
Molecular Divergence upon EGFR-TKI Resistance Could Be Dependent on the Exon Location of the Original EGFR-Sensitizing Mutation

 ,
,  , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Samples
2.2. Laboratory Procedures
2.3. Statistical Analysis
3. Results
3.1. Study Cohort
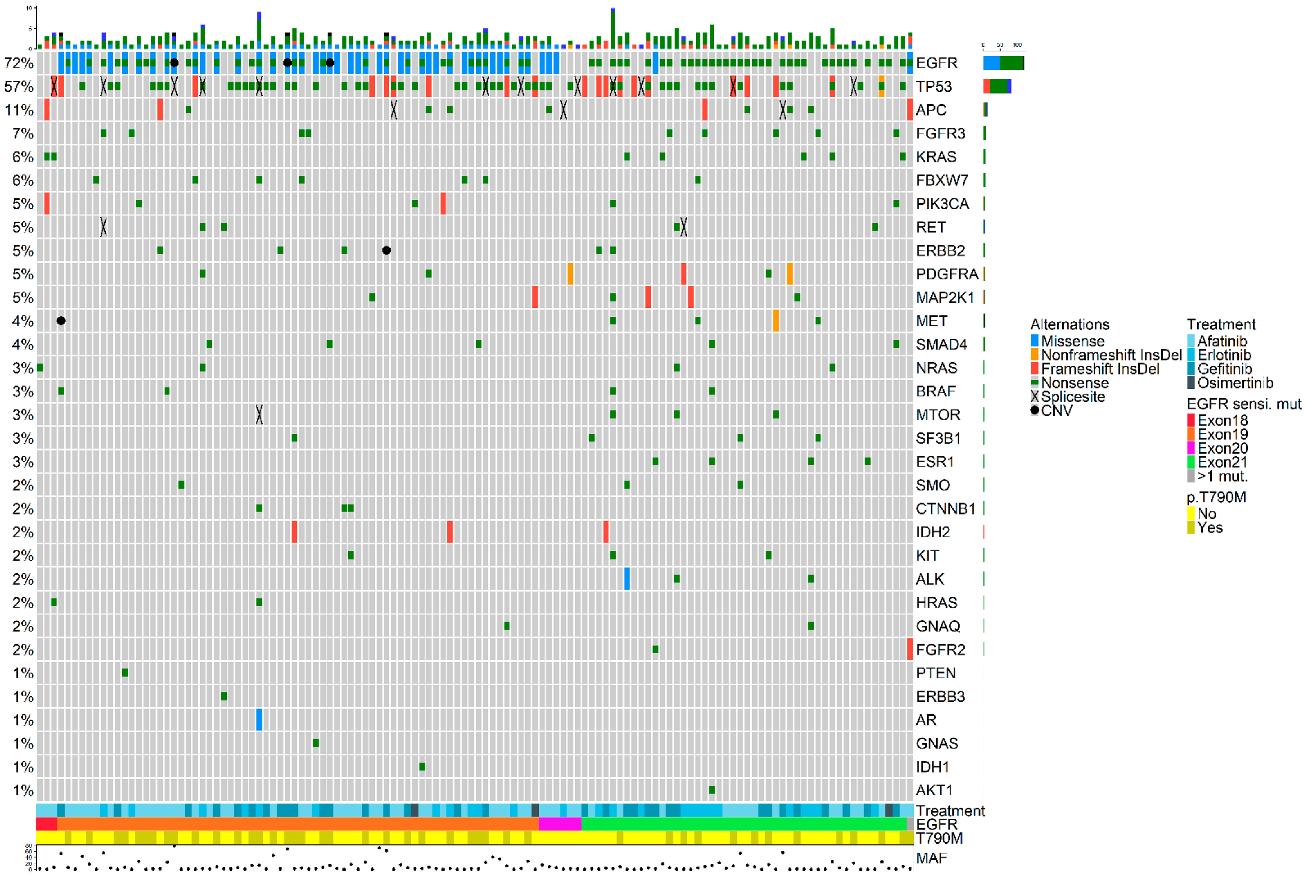
3.2. Molecular Landscape upon Disease Progression
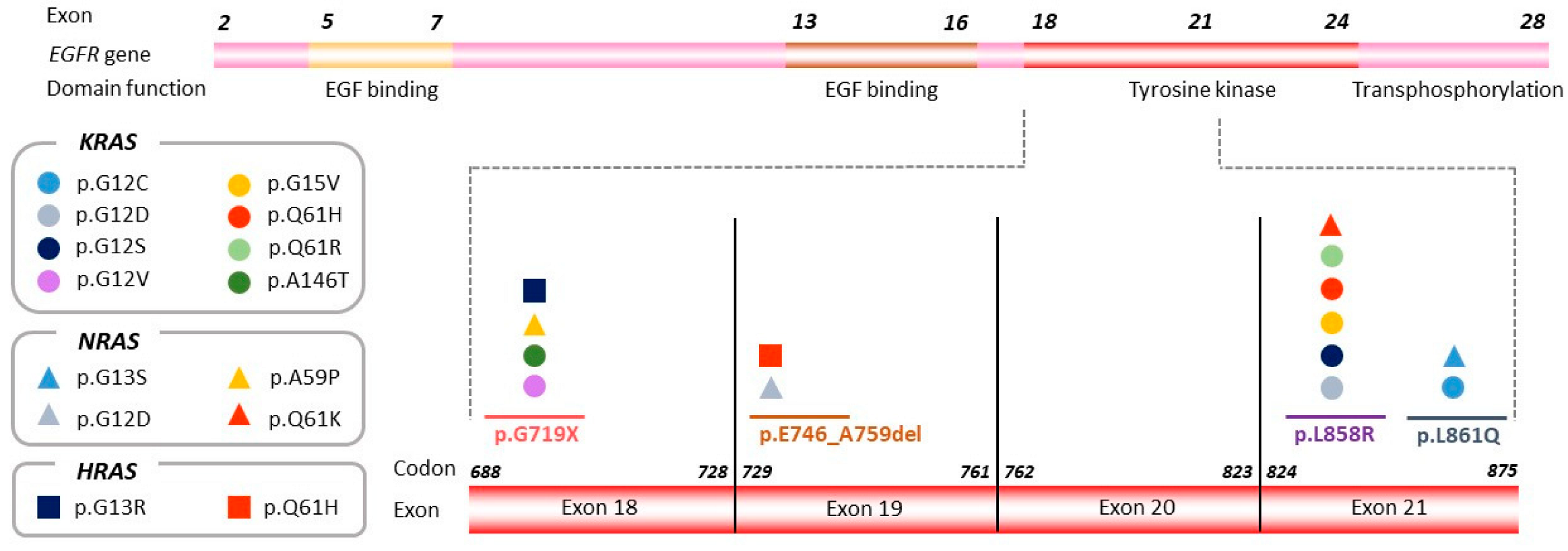
3.3. Exon Location of the Original Sensitizing Mutation Determines Distinct Molecular Profiles upon Disease Progression
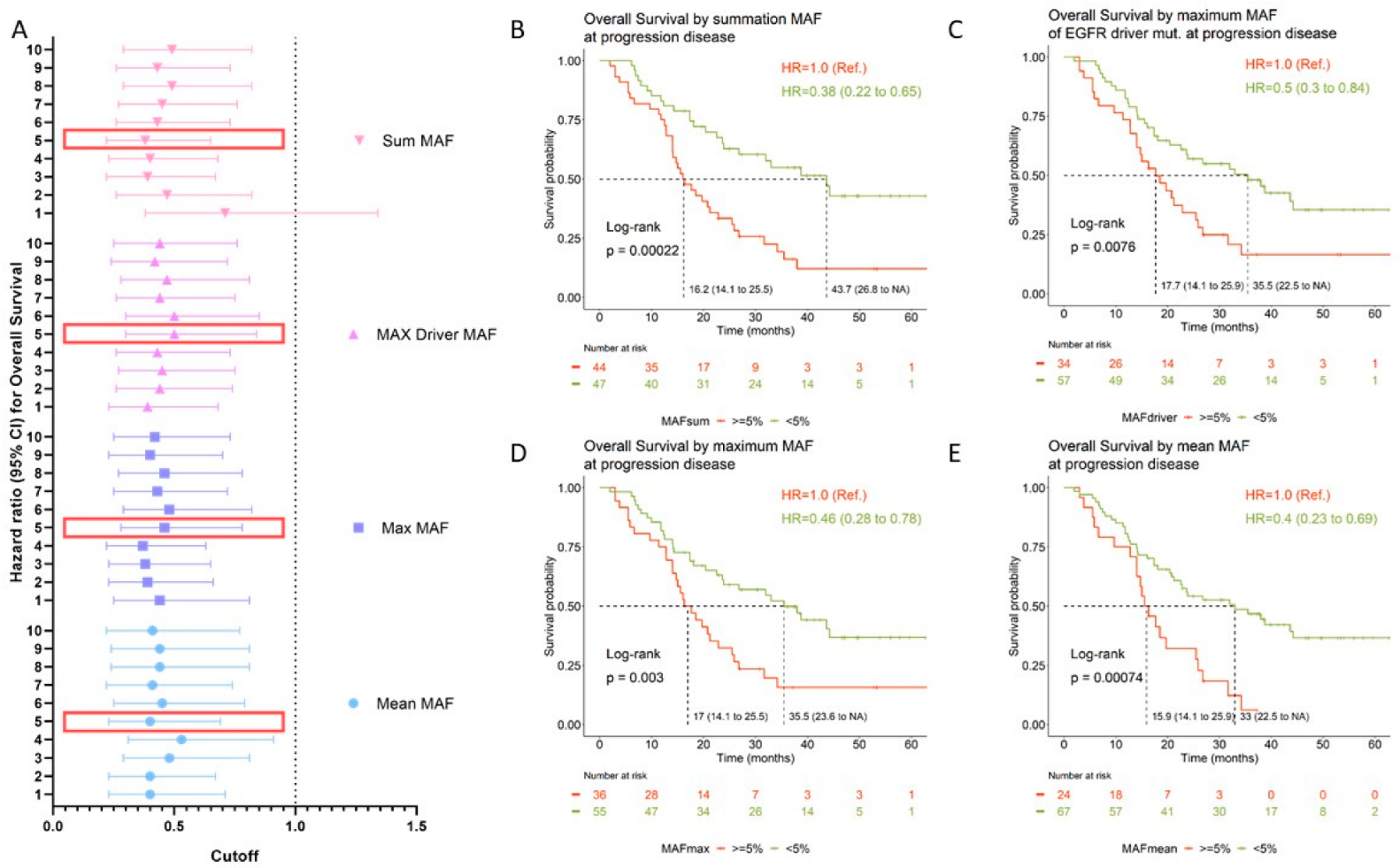
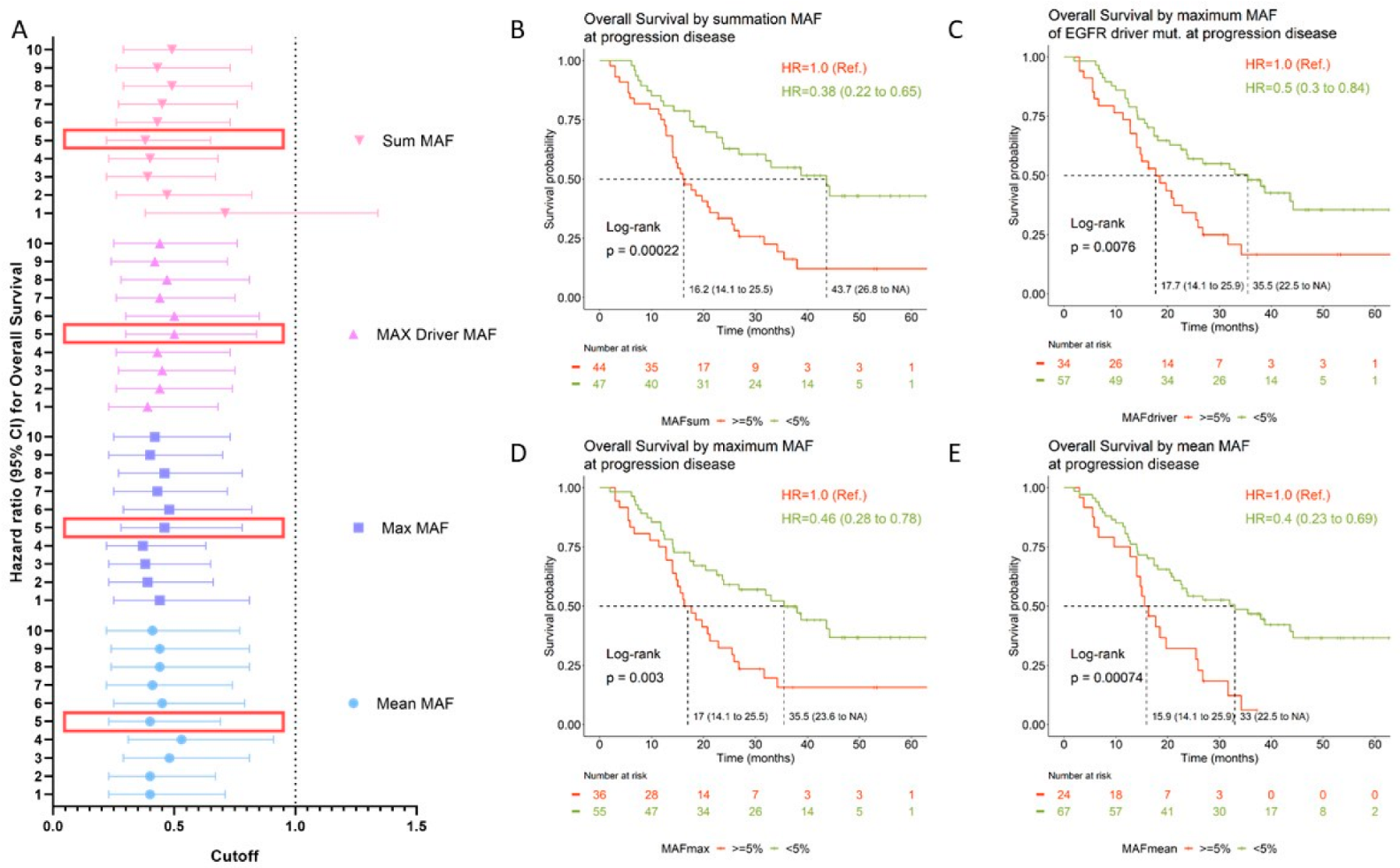
3.4. Prognostic Value of Circulating Tumor DNA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Thress, K.S.; Brant, R.; Carr, T.H.; Dearden, S.; Jenkins, S.; Brown, H.; Hammett, T.; Cantarini, M.; Barrett, J.C. EGFR Mutation Detection in CtDNA from NSCLC Patient Plasma: A Cross-Platform Comparison of Leading Technologies to Support the Clinical Development of AZD9291. Lung Cancer 2015, 90, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Aye, P.S.; McKeage, M.J.; Tin, S.T.; Khwaounjoo, P.; Elwood, J.M. Population-Based Incidence Rates and Increased Risk of EGFR Mutated Non-Small Cell Lung Cancer in Māori and Pacifica in New Zealand. PLoS ONE 2021, 16, e0251357. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.H.; Camidge, D.R.; Yang, C.T.; Zhou, J.; Guo, R.; Chiu, C.H.; Chang, G.C.; Shiah, H.S.; Chen, Y.; Wang, C.C.; et al. Safety, Efficacy, and Pharmacokinetics of Almonertinib (HS-10296) in Pretreated Patients with EGFR-Mutated Advanced NSCLC: A Multicenter, Open-Label, Phase 1 Trial. J. Thorac. Oncol. 2020, 15, 1907–1918. [Google Scholar] [CrossRef]
- Ahn, M.J.; Han, J.Y.; Lee, K.H.; Kim, S.W.; Kim, D.W.; Lee, Y.G.; Cho, E.K.; Kim, J.H.; Lee, G.W.; Lee, J.S.; et al. Lazertinib in Patients with EGFR Mutation-Positive Advanced Non-Small-Cell Lung Cancer: Results from the Dose Escalation and Dose Expansion Parts of a First-in-Human, Open-Label, Multicentre, Phase 1-2 Study. Lancet Oncol. 2019, 20, 1681–1690. [Google Scholar] [CrossRef]
- Zhou, Q.; Wu, L.; Feng, L.; An, T.; Cheng, Y.; Zhou, J.; Li, J.; Feng, J.F.; Zhang, L.; Han, B.; et al. Safety and Efficacy of Abivertinib (AC0010), a Third-Generation EGFR Tyrosine Kinase Inhibitor, in Chinese Patients with EGFR-T790M Positive Non-Small Cell Lung Cancer (NCSLC). J. Clin. Oncol. 2019, 37, 9091. [Google Scholar] [CrossRef]
- Du, X.; Yang, B.; An, Q.; Assaraf, Y.G.; Cao, X.; Xia, J. Acquired Resistance to Third-Generation EGFR-TKIs and Emerging next-Generation EGFR Inhibitors. Innovation 2021, 2, 100103. [Google Scholar] [CrossRef]
- Noronha, V.; Patil, V.M.; Joshi, A.; Menon, N.; Chougule, A.; Mahajan, A.; Janu, A.; Purandare, N.; Kumar, R.; More, S.; et al. Gefitinib Versus Gefitinib Plus Pemetrexed and Carboplatin Chemotherapy in EGFR-Mutated Lung Cancer. J. Clin. Oncol. 2020, 38, 124–136. [Google Scholar] [CrossRef]
- Nakagawa, K.; Garon, E.B.; Seto, T.; Nishio, M.; Ponce Aix, S.; Paz-Ares, L.; Chiu, C.H.; Park, K.; Novello, S.; Nadal, E.; et al. Ramucirumab plus Erlotinib in Patients with Untreated, EGFR-Mutated, Advanced Non-Small-Cell Lung Cancer (RELAY): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2019, 20, 1655–1669. [Google Scholar] [CrossRef] [Green Version]
- Bauml, J.; Cho, B.C.; Park, K.; Lee, K.H.; CHO, E.K.; Kim, D.-W.; Kim, S.-W.; Haura, E.B.; Sabari, J.K.; Sanborn, R.E.; et al. Amivantamab in Combination with Lazertinib for the Treatment of Osimertinib-Relapsed, Chemotherapy-Naïve EGFR Mutant (EGFRm) Non-Small Cell Lung Cancer (NSCLC) and Potential Biomarkers for Response. J. Clin. Oncol. 2021, 39, 9006. [Google Scholar] [CrossRef]
- Provencio, M.; Serna-Blasco, R.; Franco, F.; Calvo, V.; Royuela, A.; Auglytė, M.; Sánchez-Hernández, A.; de Julián Campayo, M.; García-Girón, C.; Dómine, M.; et al. Analysis of Circulating Tumour DNA to Identify Patients with Epidermal Growth Factor Receptor–Positive Non-Small Cell Lung Cancer Who Might Benefit from Sequential Tyrosine Kinase Inhibitor Treatment. Eur. J. Cancer 2021, 149, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Nahar, R.; Zhai, W.; Zhang, T.; Takano, A.; Khng, A.J.; Lee, Y.Y.; Liu, X.; Lim, C.H.; Koh, T.P.T.; Aung, Z.W.; et al. Elucidating the Genomic Architecture of Asian EGFR-Mutant Lung Adenocarcinoma through Multi-Region Exome Sequencing. Nat. Commun. 2018, 9, 216. [Google Scholar] [CrossRef]
- Hong, S.; Gao, F.; Fu, S.; Wang, Y.; Fang, W.; Huang, Y.; Zhang, L. Concomitant Genetic Alterations with Response to Treatment and Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Patients with EGFR-Mutant Advanced Non-Small Cell Lung Cancer. JAMA Oncol. 2018, 4, 739–742. [Google Scholar] [CrossRef] [PubMed]
- Blakely, C.M.; Watkins, T.B.K.; Wu, W.; Gini, B.; Chabon, J.J.; McCoach, C.E.; McGranahan, N.; Wilson, G.A.; Birkbak, N.J.; Olivas, V.R.; et al. Evolution and Clinical Impact of Co-Occurring Genetic Alterations in Advanced-Stage EGFR-Mutant Lung Cancers. Nat. Genet. 2017, 49, 1693–1704. [Google Scholar] [CrossRef]
- Canale, M.; Petracci, E.; Delmonte, A.; Chiadini, E.; Dazzi, C.; Papi, M.; Capelli, L.; Casanova, C.; De Luigi, N.; Mariotti, M.; et al. Impact of TP53 Mutations on Outcome in EGFR-Mutated Patients Treated with First-Line Tyrosine Kinase Inhibitors. Clin. Cancer Res. 2017, 23, 2195–2202. [Google Scholar] [CrossRef]
- Offin, M.; Chan, J.M.; Tenet, M.; Rizvi, H.A.; Shen, R.; Riely, G.J.; Rekhtman, N.; Daneshbod, Y.; Quintanal-Villalonga, A.; Penson, A.; et al. Concurrent RB1 and TP53 Alterations Define a Subset of EGFR-Mutant Lung Cancers at Risk for Histologic Transformation and Inferior Clinical Outcomes. J. Thorac. Oncol. 2019, 14, 1784–1793. [Google Scholar] [CrossRef]
- Collisson, E.A.; Campbell, J.D.; Brooks, A.N.; Berger, A.H.; Lee, W.; Chmielecki, J.; Beer, D.G.; Cope, L.; Creighton, C.J.; Danilova, L.; et al. Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar]
- Chen, Z.; Fillmore, C.M.; Hammerman, P.S.; Kim, C.F.; Wong, K.K. Non-Small-Cell Lung Cancers: A Heterogeneous Set of Diseases. Nat. Rev. Cancer 2014, 14, 535–546. [Google Scholar] [CrossRef]
- Diaz, L.A.; Bardelli, A. Liquid Biopsies: Genotyping Circulating Tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ClinGen TP53 Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines Version 1 TP53 Rule Specifications for the ACMG/AMP Variant Curation Guidelines Gene Disease (MONDO ID) Clinically Significant Transcript TP53 Li-Fraumeni Syndrome (0007903) Pathogenic Criteria Criteria Original Criteria Description Specification(S). Available online: https://www.clinicalgenome.org/affiliation/50013 (accessed on 15 August 2022).
- Provencio, M.; Torrente, M.; Calvo, V.; Pérez-Callejo, D.; Gutiérrez, L.; Franco, F.; Pérez-Barrios, C.; Barquín, M.; Royuela, A.; García-García, F.; et al. Prognostic Value of Quantitative CtDNA Levels in Non Small Cell Lung Cancer Patients. Oncotarget 2018, 9, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; Laframboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL Kinase Causes Resistance to EGFR-Targeted Therapy in Lung Cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Bivona, T.G.; Hieronymus, H.; Parker, J.; Chang, K.; Taron, M.; Rosell, R.; Moonsamy, P.; Dahlman, K.; Miller, V.A.; Costa, C.; et al. FAS and NF-ΚB Signalling Modulate Dependence of Lung Cancers on Mutant EGFR. Nature 2011, 471, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Ono, M.; Kuwano, M. Molecular Mechanisms of Epidermal Growth Factor Receptor (EGFR) Activation and Response to Gefitinib and Other EGFR-Targeting Drugs. Clin. Cancer Res. 2006, 12, 7242–7251. [Google Scholar] [CrossRef]
- Jackman, D.M.; Yeap, B.Y.; Sequist, L.V.; Lindeman, N.; Holmes, A.J.; Joshi, V.A.; Bell, D.W.; Huberman, M.S.; Halmos, B.; Rabin, M.S.; et al. Exon 19 Deletion Mutations of Epidermal Growth Factor Receptor Are Associated with Prolonged Survival in Non-Small Cell Lung Cancer Patients Treated with Gefitinib or Erlotinib. Clin. Cancer Res. 2006, 12, 3908–3914. [Google Scholar] [CrossRef]
- Yasuda, H.; Kobayashi, S.; Costa, D.B. EGFR Exon 20 Insertion Mutations in Non-Small-Cell Lung Cancer: Preclinical Data and Clinical Implications. Lancet Oncol. 2012, 13, e23–e31. [Google Scholar] [CrossRef]
- Piper-Vallillo, A.J.; Sequist, L.V.; Piotrowska, Z. Emerging Treatment Paradigms for EGFR-Mutant Lung Cancers Progressing on Osimertinib: A Review. J. Clin. Oncol. 2020, 38, 2926–2936. [Google Scholar] [CrossRef]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR Mutation and Resistance of Non-Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef]
- Arcila, M.E.; Oxnard, G.R.; Nafa, K.; Riely, G.J.; Solomon, S.B.; Zakowski, M.F.; Kris, M.G.; Pao, W.; Miller, V.A.; Ladanyi, M. Rebiopsy of Lung Cancer Patients with Acquired Resistance to EGFR Inhibitors and Enhanced Detection of the T790M Mutation Using a Locked Nucleic Acid-Based Assay. Clin. Cancer Res. 2011, 17, 1169–1180. [Google Scholar] [CrossRef]
- Chen, H.J.; Mok, T.S.; Chen, Z.H.; Guo, A.L.; Zhang, X.C.; Su, J.; Wu, Y.L. Clinicopathologic and Molecular Features of Epidermal Growth Factor Receptor T790M Mutation and C-MET Amplification in Tyrosine Kinase Inhibitor-Resistant Chinese Non-Small Cell Lung Cancer. Pathol. Oncol. Res. 2009, 15, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, N.; Azuma, K.; Sakai, K.; Hattori, S.; Kawahara, A.; Ishii, H.; Tokito, T.; Kinoshita, T.; Yamada, K.; Nishio, K.; et al. Association of EGFR Exon 19 Deletion and EGFR-TKI Treatment Duration with Frequency of T790M Mutation in EGFR-Mutant Lung Cancer Patients. Sci. Rep. 2016, 6, 36458. [Google Scholar] [CrossRef] [PubMed]
- Politi, K.; Fan, P.D.; Shen, R.; Zakowski, M.; Varmus, H. Erlotinib Resistance in Mouse Models of Epidermal Growth Factor Receptor-Induced Lung Adenocarcinoma. Dis. Model. Mech. 2010, 3, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Nanjo, S.; Arai, S.; Wang, W.; Takeuchi, S.; Yamada, T.; Hata, A.; Katakami, N.; Okada, Y.; Yano, S. MET Copy Number Gain Is Associated with Gefitinib Resistance in Leptomeningeal Carcinomatosis of EGFR-Mutant Lung Cancer. Mol. Cancer Ther. 2017, 16, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Marti, A.; Felip, E.; Matito, J.; Mereu, E.; Navarro, A.; Cedrés, S.; Pardo, N.; de Castro, A.M.; Remon, J.; Miquel, J.M.; et al. Dual MET and ERBB Inhibition Overcomes Intratumor Plasticity in Osimertinib-Resistant-Advanced Non-Small-Cell Lung Cancer (NSCLC). Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 2451–2457. [Google Scholar] [CrossRef]
- Deng, L.; Kiedrowski, L.A.; Ravera, E.; Cheng, H.; Halmos, B. Response to Dual Crizotinib and Osimertinib Treatment in a Lung Cancer Patient with MET Amplification Detected by Liquid Biopsy Who Acquired Secondary Resistance to EGFR Tyrosine Kinase Inhibition. J. Thorac. Oncol. 2018, 13, e169–e172. [Google Scholar] [CrossRef]
- Mazières, J.; Peters, S.; Lepage, B.; Cortot, A.B.; Barlesi, F.; Beau-Faller, M.; Besse, B.; Blons, H.; Mansuet-Lupo, A.; Urban, T.; et al. Lung Cancer That Harbors an HER2 Mutation: Epidemiologic Characteristics and Therapeutic Perspectives. J. Clin. Oncol. 2013, 31, 1997–2003. [Google Scholar] [CrossRef]
- Planchard, D.; Loriot, Y.; André, F.; Gobert, A.; Auger, N.; Lacroix, L.; Soria, J.C. EGFR-Independent Mechanisms of Acquired Resistance to AZD9291 in EGFR T790M-Positive NSCLC Patients. Ann. Oncol. 2015, 26, 2073–2078. [Google Scholar] [CrossRef]
- Sun, Y.; Meyers, B.A.; Czako, B.; Leonard, P.; Mseeh, F.; Harris, A.L.; Wu, Q.; Johnson, S.; Parker, C.A.; Cross, J.B.; et al. Allosteric SHP2 Inhibitor, IACS-13909, Overcomes EGFR-Dependent and EGFR-Independent Resistance Mechanisms toward Osimertinib. Cancer Res. 2020, 80, 4840–4853. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Hu, Y.; Mileham, K.F.; Husain, H.; Costa, D.B.; Tracy, P.; Feeney, N.; Sholl, L.M.; Dahlberg, S.E.; Redig, A.J.; et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients With EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol. 2018, 4, 1527–1534. [Google Scholar] [CrossRef]
- Ho, C.C.; Liao, W.Y.; Lin, C.A.; Shih, J.Y.; Yu, C.J.; Chih-Hsin Yang, J. Acquired BRAF V600E Mutation as Resistant Mechanism after Treatment with Osimertinib. J. Thorac. Oncol. 2017, 12, 567–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linardou, H.; Dahabreh, I.J.; Kanaloupiti, D.; Siannis, F.; Bafaloukos, D.; Kosmidis, P.; Papadimitriou, C.A.; Murray, S. Assessment of Somatic K-RAS Mutations as a Mechanism Associated with Resistance to EGFR-Targeted Agents: A Systematic Review and Meta-Analysis of Studies in Advanced Non-Small-Cell Lung Cancer and Metastatic Colorectal Cancer. Lancet Oncol. 2008, 9, 962–972. [Google Scholar] [CrossRef]
- Serna-Blasco, R.; Sánchez-Herrero, E.; Sanz-Moreno, S.; Rodriguez-Festa, A.; García-Veros, E.; Casarrubios, M.; Sierra-Rodero, B.; Laza-Briviesca, R.; Cruz-Bermúdez, A.; Mielgo-Rubio, X.; et al. KRAS p.G12C Mutation Occurs in 1% of EGFR-Mutated Advanced Non-Small-Cell Lung Cancer Patients Progressing on a First-Line Treatment with a Tyrosine Kinase Inhibitor. ESMO Open 2021, 6, 100279. [Google Scholar] [CrossRef] [PubMed]
- Provencio, M.; Torrente, M.; Calvo, V.; Gutiérrez, L.; Pérez-Callejo, D.; Pérez-Barrios, C.; Barquín, M.; Royuela, A.; Rodriguez-Alfonso, B.; Sotelo, M.; et al. Dynamic Circulating Tumor DNA Quantificaton for the Individualization of Non-Small-Cell Lung Cancer Patients Treatment. Oncotarget 2017, 8, 60291–60298. [Google Scholar] [CrossRef] [PubMed]
- Iwama, E.; Sakai, K.; Azuma, K.; Harada, T.; Harada, D.; Nosaki, K.; Hotta, K.; Ohyanagi, F.; Kurata, T.; Fukuhara, T.; et al. Monitoring of Somatic Mutations in Circulating Cell-Free DNA by Digital PCR and next-Generation Sequencing during Afatinib Treatment in Patients with Lung Adenocarcinoma Positive for EGFR Activating Mutations. Ann. Oncol. 2017, 28, 136–141. [Google Scholar] [CrossRef]
- Yang, M.; Forbes, M.E.; Bitting, R.L.; O’Neill, S.S.; Chou, P.C.; Topaloglu, U.; Miller, L.D.; Hawkins, G.A.; Grant, S.C.; DeYoung, B.R.; et al. Incorporating Blood-Based Liquid Biopsy Information into Cancer Staging: Time for a TNMB System? Ann. Oncol. 2018, 29, 311–323. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinicopathologic Characteristics | N = 124 |
|---|---|
| Age, mean (SD), years | 65.8 (11.1) |
| Sex, No. (%) with data | |
| Female | 77 (62.1) |
| Male | 47 (37.9) |
| Smoking, No. (%) with data | |
| Never-smokers | 71 (57.3) |
| Active smokers | 9 (7.2) |
| Former smokers | 44 (35.5) |
| ECOG-PS a, No. (%) with data | |
| 0 | 35 (28.2) |
| 1 | 37 (29.8) |
| 2 | 5 (4.0) |
| Stage, No. (%) with data | |
| IVA | 60 (48.4) |
| IVB | 64 (51.6) |
| Histology, No. (%) with data | |
| Adenocarcinoma | 116 (93.6) |
| Adenosquamous | 2 (1.6) |
| Large cell | 4 (3.2) |
| Squamous | 2 (1.6) |
| Metastases at IV stage diagnosis b, No. (%) with data | |
| Local | 88 (71) |
| Bone | 61 (49.2) |
| CNS | 18 (14.5) |
| Liver | 17 (13.7) |
| Progression site c, No. (%) with data | |
| Bone | 21 (16.9) |
| CNS | 13 (10.5) |
| Liver | 13 (10.5) |
| EGFR mutation, No. (%) with data | |
| Del19 | 68 (54.8) |
| G719X | 3 (2.4) |
| Ins20 | 6 (4.8) |
| L858R | 43 (34.7) |
| L861Q | 3 (2.4) |
| >1 mut. | 1 (0.8) |
| Treatment, No. (%) with data | |
| Afatinib | 62 (50) |
| Erlotinib | 26 (21) |
| Gefitinib | 33 (26.6) |
| Osimertinib | 3 (2.4) |
| Second line treatment d, No. (%) with data | |
| Osimertinib | 63 (50.8) |
| Others | 25 (20.2) |
| None | 8 (6.4) |
| Exitus e | |
| Yes | 59 (47.6) |
| No | 32 (25.6) |
| Sample | Gene | Coding Transcript Change | Protein Change | Functional Classification | PFS b | OS b | B | ≈3 | ≈6 | P |
|---|---|---|---|---|---|---|---|---|---|---|
| 400003 | KRAS | c.35G>T | p.G12V | Missense | 5.8 | NA | - | - | - | ● |
| 1000040 | KRAS | c.182A>G | p.Q61R | Missense | 9.7 | NA | - | - | - | ● |
| 8500002 | KRAS | c.34G>A | p.G12S | Missense | 12.3 | NA | - | - | - | ● |
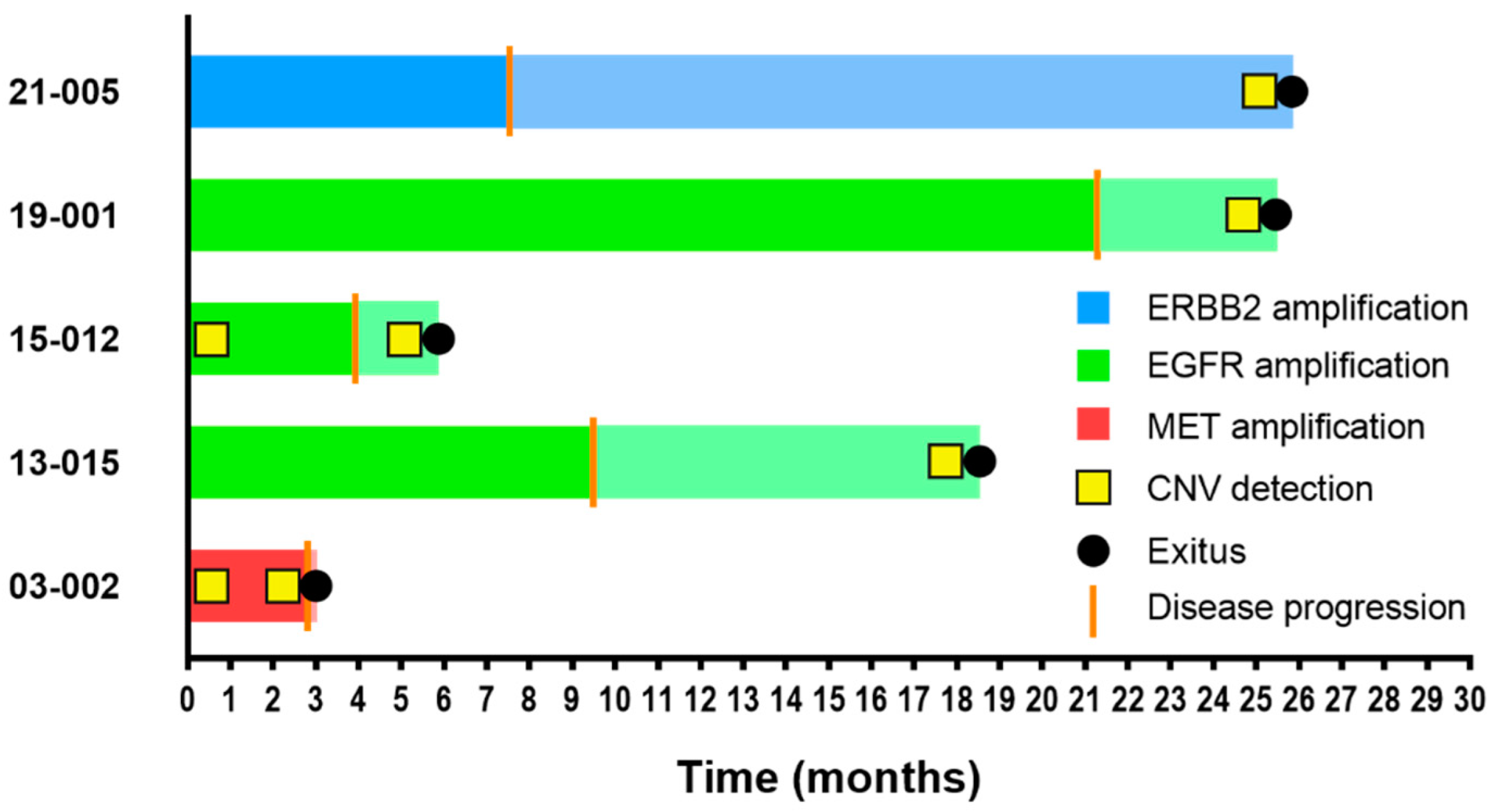
| 03-002 a | MET | CNV | CNV | CNV | 2.8 | 3 | ● | ● | ||
| 11-006 | KRAS | c.34G>T | p.G12C | Missense | 11.7 | 15.5 | ● | ● | ● | ● |
| 11-008 | PIK3CA | c.1633G>A | p.E545K | Missense | 7.7 | 15 | ● | ● | - | ● |
| 13-014 | BRAF | c.1799T>A | p.V600E | Missense | 15.8 | 34.2 | ● | ● | ● | ● |
| 13-015 a | EGFR | CNV | CNV | CNV | 9.5 | 18.5 | ● | ● | ||
| 15-012 a | EGFR | CNV | CNV | CNV | 3.9 | 5.9 | ● | ● | ||
| 19-001 a | EGFR | CNV | CNV | CNV | 21.3 | 25.5 | ● | ● | ||
| 21-005 a | ERBB2 | CNV | CNV | CNV | 7.5 | 25.9 | ● | ● | ||
| 26-004 | KRAS | c.436G>A | p.A146T | Missense | 2.8 | 14.3 | ● | - | - | ● |
| 33-001 | KRAS | c.44G>T | p.G15V | Missense | 11.9 | 30.4 | ● | ● | ● | ● |
| 36-005 | KRAS | c.183A>C | p.Q61H | Missense | 7.6 | 9.7 | ● | ● | ● | ● |
| 36-005 | KRAS | c.35G>A | p.G12D | Missense | 7.6 | 9.7 | ● | ● | ● | ● |
| 400003 | 1000040 | 8500002 | 11-006 | 26-004 | 33-001 | 36-005 | |
|---|---|---|---|---|---|---|---|
| KRAS mutation by NGS (coding/protein) | c.35G>T/p.G12V | c.182A>G/p.Q61R | c.34G>A/p.G12S | c.34G>T/p.G12C | c.436G>A/p.A146T | c.44G>T/p.G15V | c.183A>C/p.Q61H c.35G>A/p.G12D |
| MAF KRAS mutation (%) | 4.15 | 0.18 | 0.62 | 18.11 | 0.19 | 0.57 | 3.67/0.64 |
| EGFR mutation by NGS (coding/protein) | Not detected | c.2573T>G/p.L858R | c.2573T>G/p.L858R | c.2582T>A/p.L861Q | Not detected | Not detected | c.2573T>G/p.L858R |
| MAF EGFR mutation (%) | - | 0.30 | 9.77 | 2.15 | - | - | 25.63 |
| p.T790M by NGS | Not detected | Not detected | Detected | Not detected | Not detected | Not detected | Not detected |
| EGFR mutation in tumor at stage IV diagnosis | p.G719X | p.L858R | p.L858R | p.L861Q | p.G719X | p.L858R | p.L858R |
| Sex | Male | Female | Male | Female | Female | Female | Female |
| Age (years) | 74 | 76 | 74 | 68 | 58 | 79 | 52 |
| Smoking | Former smoker | Former smoker | Former smoker | Smoker | Never-smoker | Never-smoker | Never-smoker |
| Histology | Adenocar. | Adenocar. | Adenocar. | Adenocar. | Adenocar. | Adenocar. | Adenocar. |
| ECOG-PS | NA | NA | NA | 0 | 1 | 1 | 1 |
| Metastasis location at stage IV diagnosis | Thoracic and bone | Bone | Thoracic | Thoracic | Bone and CNS | Thoracic | CNS |
| Stage | IVB | IVB | IVA | IVA | IVB | IVA | IVB |
| First-line TKI | Afatinib | Gefitinib | Afatinib | Afatinib | Afatinib | Erlotinib | Afatinib |
| PFS (months) | 5.8 | 9.7 | 12.3 | 11.7 | 2.8 | 11.9 | 7.6 |
| Progression site | NA | NA | NA | CNS | Liver | NA | NA |
| Second-line treatment | None | Gefitinib | Osimertinib | None | None | Chemoth. | Osimertinib |
| Exitus | NA | NA | NA | Yes | Yes | No | Yes |
| OS (months) | NA | NA | NA | 15.5 | 14.3 | 30.4 | 9.7 |
| MAF Cutoff | Mean MAF a | Max MAF b | Driver MAF c | Sum MAF d | ||||
|---|---|---|---|---|---|---|---|---|
| HR (95%CI) | p-Value | HR (95%CI) | p-Value | HR (95%CI) | p-Value | HR (95%CI) | p-Value | |
| 1% | 0.40 (0.23–0.71) | 0.002 | 0.44 (0.25–0.81) | 0.008 | 0.39 (0.23–0.68) | 0.001 | 0.71 (0.38–1.34) | 0.295 |
| 2% | 0.40 (0.23–0.67) | 0.001 | 0.39 (0.23–0.66) | 0.001 | 0.44 (0.26–0.74) | 0.002 | 0.47 (0.26–0.82) | 0.009 |
| 3% | 0.48 (0.29–0.81) | 0.006 | 0.38 (0.23–0.65) | <0.001 | 0.45 (0.27–0.75) | 0.003 | 0.39 (0.22–0.67) | 0.001 |
| 4% | 0.53 (0.31–0.91) | 0.022 | 0.37 (0.22–0.63) | <0.001 | 0.43 (0.26–0.73) | 0.002 | 0.40 (0.23–0.68) | 0.001 |
| 5% | 0.40 (0.23–0.69) | 0.001 | 0.46 (0.28–0.78) | 0.004 | 0.50 (0.30–0.84) | 0.009 | 0.38 (0.22–0.65) | <0.001 |
| 6% | 0.45 (0.25–0.79) | 0.006 | 0.48 (0.29–0.82) | 0.007 | 0.50 (0.30–0.85) | 0.010 | 0.43 (0.26–0.73) | 0.002 |
| 7% | 0.41 (0.22–0.74) | 0.003 | 0.43 (0.25–0.72) | 0.002 | 0.44 (0.26–0.75) | 0.003 | 0.45 (0.27–0.76) | 0.003 |
| 8% | 0.44 (0.24–0.81) | 0.008 | 0.46 (0.27–0.78) | 0.004 | 0.47 (0.28–0.81) | 0.007 | 0.49 (0.29–0.82) | 0.006 |
| 9% | 0.44 (0.24–0.81) | 0.008 | 0.40 (0.23–0.70) | 0.001 | 0.42 (0.24–0.72) | 0.002 | 0.43 (0.26–0.73) | 0.002 |
| 10% | 0.41 (0.22–0.77) | 0.005 | 0.42 (0.25–0.73) | 0.002 | 0.44 (0.25–0.76) | 0.003 | 0.49 (0.29–0.82) | 0.007 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serna-Blasco, R.; Sánchez-Herrero, E.; Robado de Lope, L.; Sanz-Moreno, S.; Rodríguez-Festa, A.; Ares-Trotta, D.; Cruz-Bermúdez, A.; Franco, F.; Sánchez-Hernández, A.; Campayo, M.d.J.; et al. Molecular Divergence upon EGFR-TKI Resistance Could Be Dependent on the Exon Location of the Original EGFR-Sensitizing Mutation. Cancers 2022, 14, 4446. https://doi.org/10.3390/cancers14184446
Serna-Blasco R, Sánchez-Herrero E, Robado de Lope L, Sanz-Moreno S, Rodríguez-Festa A, Ares-Trotta D, Cruz-Bermúdez A, Franco F, Sánchez-Hernández A, Campayo MdJ, et al. Molecular Divergence upon EGFR-TKI Resistance Could Be Dependent on the Exon Location of the Original EGFR-Sensitizing Mutation. Cancers. 2022; 14(18):4446. https://doi.org/10.3390/cancers14184446
Chicago/Turabian StyleSerna-Blasco, Roberto, Estela Sánchez-Herrero, Lucía Robado de Lope, Sandra Sanz-Moreno, Alejandro Rodríguez-Festa, Dunixe Ares-Trotta, Alberto Cruz-Bermúdez, Fabio Franco, Alfredo Sánchez-Hernández, María de Julián Campayo, and et al. 2022. "Molecular Divergence upon EGFR-TKI Resistance Could Be Dependent on the Exon Location of the Original EGFR-Sensitizing Mutation" Cancers 14, no. 18: 4446. https://doi.org/10.3390/cancers14184446
APA StyleSerna-Blasco, R., Sánchez-Herrero, E., Robado de Lope, L., Sanz-Moreno, S., Rodríguez-Festa, A., Ares-Trotta, D., Cruz-Bermúdez, A., Franco, F., Sánchez-Hernández, A., Campayo, M. d. J., García-Girón, C., Dómine, M., Blasco, A., Sánchez, J. M., Oramas, J., Bosch-Barrera, J., Sala, M. Á., Sereno, M., Romero, A., & Provencio, M. (2022). Molecular Divergence upon EGFR-TKI Resistance Could Be Dependent on the Exon Location of the Original EGFR-Sensitizing Mutation. Cancers, 14(18), 4446. https://doi.org/10.3390/cancers14184446