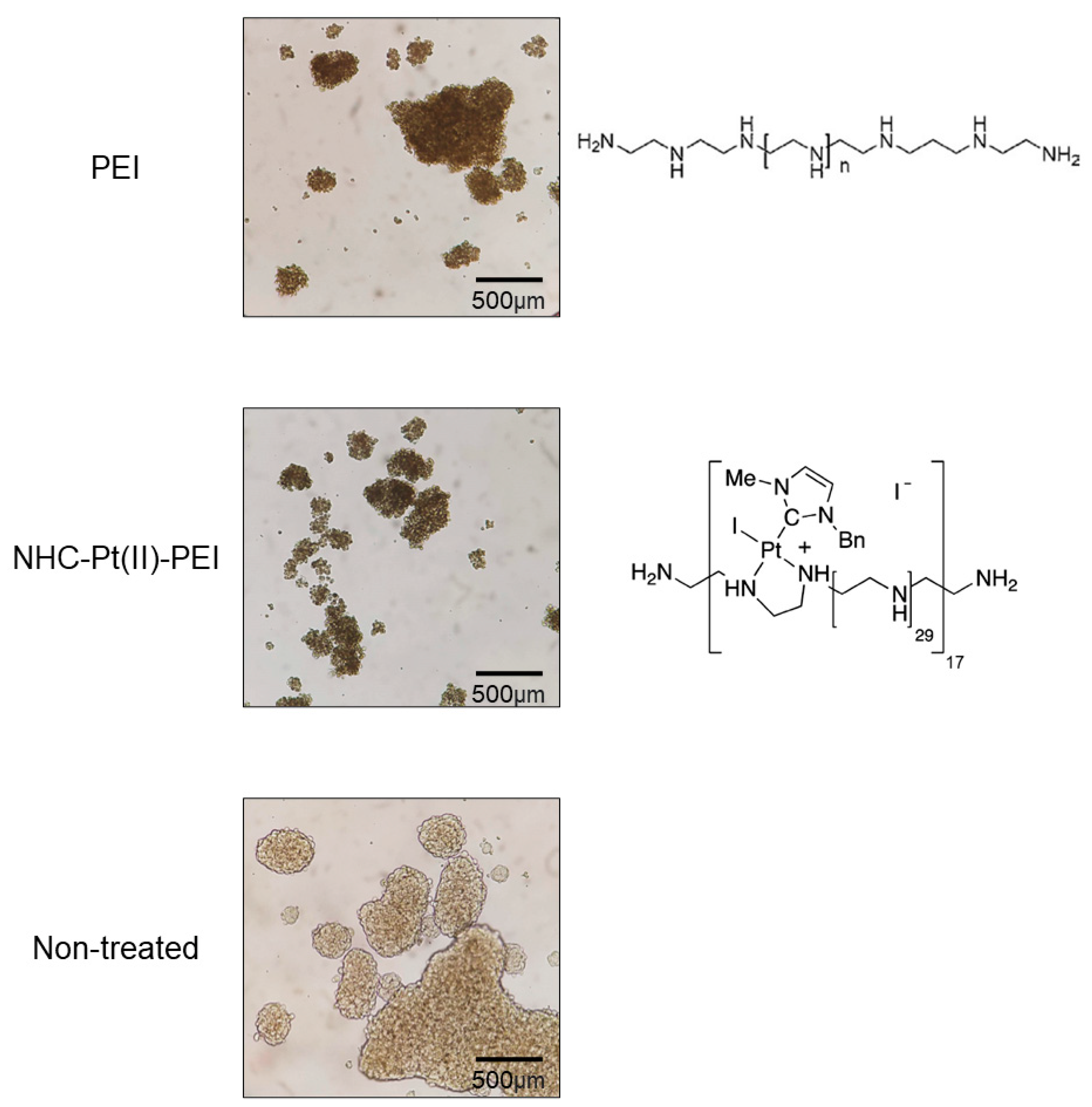
2.4. Spheroid Formation Assay
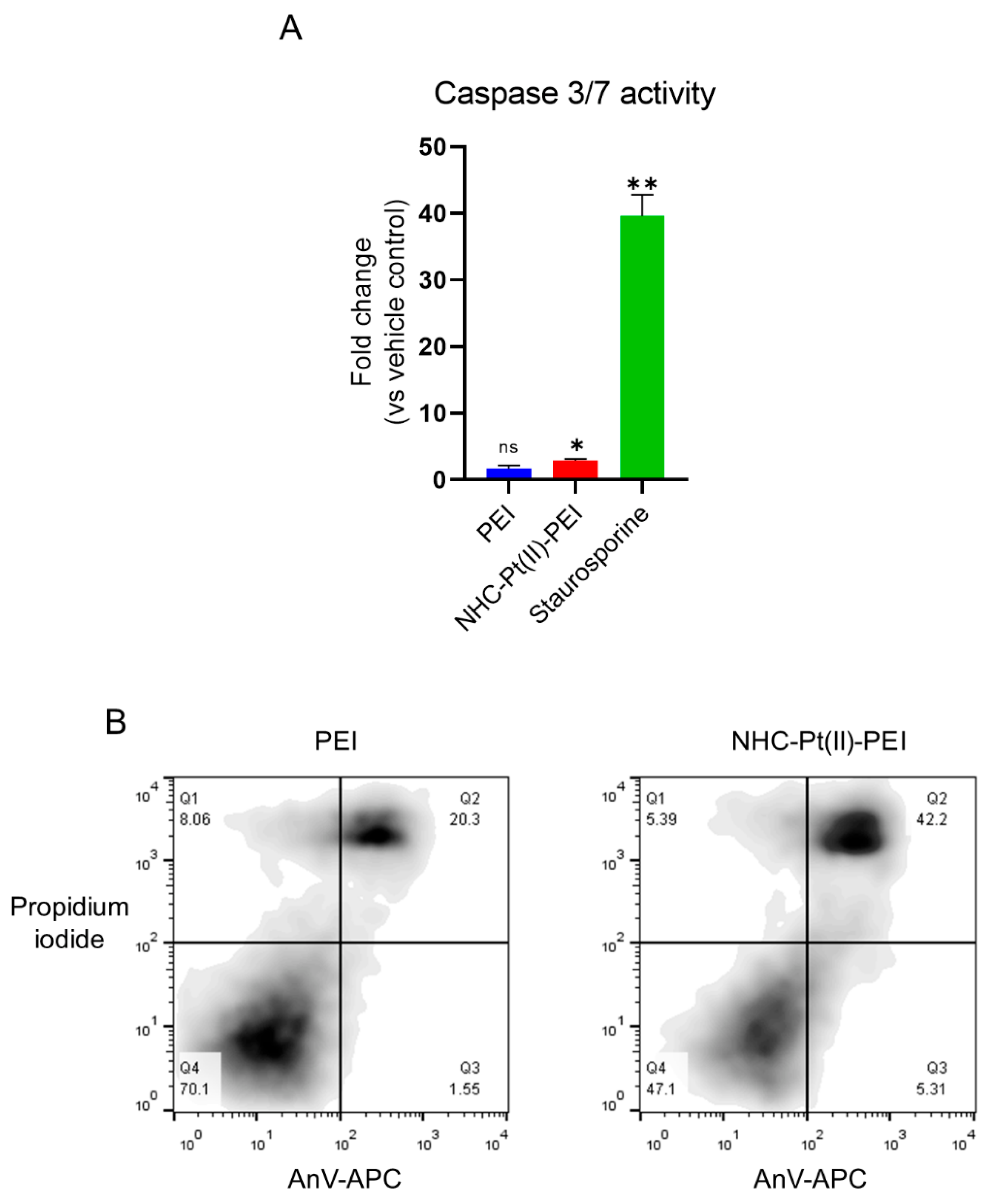
2.5. Annexin V/Propidium Iodide Assay
2.6. Caspase 3/7 Activation Assay
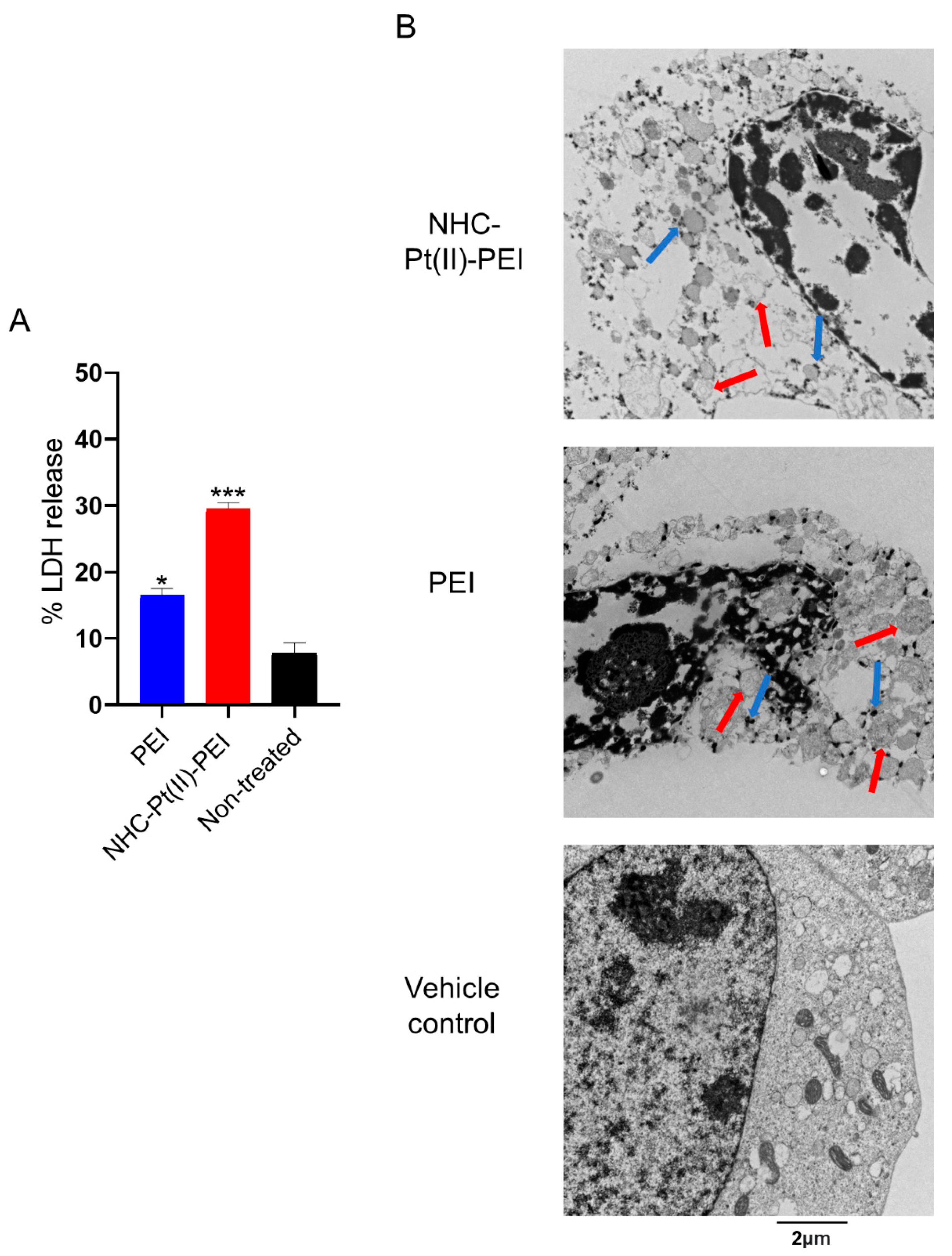
2.7. Transmission Electron Microscopy
2.8. LDH Release Assay
2.9. Western Blot
2.9.1. Cell Treatment, Lysis, and Protein Quantification
2.9.2. SDS-PAGE
2.9.3. Western Blot
2.10. Lysotracker Green Flow Cytometry
2.11. CD133 Expression
2.12. NCH421K-RAW 264.7 Macrophage Immunogenic Cell Death Co-Culture
2.13. Phagocytosis Assay
2.14. ATP Release Assay
2.15. MHC-II Expression
3. Results and Discussion
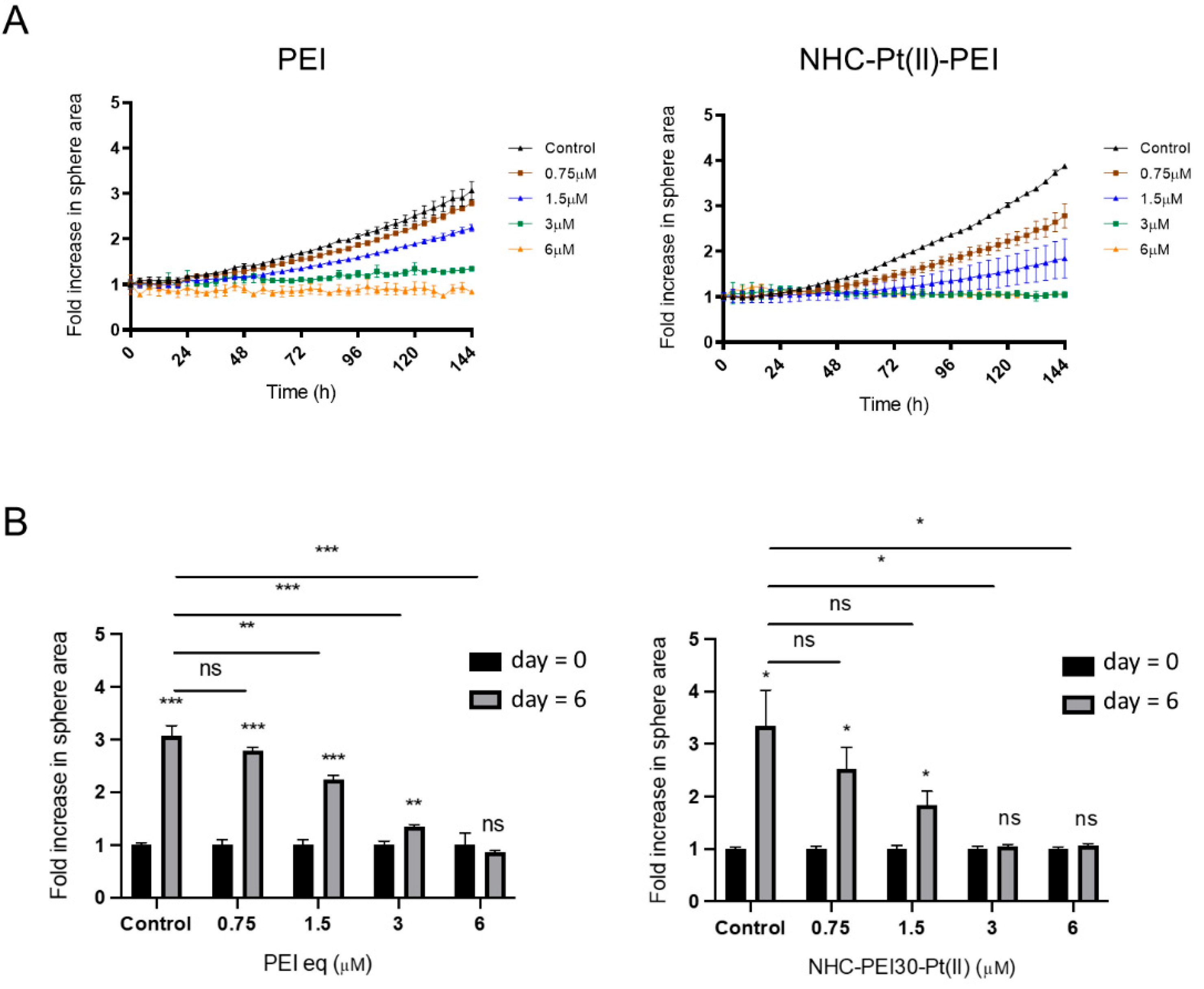
3.1. NHC-Pt(II)-PEI and PEI Decrease CSC Viability and Spheroid Formation
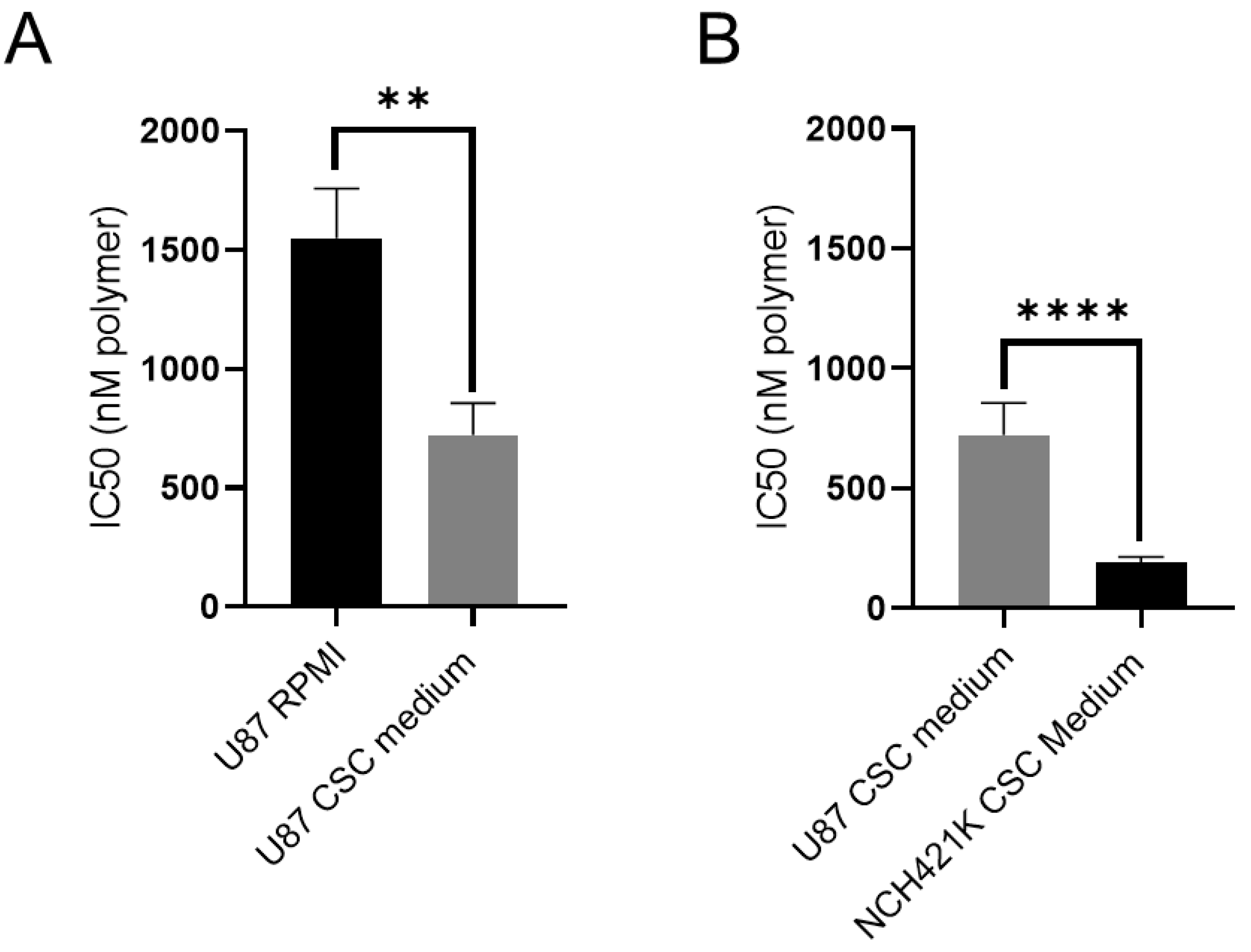
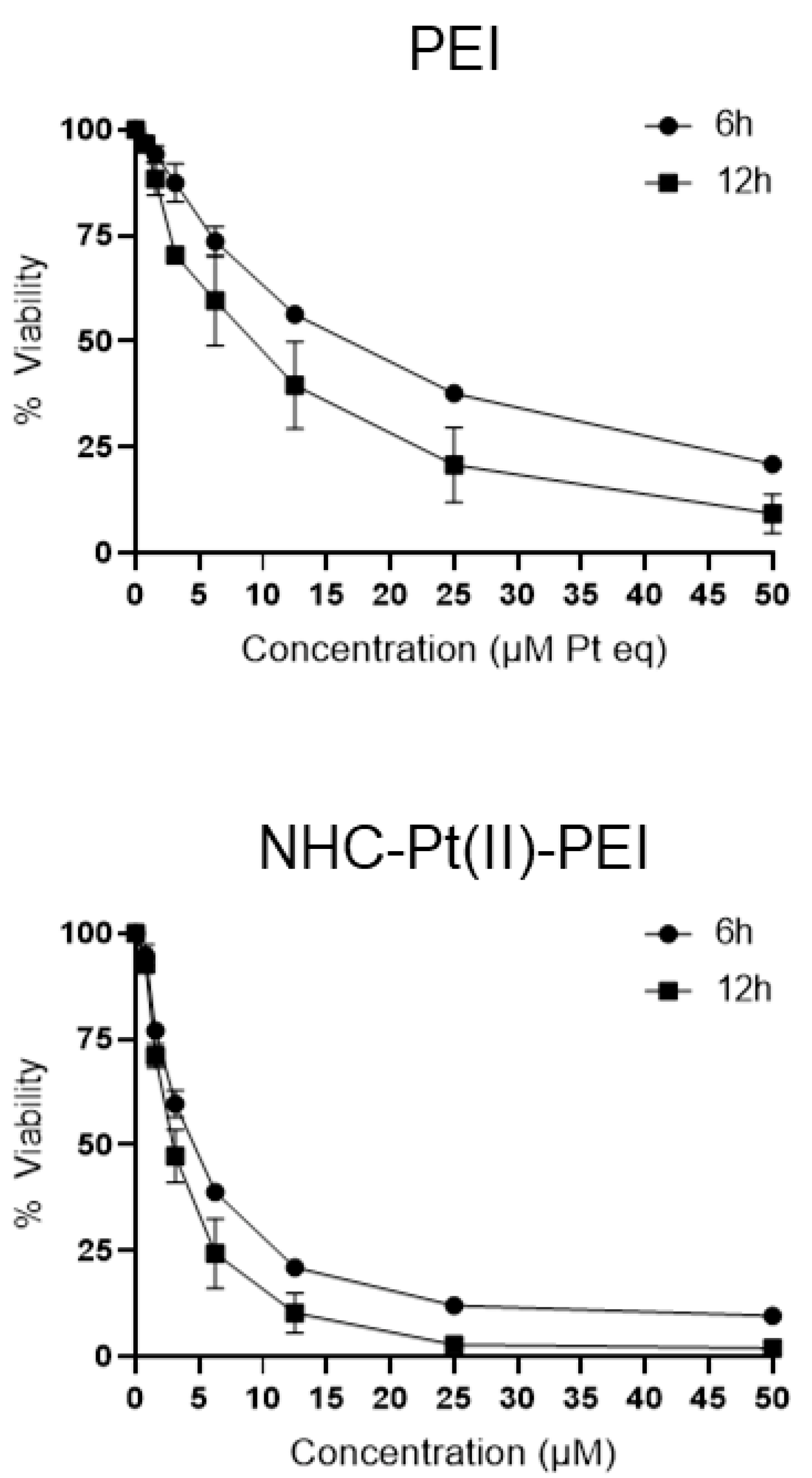
3.2. Culture Media Impacts Polymer Toxicity
3.3. NHC-Pt(II)-PEI and PEI Induce Rapid Membrane Permeabilisation and Cytoplasm Vacuolisation in GSCs
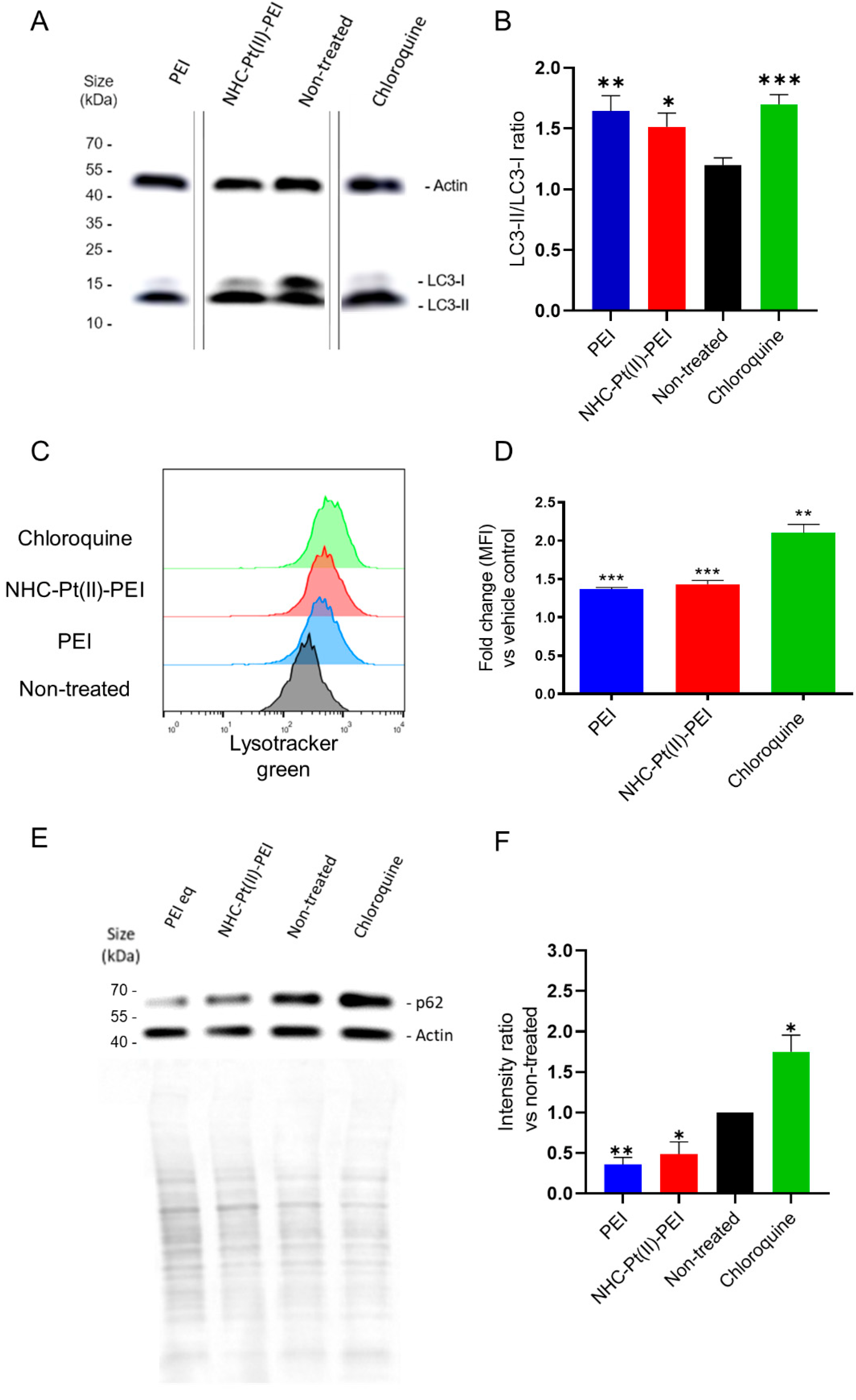
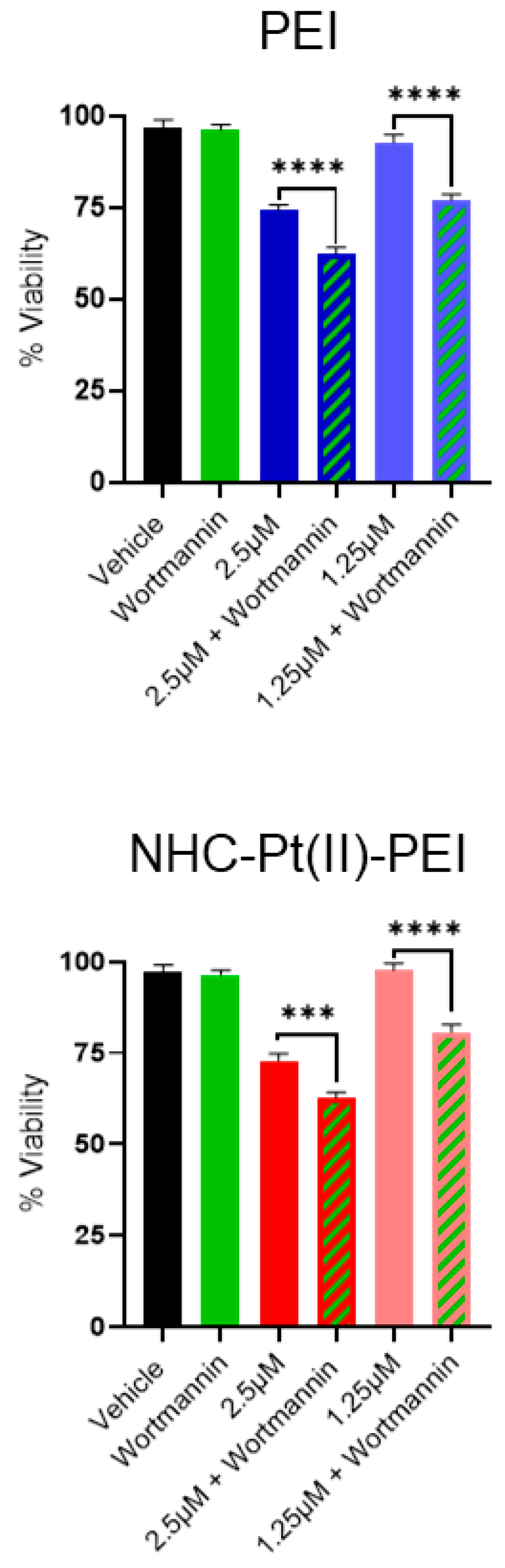
3.4. NHC-Pt(II)-PEI and PEI Induce a Protective Autophagy Response
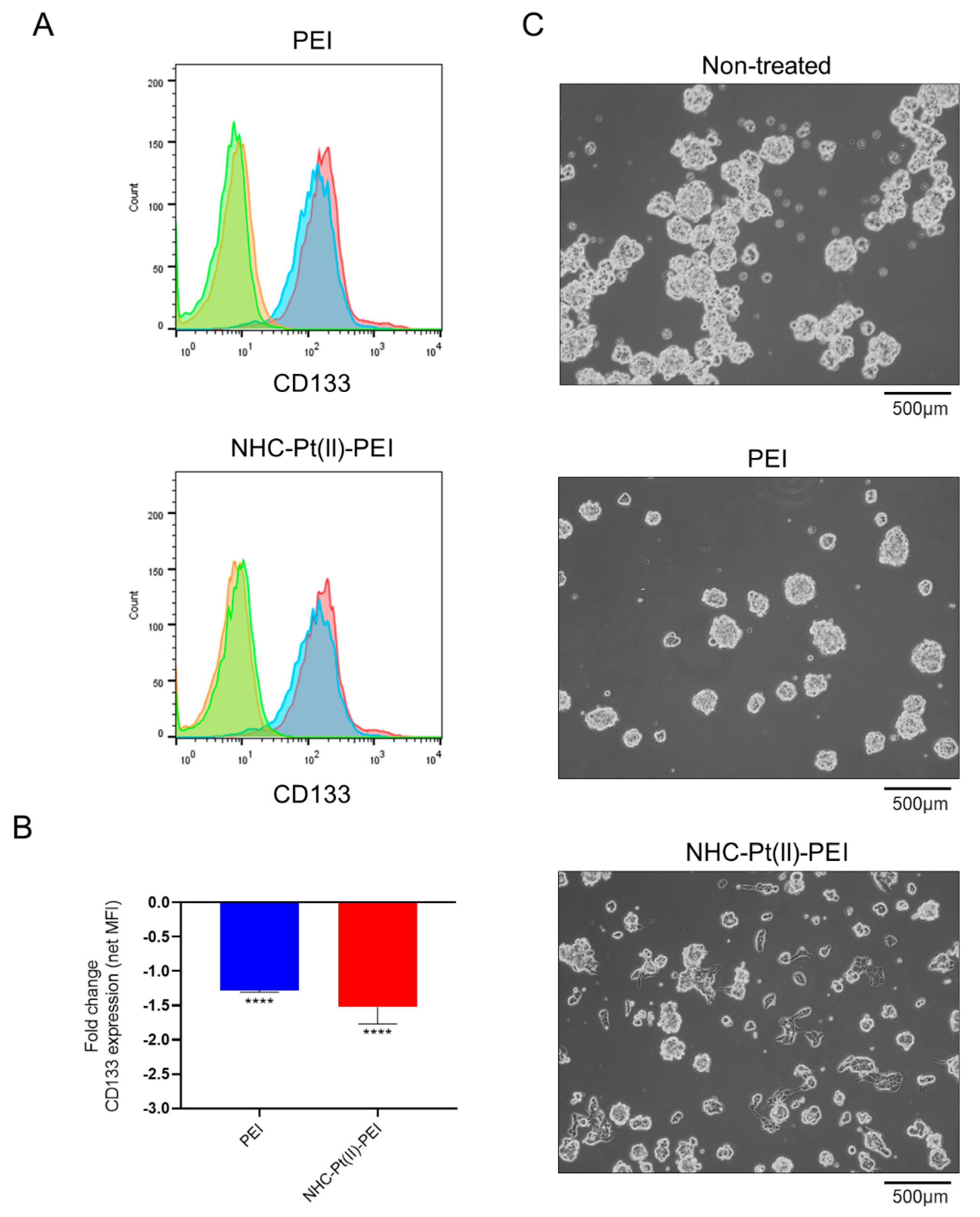
3.5. NHC-Pt(II)-PEI and PEI Treatment Reduce the Expression of CSC Marker CD133
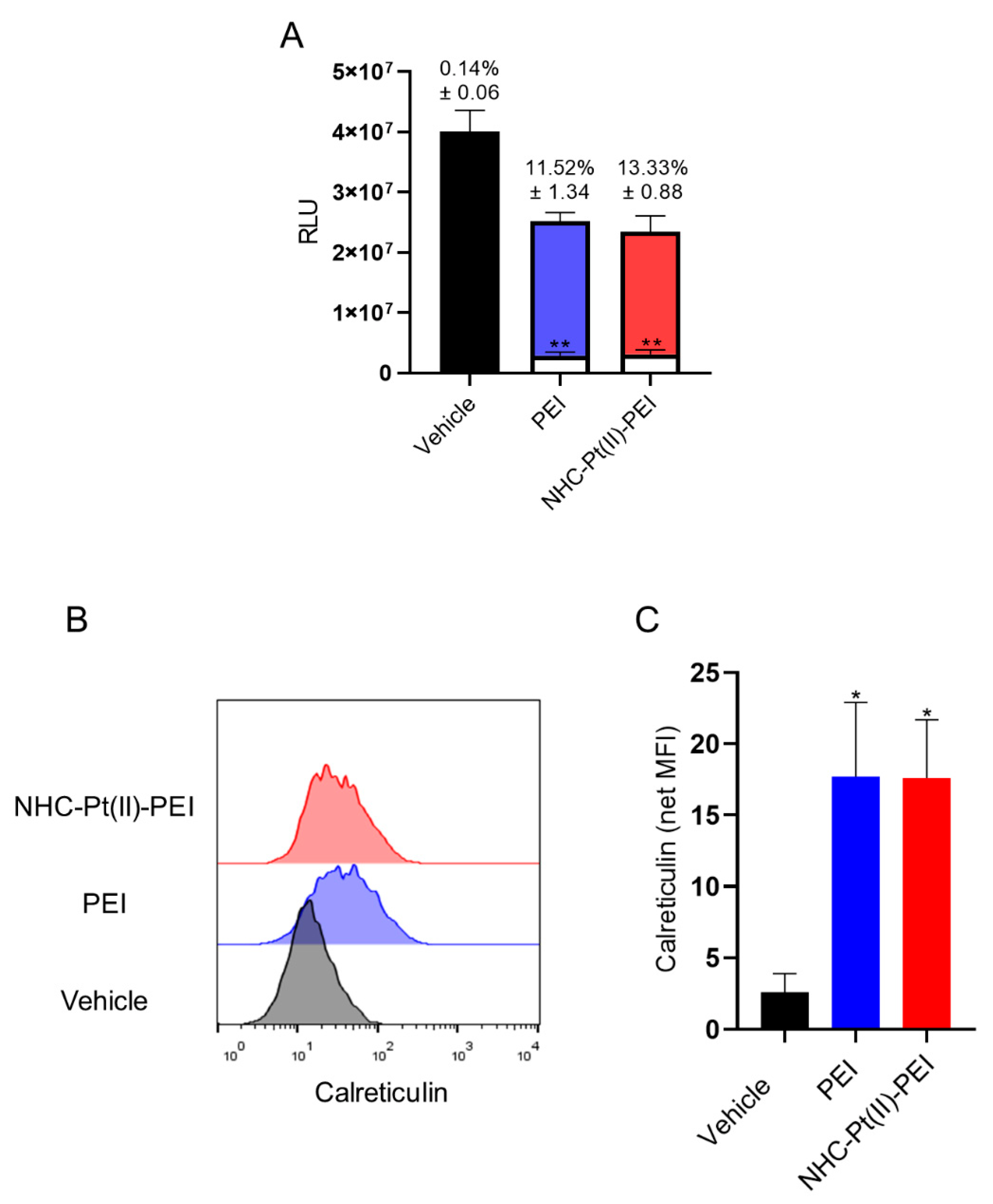
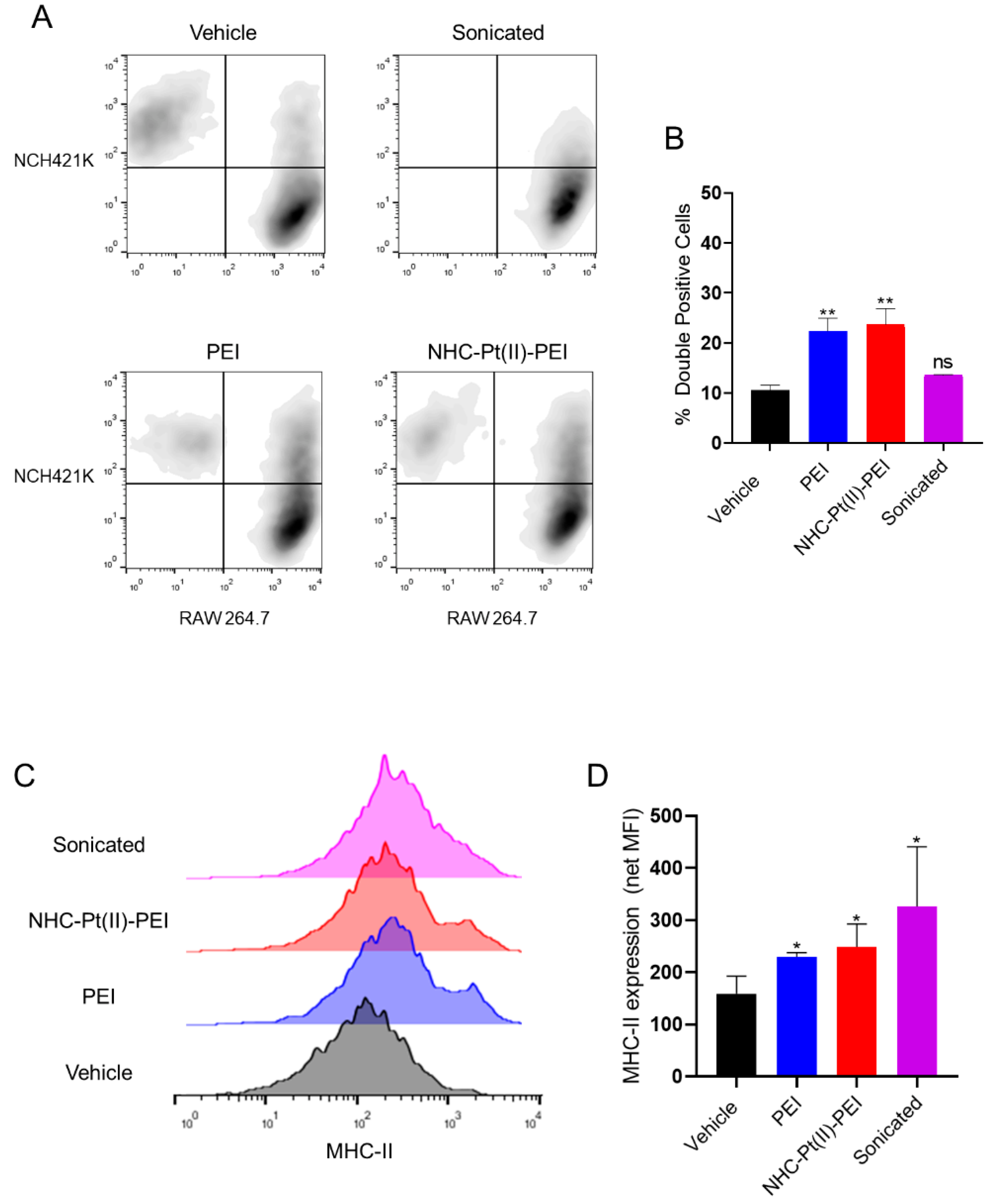
3.6. NHC-Pt(II)-PEI and PEI Cell Death Induces Phagocytosis and DAMPs Release
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AnV | Annexin V |
| APC | Allophycocyanin/Antigen-presenting cell |
| CSC | Cancer stem cell |
| DAMP | Damage associated molecular pattern |
| DER | Poly(propylene glycol) diglycidyl ether |
| DMEM | Dulbecco's Modified Eagle Medium |
| DMAE | Dimethylethanolamine |
| DPSC | Dental pulp stem cell |
| EPR | Enhanced permeability and retention effect |
| FBS | Fetal bovine serum |
| FSC | Forward scatter |
| GSC | Glioblastoma stem cell |
| ICD | Immunogenic cell death |
| LC3 | protein microtubule-associated proteins 1A/1B light chain 3 |
| LDH | Lactate dehydrogenase |
| MEM | Minimum essential medium |
| MFI | Median/Geometric mean fluorescence intensity |
| NHC | N-heterocyclic carbene |
| NSA | Nonenyl succinic anhydride |
| PBS | Dulbecco’s phosphate-buffered saline |
| PDC | Polymer–drug conjugate |
| PEG | Polyethylene glycol |
| PEI | Polyethylenimine |
| PI | Propidium iodide |
| RLU | Relative luminescence unit |
| RPMI | Roswell Park Memorial Institute |
| SEM | Standard error of the mean |
| SDS-PAGE | Sodium dodecyl sulphate–polyacrylamide gel electrophoresis |
| SSC | Side scatter |
| TEM | Transmission electron microscopy |
References
- Dilruba, S.; Kalayda, G.V. Platinum-Based Drugs: Past, Present and Future. Cancer Chemother. Pharmacol. 2016, 77, 1103–1124. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, M.; Chawla, S.; Singh, K.; Rana, P. Evaluation of Anticancer Drug Utilization and Monitoring of Adverse Drug Reaction in the Indoor Patients Receiving Cancer Chemotherapy in a Tertiary Care Hospital in New Delhi. J. Basic Clin. Pharm. 2018, 9. [Google Scholar]
- Pages, B.J.; Garbutcheon-Singh, K.B.; Aldrich-Wright, J.R. Platinum Intercalators of DNA as Anticancer Agents. Eur. J. Inorg. Chem. 2017, 2017, 1613–1624. [Google Scholar] [CrossRef]
- Dasari, S.; Bernard Tchounwou, P. Cisplatin in Cancer Therapy: Molecular Mechanisms of Action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Zhang, C.; Xu, C.; Gao, X.; Yao, Q. Platinum-Based Drugs for Cancer Therapy and Anti-Tumor Strategies. Theranostics 2022, 12, 2115–2132. [Google Scholar] [CrossRef]
- Skander, M.; Retailleau, P.; Bourrié, B.; Schio, L.; Mailliet, P.; Marinetti, A. N-Heterocyclic Carbene-Amine Pt(II) Complexes, a New Chemical Space for the Development of Platinum-Based Anticancer Drugs. J. Med. Chem. 2010, 53, 2146–2154. [Google Scholar] [CrossRef]
- Bellemin-Laponnaz, S. N-Heterocyclic Carbene Platinum Complexes: A Big Step Forward for Effective Antitumor Compounds. Eur. J. Inorg. Chem. 2020, 2020, 10–20. [Google Scholar] [CrossRef]
- Ekladious, I.; Colson, Y.L.; Grinstaff, M.W. Polymer–Drug Conjugate Therapeutics: Advances, Insights and Prospects. Nat. Rev. Drug Discov. 2019, 18, 273–294. [Google Scholar] [CrossRef]
- Wadhwa, S.; Mumper, R.J. Polymer-Drug Conjugates for Anticancer Drug Delivery. Crit. Rev. Ther. Drug Carrier Syst. 2015, 32, 215–245. [Google Scholar] [CrossRef]
- Petros, R.A.; Desimone, J.M. Strategies in the Design of Nanoparticles for Therapeutic Applications. Nat. Rev. Drug Discov. 2010, 9, 615–627. [Google Scholar] [CrossRef]
- Chekkat, N.; Dahm, G.; Chardon, E.; Wantz, M.; Sitz, J.; Decossas, M.; Lambert, O.; Frisch, B.; Rubbiani, R.; Gasser, G.; et al. N-Heterocyclic Carbene-Polyethylenimine Platinum Complexes with Potent in Vitro and in Vivo Antitumor Efficacy. Bioconjug. Chem. 2016, 27, 1942–1948. [Google Scholar] [CrossRef]
- Torchilin, V. Tumor Delivery of Macromolecular Drugs Based on the EPR Effect. Adv. Drug Deliv. Rev. 2011, 63, 131–135. [Google Scholar] [CrossRef]
- Maeda, H. Polymer Therapeutics and the EPR Effect. J. Drug Target. 2017, 25, 781–785. [Google Scholar] [CrossRef]
- Fahira, A.I.; Abdulah, R.; Barliana, M.I.; Gatera, V.A.; Amalia, R. Polyethyleneimine (PEI) as a Polymer-Based Co-Delivery System for Breast Cancer Therapy. Breast Cancer Targets Ther. 2022, 14, 71–83. [Google Scholar] [CrossRef]
- Neuberg, P.; Kichler, A. Recent Developments in Nucleic Acid Delivery with Polyethylenimines; Elsevier, 2014; Volume 88, ISBN 9780128001486. [Google Scholar]
- Xu, C.; Wang, P.; Zhang, J.; Tian, H.; Park, K.; Chen, X. Pulmonary Codelivery of Doxorubicin and SiRNA by PH-Sensitive Nanoparticles for Therapy of Metastatic Lung Cancer. Small 2015, 11, 4321–4333. [Google Scholar] [CrossRef]
- Zhou, Z.; Murdoch, W.J.; Shen, Y. A Linear Polyethylenimine (LPEI) Drug Conjugate with Reversible Charge to Overcome Multidrug Resistance in Cancer Cells. Polymer 2015, 76, 150–158. [Google Scholar] [CrossRef]
- Wantz, M.; Bouché, M.; Dahm, G.; Chekkat, N.; Fournel, S.; Bellemin-Laponnaz, S. N-Heterocyclic Carbene-Polyethyleneimine (PEI) Platinum Complexes Inducing Human Cancer Cell Death: Polymer Carrier Impact. Int. J. Mol. Sci. 2018, 19, 3472. [Google Scholar] [CrossRef]
- Neuzil, J.; Stantic, M.; Zobalova, R.; Chladova, J.; Wang, X.; Prochazka, L.; Dong, L.; Andera, L.; Ralph, S.J. Tumour-Initiating Cells vs. Cancer “stem” Cells and CD133: What’s in the Name? Biochem. Biophys. Res. Commun. 2007, 355, 855–859. [Google Scholar] [CrossRef]
- Wu, X.Z. Origin of Cancer Stem Cells: The Role of Self-Renewal and Differentiation. Ann. Surg. Oncol. 2008, 15, 407–414. [Google Scholar] [CrossRef]
- Batlle, E.; Clevers, H. Cancer Stem Cells Revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Ayob, A.Z.; Ramasamy, T.S. Cancer Stem Cells as Key Drivers of Tumour Progression. J. Biomed. Sci. 2018, 25, 1–18. [Google Scholar] [CrossRef]
- Zhao, J. Cancer Stem Cells and Chemoresistance: The Smartest Survives the Raid. Pharmacol. Ther. 2016, 160, 145–158. [Google Scholar] [CrossRef]
- Abdullah, L.; Chow, E. Chemoresistance in Cancer Stem Cells. Clin. Transl. Med. 2013, 2, 1–9. [Google Scholar] [CrossRef]
- Najafi, M.; Mortezaee, K.; Majidpoor, J. Cancer Stem Cell (CSC) Resistance Drivers. Life Sci. 2019, 234, 116781. [Google Scholar] [CrossRef]
- Tanida, I. Autophagy Basics. Microbiol. Immunol. 2011, 55, 1–11. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and Function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef]
- Virolle, T. Cancer Stem Cells in Glioblastoma. Bull. Cancer 2017, 104, 1075–1079. [Google Scholar] [CrossRef]
- Seymour, T.; Nowak, A.; Kakulas, F. Targeting Aggressive Cancer Stem Cells in Glioblastoma. Front. Oncol. 2015, 5, 1–9. [Google Scholar] [CrossRef]
- Lamb, R.; Harrison, H.; Hulit, J.; Smith, D.L.; Lisanti, M.P.; Sotgia, F. Mitochondria as New Therapeutic Targets for Eradicating Cancer Stem Cells: Quantitative Proteomics and Functional Validation via MCT1/2 Inhibition. Oncotarget 2014, 5, 11029–11037. [Google Scholar] [CrossRef]
- Jagust, P.; De Luxán-Delgado, B.; Parejo-Alonso, B.; Sancho, P. Metabolism-Based Therapeutic Strategies Targeting Cancer Stem Cells. Front. Pharmacol. 2019, 10, 1–26. [Google Scholar] [CrossRef]
- Farnie, G.; Sotgia, F.; Lisanti, M.P. High Mitochondrial Mass Identifies a Sub-Population of Stem-like Cancer Cells That Are Chemo-Resistant. Oncotarget 2015, 6, 30472–30486. [Google Scholar] [CrossRef] [PubMed]
- Skoda, J.; Borankova, K.; Jansson, P.J.; Huang, M.L.H.; Veselska, R.; Richardson, D.R. Pharmacological Targeting of Mitochondria in Cancer Stem Cells: An Ancient Organelle at the Crossroad of Novel Anti-Cancer Therapies. Pharmacol. Res. 2019, 139, 298–313. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y. CD133 as a Regulator of Cancer Metastasis through the Cancer Stem Cells. Int. J. Biochem. Cell Biol. 2019, 106, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Thapa, R.; Wilson, G.D. The Importance of CD44 as a Stem Cell Biomarker and Therapeutic Target in Cancer. Stem Cells Int. 2016, 2016, 2087204. [Google Scholar] [CrossRef]
- Garg, A.D.; Nowis, D.; Golab, J.; Vandenabeele, P.; Krysko, D.V.; Agostinis, P. Immunogenic Cell Death, DAMPs and Anticancer Therapeutics: An Emerging Amalgamation. Biochim. Biophys. Acta—Rev. Cancer 2010, 1805, 53–71. [Google Scholar] [CrossRef]
- Tesniere, A.; Panaretakis, T.; Kepp, O.; Apetoh, L.; Ghiringhelli, F.; Zitvogel, L.; Kroemer, G. Molecular Characteristics of Immunogenic Cancer Cell Death. Cell Death Differ. 2008, 15, 3–12. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, G.; Chen, Y.; Wang, H.; Hua, Y.; Cai, Z. Immunogenic Cell Death in Cancer Therapy: Present and Emerging Inducers. J. Cell. Mol. Med. 2019, 23, 4854–4865. [Google Scholar] [CrossRef]
- Pandolfi, F.; Altamura, S.; Frosali, S.; Conti, P. Key Role of DAMP in Inflammation, Cancer, and Tissue Repair. Clin. Ther. 2016, 38, 1017–1028. [Google Scholar] [CrossRef]
- Brissault, B.; Kichler, A.; Guis, C.; Leborgne, C.; Danos, O.; Cheradame, H. Synthesis of Linear Polyethylenimine Derivatives for DNA Transfection. Bioconjug. Chem. 2003, 14, 581–587. [Google Scholar] [CrossRef]
- Campos, B.; Wan, F.; Farhadi, M.; Ernst, A.; Zeppernick, F.; Tagscherer, K.E.; Ahmadi, R.; Lohr, J.; Dictus, C.; Gdynia, G.; et al. Differentiation Therapy Exerts Antitumor Effects on Stem-like Glioma Cells. Clin. Cancer Res. 2010, 16, 2715–2728. [Google Scholar] [CrossRef]
- Verreault, M.; Schmitt, C.; Goldwirt, L.; Pelton, K.; Haidar, S.; Levasseur, C.; Guehennec, J.; Knoff, D.; Labussière, M.; Marie, Y.; et al. Preclinical Efficacy of the MDM2 Inhibitor RG7112 in MDM2-Amplified and TP53 Wild-Type Glioblastomas. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 1185–1196. [Google Scholar] [CrossRef]
- Taciak, B.; Białasek, M.; Braniewska, A.; Sas, Z.; Sawicka, P.; Kiraga, Ł.; Rygiel, T.; Król, M. Evaluation of Phenotypic and Functional Stability of RAW 264.7 Cell Line through Serial Passages. PLoS ONE 2018, 13, e0198943. [Google Scholar] [CrossRef]
- Podergajs, N.; Brekka, N.; Radlwimmer, B.; Herold-Mende, C.; Talasila, K.M.; Tiemann, K.; Rajcevic, U.; Lah, T.T.; Bjerkvig, R.; Miletic, H. Expansive Growth of Two Glioblastoma Stem-like Cell Lines Is Mediated by BFGF and Not by EGF. Radiol. Oncol. 2013, 47, 330–337. [Google Scholar] [CrossRef]
- McCartin, C.; Mathieu, E.; Dontenwill, M.; Herold-Mende, C.; Idbaih, A.; Bonfiglio, A.; Mauro, M.; Fournel, S.; Kichler, A. An N-Heterocyclic Carbene Iridium(III) Complex as a Potent Anti-Cancer Stem Cell Therapeutic. Chem. Biol. Interact. 2022, 367, 110167. [Google Scholar] [CrossRef]
- Prabhakar, N.; Merisaari, J.; Le Joncour, V.; Peurla, M.; Karaman, D.Ş.; Casals, E.; Laakkonen, P.; Westermarck, J.; Rosenholm, J.M. Circumventing Drug Treatment? Intrinsic Lethal Effects of Polyethyleneimine (PEI)-Functionalized Nanoparticles on Glioblastoma Cells Cultured in Stem Cell Conditions. Cancers 2021, 13, 2631. [Google Scholar] [CrossRef]
- Knauer, N.; Arkhipova, V.; Li, G.; Hewera, M.; Pashkina, E.; Nguyen, P.-H.; Meschaninova, M.; Kozlov, V.; Zhang, W.; Croner, R.; et al. In Vitro Validation of the Therapeutic Potential of Dendrimer-Based Nanoformulations against Tumor Stem Cells. Int. J. Mol. Sci. 2022, 23, 5691. [Google Scholar] [CrossRef]
- Zhang, Q.F.; Wang, B.; Yin, D.X.; Zhang, J.; Wu, W.X.; Yu, Q.Y.; Yu, X.Q. Linear TACN-Based Cationic Polymers as Non-Viral Gene Vectors. RSC Adv. 2014, 4, 59164–59174. [Google Scholar] [CrossRef]
- Kheraldine, H.; Rachid, O.; Habib, A.M.; Al Moustafa, A.E.; Benter, I.F.; Akhtar, S. Emerging Innate Biological Properties of Nano-Drug Delivery Systems: A Focus on PAMAM Dendrimers and Their Clinical Potential. Adv. Drug Deliv. Rev. 2021, 178, 113908. [Google Scholar] [CrossRef]
- Jevprasesphant, R.; Penny, J.; Jalal, R.; Attwood, D.; McKeown, N.B.; D’Emanuele, A. The Influence of Surface Modification on the Cytotoxicity of PAMAM Dendrimers. Int. J. Pharm. 2003, 252, 263–266. [Google Scholar] [CrossRef]
- Fischer, D.; Li, Y.; Ahlemeyer, B.; Krieglstein, J.; Kissel, T. In Vitro Cytotoxicity Testing of Polycations: Influence of Polymer Structure on Cell Viability and Hemolysis. Biomaterials 2003, 24, 1121–1131. [Google Scholar] [CrossRef]
- Beyerle, A.; Irmler, M.; Beckers, J.; Kissel, T.; Stoeger, T. Toxicity Pathway Focused Gene Expression Profiling of PEI-Based Polymers for Pulmonary Applications. Mol. Pharm. 2010, 7, 727–737. [Google Scholar] [CrossRef]
- Moghimi, S.M.; Symonds, P.; Murray, J.C.; Hunter, A.C.; Debska, G.; Szewczyk, A. A Two-Stage Poly(Ethylenimine)-Mediated Cytotoxicity: Implications for Gene Transfer/Therapy. Mol. Ther. 2005, 11, 990–995. [Google Scholar] [CrossRef]
- Grandinetti, G.; Ingle, N.P.; Reineke, T.M. Interaction of Poly(Ethylenimine)-DNA Polyplexes with Mitochondria: Implications for a Mechanism of Cytotoxicity. Mol. Pharm. 2011, 8, 1709–1719. [Google Scholar] [CrossRef]
- Novikoff, A.B.; Beaufay, H.; De Duve, C. Electron Microscopy of Lysosome-Rich Fractions from Rat Liver. J. Cell Biol. 1956, 2, 179–184. [Google Scholar] [CrossRef]
- Smith, A.G.; Macleod, K.F. Autophagy, Cancer Stem Cells and Drug Resistance. J. Pathol. 2019, 247, 708–718. [Google Scholar] [CrossRef]
- Arima, Y.; Nobusue, H.; Saya, H. Targeting of Cancer Stem Cells by Differentiation Therapy. Cancer Sci. 2020, 111, 2689–2695. [Google Scholar] [CrossRef]
- El-Gowily, A.H.; Abosheasha, M.A. Differential Mechanisms of Autophagy in Cancer Stem Cells: Emphasizing Gastrointestinal Cancers. Cell Biochem. Funct. 2021, 39, 162–173. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in Mammalian Autophagy Research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef]
- Bik, E.; Mateuszuk, L.; Orleanska, J.; Baranska, M.; Chlopicki, S.; Majzner, K. Chloroquine-Induced Accumulation of Autophagosomes and Lipids in the Endothelium. Int. J. Mol. Sci. 2021, 22, 2401. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine Inhibits Autophagic Flux by Decreasing Autophagosome-Lysosome Fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef] [PubMed]
- Chikte, S.; Panchal, N.; Warnes, G. Use of LysoTracker Dyes: A Flow Cytometric Study of Autophagy. Cytom. Part A 2014, 85, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Sung, T.; Lin, N.; Abraham, R.T.; Jessen, B.A. Lysosomal Adaptation: How Cells Respond to Lysosomotropic Compounds. PLoS ONE 2017, 12, e0173771. [Google Scholar] [CrossRef] [PubMed]
- Kichler, A.; Leborgne, C.; Coeytaux, E.; Danos, O. Polyethylenimine-Mediated Gene Delivery: A Mechanistic Study. J. Gene Med. 2001, 3, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Thomas, M.; Klibanov, A.M.; Langer, R. Exploring Polyethylenimine-Mediated DNA Transfection and the Proton Sponge Hypothesis. J. Gene Med. 2005, 7, 657–663. [Google Scholar] [CrossRef]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. P62 Links the Autophagy Pathway and the Ubiqutin–Proteasome System upon Ubiquitinated Protein Degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef]
- Gao, X.; Yao, L.; Song, Q.; Zhu, L.; Xia, Z.; Xia, H.; Jiang, X.; Chen, J.; Chen, H. The Association of Autophagy with Polyethylenimine-Induced Cytotoxity in Nephritic and Hepatic Cell Lines. Biomaterials 2011, 32, 8613–8625. [Google Scholar] [CrossRef]
- Lin, C.W.; Jan, M.S.; Kuo, J.H.S.; Hsu, L.J.; Lin, Y.S. Protective Role of Autophagy in Branched Polyethylenimine (25K)-and Poly(L-Lysine) (30–70K)-Induced Cell Death. Eur. J. Pharm. Sci. 2012, 47, 865–874. [Google Scholar] [CrossRef]
- Wang, F.; Salvati, A.; Boya, P. Lysosome-Dependent Cell Death and Deregulated Autophagy Induced by Amine-Modified Polystyrene Nanoparticles. Open Biol. 2018, 4, 170271. [Google Scholar] [CrossRef]
- Pasquier, B. Autophagy Inhibitors. Cell. Mol. Life Sci. 2016, 73, 985–1001. [Google Scholar] [CrossRef]
- Wang, X.; Lee, J.; Xie, C. Autophagy Regulation on Cancer Stem Cell Maintenance, Metastasis, and Therapy Resistance. Cancers 2022, 14, 381. [Google Scholar] [CrossRef]
- Ryskalin, L.; Gaglione, A.; Limanaqi, F.; Biagioni, F.; Familiari, P.; Frati, A.; Esposito, V.; Fornai, F. The Autophagy Status of Cancer Stem Cells in Gliobastoma Multiforme: From Cancer Promotion to Therapeutic Strategies. Int. J. Mol. Sci. 2019, 20, 3824. [Google Scholar] [CrossRef]
- Zhuang, W.; Long, L.; Zheng, B.; Ji, W.; Yang, N.; Zhang, Q.; Liang, Z. Curcumin Promotes Differentiation of Glioma-Initiating Cells by Inducing Autophagy. Cancer Sci. 2012, 103, 684–690. [Google Scholar] [CrossRef]
- Lei, Y.; Zhang, D.; Yu, J.; Dong, H.; Zhang, J.; Yang, S.; Yang, S. Targeting Autophagy in Cancer Stem Cells as an Anticancer Therapy. Cancer Lett. 2017, 393, 33–39. [Google Scholar] [CrossRef]
- Hao, C.; Liu, G.; Tian, G. Autophagy Inhibition of Cancer Stem Cells Promotes the Efficacy of Cisplatin against Non-Small Cell Lung Carcinoma. Ther. Adv. Respir. Dis. 2019, 13, 1753466619866097. [Google Scholar] [CrossRef]
- Babaei, G.; Aziz, S.G.G.; Jaghi, N.Z.Z. EMT, Cancer Stem Cells and Autophagy; The Three Main Axes of Metastasis. Biomed. Pharmacother. 2021, 133, 110909. [Google Scholar] [CrossRef]
- Gamrekelashvili, J.; Greten, T.F.; Korangy, F. Immunogenicity of Necrotic Cell Death. Cell. Mol. Life Sci. 2015, 72, 273–283. [Google Scholar] [CrossRef]
- Wang, Y.; Martins, I.; Ma, Y.; Kepp, O.; Galluzzi, L.; Kroemer, G. Autophagy-Dependent ATP Release from Dying Cells via Lysosomal Exocytosis. Autophagy 2013, 9, 1624–1625. [Google Scholar] [CrossRef]
- Luo, X.; Qiu, Y.; Dinesh, P.; Gong, W.; Jiang, L.; Feng, X.; Li, J.; Jiang, Y.; Lei, Y.L.; Chen, Q. The Functions of Autophagy at the Tumour-Immune Interface. J. Cell. Mol. Med. 2021, 25, 2333–2341. [Google Scholar] [CrossRef]
- Wang, Y.J.; Fletcher, R.; Yu, J.; Zhang, L. Immunogenic Effects of Chemotherapy-Induced Tumor Cell Death. Genes Dis. 2018, 5, 194–203. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Glioblastoma Stem Cells | Non-Cancer Stem Cells | Glioma Cells | |||
|---|---|---|---|---|---|
| NCH421K | NCH644 | 3731 | DPSC | U87 | |
| Oxaliplatin | 53 ± 1.9 | 55 ± 1.6 | 55.9 ± 2 | >100 | >100 |
| Temozolomide | >100 | 97.2 ± 15 | >100 | >100 | >100 |
| NHC-Pt(II)-PEI | 2.6 ± 0.1 | 1.4 ± 0.3 | 2.1 ± 0.3 | 6.6 ± 0.9 ****, ####, xxxx | 9.3 ± 0.8 ****, ####, xxxx |
| PEI (eq) PEI (nM polymer) | 3.3 ± 0.4 (194) | 10.6 ± 0.6 (624) | 2.9 ± 0.2 (169) | 32.5 ± 0.9 ****, ####, xxxx (1912) | 26.3 ± 3.5 ****, ###, xxxx (1547) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCartin, C.; Dussouillez, C.; Bernhard, C.; Mathieu, E.; Blumberger, J.; Dontenwill, M.; Herold-Mende, C.; Idbaih, A.; Lavalle, P.; Bellemin-Laponnaz, S.; et al. Polyethylenimine, an Autophagy-Inducing Platinum-Carbene-Based Drug Carrier with Potent Toxicity towards Glioblastoma Cancer Stem Cells. Cancers 2022, 14, 5057. https://doi.org/10.3390/cancers14205057
McCartin C, Dussouillez C, Bernhard C, Mathieu E, Blumberger J, Dontenwill M, Herold-Mende C, Idbaih A, Lavalle P, Bellemin-Laponnaz S, et al. Polyethylenimine, an Autophagy-Inducing Platinum-Carbene-Based Drug Carrier with Potent Toxicity towards Glioblastoma Cancer Stem Cells. Cancers. 2022; 14(20):5057. https://doi.org/10.3390/cancers14205057
Chicago/Turabian StyleMcCartin, Conor, Candice Dussouillez, Chloé Bernhard, Eric Mathieu, Juliette Blumberger, Monique Dontenwill, Christel Herold-Mende, Ahmed Idbaih, Philippe Lavalle, Stéphane Bellemin-Laponnaz, and et al. 2022. "Polyethylenimine, an Autophagy-Inducing Platinum-Carbene-Based Drug Carrier with Potent Toxicity towards Glioblastoma Cancer Stem Cells" Cancers 14, no. 20: 5057. https://doi.org/10.3390/cancers14205057
APA StyleMcCartin, C., Dussouillez, C., Bernhard, C., Mathieu, E., Blumberger, J., Dontenwill, M., Herold-Mende, C., Idbaih, A., Lavalle, P., Bellemin-Laponnaz, S., Kichler, A., & Fournel, S. (2022). Polyethylenimine, an Autophagy-Inducing Platinum-Carbene-Based Drug Carrier with Potent Toxicity towards Glioblastoma Cancer Stem Cells. Cancers, 14(20), 5057. https://doi.org/10.3390/cancers14205057