
Antigens Expressed by Breast Cancer Cells Undergoing EMT Stimulate Cytotoxic CD8+ T Cell Immunity

 ,
,  , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Cell Lines and Cell Culture
2.3. Flow Cytometry
2.4. Antibodies
2.5. Whole Cell Vaccine and Tumor Studies
2.6. CD8 Depletion
2.7. Inducible miR-200c Model
2.8. Transient miR-200c Model
2.9. Quantitative Real-Time PCR
2.10. Cloning of EO771 to Express B7-1 (CD80)
2.11. Western Blot
2.12. RNAseq Library Preparation and Sequencing
2.13. Raw Data Quality Control and Trimming
2.14. RNAseq Alignment, Quality Control, and Transcript Quantification
2.15. RNA Variant Calling and Quality Control
2.16. WES Variant Calling
2.17. Variant Integration and Annotation
2.18. Detection of Neoantigens
2.19. Detection of Neojunctions
2.20. Intron Retention (IR) Validation
2.21. T Cell Assays
3. Results
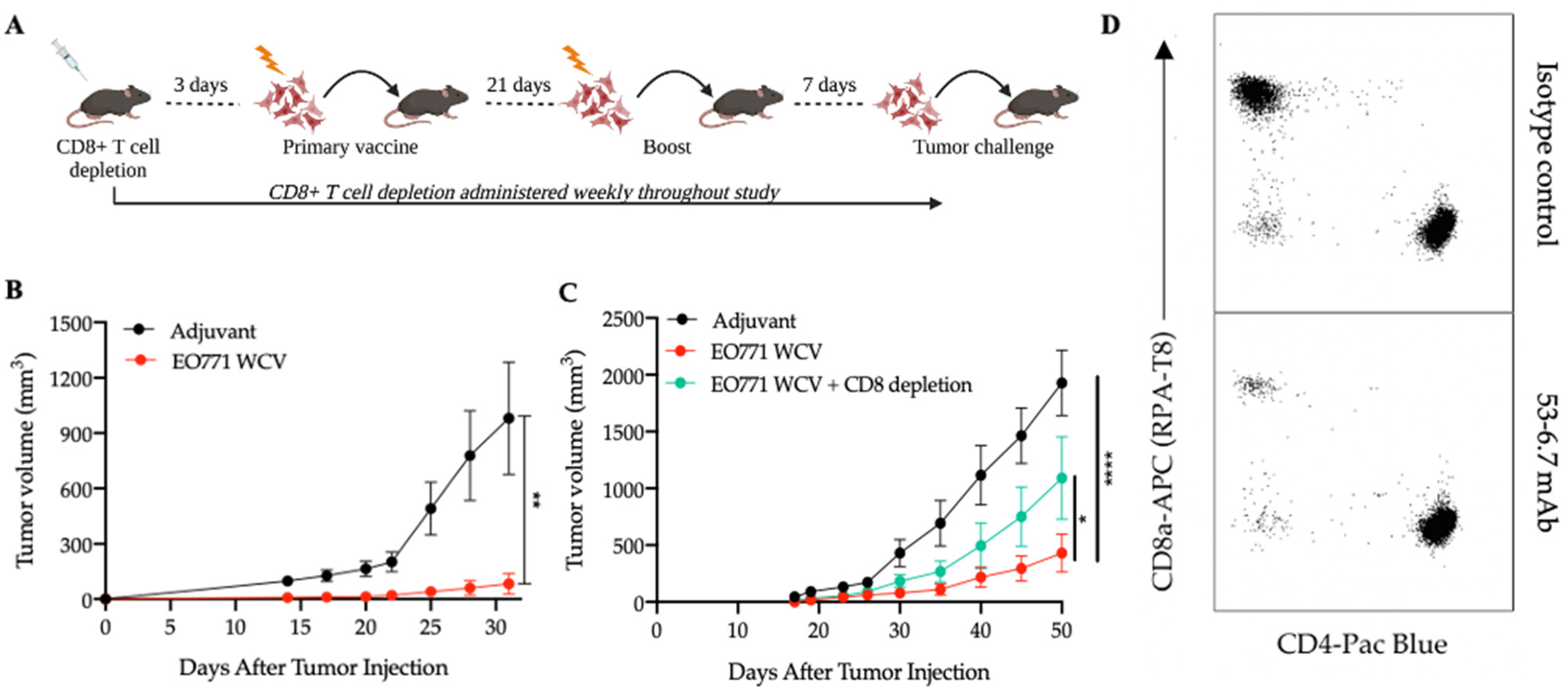
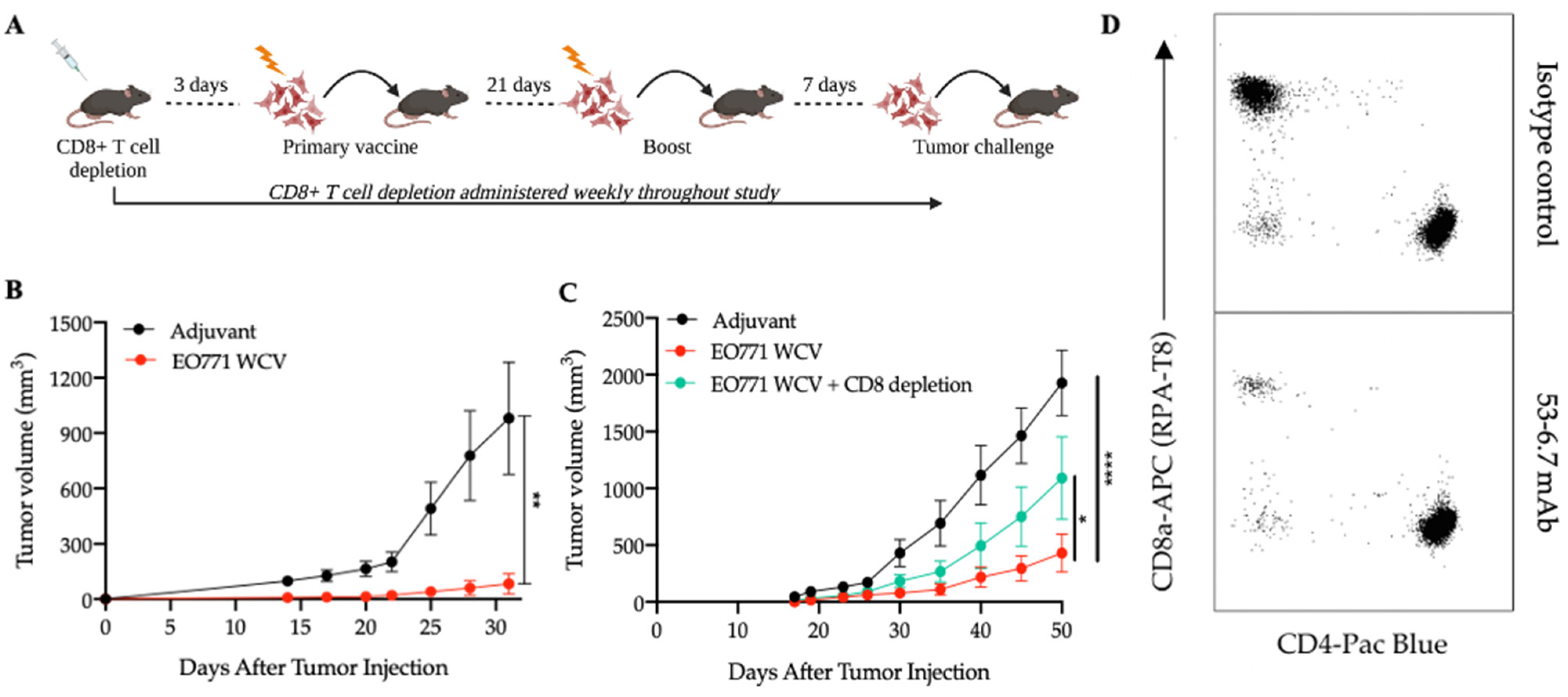
3.1. Antigens from Mesenchymal-like EO771 Whole Cell Vaccine Elicit CD8+ T Cell-Specific Antitumor Immunity
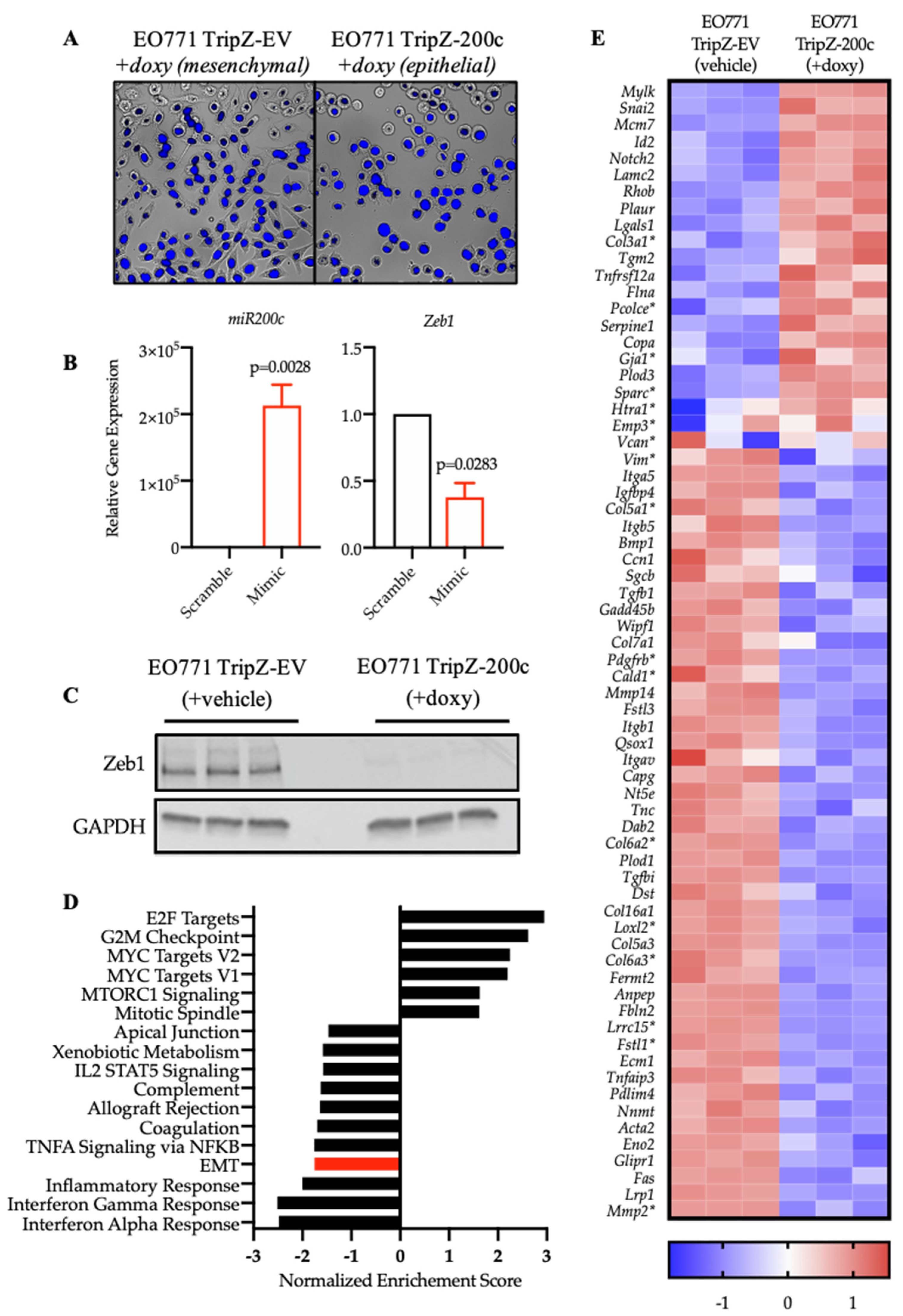
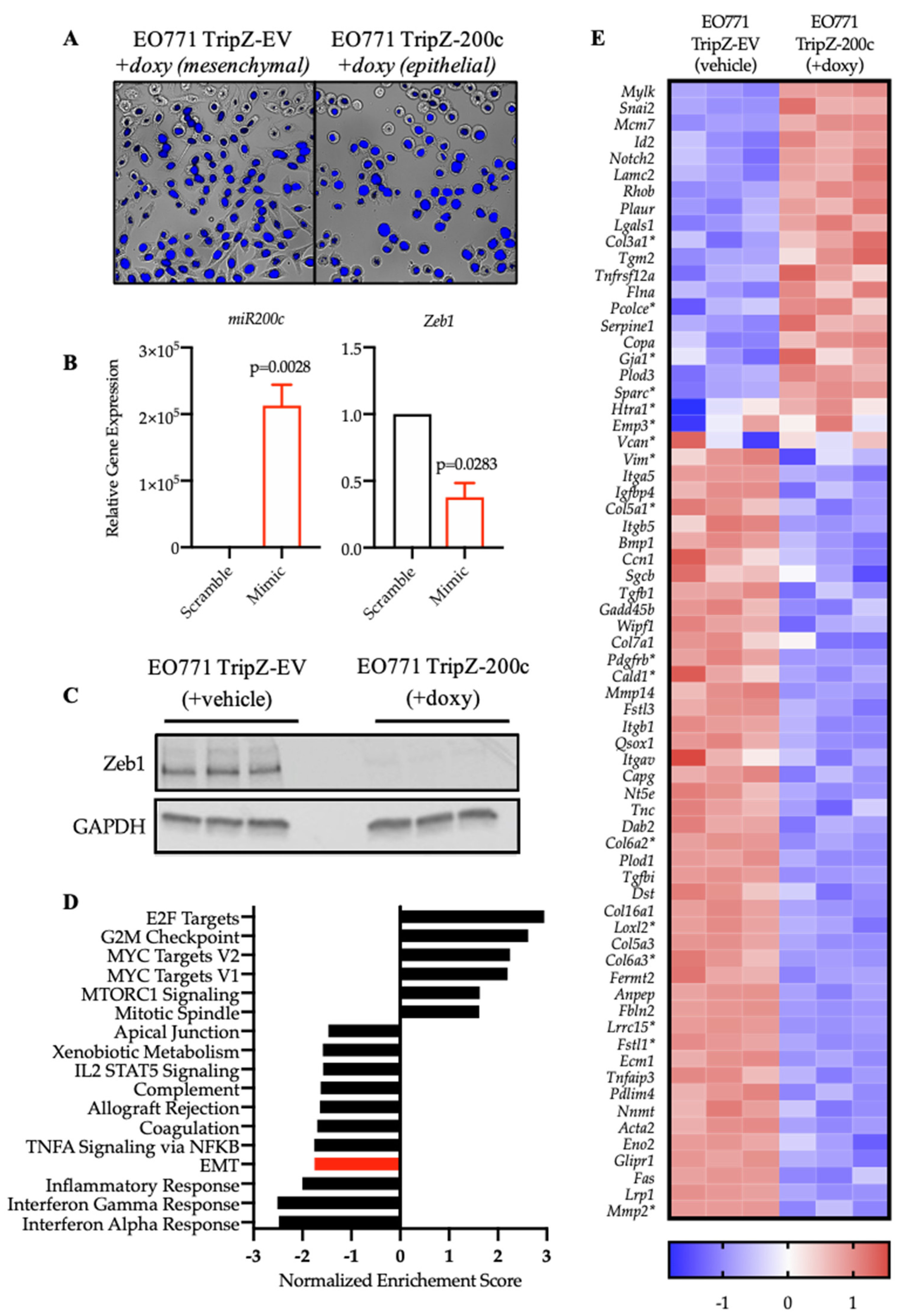
3.2. Restoration of miR-200c in Mesenchymal EO771 Cells Effectively Reverses the EMT Program
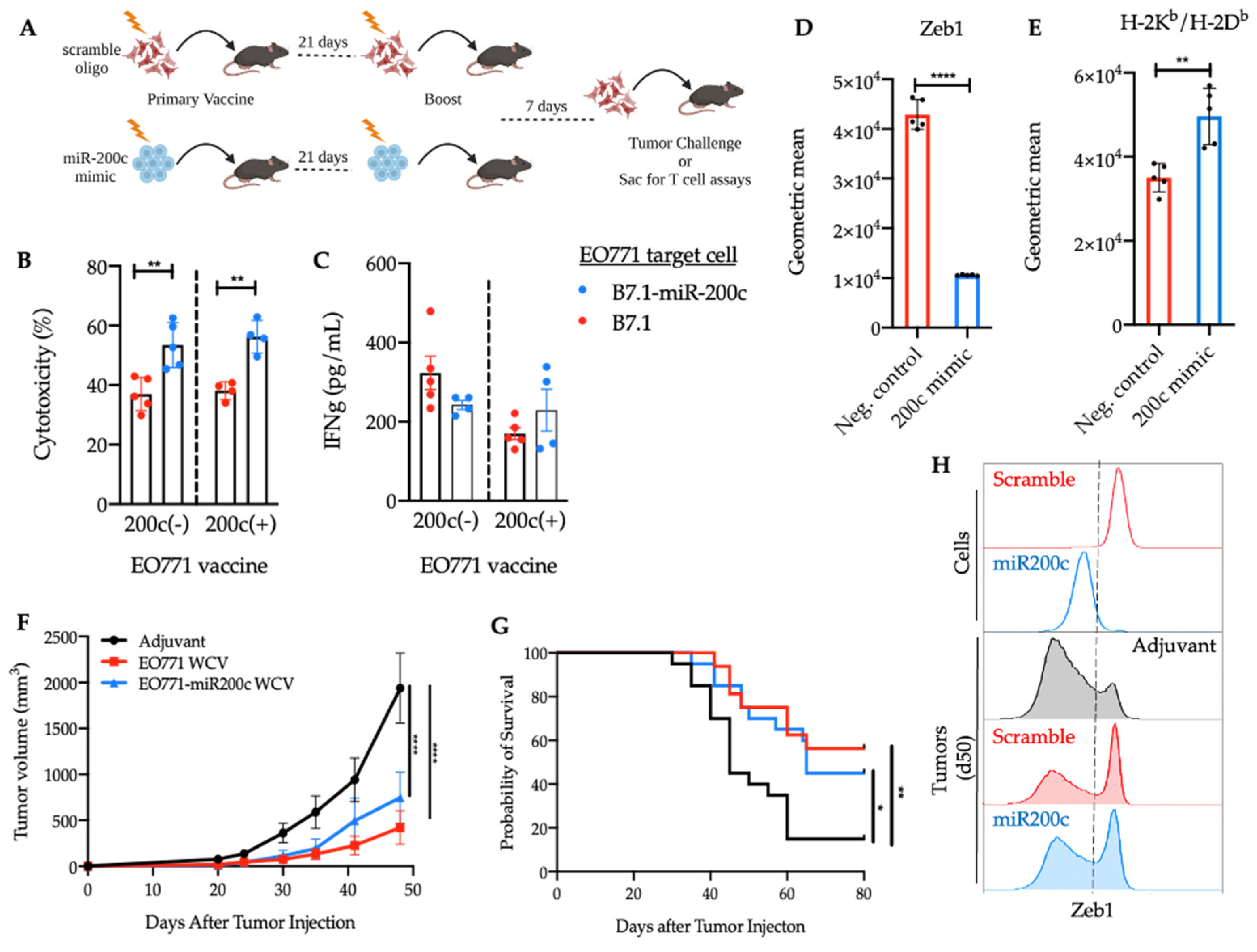
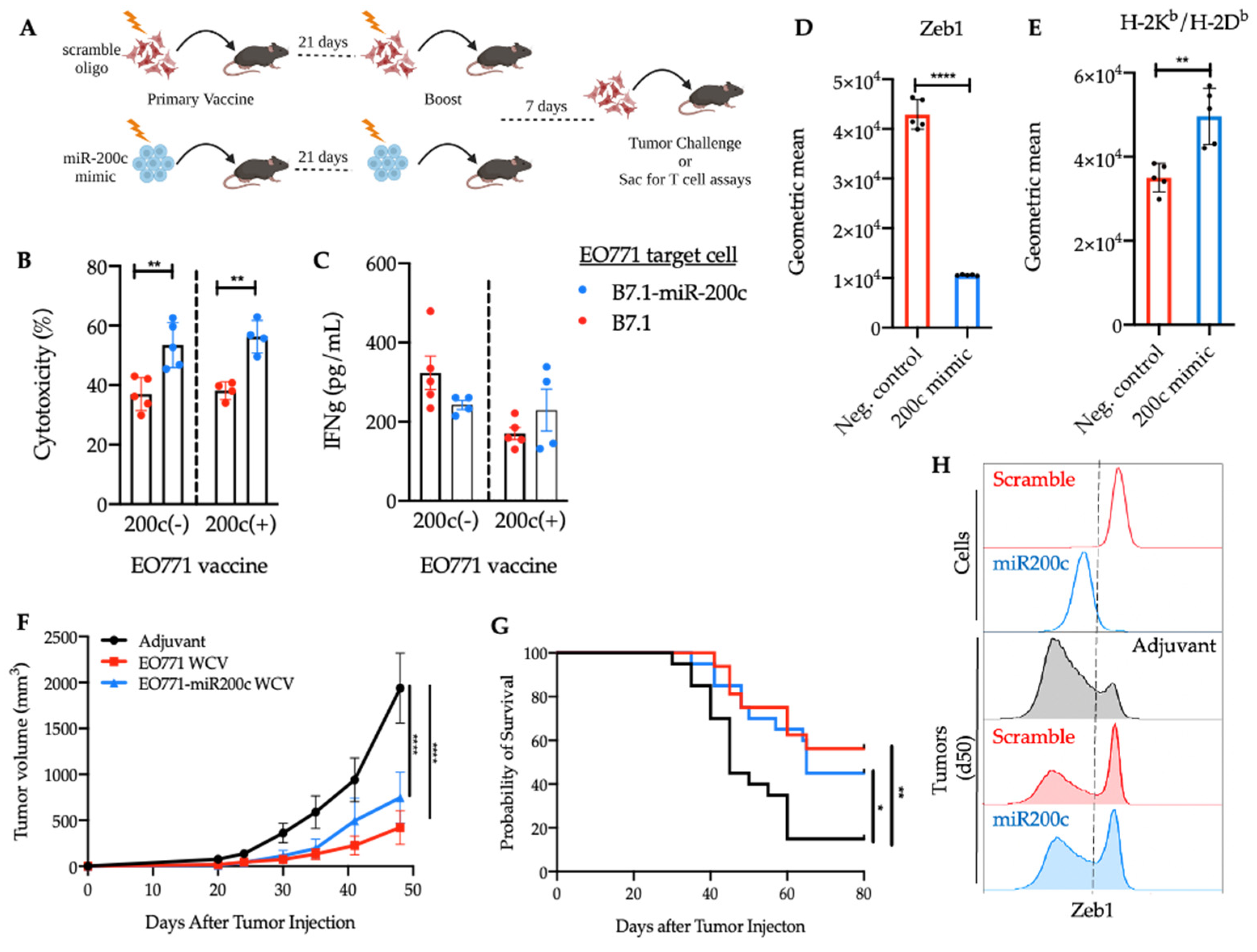
3.3. Reversal of EMT Elicited a Superior Cytotoxic Response against EO771-miR-200c Target Cells
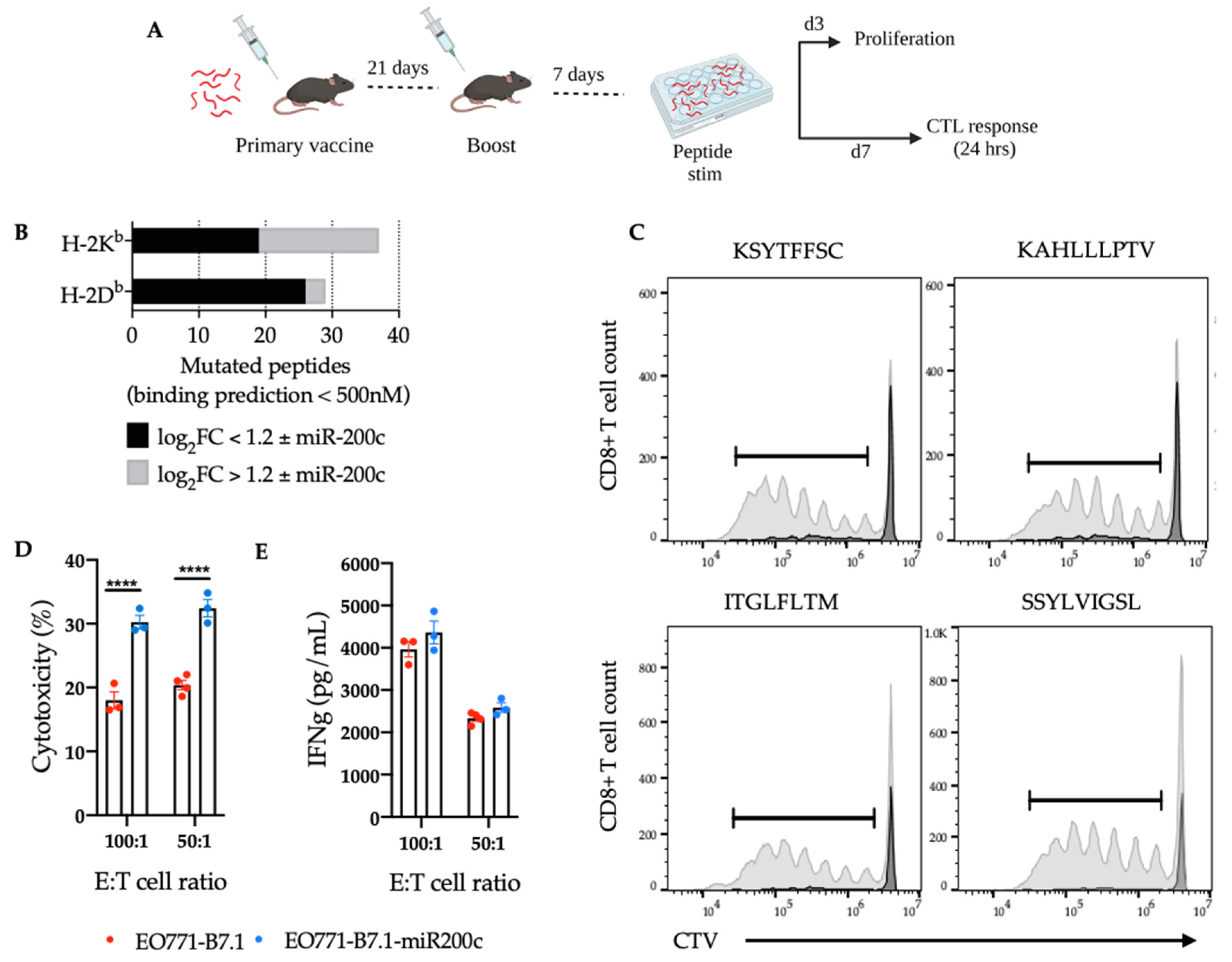
3.4. SNV-Derived Antigens Upregulated in Mesenchymal EO771 Cells Elicit Superior Cytotoxic Responses against EO771-miR-200c+ Target Cells
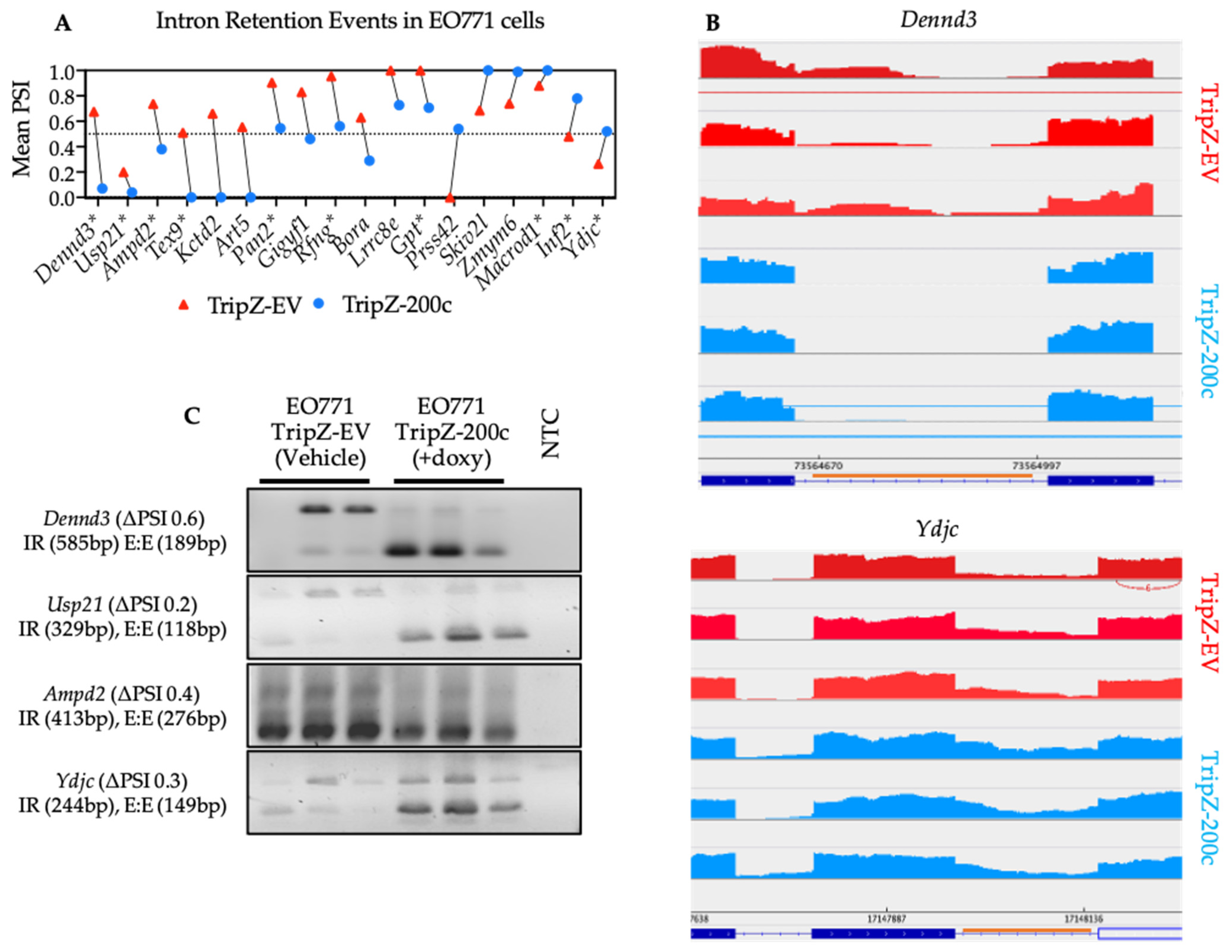
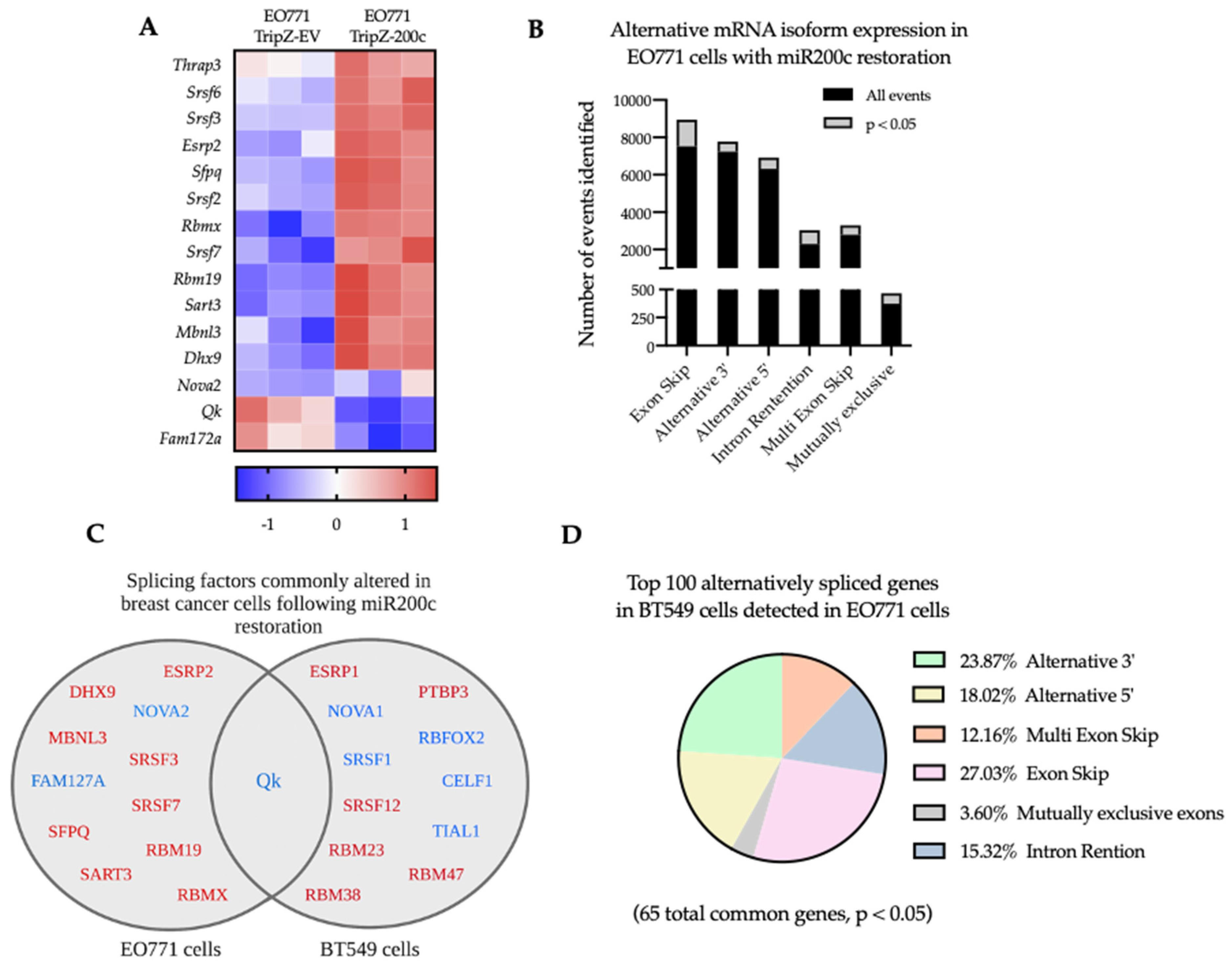
3.5. Reversal of EMT Alters RNA Splicing Factors and Splicing Events in EO771 Cells That Are Conserved in Human BT549 Breast Cancer Cells
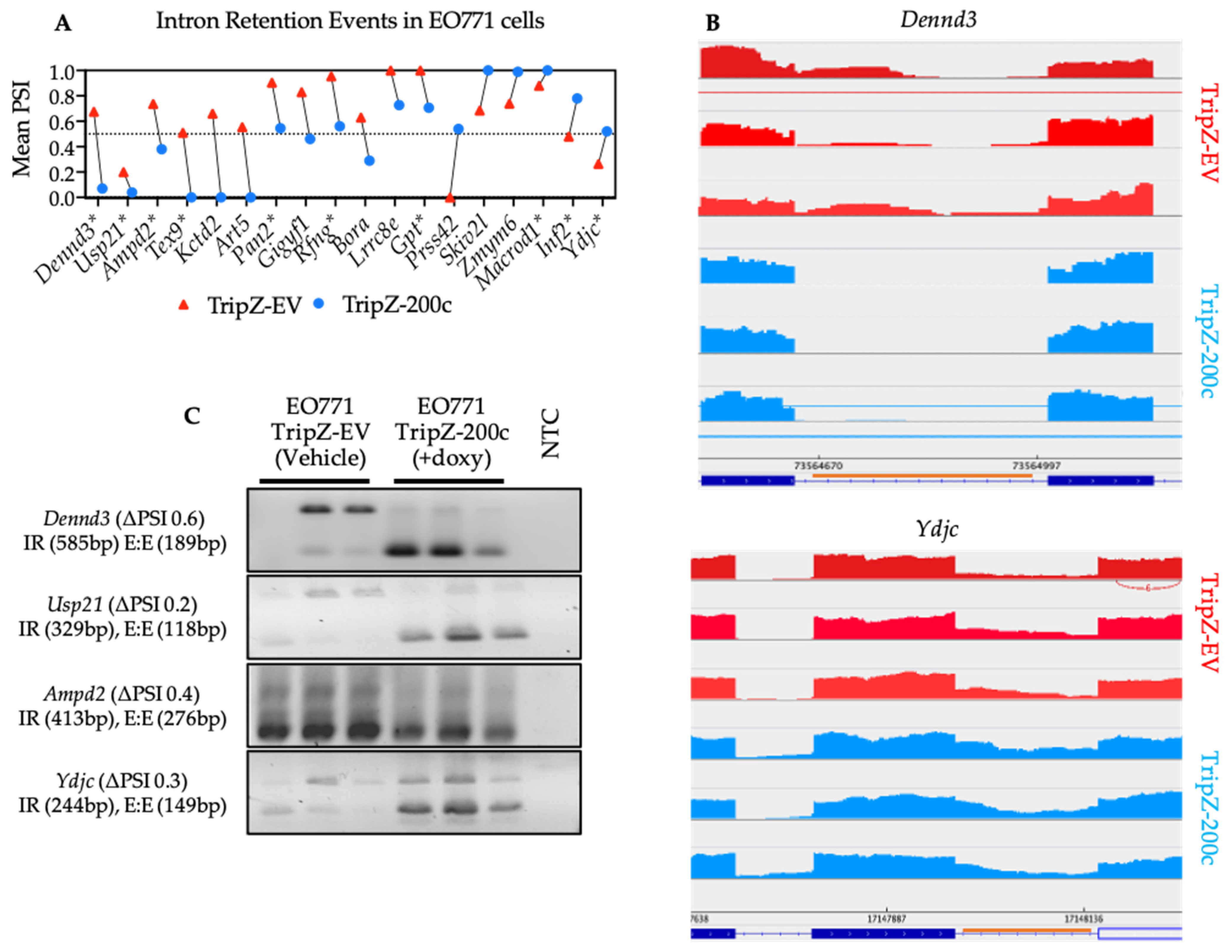
3.6. Validation of Neojunction-Derived Antigens from Alternative Splicing in EO771 Cancer Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Seyfried, T.N.; Huysentruyt, L.C. On the origin of cancer metastasis. Crit. Rev. Oncog. 2013, 18, 43–73. [Google Scholar] [CrossRef] [PubMed]
- Riggio, A.I.; Varley, K.E.; Welm, A.L. The lingering mysteries of metastatic recurrence in breast cancer. Br. J. Cancer 2021, 124, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Dafni, U.; Tsourti, Z.; Alatsathianos, I. Breast Cancer Statistics in the European Union: Incidence and Survival across European Countries. Breast Care 2019, 14, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Caswell-Jin, J.L.; Plevritis, S.K.; Tian, L.; Cadham, C.J.; Xu, C.; Stout, N.K.; Sledge, G.W.; Mandelblatt, J.S.; Kurian, A.W. Change in Survival in Metastatic Breast Cancer with Treatment Advances: Meta-Analysis and Systematic Review. JNCI Cancer Spectr. 2018, 2, pky062. [Google Scholar] [CrossRef]
- Wang, R.; Zhu, Y.; Liu, X.; Liao, X.; He, J.; Niu, L. The Clinicopathological features and survival outcomes of patients with different metastatic sites in stage IV breast cancer. BMC Cancer 2019, 19, 1091. [Google Scholar] [CrossRef]
- Ye, X.; Brabletz, T.; Kang, Y.; Longmore, G.D.; Nieto, M.A.; Stanger, B.Z.; Yang, J.; Weinberg, R.A. Upholding a role for EMT in breast cancer metastasis. Nature 2017, 547, E1–E3. [Google Scholar] [CrossRef]
- Wu, Y.; Sarkissyan, M.; Vadgama, J.V. Epithelial-Mesenchymal Transition and Breast Cancer. J. Clin. Med. 2016, 5, 13. [Google Scholar] [CrossRef]
- Kalluri, R. EMT: When epithelial cells decide to become mesenchymal-like cells. J. Clin. Investig. 2009, 119, 1417–1419. [Google Scholar] [CrossRef]
- Baum, B.; Settleman, J.; Quinlan, M.P. Transitions between epithelial and mesenchymal states in development and disease. Semin. Cell Dev. Biol. 2008, 19, 294–308. [Google Scholar] [CrossRef]
- Hilmarsdottir, B.; Briem, E.; Bergthorsson, J.T.; Magnusson, M.K.; Gudjonsson, T. Functional Role of the microRNA-200 Family in Breast Morphogenesis and Neoplasia. Genes 2014, 5, 804–820. [Google Scholar] [CrossRef]
- Peter, M.E. Let-7 and miR-200 microRNAs: Guardians against pluripotency and cancer progression. Cell Cycle 2009, 8, 843–852. [Google Scholar] [CrossRef]
- Williams, M.M.; Christenson, J.L.; O’Neill, K.I.; Hafeez, S.A.; Ihle, C.L.; Spoelstra, N.S.; Slansky, J.E.; Richer, J.K. MicroRNA-200c restoration reveals a cytokine profile to enhance M1 macrophage polarization in breast cancer. NPJ Breast Cancer 2021, 7, 64. [Google Scholar] [CrossRef]
- Rogers, T.J.; Christenson, J.L.; Greene, L.I.; O’Neill, K.I.; Williams, M.M.; Gordon, M.A.; Nemkov, T.; D’Alessandro, A.; Degala, G.D.; Shin, J.; et al. Reversal of Triple-Negative Breast Cancer EMT by miR-200c Decreases Tryptophan Catabolism and a Program of Immunosuppression. Mol. Cancer Res. 2019, 17, 30–41. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, W.; Li, B.; Stringer-Reasor, E.; Chu, C.; Sun, L.; Bae, S.; Chen, D.; Wei, S.; Jiao, K.; et al. MicroRNA-200c and microRNA- 141 are regulated by a FOXP3-KAT2B axis and associated with tumor metastasis in breast cancer. Breast Cancer Res. 2017, 19, 73. [Google Scholar] [CrossRef]
- Howe, E.N.; Cochrane, D.R.; Richer, J.K. Targets of miR-200c mediate suppression of cell motility and anoikis resistance. Breast Cancer Res. 2011, 13, R45. [Google Scholar] [CrossRef]
- Mongroo, P.S.; Rustgi, A.K. The role of the miR-200 family in epithelial-mesenchymal transition. Cancer Biol. Ther. 2010, 10, 219–222. [Google Scholar] [CrossRef]
- Dongre, A.; Rashidian, M.; Reinhardt, F.; Bagnato, A.; Keckesova, Z.; Ploegh, H.L.; Weinberg, R.A. Epithelial-to-Mesenchymal Transition Contributes to Immunosuppression in Breast Carcinomas. Cancer Res. 2017, 77, 3982–3989. [Google Scholar] [CrossRef]
- Sahin, U.; Tureci, O. Personalized vaccines for cancer immunotherapy. Science 2018, 359, 1355–1360. [Google Scholar] [CrossRef]
- Morisaki, T.; Kubo, M.; Umebayashi, M.; Yew, P.Y.; Yoshimura, S.; Park, J.H.; Kiyotani, K.; Kai, M.; Yamada, M.; Oda, Y.; et al. Neoantigens elicit T cell responses in breast cancer. Sci. Rep. 2021, 11, 13590. [Google Scholar] [CrossRef]
- Zhou, S.; Liu, S.; Zhao, L.; Sun, H.X. A Comprehensive Survey of Genomic Mutations in Breast Cancer Reveals Recurrent Neoantigens as Potential Therapeutic Targets. Front. Oncol. 2022, 12, 786438. [Google Scholar] [CrossRef]
- Kahles, A.; Lehmann, K.V.; Toussaint, N.C.; Huser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; Cancer Genome Atlas Research Network; et al. Comprehensive Analysis of Alternative Splicing Across Tumors from 8,705 Patients. Cancer Cell 2018, 34, 211–224.e6. [Google Scholar] [CrossRef]
- Kahles, A.; Ong, C.S.; Zhong, Y.; Ratsch, G. SplAdder: Identification, quantification and testing of alternative splicing events from RNA-Seq data. Bioinformatics 2016, 32, 1840–1847. [Google Scholar] [CrossRef]
- Smart, A.C.; Margolis, C.A.; Pimentel, H.; He, M.X.; Miao, D.; Adeegbe, D.; Fugmann, T.; Wong, K.K.; Van Allen, E.M. Intron retention is a source of neoepitopes in cancer. Nat. Biotechnol. 2018, 36, 1056–1058. [Google Scholar] [CrossRef]
- Shapiro, I.M.; Cheng, A.W.; Flytzanis, N.C.; Balsamo, M.; Condeelis, J.S.; Oktay, M.H.; Burge, C.B.; Gertler, F.B. An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet. 2011, 7, e1002218. [Google Scholar] [CrossRef]
- Ahonen, C.L.; Doxsee, C.L.; McGurran, S.M.; Riter, T.R.; Wade, W.F.; Barth, R.J.; Vasilakos, J.P.; Noelle, R.J.; Kedl, R.M. Combined TLR and CD40 triggering induces potent CD8+ T cell expansion with variable dependence on type I IFN. J. Exp. Med. 2004, 199, 775–784. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. Genome Project Data Processing S: The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Oikkonen, L.; Lise, S. Making the most of RNA-seq: Pre-processing sequencing data with Opossum for reliable SNP variant detection. Wellcome Open Res. 2017, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Rimmer, A.; Phan, H.; Mathieson, I.; Iqbal, Z.; Twigg, S.R.F.; Consortium, W.G.S.; Wilkie, A.O.M.; McVean, G.; Lunter, G. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat. Genet. 2014, 46, 912–918. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Tan, A.; Abecasis, G.R.; Kang, H.M. Unified representation of genetic variants. Bioinformatics 2015, 31, 2202–2204. [Google Scholar] [CrossRef]
- Kisielow, J.; Obermair, F.J.; Kopf, M. Deciphering CD4(+) T cell specificity using novel MHC-TCR chimeric receptors. Nat. Immunol. 2019, 20, 652–662. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Jurtz, V.; Paul, S.; Andreatta, M.; Marcatili, P.; Peters, B.; Nielsen, M. NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol. 2017, 199, 3360–3368. [Google Scholar] [CrossRef]
- Nielsen, M.; Lundegaard, C.; Worning, P.; Lauemoller, S.L.; Lamberth, K.; Buus, S.; Brunak, S.; Lund, O. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 2003, 12, 1007–1017. [Google Scholar] [CrossRef]
- Johnstone, C.N.; Smith, Y.E.; Cao, Y.; Burrows, A.D.; Cross, R.S.; Ling, X.; Redvers, R.P.; Doherty, J.P.; Eckhardt, B.L.; Natoli, A.L.; et al. Functional and molecular characterisation of EO771.LMB tumours, a new C57BL/6-mouse-derived model of spontaneously metastatic mammary cancer. Dis. Model. Mech. 2015, 8, 237–251. [Google Scholar] [CrossRef] [Green Version]
- Ewens, A.; Mihich, E.; Ehrke, M.J. Distant metastasis from subcutaneously grown E0771 medullary breast adenocarcinoma. Anticancer Res. 2005, 25, 3905–3915. [Google Scholar] [PubMed]
- Desch, A.N.; Gibbings, S.L.; Clambey, E.T.; Janssen, W.J.; Slansky, J.E.; Kedl, R.M.; Henson, P.M.; Jakubzick, C. Dendritic cell subsets require cis-activation for cytotoxic CD8 T-cell induction. Nat. Commun. 2014, 5, 4674. [Google Scholar] [CrossRef] [PubMed]
- Tavare, R.; McCracken, M.N.; Zettlitz, K.A.; Knowles, S.M.; Salazar, F.B.; Olafsen, T.; Witte, O.N.; Wu, A.M. Engineered antibody fragments for immuno-PET imaging of endogenous CD8+ T cells in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, 1108–1113. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, B.P. Epithelial-mesenchymal Transition—A Hallmark of Breast Cancer Metastasis. Cancer Hallm. 2013, 1, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Mak, M.P.; Tong, P.; Diao, L.; Cardnell, R.J.; Gibbons, D.L.; William, W.N.; Skoulidis, F.; Parra, E.R.; Rodriguez-Canales, J.; Wistuba, I.I.; et al. A Patient-Derived, Pan-Cancer EMT Signature Identifies Global Molecular Alterations and Immune Target Enrichment Following Epithelial-to-Mesenchymal Transition. Clin. Cancer Res. 2016, 22, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Gimmi, C.D.; Freeman, G.J.; Gribben, J.G.; Sugita, K.; Freedman, A.S.; Morimoto, C.; Nadler, L.M. B-cell surface antigen B7 provides a costimulatory signal that induces T cells to proliferate and secrete interleukin 2. Proc. Natl. Acad. Sci. USA 1991, 88, 6575–6579. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Fallarino, F.; Uyttenhove, C.; Boon, T. Tumor rejection requires a CTLA4 ligand provided by the host or expressed on the tumor: Superiority of B7-1 over B7-2 for active tumor immunization. J. Immunol. 1996, 156, 2909–2917. [Google Scholar]
- Jolly, M.K.; Somarelli, J.A.; Sheth, M.; Biddle, A.; Tripathi, S.C.; Armstrong, A.J.; Hanash, S.M.; Bapat, S.A.; Rangarajan, A.; Levine, H. Hybrid epithelial/mesenchymal phenotypes promote metastasis and therapy resistance across carcinomas. Pharmacol. Ther. 2019, 194, 161–184. [Google Scholar] [CrossRef]
- Camp, F.A.; Slansky, J.E. Implications of Antigen Selection on T Cell-Based Immunotherapy. Pharmaceuticals 2021, 14, 993. [Google Scholar] [CrossRef]
- Saito, Y.; Miranda-Rottmann, S.; Ruggiu, M.; Park, C.Y.; Fak, J.J.; Zhong, R.; Duncan, J.S.; Fabella, B.A.; Junge, H.J.; Chen, Z.; et al. NOVA2-mediated RNA regulation is required for axonal pathfinding during development. Elife 2016, 5, e14371. [Google Scholar] [CrossRef]
- Bebee, T.W.; Park, J.W.; Sheridan, K.I.; Warzecha, C.C.; Cieply, B.W.; Rohacek, A.M.; Xing, Y.; Carstens, R.P. The splicing regulators Esrp1 and Esrp2 direct an epithelial splicing program essential for mammalian development. Elife 2015, 4, e08954. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Brouckaert, O.; Wildiers, H.; Floris, G.; Neven, P. Update on triple-negative breast cancer: Prognosis and management strategies. Int. J. Womens Health 2012, 4, 511–520. [Google Scholar] [CrossRef]
- Kong, D.; Hughes, C.J.; Ford, H.L. Cellular Plasticity in Breast Cancer Progression and Therapy. Front. Mol. Biosci. 2020, 7, 72. [Google Scholar] [CrossRef]
- Chockley, P.J.; Keshamouni, V.G. Immunological Consequences of Epithelial-Mesenchymal Transition in Tumor Progression. J. Immunol. 2016, 197, 691–698. [Google Scholar] [CrossRef]
- Chakraborty, P.; Chen, E.L.; McMullen, I.; Armstrong, A.J.; Kumar Jolly, M.; Somarelli, J.A. Analysis of immune subtypes across the epithelial-mesenchymal plasticity spectrum. Comput. Struct. Biotechnol. J. 2021, 19, 3842–3851. [Google Scholar] [CrossRef]
- Yang, Q.; Zhao, J.; Zhang, W.; Chen, D.; Wang, Y. Aberrant alternative splicing in breast cancer. J. Mol. Cell Biol. 2019, 11, 920–929. [Google Scholar] [CrossRef]
- Hall, A.E.; Pohl, S.O.; Cammareri, P.; Aitken, S.; Younger, N.T.; Raponi, M.; Billard, C.V.; Carrancio, A.B.; Bastem, A.; Freile, P.; et al. RNA splicing is a key mediator of tumour cell plasticity and a therapeutic vulnerability in colorectal cancer. Nat. Commun. 2022, 13, 2791. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Mutant Peptide | Allele | Log2FC (EO771-EV/EO771-miR-200c) | Binding Affinity (nM) |
|---|---|---|---|---|
| Extl1 | VWQSFPEL | H-2Kb | 1.56 | 20.3 |
| Mt-Cytb | ITGLFLTM | H-2Kb | 2.7 | 120.2 |
| Mt-Nd5 | SSYLVIGSL | H-2Kb | 2.53 | 25.3 |
| CD46 | KSYTFFSC | H-2Kb | 2.03 | 36.2 |
| Ptprt | KAHLLLPTV | H-2Db | 1.22 | 451 |
| Gene | Neojunction-Derived Peptide | H-2Db Binding (nM) | H-2Kb Binding (nM) |
|---|---|---|---|
| Dennd3 | VTLRSQRGL | - | 420.6 |
| TTHLHSPPL | - | 172.4 | |
| SKVVSATPL | 707.4 | - | |
| RGLKNMLSA | 1704.4 | - | |
| Usp21 | ILIFTFLFL TFLFLLGYL | 6191 | 902 |
| 6003.4 | 779 | ||
| SSPSFEFPL | 382 | 13 | |
| LIFTFLFLL | 2071 | 53.6 | |
| FTFLFLLGY | - | 1175.5 | |
| CHPDFLCHL | - | 371.6 | |
| Ampd2 | VSGGYWVPL | - | 50.1 |
| Ydjc | VHVLPGTRL | - | 966.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Camp, F.A.; Brunetti, T.M.; Williams, M.M.; Christenson, J.L.; Sreekanth, V.; Costello, J.C.; Hay, Z.L.Z.; Kedl, R.M.; Richer, J.K.; Slansky, J.E. Antigens Expressed by Breast Cancer Cells Undergoing EMT Stimulate Cytotoxic CD8+ T Cell Immunity. Cancers 2022, 14, 4397. https://doi.org/10.3390/cancers14184397
Camp FA, Brunetti TM, Williams MM, Christenson JL, Sreekanth V, Costello JC, Hay ZLZ, Kedl RM, Richer JK, Slansky JE. Antigens Expressed by Breast Cancer Cells Undergoing EMT Stimulate Cytotoxic CD8+ T Cell Immunity. Cancers. 2022; 14(18):4397. https://doi.org/10.3390/cancers14184397
Chicago/Turabian StyleCamp, Faye A., Tonya M. Brunetti, Michelle M. Williams, Jessica L. Christenson, Varsha Sreekanth, James C. Costello, Zachary L. Z. Hay, Ross M. Kedl, Jennifer K. Richer, and Jill E. Slansky. 2022. "Antigens Expressed by Breast Cancer Cells Undergoing EMT Stimulate Cytotoxic CD8+ T Cell Immunity" Cancers 14, no. 18: 4397. https://doi.org/10.3390/cancers14184397
APA StyleCamp, F. A., Brunetti, T. M., Williams, M. M., Christenson, J. L., Sreekanth, V., Costello, J. C., Hay, Z. L. Z., Kedl, R. M., Richer, J. K., & Slansky, J. E. (2022). Antigens Expressed by Breast Cancer Cells Undergoing EMT Stimulate Cytotoxic CD8+ T Cell Immunity. Cancers, 14(18), 4397. https://doi.org/10.3390/cancers14184397