
Ceramide Kinase (CERK) Emerges as a Common Therapeutic Target for Triple Positive and Triple Negative Breast Cancer Cells

 , and
, and
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results
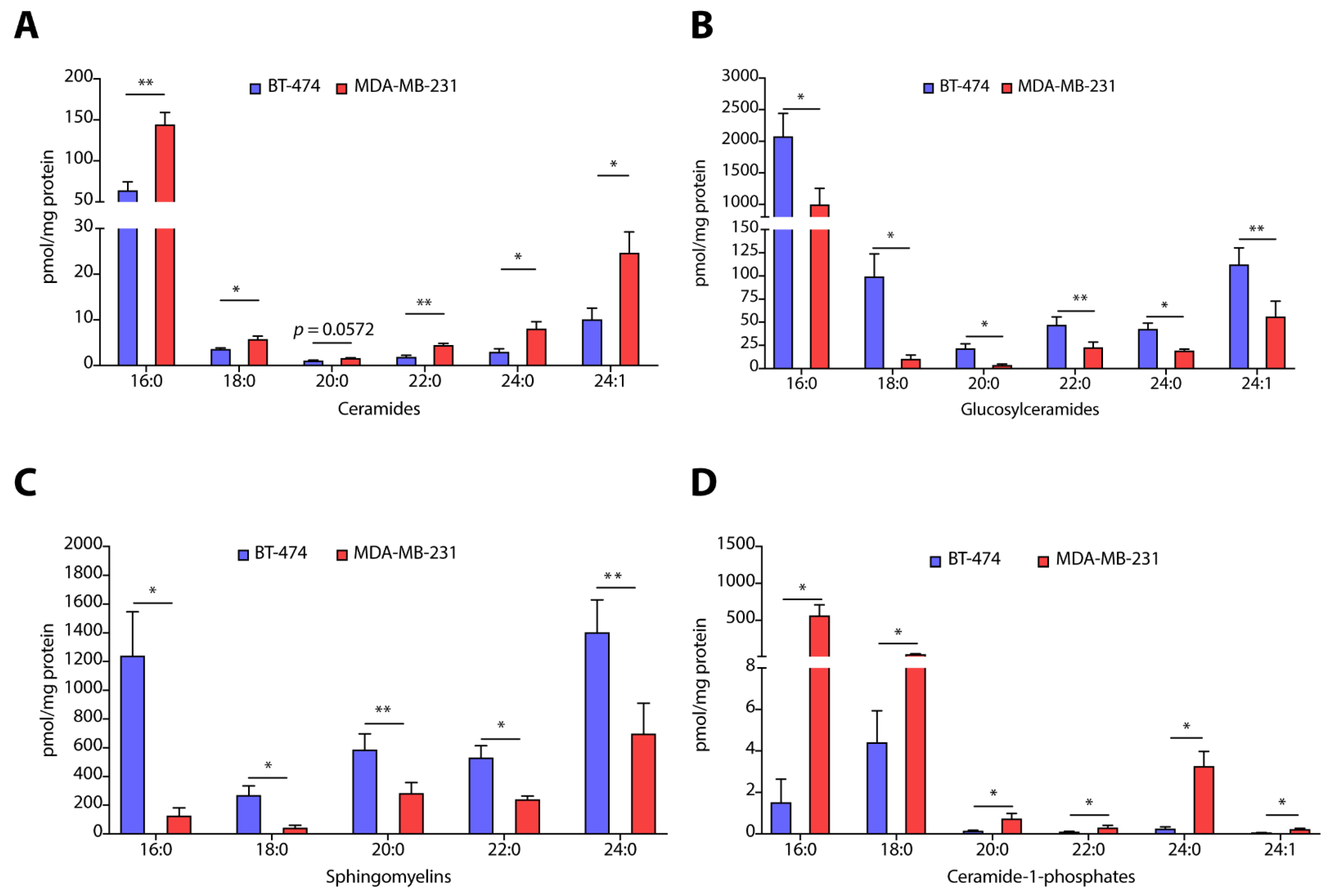
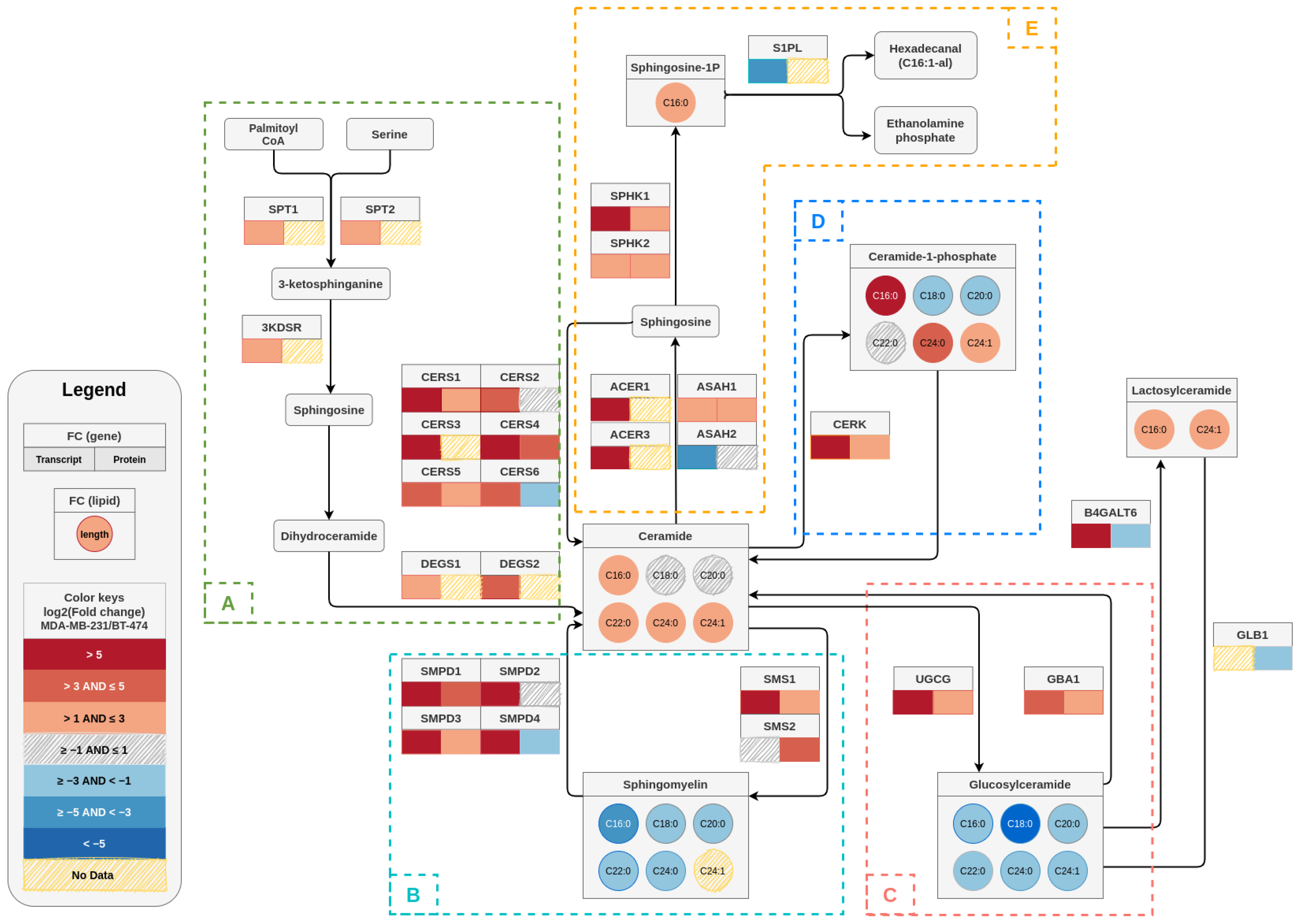
3.1. TPBC and TNBC Cell Lines Possess Unique Sphingolipid Profile
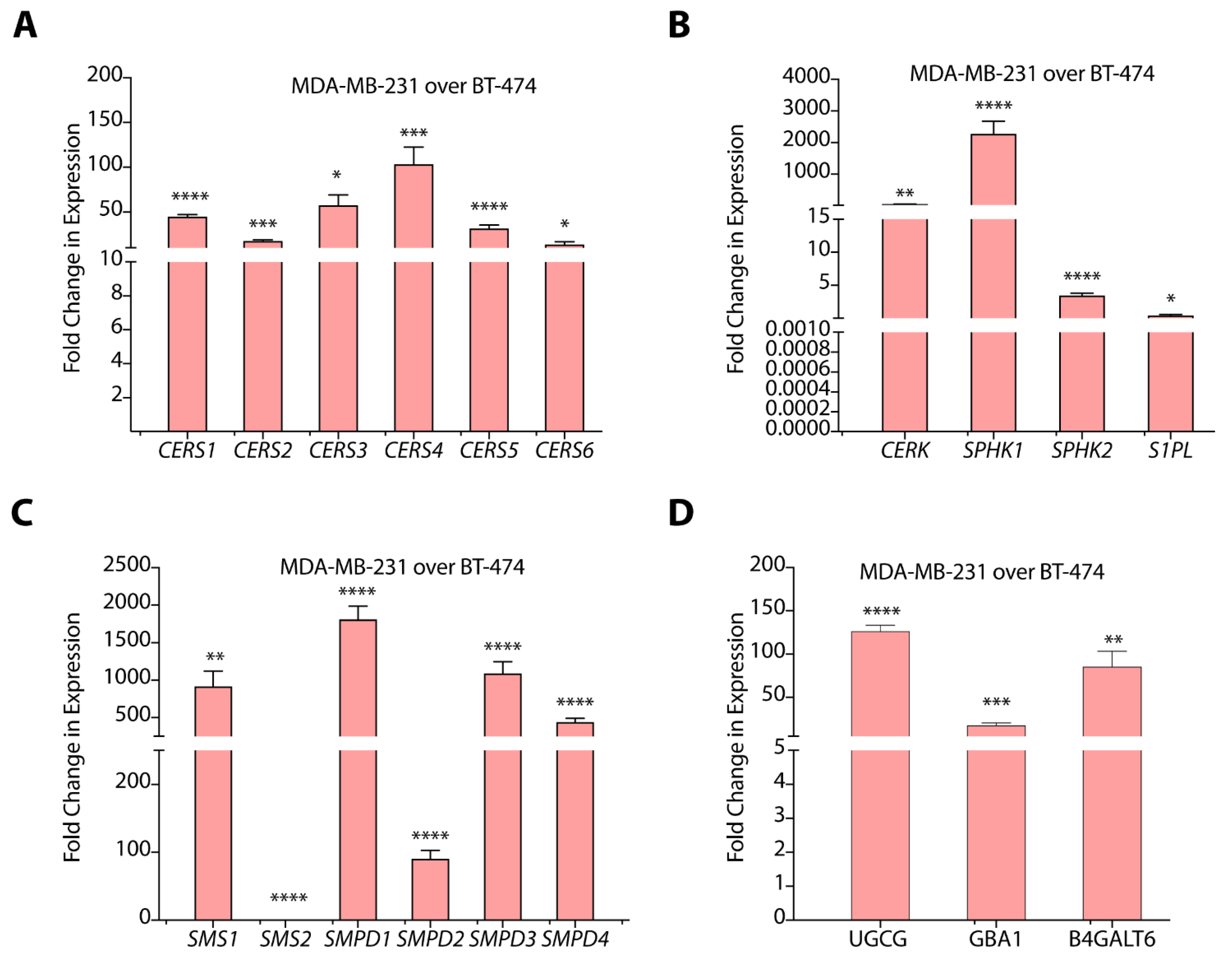
3.2. TPBC and TNBC Cells Exhibit Distinct Expression of Sphingolipid-Metabolism Genes
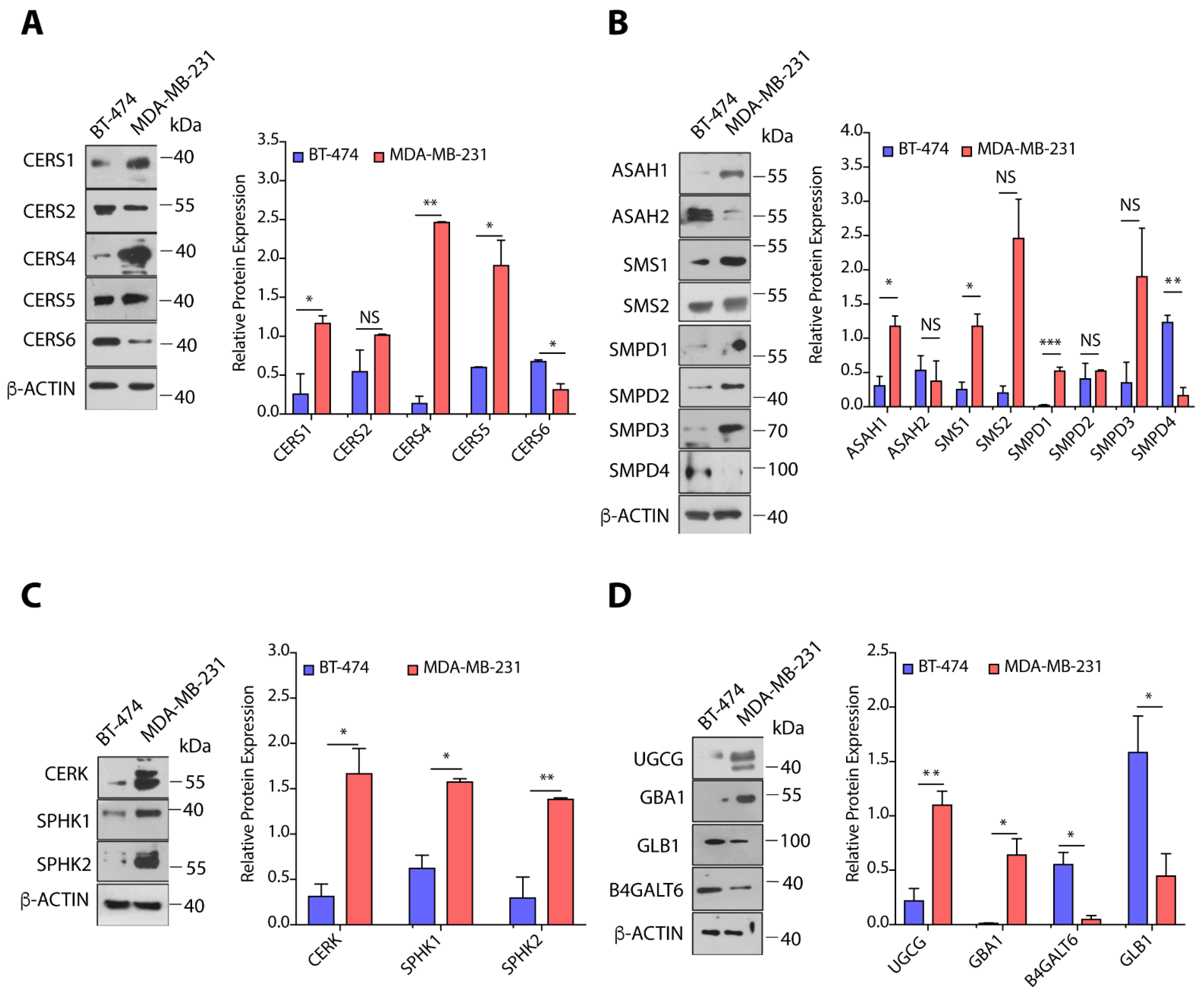
3.3. TPBC and TNBC Cells Exhibit Distinct Expression of Sphingolipid-Metabolizing Enzymes
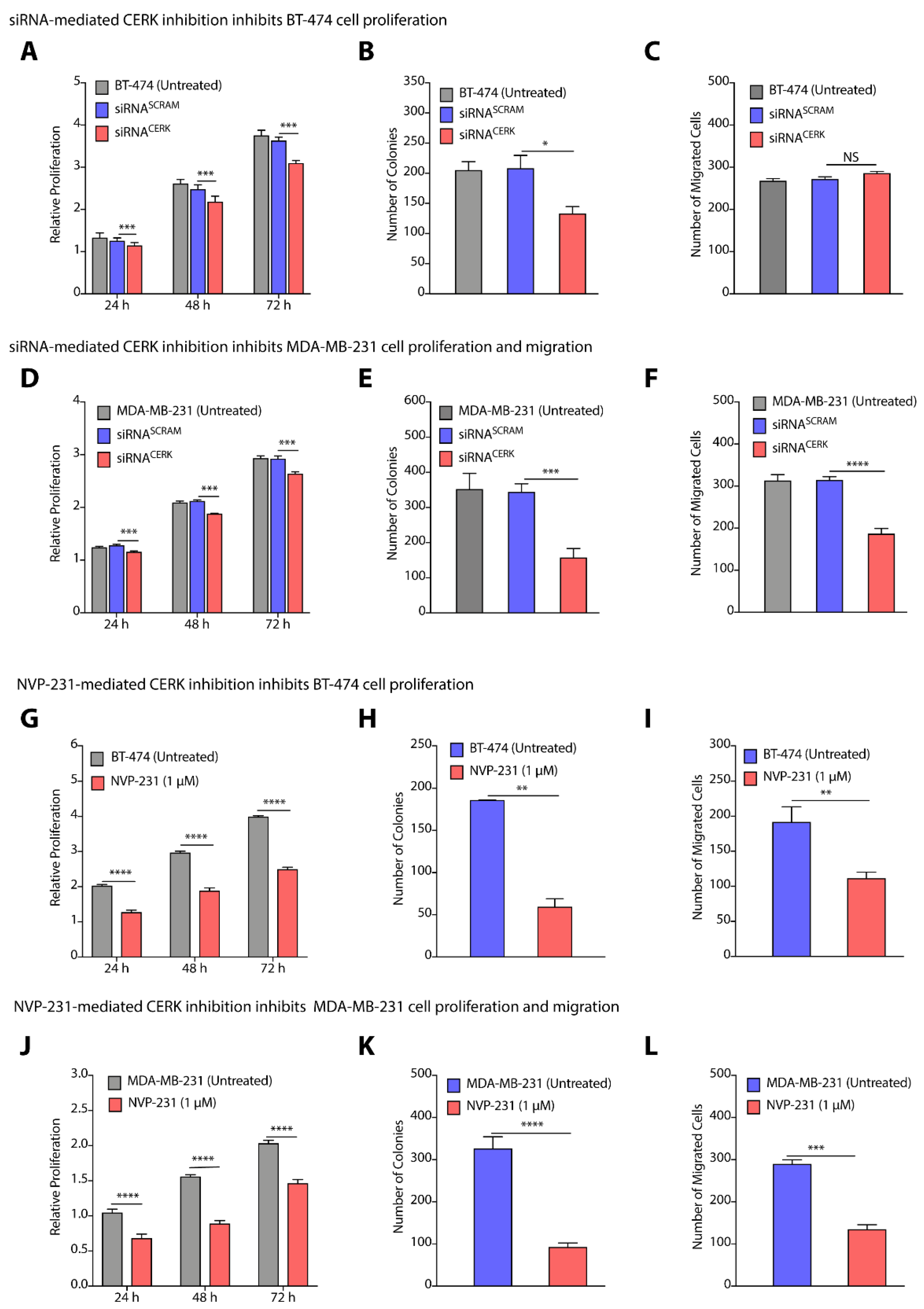
3.4. TPBC and TNBC Cell Lines Show Differential Cell Proliferation and Migration Abilities
3.5. CERK Inhibition Reduces Cell Proliferation, Colony Formation, and Migration in TPBC and TNBC Cells
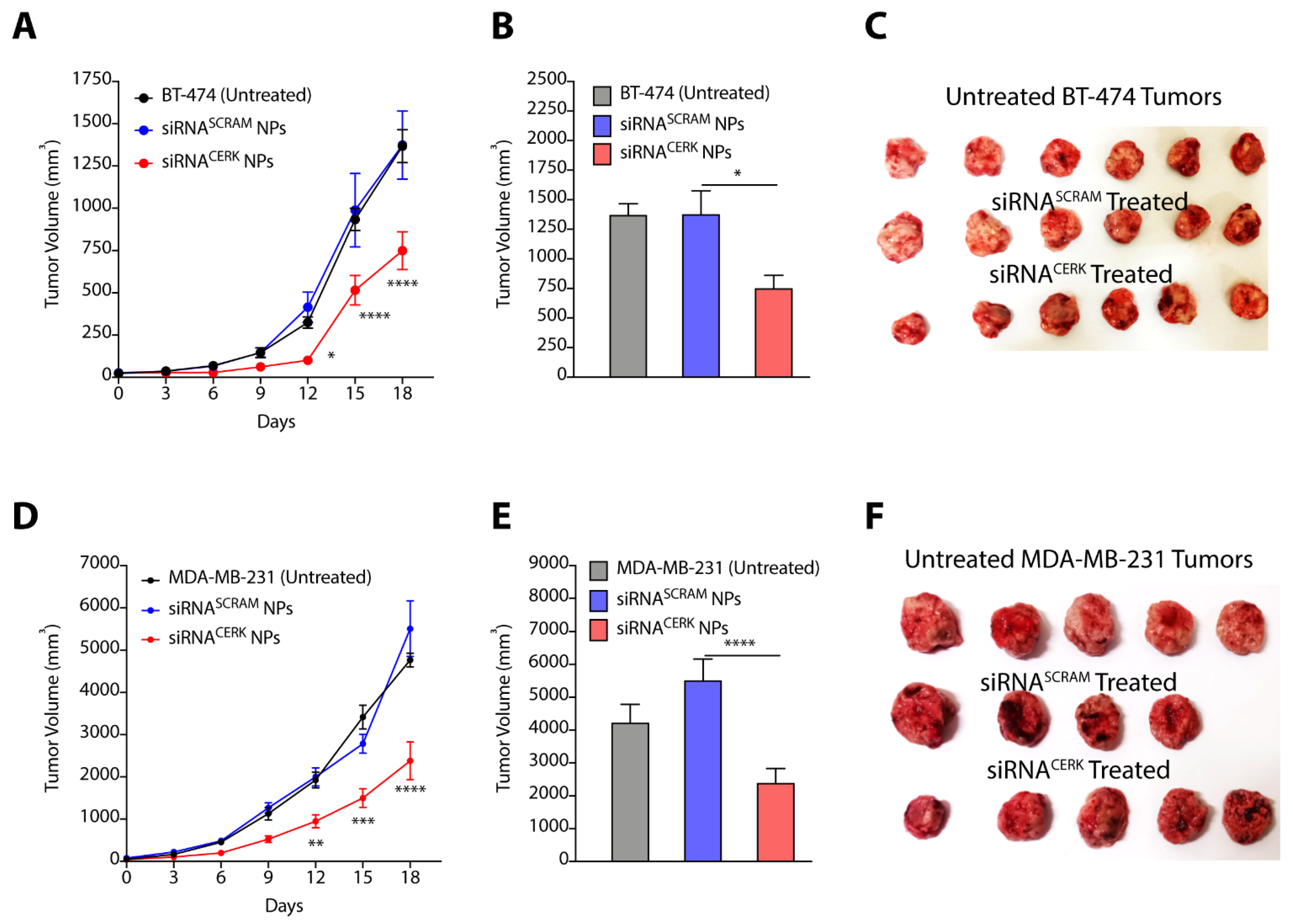
3.6. Nanoparticle-Mediated Localized Delivery of CERK siRNA Inhibits Tumor Progression
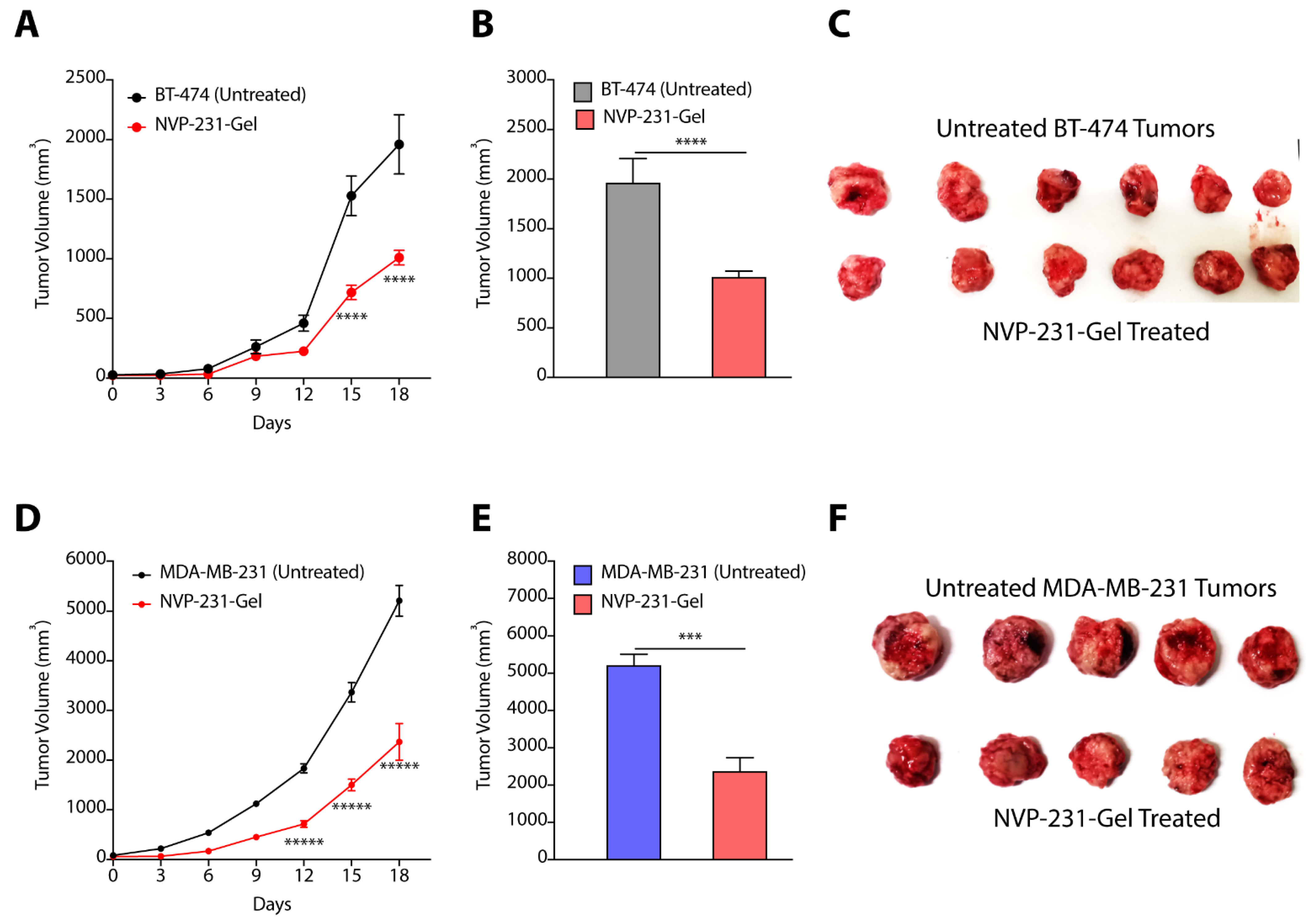
3.7. Hydrogel-Mediated Delivery of CERK Inhibitor Mitigates Tumor Progression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid Metabolic Reprogramming in Cancer Cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Xie, F.; Yang, Y.; Wang, S. Reprogramming of Fatty Acid Metabolism in Breast Cancer: A Narrative Review. Transl. Breast Cancer Res. 2021, 2, 1–9. [Google Scholar] [CrossRef]
- Ryland, L.K.; Fox, T.E.; Liu, X.; Loughran, T.P.; Kester, M. Dysregulation of Sphingolipid Metabolism in Cancer. Cancer Biol. Ther. 2011, 11, 138–149. [Google Scholar] [CrossRef]
- Ogretmen, B. Sphingolipid Metabolism in Cancer Signalling and Therapy. Nat. Rev. Cancer 2018, 18, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Principles of Bioactive Lipid Signalling: Lessons from Sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Morad, S.A.; Cabot, M.C. Ceramide-orchestrated Signalling in Cancer Cells. Nat. Rev. Cancer 2013, 13, 51–65. [Google Scholar] [CrossRef]
- Snider, J.M.; Trayssac, M.; Clarke, C.J.; Schwartz, N.; Snider, A.J.; Obeid, L.M.; Luberto, C.; Hannun, Y.A. Multiple Actions of Doxorubicin on the Sphingolipid Network Revealed by Flux Analysis. J. Lipid Res. 2019, 60, 819–831. [Google Scholar] [CrossRef]
- Patwardhan, G.A.; Liu, Y.Y. Sphingolipids and Expression Regulation of Genes in Cancer. Prog. Lipid Res. 2011, 50, 104–114. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. The Ceramide-Centric Universe of Lipid-mediated Cell Regulation: Stress Encounters of the Lipid Kind. J. Biol. Chem. 2002, 277, 25847–25850. [Google Scholar] [CrossRef]
- Del Solar, V.; Lizardo, D.Y.; Li, N.; Hurst, J.J.; Brais, C.J.; Atilla-Gokcumen, G.E. Differential Regulation of Specific Sphingolipids in Colon Cancer Cells during Staurosporine-induced Apoptosis. Chem. Biol. 2015, 22, 1662–1670. [Google Scholar] [CrossRef]
- Kolesnick, R.N.; Krönke, M. Regulation of Ceramide Production and Apoptosis. Annu. Rev. Physiol. 1998, 60, 643–665. [Google Scholar] [CrossRef]
- Janneh, A.H.; Ogretmen, B. Targeting Sphingolipid Metabolism as a Therapeutic Strategy in Cancer Treatment. Cancers 2022, 14, 2183. [Google Scholar] [CrossRef]
- Fyrst, H.; Saba, J.D. An Update on Sphingosine-1-phosphate and Other Sphingolipid Mediators. Nat. Chem. Biol. 2010, 6, 489–497. [Google Scholar] [CrossRef]
- Pyne, N.J.; Pyne, S. Sphingosine 1-phosphate and Cancer. Nat. Rev. Cancer 2010, 10, 489–503. [Google Scholar] [CrossRef]
- Payne, A.W.; Pant, D.K.; Pan, T.C.; Chodosh, L.A. Ceramide Kinase Promotes Tumor Cell Survival and Mammary Tumor Recurrence. Cancer Res. 2014, 74, 6352–6363. [Google Scholar] [CrossRef]
- Gómez-Muñoz, A. Ceramide 1-phosphate/Ceramide, a Switch between Life and Death. Biochim. Biophys. Acta Biomembr. 2006, 1758, 2049–2056. [Google Scholar] [CrossRef]
- Granado, M.H.; Gangoiti, P.; Ouro, A.; Arana, L.; González, M.; Trueba, M.; Gómez-Muñoz, A. Ceramide 1-phosphate (C1P) Promotes Cell Migration: Involvement of a Specific C1P Receptor. Cell. Signal. 2009, 21, 405–412. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Hill, R.A.; Li, Y.T. Ceramide Glycosylation Catalyzed by Glucosylceramide Synthase and Cancer Drug Resistance. Adv. Cancer Res. 2013, 117, 59–89. [Google Scholar]
- Tallima, H.; Azzazy, H.M.; El Ridi, R. Cell Surface Sphingomyelin: Key Role in Cancer Initiation, Progression, and Immune Evasion. Lipids Health Dis. 2021, 20, 1–12. [Google Scholar] [CrossRef]
- Modrak, D.E.; Gold, D.V.; Goldenberg, D.M. Sphingolipid Targets in Cancer Therapy. Mol. Cancer Ther. 2006, 5, 200–208. [Google Scholar] [CrossRef]
- Yersal, O.; Barutca, S. Biological Subtypes of Breast Cancer: Prognostic and therapeutic Implications. World J. Clin. Oncol. 2014, 5, 412. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast Cancer Intrinsic Subtype Classification, Clinical Use, and Future Trends. Am. J. Cancer Res. 2015, 5, 2929–2943. [Google Scholar] [PubMed]
- Voduc, K.D.; Cheang, M.C.; Tyldesley, S.; Gelmon, K.; Nielsen, T.O.; Kennecke, H. Breast Cancer Subtypes and the Risk of Local and Regional Relapse. Am. J. Clin. Oncol. 2010, 28, 1684–1691. [Google Scholar] [CrossRef] [PubMed]
- Vici, P.; Pizzuti, L.; Natoli, C.; Gamucci, T.; Di Lauro, L.; Barba, M.; Sergi, D.; Botti, C.; Michelotti, A.; Moscetti, L.; et al. Triple Positive Breast Cancer: A Distinct Subtype? Cancer Treat. Rev. 2015, 41, 69–76. [Google Scholar] [CrossRef]
- Wang, J.; Xu, B. Targeted Therapeutic Options and Future Perspectives for HER2-Positive Breast Cancer. Signal Transduct. Target. Ther. 2019, 4, 34. [Google Scholar] [CrossRef]
- Yin, L.; Duan, J.J.; Bian, X.W.; Yu, S.C. Triple-negative Breast Cancer Molecular Subtyping and Treatment Progress. Breast Cancer Res. 2020, 22, 1–13. [Google Scholar] [CrossRef]
- Lebert, J.M.; Lester, R.; Powell, E.; Seal, M.; McCarthy, J. Advances in the Systemic Treatment of Triple-Negative Breast Cancer. Curr. Oncol. 2018, 25, 142–150. [Google Scholar] [CrossRef]
- EP, G.A.W.; Coates, A.S.; Gelber, R.D.; Piccart-Gebhart, M.; Thürlimann, B.; Senn, H.J. Panel members. Personalizing the Treatment of Women with Early Breast Cancer: Highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer. Ann. Oncol. 2013, 24, 2206–2223. [Google Scholar]
- Purwaha, P.; Gu, F.; Piyarathna, D.W.B.; Rajendiran, T.; Ravindran, A.; Omilian, A.R.; Jiralerspong, S.; Das, G.; Morrison, C.; Ambrosone, C.; et al. Unbiased Lipidomic Profiling of Triple-Negative Breast Cancer Tissues Reveals the Association of Sphingomyelin Levels with Patient Disease-Free Survival. Metabolites 2018, 8, 41. [Google Scholar] [CrossRef]
- Cífková, E.; Holčapek, M.; Lísa, M.; Vrána, D.; Gatěk, J.; Melichar, B. Determination of Lipidomic Differences between Human Breast Cancer and Surrounding Normal Tissues using HILIC-HPLC/ESI-MS and Multivariate Data Analysis. Anal. Bioanal. Chem. 2015, 407, 991–1002. [Google Scholar] [CrossRef]
- Pani, T.; Rajput, K.; Kar, A.; Sharma, H.; Basak, R.; Medatwal, N.; Saha, S.; Dev, G.; Kumar, S.; Gupta, S.; et al. Alternative Splicing of Ceramide Synthase 2 Alters Levels of Specific Ceramides and Modulates Cancer Cell Proliferation and Migration in Luminal B Breast Cancer Subtype. Cell Death Dis. 2021, 12, 1–22. [Google Scholar] [CrossRef]
- Medatwal, N.; Dasgupta, U. Quantitation of sphingolipids in mammalian cell lines by liquid chromatography-mass spectrometry. In Analysis of Membrane Lipids; Prasad, R., Singh, A., Eds.; Springer Protocols Handbooks; Springer: New York, NY, USA, 2020; pp. 103–117. [Google Scholar]
- Yavvari, P.S.; Verma, P.; Mustfa, S.A.; Pal, S.; Kumar, S.; Awasthi, A.K.; Ahuja, V.; Srikanth, C.V.; Srivastava, A.; Bajaj, A. A Nanogel Based Oral Gene Delivery System Targeting Sumoylation Machinery to Combat Gut Inflammation. Nanoscale 2019, 11, 4970–4986. [Google Scholar] [CrossRef]
- Pal, S.; Medatwal, N.; Kumar, S.; Kar, A.; Komalla, V.; Yavvari, P.S.; Mishra, D.; Rizvi, Z.A.; Nandan, S.; Malakar, D.; et al. A Localized Chimeric Hydrogel Therapy Combats Tumor Progression Through Alteration of Sphingolipid Metabolism. ACS Cent. Sci. 2019, 5, 1648–1662. [Google Scholar] [CrossRef]
- Pastukhov, O.; Schwalm, S.; Zangemeister-Wittke, U.; Fabbro, D.; Bornancin, F.; Japtok, L.; Kleuser, B.; Pfeilschifter, J.; Huwiler, A. The Ceramide Kinase Inhibitor NVP-231 Inhibits Breast and Lung Cancer Cell Proliferation by Inducing M Phase Arrest and Subsequent Cell Death. Br. J. Pharmacol. 2014, 171, 5829–5844. [Google Scholar] [CrossRef]
- Tang, Y.; Wang, X.; Li, J.; Nie, Y.; Liao, G.; Yu, Y.; Li, C. Overcoming the Reticuloendothelial System Barrier to Drug Delivery with a “Don’t-Eat-Us” Strategy. ACS Nano 2019, 13, 13015–13026. [Google Scholar] [CrossRef]
- Pal, S.; Soni, V.; Kumar, S.; Jha, S.K.; Medatwal, N.; Rana, K.; Yadav, P.; Mehta, D.; Jain, D.; Kar, R.; et al. A Hydrogel-based Implantable Multidrug Antitubercular Formulation Outperforms Oral Delivery. Nanoscale 2021, 13, 13225–13230. [Google Scholar] [CrossRef]
- Medatwal, N.; Ansari, M.N.; Kumar, S.; Pal, S.; Jha, S.K.; Verma, P.; Rana, K.; Dasgupta, U.; Bajaj, A. Hydrogel-mediated Delivery of Celastrol and Doxorubicin Induces a Synergistic Effect on Tumor Regression via Upregulation of Ceramides. Nanoscale 2020, 12, 18463–18475. [Google Scholar] [CrossRef]
- Momin, A.A.; Park, H.; Portz, B.J.; Haynes, C.A.; Shaner, R.L.; Kelly, S.L.; Jordan, I.K.; Merrill, A.H. A Method for Visualization of “omic” Datasets for Sphingolipid Metabolism to Predict Potentially Interesting Differences. J. Lipid Res. 2011, 52, 1073–1083. [Google Scholar] [CrossRef]
- Gómez-Muñoz, A.; Kong, J.Y.; Parhar, K.; Wang, S.W.; Gangoiti, P.; González, M.; Eivemark, S.; Salh, B.; Duronio, V.; Steinbrecher, U.P. Ceramide-1-phosphate Promotes Cell Survival Through Activation of the Phosphatidylinositol 3-Kinase/Protein Kinase B Pathway. FEBS Lett. 2005, 79, 3744–3750. [Google Scholar] [CrossRef]
- Millner, A.; Running, L.; Colon-Rosa, N.; Aga, D.S.; Frasor, J.; Atilla-Gokcumen, G.E. Ceramide-1-phosphate is Involved in Therapy-Induced Senescence. ACS Chem. Biol. 2022, 17, 822–828. [Google Scholar] [CrossRef]
- Gangoiti, P.; Granado, M.H.; Arana, L.; Ouro, A.; Gomez-Muñoz, A. Activation of Protein Kinase C-A is Essential for Stimulation of Cell Proliferation by Ceramide 1-Phosphate. FEBS Lett. 2010, 584, 517–524. [Google Scholar] [CrossRef]
- Ruckhaberle, E.; Karn, T.; Rody, A.; JHanker, L.; Gatje, R.; Metzler, D.; Holtrich, U.; Kaufmann, M. Gene Expression of Ceramide Kinase, Galatosylceramide Synthase and Ganglioside GD3 Synthase is Associated with Prognosis in Breast Cancer. J. Cancer Res. Clin. Oncol. 2009, 135, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, E.W.; Green, J.J. Toward Gene Transfer Nanoparticles as Therapeutics. Adv. Healthcare Mater. 2022, 11, 2102145. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Bajaj, A. Advances in Self-assembled Injectable Hydrogels for Cancer Therapy Kumar. Biomater. Sci. 2020, 8, 2055–2073. [Google Scholar] [CrossRef] [PubMed]
- Joshi, N.; Yan, J.; Levy, S.; Bhagchandani, S.; Slaughter, K.; Sherman, N.E.; Amirault, J.; He, X.; Rui, T.S.; Valic, M.; et al. Towards an Arthritis flare-responsive Drug Delivery System. Nat. Commun. 2018, 9, 1275. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajput, K.; Ansari, M.N.; Jha, S.K.; Pani, T.; Medatwal, N.; Chattopadhyay, S.; Bajaj, A.; Dasgupta, U. Ceramide Kinase (CERK) Emerges as a Common Therapeutic Target for Triple Positive and Triple Negative Breast Cancer Cells. Cancers 2022, 14, 4496. https://doi.org/10.3390/cancers14184496
Rajput K, Ansari MN, Jha SK, Pani T, Medatwal N, Chattopadhyay S, Bajaj A, Dasgupta U. Ceramide Kinase (CERK) Emerges as a Common Therapeutic Target for Triple Positive and Triple Negative Breast Cancer Cells. Cancers. 2022; 14(18):4496. https://doi.org/10.3390/cancers14184496
Chicago/Turabian StyleRajput, Kajal, Mohammad Nafees Ansari, Somesh K. Jha, Trishna Pani, Nihal Medatwal, Somdeb Chattopadhyay, Avinash Bajaj, and Ujjaini Dasgupta. 2022. "Ceramide Kinase (CERK) Emerges as a Common Therapeutic Target for Triple Positive and Triple Negative Breast Cancer Cells" Cancers 14, no. 18: 4496. https://doi.org/10.3390/cancers14184496
APA StyleRajput, K., Ansari, M. N., Jha, S. K., Pani, T., Medatwal, N., Chattopadhyay, S., Bajaj, A., & Dasgupta, U. (2022). Ceramide Kinase (CERK) Emerges as a Common Therapeutic Target for Triple Positive and Triple Negative Breast Cancer Cells. Cancers, 14(18), 4496. https://doi.org/10.3390/cancers14184496